
 Florian Vogel1,2
Florian Vogel1,2
- 1 Lehrstuhl für Biochemie, Friedrich-Alexander Universität Erlangen-Nürnberg, Erlangen, Germany
- 2 Environmental Process Engineering, Friedrich-Alexander University Erlangen-Nürnberg Busan Campus, Busan, Republic of Korea
Many plant viruses encode for specialized movement proteins (MP) to facilitate passage of viral material to and through plasmodesmata (PD). To analyze intracellular trafficking of potato leaf roll virus (PLRV) movement protein (MP17) we performed GFP fusion experiments with distinct deletion variants of MP17. These studies revealed that the C-terminus of MP17 is essential but not sufficient for PD targeting. Interestingly, fusion of GFP to three C-terminal MP17 deletion variants resulted in the accumulation of GFP in chloroplasts. This indicates that MP17 harbors hidden plastid targeting sequences. Previous studies showed that posttranslational protein phosphorylation influences PD targeting of MP and virus spread. Analysis of MP17-derived phospho-peptides by mass spectrometry revealed four phosphorylated serine residues (S71, S79, S137, and S140). Site-directed mutagenesis of S71/S79 and S137/S140 showed that the C-terminal serine residues S137/S140 are dispensable for PD targeting. However, exchange of S71/S79 to A71/A79 abolished PD targeting of the mutated MP17 protein. To mimic phosphorylation of S71/S79 both amino acids were substituted by aspartic acid. The resulting D71/D79 variant of MP17 was efficiently targeted to PD. Further deletion analysis showed that PD targeting of MP17 is dependent on the C-terminus, phosphorylation of S71 and/or S79 and a N-terminal domain.
Introduction
Plasmodesmata (PD) provide a symplastic connection between neighboring cells in plants and thereby enable the selective transport of assimilates and macromolecules (Ding, 1998; Roberts and Oparka, 2003; Lucas et al., 2009). Many plant viruses encode for one or more movement protein (MPs), which facilitate cell-to-cell transport of viral ribonucleoprotein complexes or viral particles to and through PD by interacting with host factors thus enabling viral cell-to-cell and systemic spread (Lazarowitz and Beachy, 1999). While the structure of MPs among different plant virus families is not conserved and apparently different strategies for PD targeting are employed, it is well established that most of these proteins can increase the basal size exclusion limit (SEL) of PD and move through PD in a non-cell-autonomous manner (Wolf et al., 1989; Waigmann et al., 1994; Haywood et al., 2002; Taliansky et al., 2008). Importantly, several MPs have been shown to be phosphorylated by plasma membrane- or ER-localized kinases (Citovsky et al., 1993; Matsushita et al., 2000, 2002; Waigmann et al., 2000; Karger et al., 2003; Módena et al., 2008). Mimicking this modification of tobacco mosaic virus (TMV) MP30 by substitution of phosphorylated amino acids with aspartate severely hampered PD targeting, SEL modification, and viral spread (Waigmann et al., 2000; Karger et al., 2003).
While most studies focused on TMV MP30, little is known about the importance of posttranslational modifications of other viral MPs for PD targeting and virus infection. Here we report on PLRV–MP17, which can be functionally expressed in Arabidopsis plants (Kronberg et al., 2007). MP17 localizes to and modifies SEL of secondary branched PD in source tissues, but does not target structural dissimilar simple PD in sink tissues (Hofius et al., 2001; Kronberg et al., 2007; Vogel et al., 2007). Unlike TMV MP30, the protein is transported via the ER-Golgi network to PD (Hofius et al., 2001; Vogel et al., 2007). Previous studies showed RNA-binding and protein phosphorylation of MP17, but the phosphorylation sites have not been identified (Tacke et al., 1991, 1993; Sokolova et al., 1997). Using MP17 deletion mutants fused to C-terminal GFP we show that the C-terminal part of MP17 is essential but not sufficient for PD localization. Analyzing the in vivo protein phosphorylation of MP17 expressed in Arabidopsis by mass-spectrometry revealed four phosphorylated serines. Site-directed mutagenesis demonstrated that S71 and/or S79 are essential for PD targeting of MP17.
Materials and Methods
Cloning, Stable, and Transient Plant Transformation
The coding sequence of MP17 was fused C-terminally with six histidine residues by conventional PCR amplification (from plasmid p35S-1; Hofius et al., 2001; primer see Table 2) and inserted in pBinAR (Höfgen and Willmitzer, 1990) via Asp718/BamHI to create plasmid MP17–PMH. Transformation of Arabidopsis Columbia-0 was carried out as described (Kronberg et al., 2007).
Different mutations were introduced in the MP17 sequence by overlap PCR with specific primers (Table 2) or deletion variants were obtained by PCR with respective primers. Mutagenized DNA fragments were first subcloned into the pENTR directional TOPO vector (Invitrogen) followed by LR reaction into the destination vector pK7FWG2 using LR clonase enzyme to create C-terminal GFP fusions (Invitrogen). The resulting fusion proteins were transiently expressed in 4–6 weeks old Nicotiana benthamianavia agroinfiltration (Voinnet et al., 2003). Two days after infiltration, localization was analyzed by Confocal Laser Scanning Microscopy using a TCS SP5 (Leica; Bensheim). Propidium iodide staining and excitation/detection of emission were conducted as described (Vogel et al., 2007).
Affinity Purification of MP17:HIS
As previously described for the enrichment of MP30 in cell wall fractions (Citovsky et al., 1993), MP17 is closely attached to the cell wall/plasma membrane at PD and cannot be solubilized by mild detergents offering a method to remove soluble proteins from the cell wall/MP17:HIS fraction and minimize unspecific purification of host proteins. Thus rosettes of transgenic Arabidopsis plants were ground and homogenized in Nonidet P40 (NP40) buffer (2% NP40[v/v]; 100 mM Tris pH 8.0; 10 mM DTT; 5 mM EDTA; 150 mM NaCl; 1:100 [v/v] phosphatase inhibitor cocktail I and II; [Sigma]; protease inhibitor complex [Roche]; 5 μM E-64 [Sigma]). After centrifugation (15,000 × g, 15 min, 4°C), the resulting pellet was washed four times with NP40-buffer. Then the pellet was mixed 3:1 (w/v) with urea-buffer (8 M urea; 0.1 M NaH2PO4, 0.01 M Tris, pH 8.0) and incubated at 37°C for 45 min following centrifugation at room temperature. The corresponding eluate was used for affinity purification with Nickel-NTA (ProBond Resin; Invitrogen). After incubation (75 min at room temperature), beads were washed three times with urea-buffer at pH 6.3. Bound proteins were eluted with urea-buffer at pH 6.3 added with 100 mM EDTA.
