 Diro Terefe-Ayana
Diro Terefe-Ayana Aneela Yasmin
Aneela Yasmin Thanh Loan Le
Thanh Loan Le Helgard Kaufmann Anja Biber Astrid Kühr
Helgard Kaufmann Anja Biber Astrid Kühr Marcus Linde
Marcus Linde Thomas Debener*
Thomas Debener*- Institute for Plant Genetics, Leibniz University Hannover, Hannover, Germany
The interaction of roses with the leaf spot pathogen Diplocarpon rosae (the cause of black spot on roses) is an interesting pathosystem because it involves a long-lived woody perennial, with life history traits very different from most model plants, and a hemibiotrophic pathogen with moderate levels of gene flow. Here we present data on the molecular structure of the first monogenic dominant resistance gene from roses, Rdr1, directed against one isolate of D. rosae. Complete sequencing of the locus carrying the Rdr1 gene resulted in a sequence of 265,477 bp with a cluster of nine highly related TIR–NBS–LRR (TNL) candidate genes. After sequencing revealed candidate genes for Rdr1, we implemented a gene expression analysis and selected five genes out of the nine TNLs. We then silenced the whole TNL gene family using RNAi (Rdr1–RNAi) constructed from the most conserved sequence region and demonstrated a loss of resistance in the normally resistant genotype. To identify the functional TNL gene, we further screened the five TNL candidate genes with a transient leaf infiltration assay. The transient expression assay indicated a single TNL gene (muRdr1H), partially restoring resistance in the susceptible genotype. Rdr1 was found to localize within the muRdr1 gene family; the genes within this locus contain characteristic motifs of active TNL genes and belong to a young cluster of R genes. The transient leaf assay can be used to further analyze the rose black spot interaction and its evolution, extending the analyses to additional R genes and to additional pathogenic types of the pathogen.
Introduction
Black spot caused by the hemibiotrophic ascomycete Diplocarpon rosae Wolf is the most devastating disease that impacts field-grown roses (Drewes-Alwarez, 2003; Horst, 2008). Strategies to control the disease include the application of agrochemicals or the introgression of disease-resistance genes. Though roses are important ornamental crops with high economic importance, little progress has been made in the field of resistance breeding because genetic information about important disease-resistance traits is scarce (Debener and Linde, 2009).
Analyses of the genetic variability of both D. rosae and roses indicate that the interaction follows a so-called “gene for gene” interaction (Debener et al., 1998). To date, two monogenic dominant genes providing race-specific resistance to black spot have been genetically characterized (Debener et al., 1998; Whitaker et al., 2010a). Most published reports discuss Rdr1, which confers resistance to D. rosae isolates from races one to five and which was fine-mapped to the telomeric ends of rose chromosome 1 (Kaufmann et al., 2003; Biber et al., 2010). Rdr1 was introgressed into cultivated roses from the diploid Asian species Rosa multiflora and leads to an arrest of mycelia development 2–3 days after germination of conidia. The resistance phenotype is accompanied by a hypersensitive response (HR; Gachomo and Kotchoni, 2010), although it is difficult to separate this from cell death that occurs at later stages of compatible interactions.
Although 11 pathogenic races of Diplocarpon have been recently described, data on the diversity and population dynamics of this pathogen indicate a relatively slow spread of new pathogenic races within rose populations (Lühmann et al., 2010). This is in contrast to wind-borne pathogens, for example, powdery mildews. Therefore, monogenic disease-resistance genes are an interesting option for the development of black spot resistant rose cultivars.
The majority of R genes characterized so far encode NB–LRR proteins involved in the so-called “effector-triggered immunity” that directly or indirectly interact with pathogen-derived effector molecules (Dodds and Rathjen, 2010). There are two major groups of NB–LRR proteins: the CC–NB–LRRs (CNLs), which have an N-terminal coiled-coil domain, and the TIR–NB–LRRs (TNLs), which have an N-terminal domain with similarity to Toll and the human interleukin receptor (Eitas and Dangl, 2010). Whereas CNLs occur in both monocots and dicots, TNLs only occur in dicots, but both groups share overlapping, but not identical, signal transduction pathways. Most NB–LRR proteins are assumed to directly or indirectly recognize the presence of an effector molecule via the highly variable LRR domain. Less variability occurs within the CC/TIR and NB domains, which function in signal transduction and intermolecular communication. More than 50% of all NB–LRR coding genes occur in clusters of more or less tightly linked genes with very heterogeneous patterns of evolution characterized by either rapidly evolving type I genes or slowly evolving type II genes (McHale et al., 2006).
Though a large number of NB–LRR coding genes are found in long-lived woody perennials, only very few have been analyzed in more detail, and no NB–LRR gene has been functionally characterized in a woody perennial. It has been proposed that many of the NB–LRR gene families in various woody plants are phylogenetically young (Yang et al., 2008); however, little is known about the phylogenetic processes shaping the diversity of NB–LRRs in woody perennials compared to well-studied annuals.
Here we present a strategy for a functional test of a large number of R gene candidates from non-model species that are difficult to transform by conventional methods. Furthermore, we present data on the structure of the Rdr1 locus harboring the Rdr1 disease-resistance gene as a basis for future applications in resistance breeding in roses and studies on the evolution of the rose black spot pathosystem.
Materials and Methods
Plant Material, BAC Clones, and Fungal Isolates
The R. multiflora hybrids 88/124-46 (2n = 2x = 14) and 91/100-5 (2n = 4x = 28) carrying the Rdr1 gene (Debener et al., 1998) and the hybrid tea rose Pariser Charme (2n = 4x = 28) were cultivated under semi-controlled conditions in a greenhouse as described previously (Biber et al., 2010). Biber et al. (2010) previously constructed the BAC clones utilized in this study.
The black spot isolates DortE4 (race 6; Whitaker et al., 2010b) was maintained on leaves of the rose variety Pariser Charme as described by Debener et al. (1998). Conidia were washed from infected leaves with sterile distilled water and adjusted to defined densities with a hemocytometer.
DNA and RNA Isolation
For DNA isolation, about 70 mg of young leaf tissue was dried, and DNA was isolated using the Nucleospin® Plant II kit (Macherey-Nagel GmbH & Co. KG, Dueren, Germany) according to the manufacturer’s instructions. BAC DNA was isolated by alkaline lysis (SDS) from 500 ml of bacterial culture grown overnight according to Sambrook and Russell (2001). All small-scale plasmid isolations were carried out using Nucleospin® Plasmid kits (Macherey-Nagel GmbH & Co. KG, Dueren, Germany) according to the manufacturer’s instructions. For RNA extraction, plant tissues (30–50 mg) were frozen in liquid nitrogen and ground to a fine powder in a bead mill. Extractions were performed using the Invisorb® Spin Plant RNA Mini Kit (Invitek, Berlin, Germany) according to the manufacturer’s instructions. The contaminating DNA was removed from the extracted RNA using the DNase free kit from Ambion (Cambridgeshire, UK) as recommended by the manufacturer. The quality of both DNA and RNA was checked on agarose gels, and quantification was performed photometrically at 260 and 280 nm.
