
 Brent S. Pedersen
Brent S. Pedersen
- Department of Plant and Microbial Biology, University of California Berkeley, Berkeley, CA, USA
Whole genome duplications, or tetraploidies, are an important source of increased gene content. Following whole genome duplication, duplicate copies of many genes are lost from the genome. This loss of genes is biased both in the classes of genes deleted and the subgenome from which they are lost. Many or all classes are genes preferentially retained as duplicate copies are engaged in dose sensitive protein–protein interactions, such that deletion of any one duplicate upsets the status quo of subunit concentrations, and presumably lowers fitness as a result. Transcription factors are also preferentially retained following every whole genome duplications studied. This has been explained as a consequence of protein–protein interactions, just as for other highly retained classes of genes. We show that the quantity of conserved noncoding sequences (CNSs) associated with genes predicts the likelihood of their retention as duplicate pairs following whole genome duplication. As many CNSs likely represent binding sites for transcriptional regulators, we propose that the likelihood of gene retention following tetraploidy may also be influenced by dose–sensitive protein–DNA interactions between the regulatory regions of CNS-rich genes – nicknamed bigfoot genes – and the proteins that bind to them. Using grass genomes, we show that differential loss of CNSs from one member of a pair following the pre-grass tetraploidy reduces its chance of retention in the subsequent maize lineage tetraploidy.
Introduction
It was almost half a century ago that Ohno (1970) first proposed a role for whole genome duplications in the evolution of vertebrates just as Lewis (1951) did for duplications of individual genes two decades before Ohno. While the most recent tetraploidy in the lineage leading to humans is estimated to be half a billion years old (Kasahara, 2007), both modern and ancient whole genome duplications are abundant in flowering plants. An estimated 35% of flowering plants are polyploid relative to the baseline level for their genera (Wood et al., 2009). Arabidopsis thaliana – a species selected for its small genome – contains readily detectable evidence of two rounds of whole genome duplication within its order and a more ancient hexaploidy, all estimated to have occurred within the last 120 million years (Bowers et al., 2003; Maere et al., 2005; Paterson et al., 2010).
Whole genome duplications create two copies of every gene and all associated regulatory sequences. These duplicate genes and chromosomal segments are referred to as homeologs and homeologous throughout this paper. However they are known variously throughout the literature as ohnologs, homeologs, or syntenic paralogs. In most cases, one of the two homeologs, each now potentially redundant, is lost by fractionation. In maize the mechanism of fractionation was shown to involve short deletions by nonhomologous recombination (Woodhouse et al., 2010). Although duplicated regions are initially identical or near-identical, gene loss data from all studied tetraploidies show clear bias between duplicate chromosomal segments with one region sustaining the majority of gene copy deletion (Thomas et al., 2006; Sankoff et al., 2010; Woodhouse et al., 2010). This bias remains consistent across each pair of paleochromosomes in maize and is paralleled by differences in expression levels of duplicate genes located on homeologous paleochromosomes (Schnable et al., 2011).
While duplicate copies of many genes are lost following whole genome duplication, in some cases both copies of a gene are retained. It was initially thought that these cases were consequences of sub- or neofunctionalization. However, most researchers now embrace an entirely different explanation: duplicate genes are retained following whole genome duplication in cases where loss generates imbalance in dosage sensitive interactions of the products of those genes with other proteins encoding by duplicated genes. This explanation, a corollary of the Gene Dosage Hypothesis (Birchler et al., 2005; Veitia et al., 2008), is a powerful tool for explaining many observations regarding genes retained as duplicate copies following whole genome duplication (reviews: Birchler et al., 2007; Sémon and Wolfe, 2007; Freeling, 2009). Genes involved in forming multi-protein complexes – such as the proteasome core, ribosome components, and molecular motors – are some of the most enriched in retained duplicate copies following whole genome duplication, and any gene annotated with the molecular function GO0003700, “transcription factor activity” is particularly likely to have been retained after the most recent tetraploidy in Arabidopsis (review: Freeling, 2009). An inverse relationship has been found between genes that form local duplicates, a process that disrupts gene dosage, and genes that are retained following tetraploidy (Cannon et al., 2004; Freeling, 2009). Subfunctionalization cannot explain this result as both forms of duplication represent sources of potentially subfunctionizable genes. However the result is consistent with selection to maintain the relative dosage among many genes.