SDS-Page and Western Blotting
Proteins were precipitated for 30 min at −20°C after adding four volumes ice cold Methanol/Chloroform (2:1 v/v). Following drying, proteins were directly dissolved in 2× SDS loading buffer (Hofius et al., 2001) and separated on a 12.5% gel (acrylamide/piperazin diacrylamide) by discontinuous electrophoresis (Schmidt et al., 2006). Fixation and staining with colloidal coomassie was conducted according to the manufactures instructions (Sigma).
Proteins were separated on 12.5% (v/v) SDS-polyacrylamid gels and transferred to nitrocellulose membrane (Porablot). Immunoreaction was carried out using anti-MP17 antiserum as described before (Hofius et al., 2001).
Mass Spectrometry
MP17:HIS bands were cut with a scalpel and gel slices reduced, alkylated, and digested with trypsin (Catrein et al., 2005) using a Digest pro MS liquid handling system (Intavis AG, Germany). Following digestion, tryptic peptides were extracted from the gel pieces with 50% acetonitrile/0.1% TFA, concentrated nearly to dryness in a speed Vac vacuum centrifuge and diluted to a total volume of 30 μl with 0.1% TFA. Optionally, tryptic peptides were digested additionally after gel extraction with chymotrypsin before further processing. The samples (each 25 μl) were analyzed by a nanoHPLC system (Eksigent) coupled to a ESI LTQ Orbitrap (Thermo Fisher) mass spectrometer (Tegha-Dunghu et al., 2008). One survey scan (res: 30,000) was followed by three information dependent product ion scans in the Orbitrap (res: 15,000) and 2+, 3+, and 4+ charged ions were selected for fragmentation.
The MS/MS spectra were searched against a small protein database containing the MP17 sequence using the Mascot software (Perkins et al., 1999). The algorithm was set to use no enzyme specificity, assuming carbamidomethyl as a fixed modification of cysteine, and oxidized methionine and deamidation of asparagines and glutamine as variable modifications. Phosphorylation of serine, threonine, and tyrosine was also set to variable modification and mass tolerance was set to 1.1 or 0.1 Da for MS and MS/MS, respectively. Each phosphopeptide found by the software was further inspected by manual interpretation of the fragment spectrum.
λ-Phosophatase Incubation
Purified MP17:HIS eluate was dialyzed gradually to 50 mM Tris/Cl pH 6.8 and subsequently 10 μl were incubated with or without 400 units λ-Phosphatase (New England Biolabs, Ipswich, USA) for 60 min at 30°C. For evaluation of mobility shift, proteins were separated by SDS-PAGE (15% acrylamide/bisacrylamide) and MP17 was detected by the anti-MP17 antiserum as described above.
Alignment of Luteoviral MPs
Protein sequences of luteoviral MPs homologous to PLRV–MP17 were aligned with “ClustalW2”1. Accession numbers: AAB70926.1 (barley yellow dwarf virus); AAF62534.1 (cereal yellow dwarf virus); AAK49962.1 (beet chlorosis virus); AAM22679.1 (beet western yellows virus); AAZ57428.1 (beet mild yellowing virus); AAX08105.1 (cotton leaf roll dwarf virus); ACA04435.1 (soybean dwarf virus); ABG46340.1 (sweet potato leaf speckling virus).
Results
Deletion Analysis of MP17
To start analysis of MP17 PD targeting, we tested intracellular localization of different deletion variants of the protein (Figure 1). To this end, MP17 variants were fused at their C-terminus to GFP, transiently expressed in N. benthamiana leaf epidermal cells via Agrobacterium-mediated gene transfer and imaged by confocal microscopy. In a previous study (Hofius et al., 2001) PD localization of MP17:GFP was verified by immuno-EM localization of GFP and MP17, respectively. According to “GlobPlot”2, there are three disordered domains (Figure 1, DD I, II, and III) within MP17, which might be putative functional domains (Linding et al., 2003).
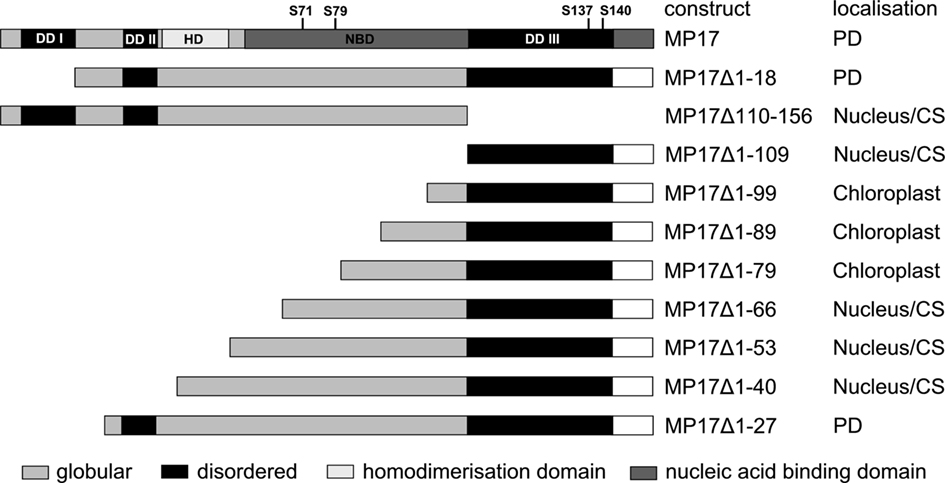
Figure 1. MP 17 deletion variants. Schematic overview of MP 17 protein structure with disordered (black) or globular domains [middle gray] according to “GlobPlot” and intracellular localization of the respective C-terminal GFP fusion proteins. The wild-type MP 17 depicts the areas of disordered domain 1 (DDI; aa 5–18), DDII (aa 30–37), DDIII (aa 110–143), the positions of the four phosphorylated serines and the putative domains for homodimerization (HD; aa 38–54) and nucleic acid binding (NBD; aa 57–156).
Therefore, we deleted separately 18 aa of the N-terminus (MP17Δ1-18) or 57 aa of the C-terminus (MP17Δ110-156), corresponding to the predicted disordered domains DD I and DD III. While MP17Δ1-18 was still targeted to PD, MP17Δ110-156 localized in the nucleus and the cytoplasm (Figure 2A). To test whether the C-terminus is sufficient for PD targeting, the C-terminus was fused to GFP (MP17Δ1-109) and tested for PD localization. As shown in Figure 2A the MP17Δ1-109 GFP fusion protein did not target to PD but accumulated in the cytoplasm and nucleus. As this might be due to incorrect folding and/or missing additional amino acids, we prolonged the C-terminal part in several steps. Surprisingly, extension with 10, 20, or 30 aa (MP17Δ1-99, MP17Δ1-89, MP17Δ1-79) resulted in chloroplast targeting of GFP (Figures 2A, B). Analyzing the N-terminus of these fusion proteins by “ChloroP” revealed the presence of predicted chloroplast transit peptides3 (Score: 0,534 for MP17Δ1-99, 0,545 for MP17Δ1-89, and 0,522 for MP17Δ1-79). Subsequent extensions abolished chloroplast targeting and led to accumulation of GFP in the cytoplasm and the nucleus. Only deletion variant MP17Δ1-27, lacking the first 27 N-terminal amino acids of MP17, was efficiently targeted to PD (Figure 2A).