Sequencing and de novo Assembly
The physical map of Rdr1 was confined to four overlapping BAC clones (Biber et al., 2010). Escherichia coli DH10B cells carrying the BAC clones were delivered in stab agar to Cogenics (Cogenics Ltd., Morrisville, NC, USA), which performed 454 FLX sequencing with 50% of a full run. Sequences were automatically clipped for adaptors and primer sequences and assembled with Newbler assembler by Cogenics (Cogenics Ltd., Morrisville, NC, USA). After the 454 sequencing and de novo assembly, the BAC clone sequences did not completely assemble into the desired large fragment. Therefore, subclones were established from DNA of each BAC clone as described in a later section. The subclones were transferred onto new 96-well microtiter plates containing stab agar and delivered to commercial sequencing services for plasmid DNA preparation and Sanger sequencing. For some of the subclones generated in the pBSKII-vector (Stratagene, La Jolla, CA, USA), plasmid DNA was prepared by a standard alkaline lysis method (Sambrook and Russell, 2001), and sequencing was performed with a Licor DNA-Analyzer Gene Read IR 4200 (LI-COR, Lincoln, NE, USA) using 6% Polyacrylamide gels (National Diagnostics, Atlanta, GA, USA). Fluorescent-labeled standard M13 primers and a Thermo Sequenase cycle sequencing kit (USB/Affymetrix, Santa Clara, CA, USA) were utilized in the sequence reaction mixtures. All of the sequences generated were assembled with SeqMan (DNASTAR, Madison, WI, USA) and ContigExpress (Invitrogen, La Jolla, CA, USA) software. The final assembly was checked by comparison of an experimentally determined restriction enzyme fingerprint for each BAC clone with that of computationally generated fingerprints.
Gene Prediction and Annotation
Bioedit (Hall, 1999) was used for viewing and editing of sequences. Gene prediction and annotation was done using the gene prediction programs FGENESH and GENSCAN (http://www.softberry.com; http://genes.mit.edu/GENSCAN.html). To complement the gene prediction programs and determine sequence similarity, the whole sequence was fragmented in silico into 1-kb fragments with 200-bp overlaps and subjected to BLASTn and BLASTx searches (Altschul et al., 1997) against the GenBank database. Furthermore, the protein domains of the gene clusters on the BAC sequence were determined using Pfam version 23.0 (Finn et al., 2008), SMART 6 (Letunic et al., 2009), and conserved domain database (CDD) v2.16 (Marchler-Bauer et al., 2007). Sequences that were identical to known genes in GenBank were assigned that gene name.
Generation of Subclones
In addition to the complete sequencing of the BAC clones, single TNL candidate genes were subcloned by either partial or complete enzymatic digestion of BAC DNAs. Five micrograms of DNA was partially digested with 4 U of Sau 3A1 for 15 min at 37°C. Restriction fragments were size-separated on 0.8% agarose gels, and bands with ∼7–12 kb size were isolated using the QIAquick gel extraction kit (Qiagen GmbH, Hilden, Germany). For complete digestion of BAC clones, 10 μg of BAC DNA was digested with 10 U of the appropriate restriction enzymes overnight at 37°C, and fragments were isolated as described above.
Fragments were cloned into the binary vector pBINPLUS (van Engelen et al., 1995) or the pBSKII-vector (Stratagene, La Jolla, CA, USA), which were pre-digested with compatible restriction enzymes and dephosphorylated with Shrimp Alkaline phosphatase (MBI Fermentas GmbH, St. Leon-Rot, Germany) according to the manufacturer’s instructions. Ligations were performed in a total volume of 10–15 μl containing 15–30 ng of dephosphorylated vector, 30–90 ng of insert, ATP to a final concentration of 1 mM, 1/10 volume of buffer, and 2.5 Weiss units of T4 DNA ligase and were incubated at 14°C overnight.
One to two microliters of the ligation reaction mixture was mixed with 40 μl of electro-competent E. coli (DH10B) or Agrobacterium cells, and the cells were pulsed using BioRad Micropulser with the EC2 program (2.5 kV for 0.2 cm cuvettes, 25 μFD, 400 Ω, Pulse time 8–12 ms) and further processed according to Sambrook and Russell (2001). Positive clones were picked and confirmed through PCR using general primers for the TNL or the standard M13 primers (Table A2 in Appendix). Some of the subclones were transferred onto new 96-well microtiter plates containing stab agar, delivered to sequencing companies for professional sequencing and then used in the complete assembly of the BAC clones as described earlier.
SSR-PCR for Clone Verification
Verification of the TNL subclones and analyses of cDNA samples for the expression of individual genes was conducted with primer pair Rd1LRR_F and Rd1LRR_R as described in Terefe and Debener (2010). Rd1LRR primers are markers based on simple sequence repeats (SSR) in rose TNLs.
Expression Analyses
Total RNA (300–500 ng) was used to synthesize first-strand cDNA using random primers and the high-capacity cDNA reverse transcription kit (Applied Biosystems, Karlsruhe, Germany). The quality of the cDNA and potential contaminations with genomic DNA was checked via RT-PCR using primer pairs for rose actin, which flank an intron. To evaluate the expression of individual TNL candidate genes, PCR was performed on first-strand cDNA with the specific primer pairs listed in Tables A1 and A2 in Appendix.
Transient Expression Assay, Gene Silencing, and Statistical Analysis
Agroinfiltrations into rose leaves were performed as described in Yasmin and Debener (2010) with minor modifications. The leaves of rose varieties Pariser Charme and 91/100-5 were infiltrated by Agrobacterium suspensions harboring single TNLs at OD600 = 1.5 and challenged by D. rosae (races 6 and 7) at the same time as TNL infiltration. The spore concentrations were adjusted to 5 × 102–105 conidia/ml as described by Dohm et al. (2001). Infiltrated samples were incubated at 22°C in the dark, and the samples were collected for fluorescence microscopy 4 days after infiltration/inoculation. Inoculated leaves were stained with Aniline blue after clearing in 1 M KOH solution for 15 min at 121°C as described by Hood and Shew (1996). The samples were examined under a fluorescent microscope (Zeiss epifluorescence microscope-excitation 450–490 nm, diachronic mirror 510 nm, barrier 520 nm), and fungal growth was scored as the number of hyphal clusters per infiltration site. Hyphal clusters are defined as networks of branched hyphae that develop after germination of conidia within the infiltrated area.
A double-stranded cDNA sequence of 1098 bp from the most conserved exon 2 region of the TNLs was inserted between two 35S inverted promoters in a p9U10–RNAi vector to trigger the production of an Rdr1–RNAi effect, resulting in gene silencing (DNA-Cloning Services e.K., Hamburg, Germany). Agrobacterium suspensions harboring the Rdr1–RNAi were infiltrated into leaves of Pariser Charme and 91/100-5 varieties and challenged by D. rosae (race 6). Further analysis and data collections were made as previously described.
Statistical evaluation of the infiltration experiments was conducted with the R-software package (R Development Core Team, 2009). For each experiment, 20 infiltration sites were recorded, and tests for differences between treatments were conducted with the Kruskal Wallis and the Wilcoxon procedures. In those cases where the Kruskal Wallis test indicated significant differences among treatments, pair-wise comparisons were conducted with the Wilcoxon rank sum test.
Results
The Rdr1 Locus Carries Nine Highly Similar TNL Genes
The black spot resistance gene Rdr1 is a monogenic dominant gene introgressed into diploid garden roses from R. multiflora and had been previously localized at the telomeric end of rose linkage group 1. High-resolution mapping confined the region around Rdr1 to an interval defined by four overlapping BAC clones (Biber et al., 2010). These four clones (clones 29O3, 94G8, 20F5, and 69E24) were submitted to next generation sequencing in pairs of two overlapping clones through a professional sequencing service. A total of 467,700 sequences from 50% of a 454 GS FLX run provided 227,395 sequences, with an average length of 241 bp and a theoretical sequence coverage of 137 times. These sequences were assembled by the Newbler software into 128 contigs. Highly similar repeats present on the contig made a reliable and complete assembly based on the 454 sequences alone impossible; therefore, we generated 550 additional sequence reads, using the Sanger method, from completely digested and partially digested subclones of more than 2 kb and from fragments generated by primer walking to fill the remaining gaps. This resulted in a complete sequence of 265,477 bp with an average GC content of 39%. The complete 265, 477-bp has been deposited in the GenBank database under accession number HQ455834.