Genes encoding transcription factors are not typical genes. The gene dosage hypothesis is generally discussed as applying to interactions between or among gene products. There is no reason why protein–DNA interactions, such as those between a transcription factor and its binding site, might not also be subject to dosage constraints. Known transcription factor binding sites tend to be short and are represented at many sites throughout the genome. Only a small fraction of these are biologically relevant (as reviewed Wray et al., 2003); even in prokaryotes, finding functional motifs computationally is extraordinarily challenging (Salama and Stekel, 2010). Rather than attempt to predict which binding sites are functionally relevant ab initio, it is possible to use comparative genomics to discover which non-coding sequences surrounding a gene are likely to function. Functional regions are expected show lower base pair substitution rates than functionless sequences. Data in animals (Miller et al., 2004) and plants (Freeling and Subramaniam, 2009) support this. By comparing the non-coding sequence surrounding orthologous or homeologous plant genes, we can identify conserved regions termed conserved non-coding sequences (CNSs) a procedure sometimes referred to as “phylogenetic footprinting.” Previous studies comparing orthologous genes between maize and rice (Inada et al., 2003) and homeologous duplicated genes in Arabidopsis (Thomas et al., 2006) found that genes with many associated CNSs tend to encode transcription factors, particularly those expressed in response to external stimuli. Very CNS-rich genes have been called “bigfoot genes” (Thomas et al., 2006).
Identification of CNSs requires comparing pairs of orthologous – diverged by speciation – or homeologous – diverged by whole genome duplication – genes within a critical window of sequence divergence.This interval for the grasses is marked in gray on Figure 1. Non-coding sequences surrounding recently diverged genes will show sequence conservation even in the absence of purifying selection for function, while functional non-coding sequences will sometimes fall below the limits of detectability, especially if the divergence times are too great. No species with a sequenced genome is a suitable evolutionary distance from Arabidopsis for CNS detection. Therefore, CNSs in Arabidopsis were identified by comparing the non-coding sequences surrounding retained homeologous genes (Freeling et al., 2007). As a result, all Arabidopsis genes with associated CNSs, by definition, were retained as a homeologous pair following the most recent whole genome duplication in the Arabidopsis lineage and obviously do not represent a useful system for studying any possible correlation between CNS content and retainability.
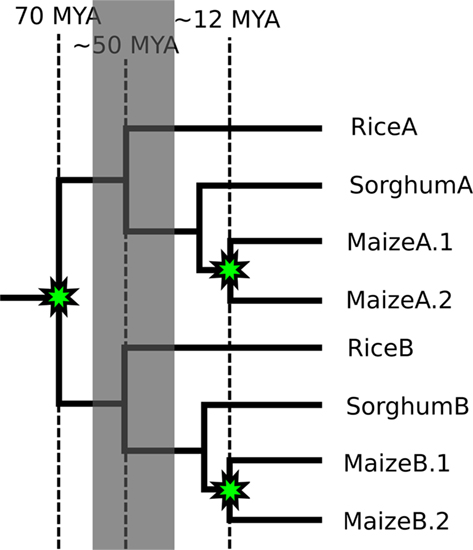
Figure 1. A phylogenetic tree showing the relationships of homeologous and orthologous regions of the genomes of maize, sorghum, and rice. Genomic relationships between the species rice, sorghum, and maize. Nodes marked with stars represent divergence by whole genome duplication. All other nodes represent divergence by speciation.
The grasses provide a model system in which to test our question: Does CNS-richness correlate with an increased tendency to have both duplicate copies retained following a whole genome duplication? In other words, are some genes retained following tetraploidy, not because their protein products are involved in dosage sensitive interactions, but because their own cis-regulatory sequences (promoters, enhancers, locus control regions, insulators, etc.) are the target of dosage sensitive transcription factors? The genomes of all grass species studied to date contain a core gene set that is maintained in a well-conserved syntenic order (Bennetzen and Freeling, 1993; Moore et al., 1995) making the identification of true orthologs and homeologs, as well as the predicted locations of deleted genes, possible. The pre-grass lineage experienced a whole genome duplication an estimated 50–70 million years ago (Vandepoele et al., 2003; Paterson et al., 2004; Yu et al., 2005). The grasses have since radiated into a few deep tribal lineages, three of which are represented by at least one species with a published genome sequence (Figure 1). The first plant CNSs described were identified by comparing orthologous rice and maize genes (Kaplinsky et al., 2002; Guo and Moose, 2003; Inada et al., 2003). Sorghum and rice share the same divergence as rice and maize and are ideally spaced for the discovery of CNSs between orthologous genes. As neither species has experienced a whole genome duplication since the two lineages diverged, the CNS-richness of individual genes can be quantified while independently quantifying that gene’s history of retention or loss following the whole genome duplication preceding the grass radiation.