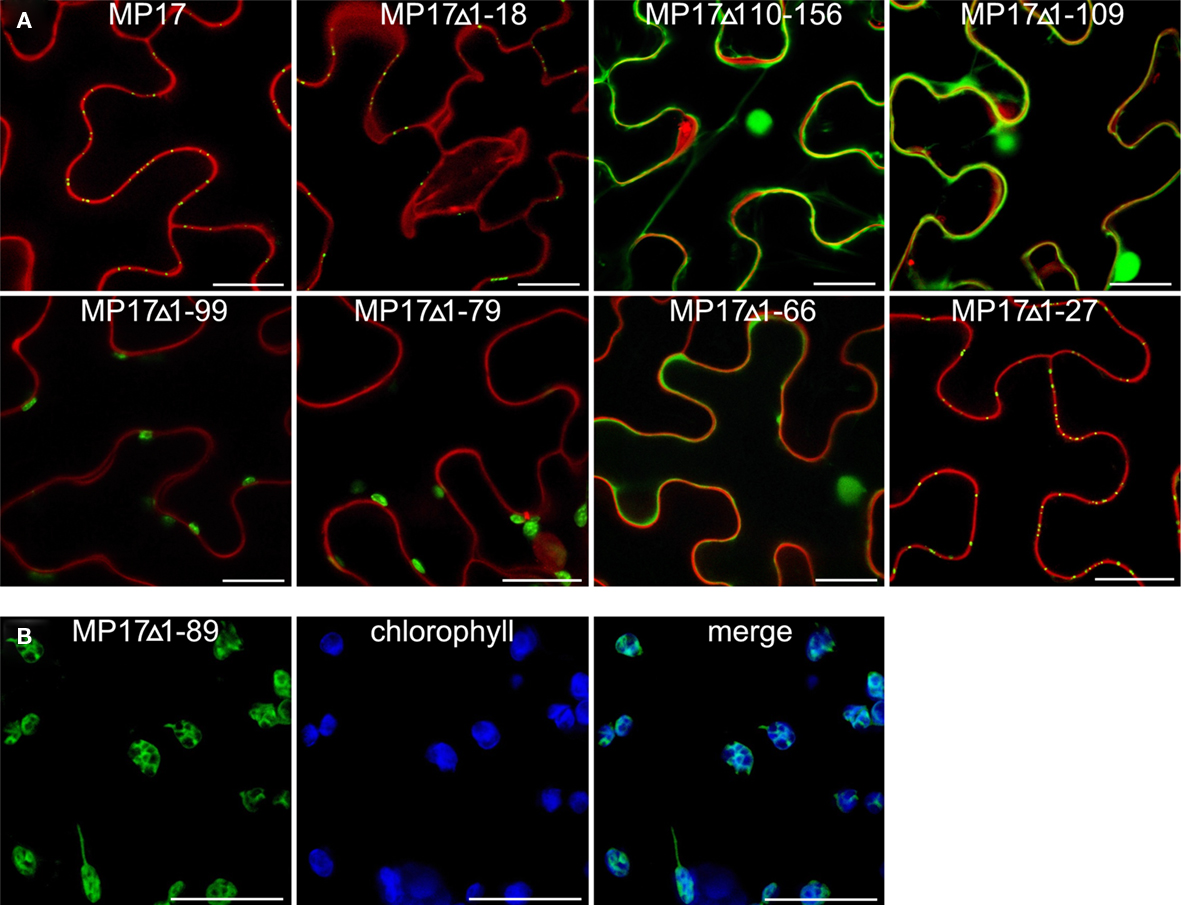
Figure 2. Intracellular localization of MP17 deletions. Confocal images of MP 17 deletion variants fused to C-terminal GFP after transient expression in N. benthamiana; (A) superimpositions of MP17:GFP fluorescence in green with propidium iodide stained cell walls/nuclei in red in epidermal cells; (B) single images and merge (right) of green MP17:GFP fluorescence and blue chlorophyll auto fluorescence in mesophyll cells; bars represents 20 μm.
As this mutant protein still contains many putative phosphorylation sites, which are potential targets for various protein kinases, we next went for direct identification of phosphorylated residues by mass-spectrometry.
In vivo Phosphorylation of MP17 in Arabidopsis
MP17 was fused to a C-terminal 6× histidine-tag (MP17:HIS, Figure 3A) to enable affinity purification and subsequently introduced into Arabidopsis thaliana by Agrobacterium-mediated gene transfer. Following transformation one Arabidopsis line (AtPMH2) showing high expression of the fusion protein, as confirmed by Western analysis (Figure 4C), was selected for further analysis. The fusion protein was detectable in SDS-PAGE at 21–22 kDa (Figure 4C) in contrast to a calculated mass4 of 18.2 kDa.

Figure 3. Stable expression of MP17:HIS in Arabidopsis. (A) Schematic drawing of the MP17:HIS fusion construct containing C-terminal six histidine residues; (B) ATPMH2 plants (right) showing chlorotic source leaves and stunted growth as compared to wild type (left); bar shows 2 cm.