By analyses with BLAST, Pfam, and SMART6, 47 genes were predicted from the sequence, 40 of which had significant matches to GenBank entries (Figure 1; Table 1). Among these 40 predicted genes, nine sequences distributed over 204 kb had highly significant similarities to resistance genes of the TIR–NBS–LRR (TNL) type. Among the other sequences we found 10 transposable-elements and sequences with similarities to transcription factor B3, AAA type ATPases, and protein kinases (Table 1). However, because the protein kinases are located beyond the recombination breakpoints present on the contig, the nine TNL sequences remained the most likely candidates for Rdr1. We therefore designated these genes as muRdr1A to muRdr1I, in which “mu” stands for the species name of the source of the sequence (R. multiflora) and “A” to “I” indicates the order of the Rdr1 TNL on the sequenced contig. The predicted amino acid sequence for the nine TNLs has been deposited in the GenBank database under accession numbers AEE43925 to AEE43933. As shown previously, a microsatellite marker was found in the expressed part of exon 4 of muRdr1A, C, and H, and this marker co-segregates with the functional Rdr1 gene without recombination (Terefe and Debener, 2010; Figure 1).
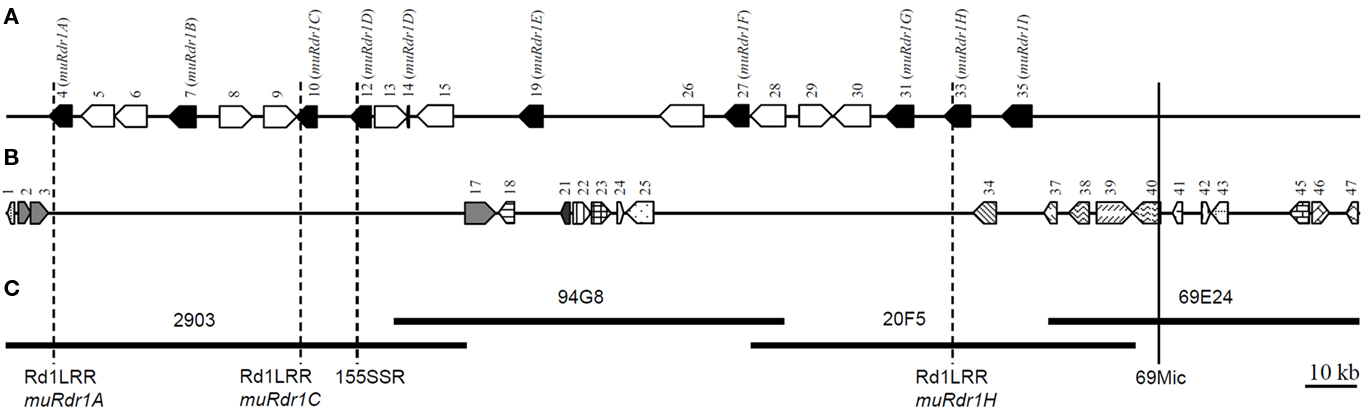
Figure 1. Physical positions of the Rdr1 gene region and schematic representation of positions of predicted genes in the R. multiflora genotype. (A) Nine TNLs (black pentagon) and 10 transposable-elements (unfilled pentagon). (B) Other genes distributed along the Rdr1 region. (C) The completely sequenced four overlapping BAC clones carrying the Rdr1 gene. The broken-line indicates Rdr1-linked SSR markers. The vertical unbroken line indicates recombination break point (69Mic) on the right side of the region. Numbers on each pentagon refer to the genes as described in Table 1. Predicted genes without any similarity in the GenBank database are not represented.

Table 1. List of putative genes on the 265,477-bp contig around the Rdr1 locus in the R. multiflora genotype as determined by FGENESH and GENSCANa.
Based on the predicted transcriptional start and stop codons, the size of the nine muRdr1 genes, including introns, varied from 4085 to 5920 bp. Only muRdr1D differed from this due to a transposable-element insertion of 6957 bp within intron 1, resulting in a total length of 11,590 bp. Six of the nine muRdr1 genes (A, C, D, E, F, H) have four exons and three introns, two (muRdr1B and G) have two additional introns and one (muRdr1I) has only three exons and two introns (Figure 2). The intron–exon structure reflects the domain structure of typical TNL proteins in that the first exon contains the TIR domain, the second exon contains the NBS domain and the forth (or in case of TNL–muRdr1I, the third exon) contains an LRR domain. The most closely related TNL sequences from other species, with a proven function as resistance genes, are CMR1 from Phaseolus vulgaris (Seo et al., 2006) and the N gene from tobacco (Whitham et al., 1994), with 44 and 36% sequence identity at the amino acid level, respectively. The overall amino acid identity among the nine members of the muRdr1 TNL-family was between 78 and 99% (Table 2). Conserved within all of the nine muRdr1 genes were motifs characteristic for functional TNL genes; for example the Kinase 1a (P-loop), kinase 2, and Kinase 3a motifs of the NBS domain (Traut, 1994; van Ooijen et al., 2008) as well as motifs 2, 3, 4, and 5 and the HD motif (Hammond-Kosack and Jones, 1997; Figures 3A,B). This confirms their putative function as TNL resistance genes; however, none of the nine genes could be excluded as the functional Rdr1 gene based on a lack of conservation in these motifs.

Figure 2. The structure of the nine TNLs. The majority of the TNLs are characterized by four exons and three introns. The fourth TNL (muRdr1D) was interrupted by a 6957-bp transposable-element at intron 1.
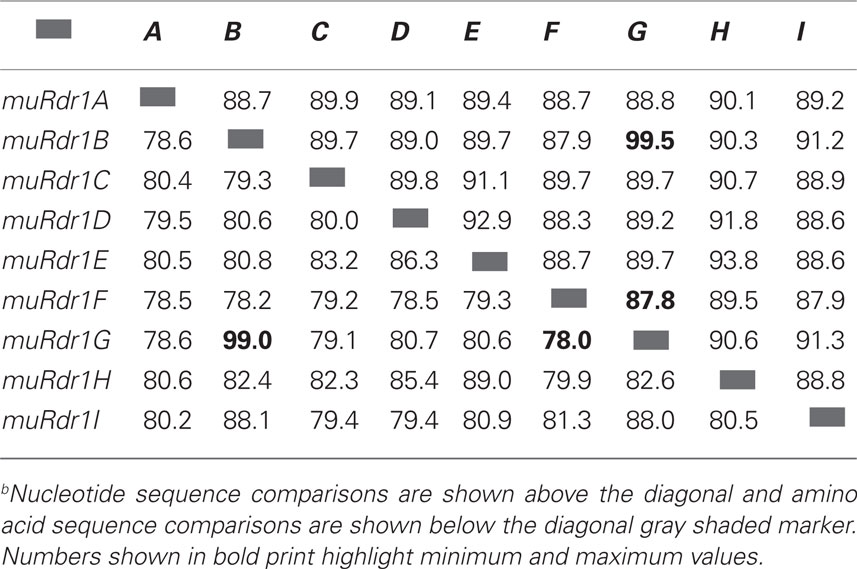
Table 2. Nucleotide and amino acid sequence similarity of the nine muRdr1–TNLsb.