The Andropogoneae, a tribe of the grasses, contain two species with sequenced genomes: sorghum and maize. The maize lineage experienced a second whole genome duplication (Gaut and Doebley, 1997) contemporaneous with its divergence from the sorghum lineage, while the sorghum lineage has remained unduplicated since the pre-grass tetraploidy (Swigon?ová et al., 2004). Ongoing fractionation in the maize genome provides a second dataset to test predictions about dosage–sensitivity made using comparisons of rice–sorghum orthologs and homeologs (Woodhouse et al., 2010; Schnable et al., 2011). The phylogenetic relationships of genome segments between rice, sorghum, and maize are summarized in Figure 1. The availability of these grass genome sequences and their relationships allow us to evaluate the role CNSs – and the regulatory sequences they mark – play in gene retention following tetraploidy and, presumably, in dose–sensitivity.
Materials and Methods
CNS Discovery
The evolutionary distance between the genomes of rice and sorghum places them within the interval for CNS discovery (as reviewed Freeling and Subramaniam, 2009). Using the CNS Discovery Pipeline (Woodhouse et al., 2010) version 2, 48,744 total CNSs (all strictly syntenic) were identified near 16,013 pairs of rice TIGR5 – sorghum JGI1.4 orthologs. CNSs were associated with the rice–sorghum gene pair separated by the smallest number of intervening genes. When there was a tie between the gene pairs up and downstream of the CNS, the CNS was assigned to the gene separated by the least physical distance. This list is called the Os-Sb gene list, v2. B. Pedersen Freeling Lab, 2009, and is included as Datasheet S1 in Supplementary Material.
Identification of Orthologous and Homeologous Syntenic Segments for Use in These Experiments
Inter- and intra-species blocks of collinear homologous genes were identified using the online tool SynMap (Lyons et al., 2008b) and enlarged using the merge function of the QuotaAlign algorithm enabled within SynMap. Collinear blocks were classified as either homeologous or orthologous based on analysis of aggregate synonymous substitution rates between all homologous gene pairs within a block of collinear genes, as previously described (Schnable et al., 2011).
Classification of Maize Retention
For each orthologous rice–sorghum gene pair we identified two orthologous locations within the maize genome. An orthologous maize gene was considered to be present either if a gene present at the predicted orthologous location matched against the rice and sorghum orthologs, or if a LASTZ (Harris, 2007) search of the region identified a putative unannotated gene or gene fragment similar to the rice and sorghum orthologs.
Results
Sorghum–Rice CNSs Obtained in Automated Fashion and Sorted to Their Nearest Gene
An automated pipeline compared the genomes of Japonica rice and sorghum for orthologous genes (Woodhouse et al., 2010). These published methods also include methods for the automated discovery of CNSs. Using these orthologous genes as syntenic anchors, CNSs conserved within, upstream and downstream of orthologous rice and sorghum genes were identified (see Materials and Methods and Data Sheet S1 in Supplementary Material) The single most CNS-rich gene in the sorghum genome is the myb transcription factor gene Sb01g037110 (Figure 2). This gene’s non-coding space covers about 30 kb in sorghum, and 70 kb in the longest of the maize homeologs. The GEvo comparison panel (Lyons et al., 2008a) shown in Figure 2 – derived from the CoGe software suite – is an example of how we check the results of our automated pipeline while tuning the parameters for optimum CNS discovery between different pairs of species. Every pair of rice–sorghum orthologous genes has an associated GEvo link included in Datasheet S1 of Supplementary Material, allowing any researcher to visually proof the accuracy of our automated CNS identification pipeline.