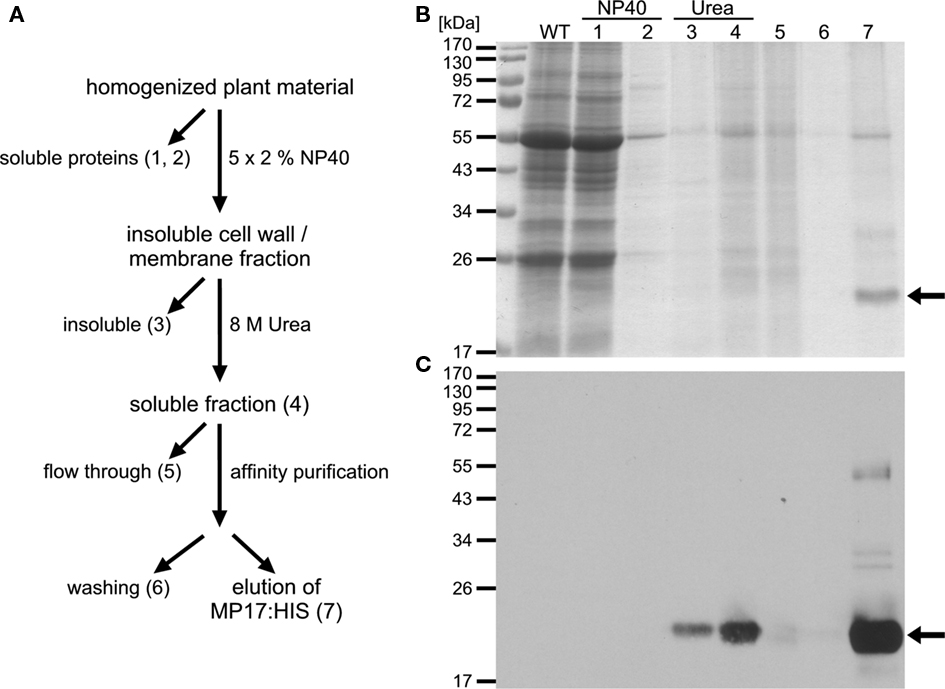
Figure 4. Affinity purification of MP17:HIS. (A) Purification strategy of MP17:HIS; ATPMH2 rosettes were homogenized in NP40-buffer and soluble (1) and insoluble fractions were separated by centrifugation; the insoluble fraction was washed four times to reduce unspecific binding (last supernatant = 2); the insoluble fraction was subsequently extracted with urea and the urea insoluble (3) and soluble fraction (4) were separated by centrifugation; the soluble fraction was used for affinity purification on Nickel-NTA resin; fraction 5 represents the flow through and fraction 6 the last wash; bound proteins were eluted with EDTA (7); (B) Aliquots of the indicated purification steps (1–7) were separated by SDS-PAGE and stained with colloidal coomassie; (C) MP17:HIS (arrows) was detected by immuno blotting using anti-MP17-antiserum; WT = wild type protein extract.
AtPMH2 plants were characterized by stunted growth and chlorotic source leaves (Figure 3B). This phenotype is reminiscent of MP17 transgenic tobacco or Arabidopsis plants (Herbers et al., 1997; Hofius et al., 2001; Kronberg et al., 2007), confirming functionality of the MP17:HIS fusion protein.
For purification of MP17:HIS, soluble proteins were removed using a mild detergent and subsequently MP17:HIS was solubilized with 8 M Urea and affinity purified (Figures 4A–C). To verify in vivo phosphorylation of MP17:HIS, the protein was incubated with λ-Phosphatase, and the shift in mobility was analyzed by SDS-PAGE. As seen in Figure 5, phosphatase treated MP17:HIS migrates faster in SDS-PAGE, indicating in vivophosphorylation of the protein (Figure 5).
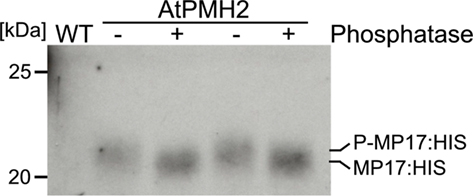
Figure 5. Treatment of MP17:HIS with λ-Phosphatase. Purified MP17:HIS was incubated with (+) or without (−) λ-Phosphatase and gel migration was monitored by SDS-PAGE following immunological detection of MP17:HIS; probes are duplicates; WT, wild type crude extract.
Identification of Phosphorylation Sites by MS/MS
Following SDS-PAGE, MP17:HIS was cut out of the gel and in gel digested with trypsin. Extracted peptides were analyzed by nano LC-ESI-MS/MS. Data evaluation with “MASCOT”5 identified 16 unique MP17-specific peptides, of which three showed phosphorylation at different serine residues – S71, S79, and S137 (see Table 1). As no peptides corresponding to the N-terminal part of MP17 were identified due to the lack of trypsin cleavage sites, extracted peptides after trypsin digestion were subjected to an additional cleavage with chymotrypsin. This yielded 20 additional peptides, giving a total peptide coverage of 73.3% of the MP17 polypeptide. Analysis of these peptides confirmed the three initially identified phosphorylation sites, along with a fourth one at S140 (Figure 1).
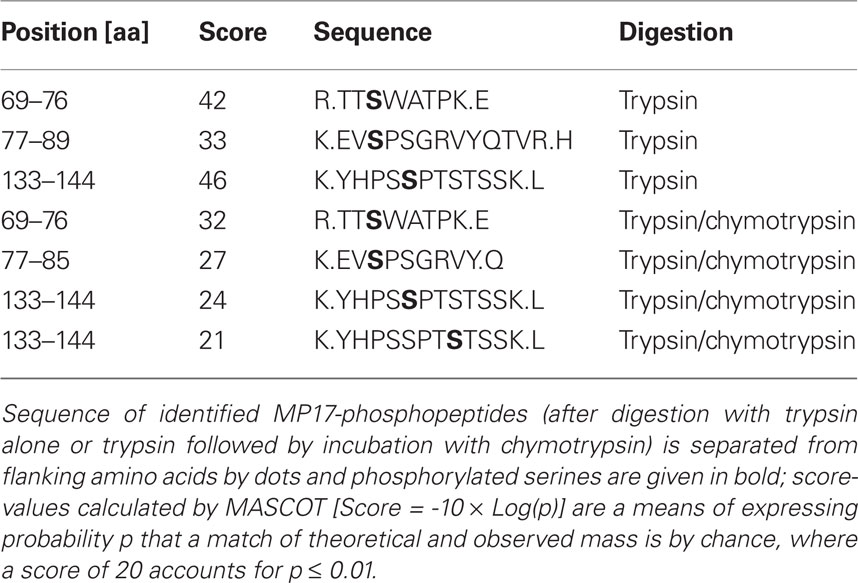
Table 1. Phosphorylated MP17 peptides.

Table 2. Primer used in this study.
Functional Analysis of Phosphorylated Serines
In order to analyze the importance of these phosphorylation sites for PD targeting of MP17, we exchanged the respective serine residues to alanine. However, there are not only two pairs of serines close to each other but also additional putative phosphorylation sites (serine, threonine) in their vicinity which could potentially be phosphorylated instead of the detected modified residues when exchanging these one by one. Thus we swapped all serines/threonines neighboring the two phosphorylation site pairs to alanine as depicted in Figure 6A. Alanine substitution at the C-terminal pair had no influence on PD targeting (MP17A136-143; Figure 6C). In contrast, alanine exchanges at the center pair abolished PD localization and resulted in highly movable aggregates in the cytoplasm (MP17A69-81; Figure 6B; Movie S1 in Supplementary Material). The same was observed for exchanges of both pairs (MP17Aall; Figure 6D).