Figure 3. The muRdr1 protein sequence and conserved motifs. (A) muRdr1H protein sequence. Conserved motifs are indicated as bold letters and the 14 imperfect leucine-rich repeats (LRRs) are underlined. (B) Alignments of kinase motifs in the conserved region for the nine TNLs and three previously described resistance genes, RPP5 (Arabidopsis, accession number: AAF08790), N (tobacco, accession number: AAA50763), and L6 (flax, accession number: AAA91022).
Five of the Nine TNLs are Expressed in Leaves and Petals
Previous analyses had shown that the Rdr1 gene is active and confers resistance in both rose leaves and petals (Debener et al., 1998); therefore, we analyzed expression of all nine TNLs in different rose organs, as non-expressed copies of the muRdr1 gene family can be excluded as likely Rdr1 candidates. PCR primers were designed based on the variable parts of exons three and four (Table A1 in Appendix) and tested for specificity with cloned individual members of the gene family. In addition, a primer pair for an expressed SSR marker in exon 4 was used (Terefe and Debener, 2010). RT-PCR was conducted on cDNA from leaves and petals of genotypes 88/124-46 and 91/100-5, which both express Rdr1. In summary, muRdr1A, C, G, H, and I were expressed in both leaves and petals, whereas muRdr1D, E, and F expression was undetectable. One of the TNL genes, muRdr1B, was not detectable in petals of genotype 88/124-46 and not at all in genotype 91/100-5 (Table 3). Therefore, muRdr1B, D, E, and F were excluded as likely candidates for Rdr1.
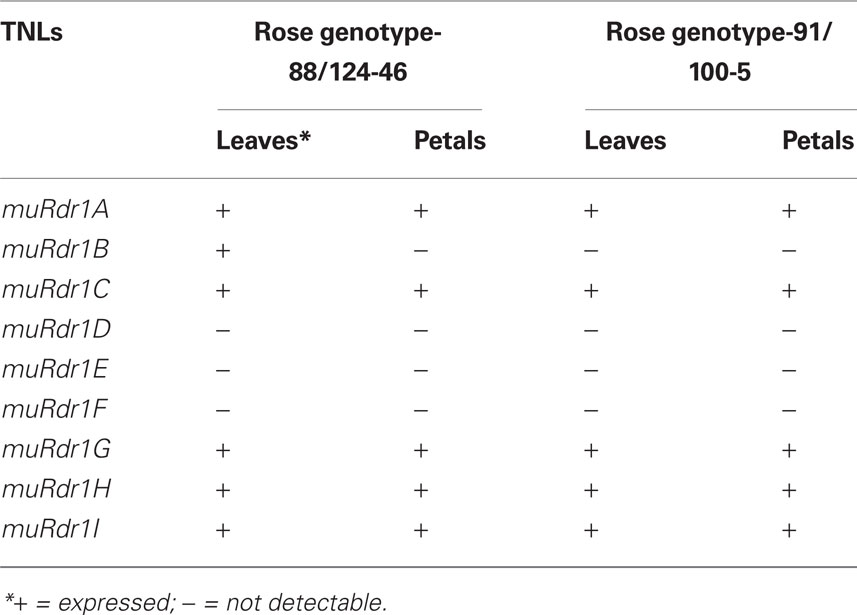
Table 3. Expression profiles of single muRdr1 TNLs in a homologous system.
Rdr1 is a Member of the muRdr1 Gene Family
To get additional information on the role of the muRdr1 genes, we performed a transient silencing experiment of the whole gene family. To accomplish this, we used a construct based on the vector p9U10–RNAi (DNA-Cloning Services e.K., Hamburg, Germany). We used a 1098-bp fragment from the highly conserved NBS region of exon 2 of muRdr1H which shares an overall similarity of 86–96% to all other muRdr1 genes (local similarities over stretches > 100 bp were more than 95%). This recently designed vector induces RNAi-based gene silencing by inducing the transcription of large inserts directed by two flanking 35S promoters in opposite orientations.
Stable transformation in rose is feasible but suffers from low efficiency and a long period before inoculation-ready plants can be obtained (Dohm, 2003). As previous experiments had shown that the rose/black spot interaction remains unaltered if conidia are infiltrated into rose tissues at the same time as Agrobacteria (unpublished data), transient infiltration experiments were conducted to silence the muRdr1 gene family.
Using the rose genotypes Pariser Charme and 91/100-5, we exposed tissues either with only conidia of DortE4 (compatible interaction with Pariser Charme, incompatible interaction with 91/100-5), with conidia of DortE4 together with a control plasmid expressing a GUS-intron or with conidia of the black spot isolate DortE4 together with Rdr1–RNAi, the silencing construct. In agreement with earlier experiments, the resistant genotype 91/100-5 did not allow growth of hyphal clusters except in 1 out of 20 infiltration sites; the one exception showed only a single cluster (Figure 4). The co-infiltration with Agrobacteria harboring the GUS-intron control plasmid allowed a small number of clusters to grow; however, these clusters had only a few hyphae and were distinct in morphology from those of the clusters growing in fully susceptible conditions (Figure 5A). In contrast, the infiltration of conidia together with the Rdr1–RNAi construct into 91/100-5, increased the number of hyphal clusters fivefold (P = 2.2e−16) compared to the GUS-control (Figures 4 and 5B). Furthermore, these clusters most closely resembled those of fully compatible interactions. The numbers of hyphal clusters were comparable to those found in experiments performed with the susceptible genotype Pariser Charme infiltrated with only conidia or conidia in combination with the GUS-construct in the same experiment (Figure 4). To our surprise, the infiltration of the Rdr1–RNAi construct together with conidia of DortE4 into the highly susceptible genotype Pariser Charme more than doubled the number of hyphal clusters compared to the GUS-intron control plasmid (P = 2.4e−5), indicating a significant increase in susceptibility (Figure 4).
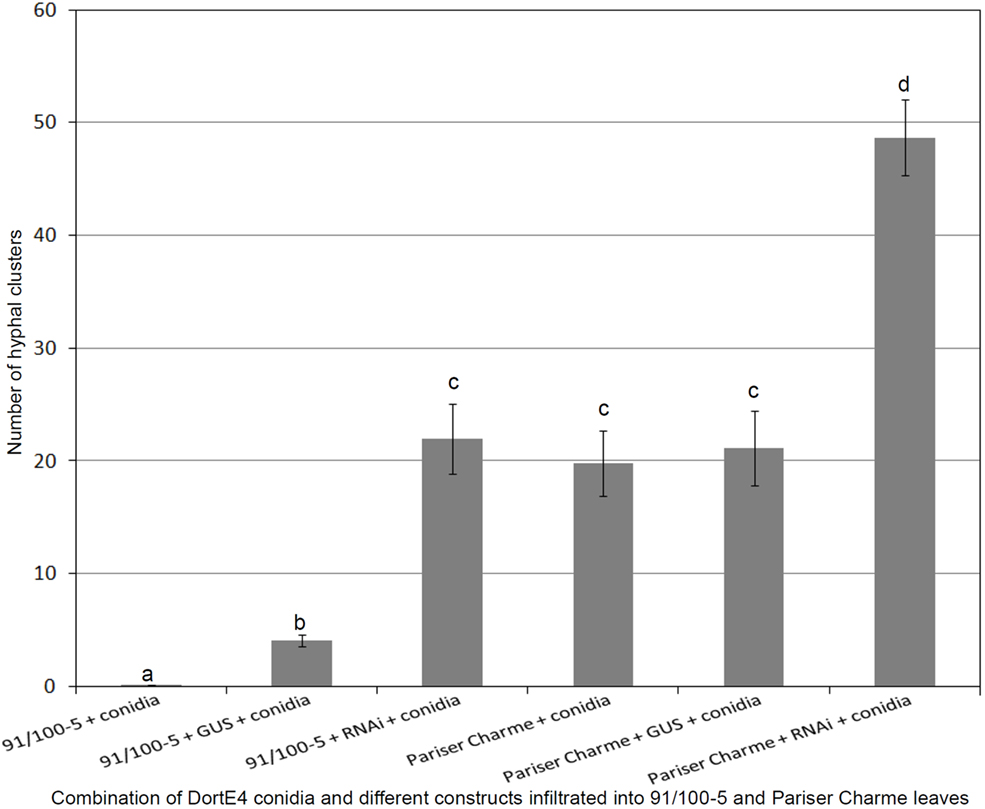
Figure 4. Results of the transient silencing experiment in genotype 91/100-5 and Pariser Charme rose leaves. Values are averages for 20 replications each ± SEM. The number of hyphal clusters are significantly increased in 91/100-5 leaves carrying Rdr1–RNAi construct. Results with same letters are not significantly different (P < 0.05).