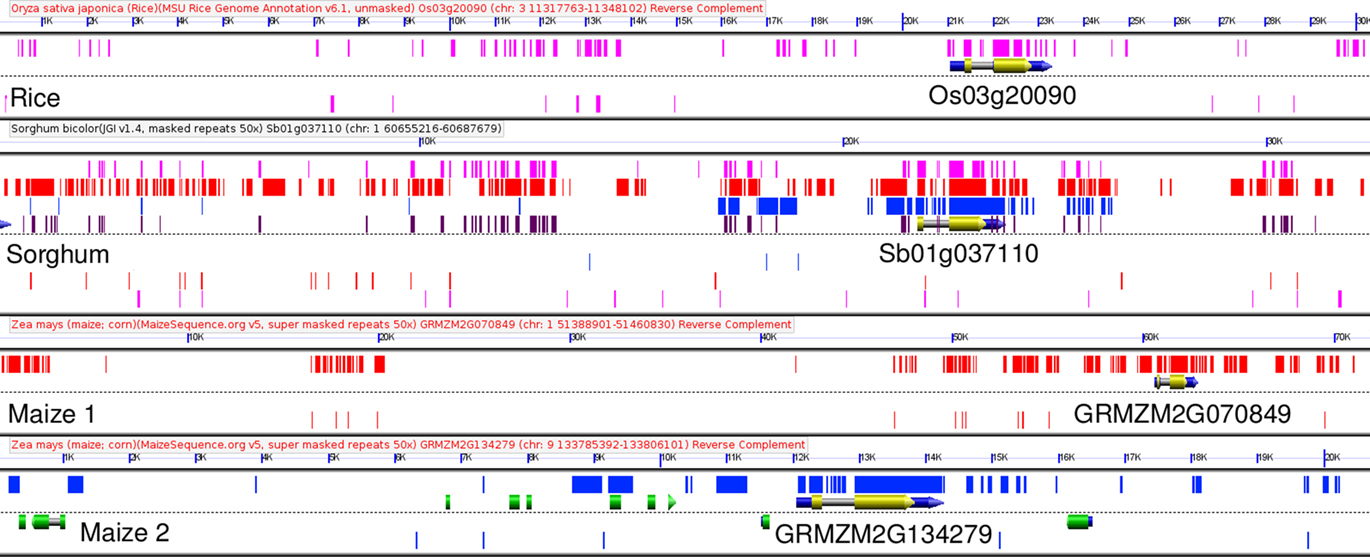
Figure 2. GEvo comparison of Sb01g037110 to conserved syntenic orthologs in rice, and maize. Relationship between myb transcription factor gene Sb01g037110, the single most CNS rich gene in sorghum, and its syntenically retained orthologs in rice and maize. Exons of the genes in the orthologous group containing this gene are marked in yellow, exons of all other genes are marked in green. Sequences identified as homologous by blastn between sorghum and rice are identified by purple rectangles. Sequences annotated as conserved non-coding sequences by the CNS-PIPELINE version 1 are marked in dark brown on the sorghum track, second from the top. Blastn hits between and maize1/maize2 are marked with red and blue rectangles respectively. This graphic was generated using GEvo, part of the CoGe toolkit (Lyons et al., 2008a). An interactive version of this experimental result can be regenerated by visiting the following link: http://genomevolution.org/r/2bgw.
CNS Counts and Retention from the Pre-Grass Tetraploidy
We first asked if genes with greater numbers of associated CNSs were more likely to possess a retained homeologous copy from the pre-grass whole genome duplication than genes with fewer or no associated CNSs. Figure 3 reports the percent of genes with a retained pre-grass homeolog in rice, binned by number of associated CNSs. Genes not retained at syntenic locations between rice and sorghum are excluded from the analysis as it is not possible to annotate CNSs for these genes. The data show a rise in the percent of genes with a retained homeologous gene from the pre-grass whole genome duplication as the number of associated CNSs increases. This trend is continuous over a range from 0 to 15 CNSs. The smallest bin in Figure 3 contains 230 genes (>15 CNSs and 33% retention). Six of the 15 rice–sorghum gene pairs with >28 CNSs possess a retained homeolog (40% retention) and 25 of the 56 gene pairs with 22–28 CNSs possess a retained homeolog (45% retention). There is an obvious positive correlation between CNS-richness and retention of duplicate gene copies post-tetraploidy.
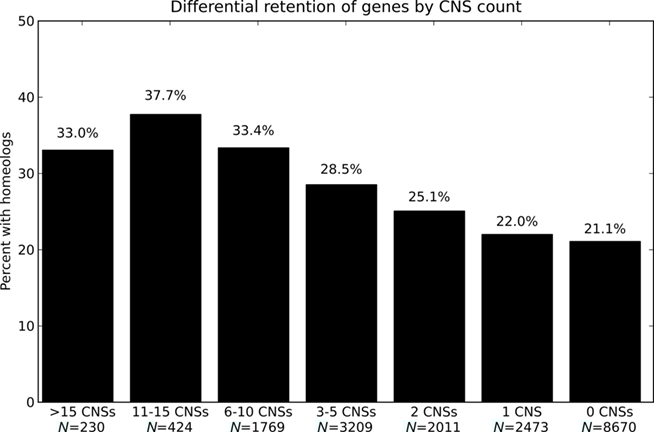
Figure 3. Odds of possessing a retained homeolog for genes with different numbers of associated CNSs. Odds of possessing a retained homeologous gene from the pregrass whole genome duplication for genes with different numbers of associated CNSs.
There are many gene categories – especially those encoding ancient components like ribosomal proteins or motor proteins – that are significantly over-retained and are conspicuously low in CNSs (Thomas et al., 2006). Dose sensitive product–product binding into large heterogenous complexes is certainly adequate to explain many categories of over-retained genes. The large collection of genes encoding transcription factors are, on average, both CNS-rich and over-retained (Freeling, 2009). So, not only is our positive correlation of CNS-richness with retention not universal to all gene groups, it is also possible that it is a mere reflection of the fact that transcription factors are both CNS-rich and highly retained following tetraploidy and not an effect of CNSs themselves. We attempted a crude experiment to test this trivial explanation.