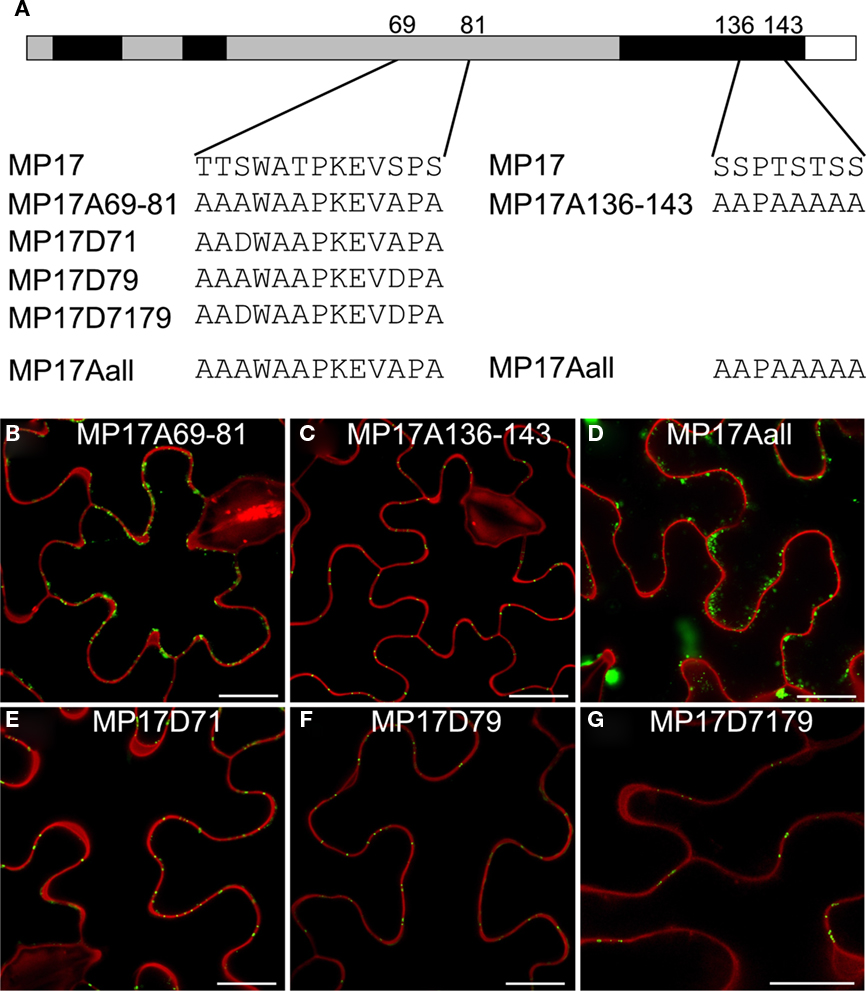
Figure 6. Impact of MP17 phosphorylation on PD targeting. (A) Amino acid changes introduced in the mutated MP 17 versions compared to wild type MP 17, position of exchanged peptides is indicated within the MP 17 polypeptide; (B–G) confocal images of the respective MP17:GFP variants transiently expressed in N. benthamiana epidermis cells; displayed are superimpositions of propidium iodide stained cell walls (red) and MP17:GFP fluorescence (green); bars represent 20 μm.
Consequently, we substituted specifically S71, S79 or both on basis of the alanine-swap variant MP17Aall to aspartate (Figure 6A) to mimic phosphorylation as already done for MP analysis before (Waigmann et al., 2000). Aspartate substitution of either S71 (MP17D71, Figure 6E) or S79 (MP17D79, Figure 6F) was sufficient to restore PD targeting of MP17. Consequently, restoration of PD localization was also observed when substituting both S71 and S79 to aspartate (MP17D7179, Figure 6G). Thus a negative charge generated by phosphorylation at S71 or S79 in this central region seems to be essential for MP17 PD localization.
Possibly, the truncated MP17Δ110-156 lacking the C-terminus might not be properly folded and therefore might not be a substrate for protein kinase(s), which would explain its inability to target PD due to its missing phosphorylation at S71/S79 rather than due to an essential role of the C-terminus itself. To test this possibility, we introduced an aspartate substitution at S71 in the MP17Δ110-156 deletion variant (MP17Δ110-156D71). CLSM analysis of the resulting protein revealed accumulation in the nucleus and the cytoplasm (Figure 7).
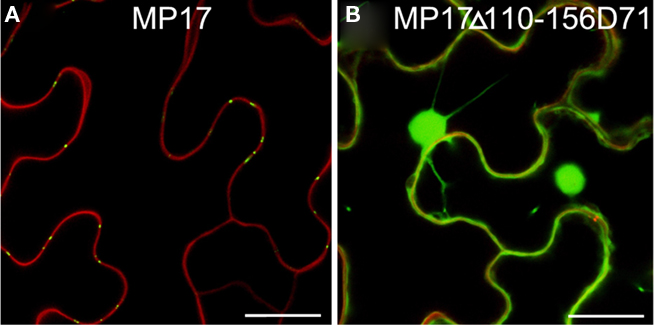
Figure 7. Substitution of S71 by aspartate does not compensate loss of C-terminus. Confocal images of MP17 (A) or MP17Δ110-156D71 (B)fusions in N. benthamiana epidermis cells; displayed are superimpositions of propidium iodide stained cell walls (red) and MP17:GFP fluorescence (green); bars represent 20 μm.
Because phosphorylation at S137/S140 is not essential for PD targeting, we checked the presence of all four serines in other luteoviral MPs. Sequence alignment of MP17 with MPs of eight additional luteoviruses showed that S71 and S79 are highly conserved corresponding to their importance (Figure 8). In contrast, S137 is unique for PLRV–MP17, while S140 is present only in some but not all luteoviral MPs.

Figure 8. Sequence alignment of MPs of different luteoviruses. Phosphorylation sites identified in PLRV–MP17 are highlighted in yellow.
Discussion
Viral MP perform – same as many other viral proteins – different tasks. They not only bind the viral genome and facilitate PD targeting of the respective ribonucleoprotein complexes, but also modify the plasmodesmal SEL to enable viral cell-to-cell and systemic spread (Lucas, 2006). Extensive studies using various techniques have been performed on the model protein TMV MP30. This includes early studies on the ability of MP30 deletion mutants to target PD and ranges to functional analysis of mutants obtained by DNA shuffling or recent data concerning in vivo oligomerization of MP30 using fluorescence lifetime imaging microscopy (FLIM; Berna et al., 1991; Gillespie et al., 2002; Boutant et al., 2010).