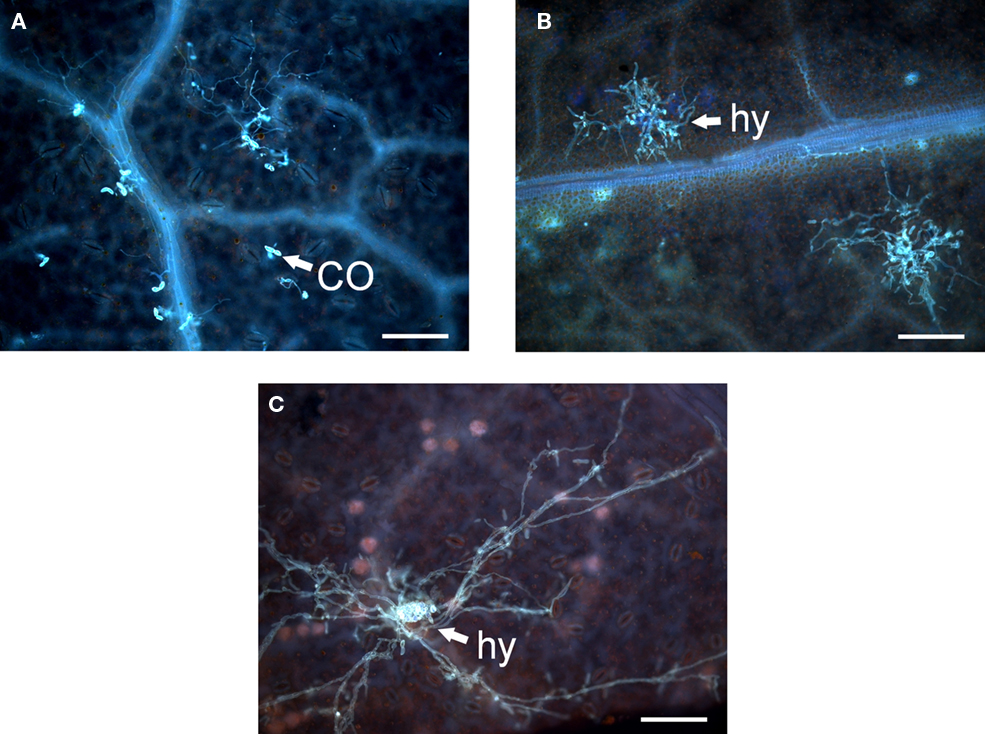
Figure 5. Infiltration of rose leaves with black spot conidia or black spot conidia and Agrobacteria containing various constructs. (A) Resistant genotype 91/100-5 infiltrated with conidia (CO) of Dort E4 and Agrobacterium strain GV3101 harboring pBIN19::GUS-intron. (B) Infiltration of 91/100-5 with conidia of Dort E4 and Agrobacterium strain GV3101 harboring Rdr1–RNAi displaying a compatible interaction. (C) Infiltration of Pariser Charme with conidia of Dort E4 displaying a compatible interaction with a hyphal cluster (hy) and young acervulus in the middle. The scale bar indicates 100 μm.
One of the muRdr1 TNL Genes Partially Complements a Susceptible Genotype in a Transient Assay
To identify the active resistance gene among the muRdr1 family members, we performed several transient infiltration experiments to test for complementation of susceptibility to DortE4 by individual muRdr1 TNLs. To accomplish this, we subcloned each of the genes except muRdr1D into the pBIN + binary plant transformation vector. In addition to the putative coding region, more than 3 kb of genomic DNA upstream of the putative ATG and more than 900 bp downstream of the putative stop codon were included in the subclones to allow expression under the native promoters. The muRdr1D is interrupted by 6957-bp transposable-element insertion within intron 1. As a result, we speculated muRdr1D as non-functional and excluded from subcloning and functional analysis.
All eight muRdr1 TNLs were transformed into Agrobacterium strain GV 3101 and transiently expressed in leaves of Nicotiana benthamiana (Wroblewski et al., 2005; Yasmin and Debener, 2010). Expression was analyzed by RT-PCR on cDNA generated from infiltrated leaf areas with PCR primer pairs from the first and last exons. The cDNA from leaves infiltrated with GUS controls did not produce a PCR signal. In contrast to the expression analyses in roses, all eight subclones produced PCR fragments with sizes as expected for expressed sequences (data not shown). Because no tobacco ortholog with sufficient similarity could be detected, this indicates heterologous expression of all eight muRdr1 genes tested.
Leaves of the susceptible rose genotype Pariser Charme were infiltrated with only conidia or conidia and Agrobacteria carrying control plasmid; the conidia germinated and formed dense clusters of hyphae after 4 days (Figure 5C). In nine independent experiments, conidia of the race six isolate DortE4 were co-infiltrated at low densities (5 × 102 conidia per ml) into the susceptible rose genotype Pariser Charme with Agrobacteria harboring different muRdr1 TNLs (Figure 6). Conidial germination and hyphal clusters occurred in all experiments including muRdr1 genes and no significant differences in the morphology of the hyphal clusters could be observed between treatments. However, in all nine experiments, muRdr1H led to a significant reduction of the number of hyphal clusters compared to the controls and the other muRdr1 TNL genes. On average, the number of hyphal clusters was reduced to 54% of the number of hyphal clusters present in the control experiments (Figure 6).
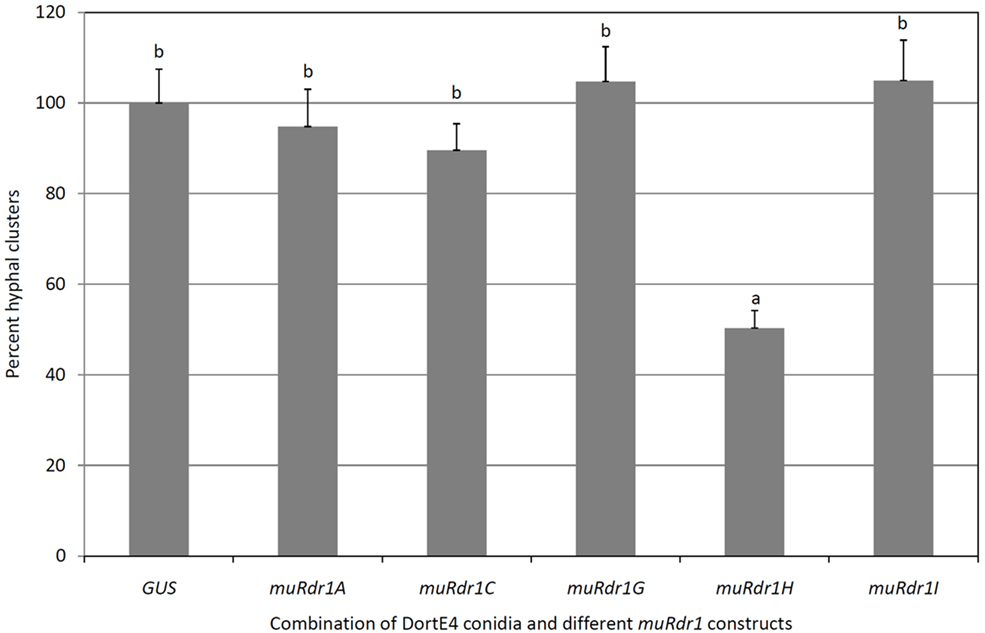
Figure 6. Transient complementation disease assay with individual muRdr1 TNLs and DortE4. This graph represents average data of nine independent experiments ± SEM. Highly significant reduction in the percent hyphal cluster is observed in Pariser Charme rose plants infiltrated with muRdr1H than the other TNLs. Results with same letters are not significantly different (P < 0.05).