We asked: For individual transcription factor gene families – each acting in complexes we assume to be of equivalent molecular complexity/connectivity – were CNS-rich genes retained from the pre-grass tetraploidy at a frequency significantly higher than the frequency for homologous CNS-poor genes? From the 1923 entries in the Database of Rice (Japonica) Transcription Factors in 2009 (http://drtf.cbi.pku.edu.cn/) we identified families with ≥6 members in rice (discounting tandem duplicates and genes not conserved as syntenic orthologs in sorghum). The orthologously paired members of each family were ranked by number of CNSs. If the bin had the minimum number of genes, 6–10, the one most CNS-rich and the one least CNS-rich gene were evaluated for whether or not they had a pre-grass homeolog (i.e., were retained). For families with greater than the minimum number of genes, the total orthologous pair gene count was divided by 10, and that number was sampled from the most CNS-rich and the most CNS-poor ends of the distribution. In this way, each transcription factor family data point was weighted by its total sorghum–rice orthologous pair count.
One hundred sixty-eight CNS-rich TF genes were paired with 168 CNS-poor genes from the same family. Overall 60% of these genes possessed a retained homeolog from the pre-grass tetraploidy. CNS-rich transcription factor genes possessed a retained duplicate copy in 75% of cases while only 45% of the CNS-poor members of the same families possessed retained duplicate copies. This distribution is significantly different from our null hypothesis of 60% retention in both groups of genes with a p-value of 0.006 (Chi-square test df = 1). However, the tenuous nature of our assumption that transcription factors of the same family should, on average, engage in complexes of equivalent complexity precludes any clean conclusion.
Differential Retention of Pre-Grass Homeologs in the Subsequent Maize Tetraploidy
The addition of the maize genome to the collection of grasses with sequenced genomes, and the second whole genome duplication found in that lineage (Figure 1), permits a more controlled experiment. An organism possesses two copies of every gene at the moment of whole genome duplication. Even if the whole genome duplication is the result of a wide cross (allotetraploidy) each duplicate copy possesses near-identical regulatory sequence, and encodes a protein with near-identical function that participates in a near-identical set of potentially dose–sensitive interactions within the cell. Specific regulatory sequences may be deleted from the promoters of either gene copy over evolutionary time – likely by the same short deletion mechanism observed to remove duplicate gene copies following the most recent tetraploidy in maize (Woodhouse et al., 2010). The expectation is that homeologous gene pairs from the pre-grass duplication will often possess unequal numbers of associated CNSs (Figure 4). This expectation was met.
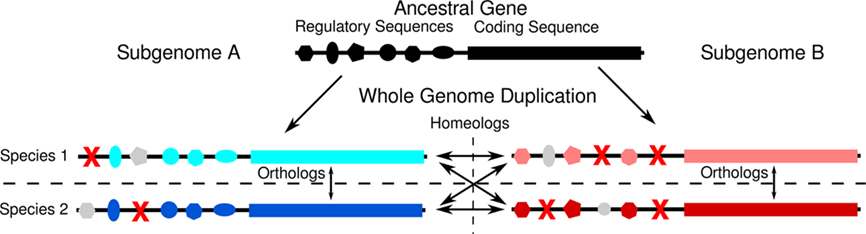
Figure 4. Model for how duplicated genes come to possess different numbers of CNSs. A hypothetical example of how regulatory sequences of duplicate genes might evolve following whole genome duplication. The original whole genome duplication creates two homeologous copies of an ancestral gene, both of which evolve separately in two species, rice and sorghum that arose from the original tetraploid species. Red X’s mark deleted sequences. Gray shapes represent intact regulatory elements which will not be identified as CNSs by comparing orthologous genes between species 1 and 2 because they are no longer shared between the two species. In this example, the genes located in subgenome A has retained more regulatory elements in both species than have the homeologous genes in subgenome B. As a result the genes in subgenome A possesses four orthologous CNSs, while the gene in subgenome B possess only three.
Homeologous genes resulting from whole genome duplication start out possessing the same functions and interaction partners; this provided a more precise control for gene function than simply belonging to the same gene family. The behavior of these genes in the subsequent maize whole genome duplication – whether one of the two new duplicates is lost or both are retained – provides a read-out of differences in dose–sensitivity which accumulated since the two genes diverged following the pre-grass tetraploidy. Using a dataset of 497 homeologous pairs of genes conserved in both rice and sorghum where the most CNS-rich rice–sorghum gene pair possessed at least five CNSs (Datasheet S2 of Supplementary Material), we tested whether or not duplicated genes were retained at different rates in a subsequent tetraploidy (maize) when they possessed different numbers of CNSs. We identified the two syntenic orthologous locations in the reduplicated maize genome for each sorghum gene. We then classified each sorghum gene as (1) retained, with orthologous genes present at both orthologous location in the maize genome (2) fractionated, with an orthologous gene present at one of the two orthologous location in the maize genome, but deleted from the second or (3) completely lost. Data for all 497 gene lineages are reported in Table 1. Genes with more associated CNSs are more likely to be retained as a homeologous pair in maize (282 cases, 56.7%) than their less CNS-rich homeologs (217 cases, 43.7%). These numbers are significantly different from the 1:1 ratio (p = 0.0036 chi-square test, df = 1) expected if CNS-richness did not impact dose–sensitivity, and are in agreement with our hypothesis that CNS-richness per se confers a significantly greater chance of duplicate gene retention following tetraploidy.