In contrast, there is little information about functional domains in the PLRV–MP17. Early deletion analysis confirmed that the C-proximal part (aa 57–156) confers nucleic acid binding activity and site directed mutagenesis revealed a putative N-terminal amphipathic alpha-helix (aa 38–54) essential for MP17 in vivo dimerization (Tacke et al., 1991, 1993; Karger et al., 2003). However, functional analysis of MP17 PD targeting has not been described until now.
PD Targeting of MP17 Deletion Mutants
Based on deletion analysis we were able to demonstrate that the C-terminus (aa 110–156) of PLRV–MP17 is essential but not sufficient for PD targeting. PD targeting motifs have been tested before in a similar way. For example the transmembrane domain of plasmodesmata localized protein 1a (PDLP1a) enables PD localization of a respective YFP fusion protein (Thomas et al., 2008). In contrast, a short sequence within pumpkin phloem heat shock cognate 70-1 (HSC70-1) essential for cell-to-cell transport is not sufficient to enable transport through PD when fused to GFP but in a heterochimeric fusion with human heat shock protein 70 (Aoki et al., 2002), indicating the need for an additional motif and/or correct secondary structure.
Subsequent deletions revealed that the shortest version of MP17 still targeting PD lacked only the first 27 aa. This might either indicate that shorter deletion variants are not properly folded or rather a second targeting signal within DD II (aa 30–37) is necessary for PD localization.
Surprisingly, our deletion analysis revealed also hidden chloroplast transit peptides. Whether these structural features have biological significance remains unknown. Testing the presence of potential chloroplast transit peptides at the N-terminus of 4002 E. coli K12 proteins revealed 163 candidates with scores between 0.5 and 0.56 (Heinrich Sticht, personal communication). This finding might indicate widespread occurrence of potential targeting signals and suggest no specific role for virus MP.
Phosphorylation of MP17 is Essential for PD Localization
Phosphorylation of viral MPs has been shown in the past to influence their function and localization (Lucas and Lee, 2004). This is not limited to MPs, as illustrated by the finding that phosphorylation of the potato virus a capsid protein is essential for virus systemic spread (Ivanov et al., 2003). Moreover, also a pumpkin non cell autonomous protein, Cucurbita maxima phloem protein 16 (CMPP16), needs to be phosphorylated in order to modify plasmodesmal SEL (Taoka et al., 2007), indicating a general mechanism involving phosphorylation of proteins trafficking within the plasmodesmal pathway.
By mass spectrometry we identified four phosphorylation sites. Mutant analysis revealed that two of these (S71 and S79) are essential for PD targeting, while mutation of the C-terminal phosphorylation sites did not alter PD targeting of the mutant protein. Similar studies have been carried out with TMV MP30 and tomato mosaic virus MP to analyze the importance of phosphorylation for MP functionality and PD targeting (Kawakami et al., 1999; Waigmann et al., 2000; Karger et al., 2003; Trutnyeva et al., 2005). Based on these results we can conclude that PD targeting of MP17 required at least two domains, the C-terminus and the central phosphorylation sites.
Disruption of S71/S79 phosphorylation by alanine substitution lead to the formation of MP17:GFP aggregates of various size moving within the cytoplasm and also to/along the cell periphery (MP17A69-81, Movie S1 in Supplementary Material). This might imply that transport to PD is not completely impaired and instead S71/S79 phosphorylation is essential for stable PD association. The nature of the observed aggregates is unknown. However, they vary in size and shape and directional trafficking could not be observed. Similar structures have been described by Grefen et al. (2010) comparing the localization of SYP121–GFP fusion proteins expressed under control of the 35S-CaMV promoter. Since these structures were not observed if the same fusion protein was expressed under control of the UBQ10 promoter, the authors conclude that the observed mobile punctuated structures may represent inclusion bodies caused by the strong overexpression using the 35S-CaMV promoter. Therefore, we believe that it is unlikely that the aggregates observed in our study represent structures of the normal PD trafficking route of MP17:GFP.
In agreement with the detection and functional requirement of S71/S79, MP17 was described before to be phosphorylated in the region of aa 55–95. These results were obtained in vitro by expression of truncated versions of MP17 in E. coli and subsequent incubation with potato membrane preparations or protein kinases (Sokolova et al., 1997). However, Sokolova et al. (1997) detected no in vitro phosphorylation in other parts of the polypeptides including S137/S140.
Detailed analysis of MP30 implicates that initial phosphorylation increases its ability to target PD, while subsequent phosphorylation, possibly during PD passage, leads to its dissociation from PD (Trutnyeva et al., 2005). In addition, it was reported that TMV MP30 ribonucleoprotein complexes are translatable in vivo, but not in vitro unless they are phosphorylated (Karpova et al., 1997, 1999). This might indicate that passage through PD and concomitant phosphorylation contribute to dissociation of the MP from the viral RNA genome, which in turn enables translation of viral RNA (Karpova et al., 1999; Waigmann and Citovsky, 2001; Trutnyeva et al., 2005). Thus there may be sequential phosphorylation of viral MPs, first enabling cell-to-cell transport of viral ribonucleoprotein complexes and second releasing viral RNA in an adjacent cell. Removing the MP from PD allows the host to regain control over plasmodesmal transport (Trutnyeva et al., 2005). In accordance with this, all four phosphorylated residues of MP17 are present within the previously identified nucleic acid binding domain of the protein (Tacke et al., 1991; Figure 1).
As phosphorylation of MP17 deletion variants was tested with membrane preparations by Sokolova et al. (1997), phosphorylation of S137 and S140 may have been overlooked since their phosphorylation might occur during PD-mediated transport, which could not be assayed in vitro. This idea is supported by bioinformatic analysis which predicts S140 to be a target of a Casein-kinase I – type kinase (consensus S/T/YPX1–2S/T), which targets serine/threonine when preceded N-terminally by another phosphorylated residue (serine/threonine/tyrosine, which would be supplied by phosphorylation of S137) and spaced by one or two amino acids (Gross and Anderson, 1998). Interestingly, an Arabidopsis kinase of this type, PLASMODESMATA ASSOCIATED PROTEIN KINASE 1, was localized to PD and shown to phosphorylate TMV MP30 along with other non cell autonomous proteins(Lee et al., 2005).
In accordance with the idea of a sequential phosphorylation of viral MPs, our results support the theory that this is also true for MP17. First, MP17 is phosphorylated at S71/79 to enable PD targeting. Apparently, also the C-terminal aa 110–156 and a N-terminal domain (possibly within DD II of aa 30–37) are essential for PD targeting, but act only in conjunction with phosphorylation of serines S71/S79.