The Effect of muRdr1H is Specific for the Interaction with Race 6 Isolates of Black Spot
The same experimental strategy was applied using the race 7 isolate R6, which is able to infect genotypes carrying the Rdr1 resistance gene (Whitaker et al., 2010b). In three independent experiments, muRdr1H had no effect on the infection process compared to the control plasmid (Figure 7), indicating that the effect of muRdr1H on the growth of isolate DortE4 is specific for this pathogen.
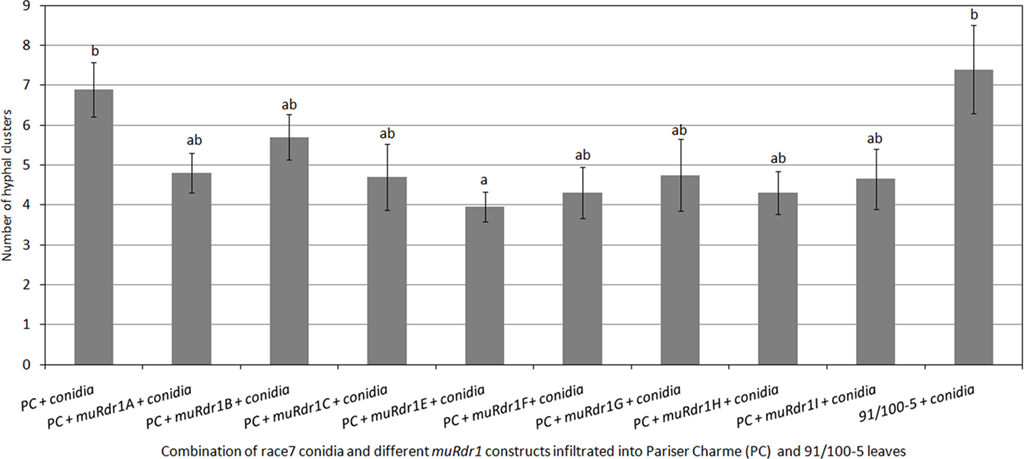
Figure 7. Transient complementation disease assay with individual muRdr1 TNLs and race7. This graph represents average data of three independent experiments ± SEM. No significant difference in the number of hyphal clusters is observed between the eight muRdr1 TNLs. There is also no significant difference between muRdr1H infiltrated rose cultivar and the non-infiltrated. Results with same letters are not significantly different (P < 0.05).
Discussion
In this study we show that the black spot resistance gene Rdr1 is a member of a highly conserved family of TNL (TIR-NBS-LRR) resistance genes.
The copy number of resistance gene paralogs may vary between species. Copy numbers of Hcr9 ranging from one to five were observed in tomato (Parniske and Jones, 1999); 12–32 copies of RGC2 were estimated in lettuce (Kuang et al., 2004); 8–10 copies of RPP5 were found in Arabidopsis (Noël et al., 1999) and four Vf paralogs were detected in apple (Xu and Korban, 2002). The nine muRdr1 paralogs obtained from R. multiflora are within the range already shown for other plant species. The observed variation in copy number may be a result of a birth-and-death model of resistance gene evolution as reviewed in Michelmore and Meyers (1998), with this model being suspected to occur for lettuce RGC2 (Kuang et al., 2004).
The muRdr1 genomic sequence size varied from 4085 bp to 5920 bp. This observation is within the size range of previously reported NBS–LRR genes; for example, resistance gene analog (RGA) 1 and RGA2 genes in wheat had lengths of 3905 and 4766 bp, respectively (Wicker et al., 2001). The hypothesis that muRdr1 is a hotspot for resistance genes is supported by the fact that repetitive transposable-elements, similar to the unusually large repetitive transposable-elements distributed in the muRdr1 region, are reported along resistance genes in wheat (Wicker et al., 2001), in potato (Kuang et al., 2005), in tomato (Parniske and Jones, 1999), in Arabidopsis (Noël et al., 1999), in maize (Sun et al., 2001), in rice (Song et al., 1997), and in lettuce (Meyers et al., 1998).
Rdr1 is the first disease-resistance gene isolated from roses and also the first gene isolated from roses via positional cloning. These advances were possible because our transient expression assay allowed us to assay a large number of candidate genes within a relatively short period of time.
Although roses have some characteristics that make them an ideal model for woody perennials, such as a generation time of less than a year and a relatively small genome size of around 500 Mb (Debener and Linde, 2009), genetic complementation experiments are very difficult due to low transformation rates and a lengthy transformation process (Dohm, 2003). Therefore, the transient assay that we employed in this study offers an alternative to stable transformation experiments in that it is much faster (a single experiment takes around 1 week) and circumvents problems in the analyses of single transgenic plants, such as positional and copy number effects. Furthermore, this assay makes possible the characterization of a large number of candidate genes that would be impossible to screen via stable transformation at transformation rates of around 3% (Dohm et al., 2002).
The susceptibility of leaf areas infiltrated with the muRdr1H gene was reduced to 54% compared to control infiltrations. However, in all of the nine biological replicates, a pair-wise comparison between areas infiltrated with muRdr1H and those infiltrated with a control plasmid revealed a statistically significant reduction of mycelia colonies formed after co-infiltration of fungal conidia and Agrobacteria. This partial reduction of infection is most probably caused by the much lower efficiency of transient transformation of rose leaf cells compared to results obtained with N. benthamiana (Yasmin and Debener, 2010). Furthermore, we expressed the Rdr1 candidate genes under their natural promoters, which induce very low levels of expression in the natural context, comparable to the majority of the NB–LRR coding genes characterized so far (for example: Wroblewski et al., 2007). The reduction of infection observed in our assays with the functional Rdr1 gene is comparable to the reduction of infection as observed in the transient leaf assays employed to analyze dominant resistance genes in cereals (Yahiaoui et al., 2004; Douchkov et al., 2005; Keller et al., 2005). In these assays, the function of resistance gene candidates was tested in transient assays of particle-bombarded epidermal cells. Here, the function of a candidate gene is determined by a statistically significant reduction in the number of epidermal cells with developed haustoria as observed in inoculated transgenic cells.
The transient silencing of the muRdr1 TNL-family gene revealed loss of resistance in the naturally resistant plant. This is an indication that the resistance gene Rdr1 is indeed the TNL type resistance gene as reported in the majority of other plant species (Sanseverino et al., 2009). The complementation analysis using the individual muRdr1 TNL-construct further confined the Rdr1 gene to the muRdr1H. Sequence analysis of the muRdr1H indicated conservation of protein motifs typical for plant resistance genes (Traut, 1994; Hammond-Kosack and Jones, 1997; van Ooijen et al., 2008). We started cloning muRdr1H homologs from several susceptible rose species including the parent of the cross which led to the identification of the Rdr1 locus. Preliminary sequence analysis revealed clear polymorphisms along the complete gene regions with a maximum amino acid sequence similarity of 87% (data not shown). Therefore, a single mutation explaining the difference between the susceptible and the resistant genotype cannot be identified among the numerous differences. More extensive analyses will be done in the future, which may lead to a detection of functional mutation responsible for the loss of resistance in the susceptible rose species.