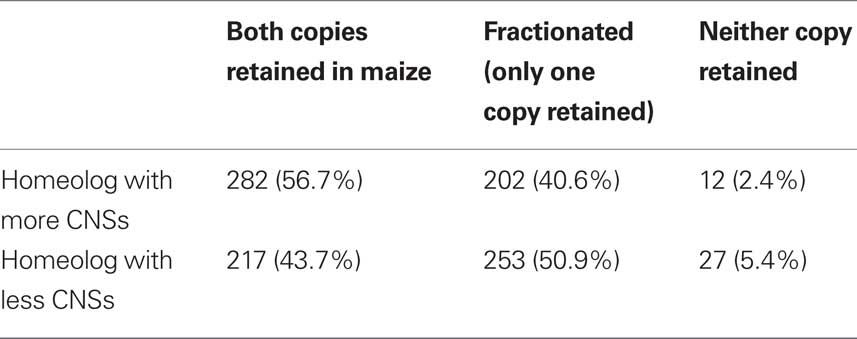
Table 1. Retention or fractionation state in maize of CNS-rich genes and their less CNS-rich homeologs.
Discussion
CNS-Richness and Duplicate Retention Following Tetraploidy
As documented in the Introduction, over-retention of genes (as post-tetraploidy gene pairs) encoding proteins of ribosomes, proteasomes, motors, and cell walls certainly make sense in light of dose–sensitive protein–protein interactions. Transcription factor genes encode proteins that sometimes function in complex multi-protein units as well, so perhaps protein–protein interactions explain the over-retention of this very large category of genes. However this is not the only possible explanation. High-level or upstream transcription factors tend to be under tight regulatory control, and the anchor sequences that act in cis on such genes are often involved in complex interactions involving proteins and multi-protein complexes; an example of this in animals is the “enhanceosome” complex (Levine, 2010). We hypothesized that protein–DNA interactions should be sensitive to the concentration of all players including the protein binding sites located in the cis-regulatory regions of the gene encoding such an upstream transcription factor.
This report presents three primary results. (1) Grass genes associated with many CNSs tend to possess homeologous duplicates retained over the ∼70 million years since the pre-grass tetraploidy (Figure 3). (2) Within individual transcription factor gene families, the most CNS-rich members are significantly more likely to possess retained duplicate copies than the least CNS-rich members. (3) Looking at copies of the same genes from the pre-grass tetraploidy, the less CNS-rich copy is significantly less likely to have both duplicate copies retained in a second round of whole genome duplication in the maize lineage (Table 1).
The concentration of the DNA binding sites and the concentration of the proteins that bind them would tend to have evolutionarily preferred stoichiometries such that fractionation (deletion) of a copy of the gene would be selectively negative because this changes the relative concentration of binding sites and binding proteins. While our results are consistent with and support our hypothesis, our explanation is not proved. There is at least one alternative explanation for our data. It is possible that the deletion of the regulatory sequences identified by CNSs reduces the contexts – tissue/organ/cell types, developmental time points, responses to stimuli – in which a gene is expressed. If a gene only participates in dose–sensitive protein–protein interaction in some specific expression contexts, the loss of CNSs could conceivably reduce the opportunities for the resulting protein to continue participating in dose–sensitive interactions and this could eliminate the selective cost associated with the loss of a duplicate gene copy. Without a detailed gene expression atlases for maize and its outgroup sorghum it is impossible to definitively rule out this alternative.
Conclusion
The supposition that the over-retention of transcription factor genes following whole genome duplications is the result of dose sensitive protein–protein interactions is an extrapolation from better-known CNS-poor gene categories such as genes encoding ribosomal proteins and is not directly supported for genes encoding transcription factors. Gene dosage effects are clearly the best single explanation for the changes that occur to gene content following whole genome duplication. However, the theoretical mechanisms explaining gene dosage should be broadened from its current focus on the concentration of protein products (Veitia, 2010) to include, for transcription factors at least, the concentration of cis-acting protein binding sequences associated with genes themselves.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research supported by a grant from the National Science Foundation to Michael Freeling (DBI 0701871) and the Chang-Lin Tien Graduate Fellowship to James C. Schnable. We thank members of our lab and previous postdocs Haibao Tang and Eric Lyons for ongoing discussions.