In contrast, phosphorylation at S137/S140 might occur later at/in PD causing the breakup of the ribonucleoprotein complex formed by MP17 and its cargo, the viral RNA genome. Based on sequence homology, S71/S79 are highly conserved among luteoviral MPs, but S140 is only present in some of the tested MPs and S137 is unique for MP17. If the assumption that S137/S140 are phosphorylated at/in PD and are essential for viral spread, this could indicate that these modifications are involved in determining host specificity. In accordance with this hypothesis, mimicking phosphorylation of TMV MP30 negatively effected its ability to modulate plasmodesmal SEL in Nicotiana tabacum, but not in N. benthamiana(Waigmann et al., 2000).
Conclusion
In summary, MP17 localization depends not on one distinct domain but is facilitated by the co-operation of three parts of the protein indicating a complex folding and/or a highly sophisticated domain architecture within the MP17 protein.
Further analysis of MP17 deletion mutants as well as variants with alanine/aspartate substitutions regarding SEL modification and RNA binding activity will give new insights in MP17 functional domain organization in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Georg Kreimer for excellent help with gel and sample preparation, Heinrich Sticht for help with the bioinformatics analysis of protein sequences, Anja Wegele for plasmid construction and Thomas Ruppert and Margit Ellis for commercial MS/MS-analysis at the ZMBH Heidelberg (Germany). This work is part of Collaborative Research Centre 796, sub-project C2, funded by Deutsche Forschungsgemeinschaft.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Plant_Traffic_and_Transport/10.3389/fpls.2011.00018/abstract
Footnotes
References
Aoki, K., Kragler, F., Xoconostle-Cazares, B., and Lucas, W. J. (2002). A subclass of plant heat shock cognate 70 chaperones carries a motif that facilitates trafficking through plasmodesmata. Proc. Natl. Acad. Sci. U.S.A. 99, 16342–16347.
Berna, A., Gafny, R., Wolf, S., Lucas, W. J., Holt, C. A., and Beachy, R. N. (1991). The TMV movement protein: role of the C-terminal 73 amino acids in subcellular localization and function. Virology 182, 682–689.
Boutant, E., Didier, P., Niehl, A., Mely, Y., Ritzenthaler, C., and Heinlein, M. (2010). Fluorescent protein recruitment assay for demonstration and analysis of in vivo protein interactions in plant cells and its application to Tobacco mosaic virus movement protein. Plant J. 62, 171–177.
Catrein, I., Herrmann, R., Bosserhoff, A., and Ruppert, T. (2005). Experimental proof for a signal peptidase I like activity in Mycoplasma pneumoniae, but absence of a gene encoding a conserved bacterial type I SPase. FEBS J. 272, 2892–2900.
Citovsky, V., Mclean, B. G., Zupan, J. R., and Zambryski, P. (1993). Phosphorylation of tobacco mosaic virus cell-to-cell movement protein by a developmentally regulated plant cell wall-associated protein kinase. Genes Dev. 7, 904–910.
Ding, B. (1998). Intercellular protein trafficking through plasmodesmata. Plant Mol. Biol. 38, 279–310.
Gillespie, T., Boevink, P., Haupt, S., Roberts, A. G., Toth, R., Valentine, T., Chapman, S., and Oparka, K. J. (2002). Functional analysis of a DNA-shuffled movement protein reveals that microtubules are dispensable for the cell-to-cell movement of tobacco mosaic virus. Plant Cell 14, 1207–1222.
Grefen, C., Donald, N., Hashimoto, K., Kudla, J., Schumacher, K., and Blatt, M. R. (2010). A ubiquitin-10 promoter-based vector set for fluorescent protein tagging facilitates temporal stability and native protein distribution in transient and stable expression studies. Plant J. 64, 355–365.
Gross, S. D., and Anderson, R. A. (1998). Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell. Signal. 10, 699–711.
Haywood, V., Kragler, F., and Lucas, W. J. (2002). Plasmodesmata: pathways for protein and ribonucleoprotein signaling. Plant Cell 14, S303–S325.
Herbers, K., Tacke, E., Hazirezaei, M., Krause, K. P., Melzer, M., Rohde, W., and Sonnewald, U. (1997). Expression of a luteoviral movement protein in transgenic plants leads to carbohydrate accumulation and reduced photosynthetic capacity in source leaves. Plant J. 12, 1045–1056.
Höfgen, R., and Willmitzer, L. (1990). Biochemical and genetic analysis of different patatin isoforms expressed in various organs of potato (Solanum tuberosum ). Plant Sci. 66, 221–230.
Hofius, D., Herbers, K., Melzer, M., Omid, A., Tacke, E., Wolf, S., and Sonnewald, U. (2001). Evidence for expression level-dependent modulation of carbohydrate status and viral resistance by the potato leafroll virus movement protein in transgenic tobacco plants. Plant J. 28, 529–543.
Ivanov, K. I., Puustinen, P., Gabrenaite, R., Vihinen, H., Ronnstrand, L., Valmu, L., Kalkkinen, N., and Makinen, K. (2003). Phosphorylation of the potyvirus capsid protein by protein kinase CK2 and Its relevance for virus infection. Plant Cell 15, 2124–2139.
Karger, E. M., Frolova, O. Y., Fedorova, N. V., Baratova, L. A., Ovchinnikova, T. V., Susi, P., Makinen, K., Ronnstrand, L., Dorokhov, Y. L., and Atabekov, J. G. (2003). Dysfunctionality of a tobacco mosaic virus movement protein mutant mimicking threonine 104 phosphorylation. J. Gen. Virol. 84, 727–732.
Karpova, O. V., Ivanov, K. I., Rodionova, N. P., Dorokhov, Y. L., and Atabekov, J. G. (1997). Nontranslatability and dissimilar behavior in plants and protoplasts of viral RNA and movement protein complexes formed in vitro. Virology 230, 11–21.
Karpova, O. V., Rodionova, N. P., Ivanov, K. I., Kozlovsky, S. V., Dorokhov, Y. L., and Atabekov, J. G. (1999). Phosphorylation of Tobacco mosaic virus movement protein abolishes its translation repressing ability. Virology 261, 20–24.
Kawakami, S., Padgett, H. S., Hosokawa, D., Okada, Y., Beachy, R. N., and Watanabe, Y. (1999). Phosphorylation and/or presence of serine 37 in the movement protein of tomato mosaic tobamovirus is essential for intracellular localization and stability in vivo. J. Virol. 73, 6831–6840.
Kronberg, K., Vogel, F., Rutten, T., Hajirezaei, M. R., Sonnewald, U., and Hofius, D. (2007). The silver lining of a viral agent: increasing seed yield and harvest index in Arabidopsis by ectopic expression of the potato leaf roll virus movement protein. Plant Physiol. 145, 905–918.
Lazarowitz, S. G., and Beachy, R. N. (1999). Viral movement proteins as probes for intracellular and intercellular trafficking in plants. Plant Cell 11, 535–548.