The identification of Rdr1 and the analyses of expression of the family members were complicated by the highly duplicated nature of the Rdr1 gene family and a high degree of conservation among the individual members. In several independent experiments, we found five members of the muRdr1 family expressed in leaves and petals, whereas all members were expressed in tobacco leaves. The most likely reason for this is a specific down regulation of individual family members in roses by either regulatory elements missing in our genomic clones or other mechanism such as siRNA, a phenomenon already described for a cluster of R genes in Arabidopsis (Yi and Richards, 2007). However, further experimental evidence is necessary to analyze these observations.
Most plant R genes exist in clusters (Hulbert et al., 2001). The different R gene paralogs in the same cluster may confer resistance to multiple pathogens or distinct isolates of a single pathogen (van der Vossen et al., 2000). Because five of the nine Rdr1 paralogs from R. multiflora are expressed in leaves and flowers, we may also be able to identify additional R genes within the muRdr1 cluster with transient assays involving additional fungal isolates. In a recent update on the number of physiological races of the black spot fungus, a minimum of 11 races was characterized (Whitaker et al., 2010b). In addition, we previously identified rose germplasm resistant to several isolates of black spot as well as to various isolates of Peronospora sparsa, which causes downy mildew in roses, and Podosphaera pannosa, which causes powdery mildew on roses (Schulz et al., 2009; Whitaker et al., 2010b). Based on a screen of a number of diploid and tetraploid roses with an SSR marker located within the last exon (Terefe and Debener, 2010), and on the complete sequence of an orthologous contig from a genotype of R. rugosa (Terefe et al., 2010), we know that the Rdr1 gene family is comprised of between 5 and 20 members in different rose genomes. Because some of these genotypes are resistant to race five isolates, it is now possible to analyze the role of Rdr1 orthologs and paralogs in this and other interactions based on the transient assay. We now have a tool at hand with which we will be able to analyze the evolution of plant pathogen interactions in a non-model species with a combination of life history traits of both partners (long-lived woody perennial host and mostly asexually reproducing pathogen with moderate gene flow rates) that has, to date, not been studied.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the German Research Foundation (DFG) grant number DE 511/4-1 and DE 511/4-2. Aneela Yasmin was supported by a scholarship from German Academic Exchange Service (DAAD). We thank Ingrid Robotta for excellent technical assistance. We also thank Maik Klie for assisting in the BAC-subcloning and screening experiments.
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Biber, A., Kaufmann, H., Linde, M., Spiller, M., Terefe, D., and Debener, T. (2010). Molecular markers from a BAC contig spanning the Rdr1 locus: a tool for marker-assisted selection in roses. Theor. Appl. Genet. 120, 765–773.
Debener, T., Drevez-Alvarez, R., and Rockstroh, K. (1998). Identification of five physiological races of black spot, Diplocarpon rosae Wolf, on roses. Plant Breed. 117, 267–270.
Debener, T., and Linde, M. (2009). Exploring complex ornamental genomes: the rose as a model plant. CRC Crit. Rev. Plant Sci. 28, 267–280.
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 11, 539–548.
Dohm, A. (2003). “Biotechnologies for breeding: genetic transformation,” in Encyclopedia of Rose Sciences, eds A. V. Roberts, T. Debener, and S. Gudin (Oxford: Elsevier Academic Press), 15–25.
Dohm, A., Ludwig, C., Schilling, D., and Debener, T. (2001). Transformation of roses with genes for antifungal proteins. Acta Hortic. 547, 27–33.
Dohm, A., Ludwig, C., Schilling, D., and Debener, T. (2002). Transformation of roses with genes for antifungal proteins to reduce their susceptibility to fungal diseases. Acta Hortic. 572, 105–111.
Douchkov, D., Nowara, D., Zierold, U., and Schweizer, P. (2005). A high-throughput gene-silencing system for the functional assessment of defense-related genes in barley epidermal cells. Mol. Plant Microbe Interact. 18, 755–761.
Drewes-Alwarez, R. (2003). “Black spot,” in Encyclopedia of Rose Sciences, eds A. V. Roberts, T. Debener, and S. Gudin (Oxford: Elsevier Academic Press), 148–153.
Eitas, T. K., and Dangl, J. L. (2010). NB-LRR proteins: pairs, pieces, perception, partners, and pathways. Curr. Opin. Plant Biol. 13, 472–477.
Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut, J. S., Hotz, H. R., Ceric, G., Forslund, K., Eddy, S. R., Sonnhammer, E. L., and Bateman, A. (2008). The Pfam protein families database. Nucleic Acids Res. 36, D281–D288.
Gachomo, E. W., and Kotchoni, S. O. (2010). Microscopic and biochemical evidence of differentially virulent field isolates of Diplocarpon rosae causing black spot disease of roses. Plant Physiol. Biochem. 48, 167–175.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hammond-Kosack, K. E., and Jones, J. D. G. (1997). Plant disease resistance genes. Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 575–607.
Hood, M. E., and Shew, H. D. (1996). Applications of KOH aniline blue fluorescence in the study of plant-fungal interactions. Phytopathology 86, 704–708.
Hulbert, S. C., Webb, C. A., Smith, S. M., and Sun, Q. (2001). Resistance gene complexes: evolution and utilization. Annu. Rev. Phytopathol. 39, 285–312.
Kaufmann, H., Mattiesch, L., Lorz, H., and Debener, T. (2003). Construction of a BAC library of Rosa rugosa Thunb. and assembly of a contig spanning Rdr1, a gene that confers resistance to black spot. Mol. Genet. Genomics 268, 666–674.
Keller, B., Feuillet, C., and Yahiaoui, N. (2005). Map-based isolation of disease resistance genes from bread wheat: cloning in a supersize genome. Genet. Res. 85, 93–100.
Kuang, H., Wei, F., Marano, M. R., Wirtz, U., Wang, Z., Liu, J., Shum, W. P., Zaborsky, J., Tallon, L. J., Rensink, W., Lobst, S., Zhang, P., Tornqvist, C. E., Tek, A., Bamberg, J., Helgeson, J., Fry, W., You, F., Luo, M. C., Jiang, J., Buell, C. R., and Baker, B. (2005). The R1 resistance gene cluster contains three groups of independently evolving, type I R1 homologues and shows substantial structural variation among haplotypes of Solanum demissum. Plant J. 44, 37–51.
Kuang, H., Woo, S. S., Meyers, B. C., Nevo, E., and Michelmore, R. W. (2004). Multiple genetic processes result in heterogeneous rates of evolution within the major cluster disease resistance genes in lettuce. Plant Cell 16, 2870–2894.
Letunic, I., Doerks, T., and Bork, P. (2009). SMART 6: recent updates and new developments. Nucleic Acids Res. 37, D229–D232.
Lühmann, A. K., Linde, M., and Debener, T. (2010). Genetic diversity of Diplocarpon rosae: implications on practical breeding. Acta Hortic. 870, 157–162.
Marchler-Bauer, A., Anderson, J. B., Derbyshire, M. K., DeWeese-Scott, C., Gonzales, N. R., Gwadz, M., Hao, L., He, S., Hurwitz, D. I., Jackson, J. D., Ke, Z., Krylov, D., Lanczycki, C. J., Liebert, C. A., Liu, C., Lu, F., Lu, S., Marchler, G. H., Mullokandov, M., Song, J. S., Thanki, N., Yamashita, R. A., Yin, J. J., Zhang, D., and Bryant, S. H. (2007). CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 35, D237–D240.