Supplementary Material
Datasheet 1 and 2 for this article can be found online at http://www.frontiersin.org/Plant_Genetics_and_Genomics/10.3389/fpls.2011.00002/abstract
References
Bennetzen, J. L., and Freeling, M. (1993). Grasses as a single genetic system: genome composition, collinearity and compatibility. Trends Genet. 9, 259–261.
Birchler, J. A., Riddle, N. C., Auger, D. L., and Veitia, R. A. (2005). Dosage balance in gene regulation: biological implications. Trends Genet. 21, 219–226.
Birchler, J. A., Yao, H., and Chudalayandi, S. (2007). Biological consequences of dosage dependent gene regulatory systems. Biochim. Biophys. Acta 1769, 422–428.
Bowers, J. E., Chapman, B. A., Rong, J., and Paterson, A. H. (2003). Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438.
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., and May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4, 10. doi: 10.1186/1471-2229-4-10
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453.
Freeling, M., Rapaka, L., Lyons, E., Pedersen, B., and Thomas, B. C. (2007). G-boxes, bigfoot genes, and environmental response: characterization of intragenomic conserved noncoding sequences in Arabidopsis. Plant Cell 19, 1441–1457.
Freeling, M., and Subramaniam, S. (2009). Conserved noncoding sequences (CNSs) in higher plants. Curr. Opin. Plant Biol. 12, 126–132.
Gaut, B. S., and Doebley, J. F. (1997). DNA sequence evidence for the segmental allotetraploid origin of maize. Proc. Natl. Acad. Sci. U.S.A. 94, 6809–6814.
Guo, H., and Moose, S. P. (2003). Conserved noncoding sequences among cultivated cereal genomes identify candidate regulatory sequence elements and patterns of promoter evolution. Plant Cell 15, 1143–1158.
Harris, R. S. (2007). Improved Pairwise Alignment of Genomic Data. Available at: http://www.bx.psu.edu/∼rsharris/rsharris_phd_thesis_2007.pdf
Inada, D. C., Bashir, A., Lee, C., Thomas, B. C., Ko, C., Goff, S. A., and Freeling, M. (2003). Conserved noncoding sequences in the grasses. Genome Res. 13, 2030–2041.
Kaplinsky, N. J., Braun, D. M., Penterman, J., Goff, S. A., and Freeling, M. (2002). Utility and distribution of conserved noncoding sequences in the grasses. Proc. Natl. Acad. Sci. U.S.A. 99, 6147–6151.
Levine, M. (2010). Transcriptional enhancers in animal development and evolution. Curr. Biol. 20, R754–R763.
Lewis, E. B. (1951). Pseudoallelism and gene evolution. Cold Spring Harb. Symp. Quant. Biol. 16, 159–174.
Lyons, E., Pedersen, B., Kane, J., Alam, M., Ming, R., Tang, H., Wang, X., Bowers, J., Paterson, A., Lisch, D., and Freeling, M. (2008a). Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148, 1772–1781.
Lyons, E., Pedersen, B., Kane, J., and Freeling, M. (2008b). The value of nonmodel genomes and an example using SynMmap within CoGe to dissect the hexaploidy that predates the rosids. Trop. Plant Biol. 1, 181–190.
Maere, S., De Bodt, S., Raes, J., Casneuf, T., Van Montagu, M., Kuiper, M., and Van de Peer, Y. (2005). Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 5454–5459.
Miller, W., Makova, K. D., Nekrutenko, A., and Hardison, R. C. (2004). Comparative genomics. Annu. Rev. Genomics Hum. Genet. 5, 15–56.
Moore, G., Devos, K. M., Wang, Z., and Gale, M. D. (1995). Cereal genome evolution. Grasses, line up and form a circle. Curr. Biol. 5, 737–739.
Paterson, A. H., Bowers, J. E., and Chapman, B. A. (2004). Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. U.S.A. 101, 9903–9908.
Paterson, A. H., Freeling, M., Tang, H., and Wang, X. (2010). Insights from the comparison of plant genome sequences. Annu. Rev. Plant Biol. 61, 349–372.
Salama, R. A., and Stekel, D. J. (2010). Inclusion of neighboring base interdependencies substantially improves genome-wide prokaryotic transcription factor binding site prediction. Nucleic Acids Res. 38, e135.
Sankoff, D., Zheng, C., and Zhu, Q. (2010). The collapse of gene complement following whole genome duplication. BMC Genomics 11, 313. doi: 10.1186/1471-2164-11-313
Schnable, J. C., Springer, N. M., and Freeling, M. (2011). Biased gene loss following the maize tetraploidy reflects genome dominance and ongoing selection. Proc. Natl. Acad. Sci. U.S.A. doi: 10.1073/pnas.1101368108. [Epub ahead of print].