Lee, J.-Y., Taoka, K.-I., Yoo, B.-C., Ben-Nissan, G., Kim, D.-J., and Lucas, W. J. (2005). Plasmodesmal-associated protein kinase in tobacco and Arabidopsis recognizes a subset of non-cell-autonomous proteins. Plant Cell 17, 2817–2831.
Linding, R., Russell, R. B., Neduva, V., and Gibson, T. J. (2003). GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 31, 3701–3708.
Lucas, W. J. (2006). Plant viral movement proteins: agents for cell-to-cell trafficking of viral genomes. Virology 344, 169–184.
Lucas, W. J., Ham, B. K., and Kim, J. Y. (2009). Plasmodesmata – bridging the gap between neighboring plant cells. Trends Cell Biol. 19, 495–503.
Lucas, W. J., and Lee, J.-Y. (2004). Plasmodesmata as a supracellular control network in plants. Nat. Rev. Mol. Cell Biol. 5, 712–726.
Matsushita, Y., Hanazawa, K., Yoshioka, K., Oguchi, T., Kawakami, S., Watanabe, Y., Nishiguchi, M., and Nyunoya, H. (2000). In vitro phosphorylation of the movement protein of tomato mosaic tobamovirus by a cellular kinase. J. Gen. Virol. 81, 2095–2102.
Matsushita, Y., Yoshioka, K., Shigyo, T., Takahashi, H., and Nyunoy, H. (2002). Phosphorylation of the movement protein of cucumber mosaic virus in transgenic tobacco plants. Virus Genes 24, 231–234.
Módena, N. A., Zelada, A. M., Conte, F., and Mentaberry, A. (2008). Phosphorylation of the TGBp1 movement protein of Potato virus X by a Nicotiana tabacum CK2-like activity. Virus Res. 137, 16–23.
Perkins, D. N., Pappin, D. J., Creasy, D. M., and Cottrell, J. S. (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567.
Roberts, A. G., and Oparka, K. J. (2003). Plasmodesmata and the control of symplastic transport. Plant Cell Environ. 26, 103–124.
Schmidt, M., Gessner, G., Luff, M., Heiland, I., Wagner, V., Kaminski, M., Geimer, S., Eitzinger, N., Reissenweber, T., Voytsekh, O., Fiedler, M., Mittag, M., and Kreimer, G. (2006). Proteomic analysis of the eyespot of Chlamydomonas reinhardtii provides novel insights into its components and tactic movements. Plant Cell 18, 1908–1930.
Sokolova, M., Prufer, D., Tacke, E., and Rohde, W. (1997). The potato leafroll virus 17K movement protein is phosphorylated by a membrane-associated protein kinase from potato with biochemical features of protein kinase C. FEBS Lett. 400, 201–205.
Tacke, E., Prufer, D., Schmitz, J., and Rohde, W. (1991). The potato leafroll luteovirus 17K protein is a single-stranded nucleic acid-binding protein. J. Gen. Virol. 72(Pt 8), 2035–2038.
Tacke, E., Schmitz, J., Prufer, D., and Rohde, W. (1993). Mutational analysis of the nucleic acid-binding 17 kDa phosphoprotein of potato leafroll luteovirus identifies an amphipathic alpha-helix as the domain for protein/protein interactions. Virology 197, 274–282.
Taliansky, M., Torrance, L., and Kalinina, N. O. (2008). Role of plant virus movement proteins. Plant Virol. Protoc. 451, 33–54.
Taoka, K.-I., Ham, B.-K., Xoconostle-Cazares, B., Rojas, M. R., and Lucas, W. J. (2007). Reciprocal phosphorylation and glycosylation recognition motifs control NCAPP1 interaction with pumpkin phloem proteins and their cell-to-cell movement. Plant Cell 19, 1866–1884.
Tegha-Dunghu, J., Neumann, B., Reber, S., Krause, R., Erfle, H., Walter, T., Held, M., Rogers, P., Hupfeld, K., Ruppert, T., Ellenberg, J., and Gruss, O. J. (2008). EML3 is a nuclear microtubule-binding protein required for the correct alignment of chromosomes in metaphase. J. Cell. Sci. 121, 1718–1726.
Thomas, C. L., Bayer, E. M., Ritzenthaler, C., Fernandez-Calvino, L., and Maule, A. J. (2008). Specific targeting of a plasmodesmal protein affecting cell-to-cell communication. PLoS Biol. 6, e7. doi: 10.1371/journal.pbio.0060007
Trutnyeva, K., Bachmaier, R., and Waigmann, E. (2005). Mimicking carboxyterminal phosphorylation differentially effects subcellular distribution and cell-to-cell movement of Tobacco mosaic virus movement protein. Virology 332, 563–577.
Vogel, F., Hofius, D., and Sonnewald, U. (2007). Intracellular trafficking of Potato leafroll virus movement protein in transgenic Arabidopsis. Traffic 8, 1205–1214.
Voinnet, O., Rivas, S., Mestre, P., and Baulcombe, D. (2003). An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J. 33, 949–956.
Waigmann, E., Chen, M. H., Bachmaier, R., Ghoshroy, S., and Citovsky, V. (2000). Regulation of plasmodesmal transport by phosphorylation of tobacco mosaic virus cell-to-cell movement protein. EMBO J. 19, 4875–4884.
Waigmann, E., and Citovsky, V. (2001). Phosphorylation of viral movement proteins – regulation of cell-to-cell trafficking: response. Trends Microbiol. 9, 8.
Waigmann, E., Lucas, W. J., Citovsky, V., and Zambryski, P. (1994). Direct functional assay for tobacco mosaic virus cell-to-cell movement protein and identification of a domain involved in increasing plasmodesmal permeability. Proc. Natl. Acad. Sci. U.S.A. 91, 1433–1437.
Keywords: MP17, PLRV, phosphorylation, posttranslational modification, intracellular trafficking, plasmodesmata
Citation: Link K, Vogel F and Sonnewald U (2011) PD trafficking of potato leaf roll virus movement protein in Arabidopsis depends on site-specific protein phosphorylation. Front. Plant Sci. 2:18. doi: 10.3389/fpls.2011.00018
Received: 24 March 2011; Accepted: 27 May 2011;
Published online: 06 June 2011.
Edited by:
Gerald Alan Berkowitz, University of Connecticut, USAReviewed by:
Maria Cristina Bonza, University of Milano, ItalyRemko Offringa, Leiden University, Netherlands
Copyright: © 2011 Link, Vogel and Sonnewald. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Uwe Sonnewald, Lehrstuhl für Biochemie, Department Biologie, Staudtstr. 5, 91058 Erlangen, Germany. e-mail:dXNvbm5lQGJpb2xvZ2llLnVuaS1lcmxhbmdlbi5kZQ==