McHale, L., Tan, X. P., Koehl, P., and Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 212.
Meyers, B. C., Chin, D. B., Shen, K. A., Sivaramakrishnan, S., Lavelle, D. O., Zhang, Z., and Michelmore, R. W. (1998). The major resistance gene cluster in lettuce is highly duplicated and spans several megabases. Plant Cell 10, 1817–1832.
Michelmore, R. W., and Meyers, B. C. (1998). Clusters of resistance genes in plants evolve by divergent selection and a birth-and death process. Genome Res. 8, 1113–1130.
Noël, L., Moores, T. L., van der Biezen, E. A., Parniske, M., Daniels, M. J., Parker, J. E., and Jones, J. D. G. (1999). Pronounced intraspecific haplotype divergence at the RPP5 complex disease resistance locus of Arabidopsis. Plant Cell 11, 2099–2111.
Parniske, M., and Jones, J. D. G. (1999). Recombination between diverged clusters of the tomato Cf-9 plant disease resistance gene family. Proc. Natl. Acad. Sci. U.S.A. 96, 5850–5855.
R Development Core Team. (2009). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd Edn. Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
Sanseverino, W., Roma, G., De Simone, M., Faino, L., Melito, S., Stupka, E., Frusciante, L., and Ercolano, M. R. (2009). PRGdb: a bioinformatics platform for plant resistance gene analysis. Nucleic Acids Res. 38, D814–D821.
Schulz, D. F., Linde, M., Blechert, O., and Debener, T. (2009). Evaluation of genus Rosa germplasm for resistance to black spot, downy mildew and powdery mildew. Eur. J. Hortic. Sci. 74, 1–9.
Seo, Y. S., Rojas, M. R., Lee, J. Y., Lee, S. W., Jeon, J. S., Ronald, P., Lucas, W. J., and Gilbertson, R. L. (2006). A viral resistance gene from common bean functions across plant families and is upregulated in a non-virus-specific manner. Proc. Natl. Acad. Sci. U.S.A. 103, 11856–11861.
Song, W. Y., Pi, L. Y., Wang, G. L., Gardner, J., Holsten, T., and Ronald, P. C. (1997). Evolution of the rice Xa21 disease resistance gene family. Plant Cell 9, 1279–1287.
Sun, Q., Collins, N. C., Ayliffe, M., Smith, S. M., Drake, J., Pryor, T., and Hulbert, S. H. (2001). Recombination between paralogues at the rp1 rust resistance locus in maize. Genetics 158, 423–438.
Terefe, D., Biber, A., Yasmin, A., Kaufmann, H., and Debener, T. (2010). Comparative genomic analysis of sequences around the Rdr1 locus in resistant and susceptible rose genotypes. Acta Hortic. 870, 197–204.
Terefe, D., and Debener, T. (2010). An SSR from the LRR region of the rose Rdr1 gene family is a useful RGA marker for roses and other Rosaceae. Plant Breed. 130, 291–293.
Traut, T. W. (1994). The functions and consensus motifs of nine types of peptide segments that form different types of nucleotide-binding sites. Eur. J. Biochem. 222, 9–19.
van der Vossen, E. A., Van der Voort, J. N., Kanyuka, K., Bendahmane, A., Sandbrink, H., Baulcombe, D. C., Bakker, J., Stiekema, W. J., and Klein-Lankhorst, R. M. (2000). Homologues of a single resistance-gene cluster in potato confer resistance to distinct pathogens: a virus and a nematode. Plant J. 23, 567–576.
van Engelen, F. A., Molthoff, J. W., Conner, A. J., Nap, J. P., Pereira, A., and Stiekema, W. J. (1995). pBINPLUS: an improved plant transformation vector based on pBIN19. Transgenic Res. 4, 288–290.
van Ooijen, G., Mayr, G., Kasiem, M. M. A., Albrecht, M., Cornelissen, B. J. C., and Takken, F. L. W. (2008). Structure-function analysis of the NB-ARC domain of plant disease resistance proteins. J. Exp. Bot. 59, 1383–1397.
Whitaker, V. M., Bradeen, J. M., Debener, T., Biber, A., and Hokanson, S. C. (2010a). Rdr3, a novel locus conferring black spot disease resistance in tetraploid rose: genetic analysis, LRR profiling, and SCAR marker development. Theor. Appl. Genet. 120, 573–585.
Whitaker, V. M., Debener, T., Roberts, A. V., and Hokanson, S. C. (2010b). A standard set of host differentials and unified nomenclature for an international collection of Diplocarpon rosae races. Plant Pathol. 59, 745–752.
Whitham, S., Dineshkumar, S. P., Choi, D., Hehl, R., Corr, C., and Baker, B. (1994). The product of the tobacco mosaic virus resistance gene N-similarity to Toll and the interleukin-1 receptor. Cell 78, 1101–1115.
Wicker, T., Stein, N., Albar, L., Feuillet, C., Schlagenhauf, E., and Keller, B. (2001). Analysis of a contiguous 211 kb sequence in diploid wheat (Triticum monococcum L.) reveals multiple mechanisms of genome evolution. Plant J. 26, 307–316.
Wroblewski, T., Piskurewicz, U., Tomczak, A., Ochoa, O., and Michelmore, R. W. (2007). Silencing of the major family of NBS-LRR-encoding genes in lettuce results in the loss of multiple resistance specificities. Plant J. 51, 803–818.
Wroblewski, T., Tomczak, A., and Michelmore, R. (2005). Optimization of Agrobacterium-mediated transient assays of gene expression in lettuce, tomato and Arabidopsis. Plant Biotechnol. J. 3, 259–273.
Xu, M., and Korban, S. S. (2002). A cluster of four receptor-like genes resides in the Vf locus that confers resistance to apple scab disease. Genetics 162, 1995–2006.
Yahiaoui, N., Srichumpa, P., Dudler, R., and Keller, B. (2004). Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant J. 37, 528–538.
Yang, S., Zhang, X., Yue, J. X., Tian, D., and Chen, J. Q. (2008). Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol. Genet. Genomics 280, 187–198.
Yasmin, A., and Debener, T. (2010). Transient gene expression in rose petals via Agrobacterium infiltration. Plant Cell Tissue Organ Cult. 102, 245–250.
Keywords: Rosa, woody plant, Diplocarpon rosae, transient assay, Rdr1, disease
Citation: Terefe-Ayana D, Yasmin A, Le TL, Kaufmann H, Biber A, Kühr A, Linde M and Debener T (2011) Mining disease-resistance genes in roses: functional and molecular characterization of the Rdr1 locus. Front. Plant Sci. 2:35. doi: 10.3389/fpls.2011.00035
Received: 17 June 2011;
Accepted: 18 July 2011;
Published online: 01 August 2011.
Edited by:
Diego Rubiales, Consejo Superior de Investigaciones Cientificas, SpainReviewed by:
Anna Maria Mastrangelo, CRA-Centro di Ricerca per la Cerealicoltura, ItalyNoel Ferro Diaz, University of Bonn, Germany
Teresa Millan, Universidad de Cordoba, Spain
Copyright: © 2011 Terefe-Ayana, Yasmin, Le, Kaufmann, Biber, Kühr, Linde and Debener. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Thomas Debener, Institute for Plant Genetics, Leibniz University Hannover, Herrenhäuser Str. 2, D-30419 Hannover, Germany. e-mail:ZGViZW5lckBnZW5ldGlrLnVuaS1oYW5ub3Zlci5kZQ==