Sémon, M., and Wolfe, K. H. (2007). Consequences of genome duplication. Curr. Opin. Genet. Dev. 17, 505–512.
Swigon?ová, Z., Lai, J., Ma, J., Ramakrishna, W., Llaca, V., Bennetzen, J. L., and Messing, J. (2004). Close split of sorghum and maize genome progenitors. Genome Res. 14, 1916–1923.
Thomas, B. C., Pedersen, B., and Freeling, M. (2006). Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose–sensitive genes. Genome Res. 16, 934–946.
Vandepoele, K., Simillion, C., and Van de Peer, Y. (2003). Evidence that rice and other cereals are ancient aneuploids. Plant Cell 15, 2192–2202.
Veitia, R. A. (2010). A generalized model of gene dosage and dominant negative effects in macromolecular complexes. FASEB J. 24, 994–1002.
Veitia, R. A., Bottani, S., and Birchler, J. A. (2008). Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet. 24, 390–397.
Wood, T. E., Takebayashi, N., Barker, M. S., Mayrose, I., Greenspoon, P. B., and Rieseberg, L. H. (2009). The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. U.S.A. 106, 13875–13879.
Woodhouse, M. R., Schnable, J. C., Pedersen, B. S., Lyons, E., Lisch, D., Subramaniam, S., and Freeling, M. (2010). Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol. 8, e1000409. doi: 10.1371/journal.pbio.1000409
Wray, G. A., Hahn, M. W., Abouheif, E., Balhoff, J. P., Pizer, M., Rockman, M. V., and Romano, L. A. (2003). The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20, 1377–1419.
Yu, J., Wang, J., Lin, W., Li, S., Li, H., Zhou, J., Ni, P., Dong, W., Hu, S., Zeng, C., Zhang, J., Zhang, Y., Li, R., Xu, Z., Li, S., Li, X., Zheng, H., Cong, L., Lin, L., Yin, J., Geng, J., Li, G., Shi, J., Liu, J., Lv, H., Li, J., Wang, J., Deng, Y., Ran, L., Shi, X., Wang, X., Wu, Q., Li, C., Ren, X., Wang, J., Wang, X., Li, D., Liu, D., Zhang, X., Ji, Z., Zhao, W., Sun, Y., Zhang, Z., Bao, J., Han, Y., Dong, L., Ji, J., Chen, P., Wu, S., Liu, J., Xiao, Y., Bu, D., Tan, J., Yang, L., Ye, C., Zhang, J., Xu, J., Zhou, Y., Yu, Y., Zhang, B., Zhuang, S., Wei, H., Liu, B., Lei, M., Yu, H., Li, Y., Xu, H., Wei, S., He, X., Fang, L., Zhang, Z., Zhang, Y., Huang, X., Su, Z., Tong, W., Li, J., Tong, Z., Li, S., Ye, J., Wang, L., Fang, L., Lei, T., Chen, C., Chen, H., Xu, Z., Li, H., Huang, H., Zhang, F., Xu, H., Li, N., Zhao, C., Li, S., Dong, L., Huang, Y., Li, L., Xi, Y., Qi, Q., Li, W., Zhang, B., Hu, W., Zhang, Y., Tian, X., Jiao, Y., Liang, X., Jin, J., Gao, L., Zheng, W., Hao, B., Liu, S., Wang, W., Yuan, L., Cao, M., McDermott, J., Samudrala, R., Wang, J., Wong, G. K.-S., and Yang, H. (2005). The genomes of Oryza sativa: a history of duplications. PLoS Biol. 3, e38. doi: 10.1371/journal.pbio.0030038
Keywords: conserved non-coding sequence, polyploidy, fractionation, gene dosage, gene regulation
Citation: Schnable JC, Pedersen BS, Subramaniam S and Freeling M (2011) Dose–sensitivity, conserved non-coding sequences, and duplicate gene retention through multiple tetraploidies in the grasses. Front. Plant Sci. 2:2. doi: 10.3389/fpls.2011.00002
Received: 09 February 2011;
Paper pending published 17 February 2011;
Accepted: 19 February 2011;
Published online: 10 March 2011.
Edited by:
Scott Jackson, Purdue University, USAReviewed by:
Randy Shoemaker, United States Department of Agriculture, USAMingsheng Chen, Chinese Academy of Sciences, China
Copyright: © 2011 Schnable, Pedersen, Subramaniam and Freeling. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Michael Freeling, Department of Plant and Microbial Biology, University of California-Berkeley, 111 Koshland Hall, Berkeley, CA 94720, United States of America e-mail:ZnJlZWxpbmdAYmVya2VsZXkuZWR1