 Takayuki Tohge1*
Takayuki Tohge1* Tabea Mettler1
Tabea Mettler1 Stéphanie Arrivault1
Stéphanie Arrivault1 Adam James Carroll2 Mark Stitt1
Adam James Carroll2 Mark Stitt1 Alisdair R. Fernie1
Alisdair R. Fernie1
- 1 Max-Planck-Institute for Molecular Plant Physiology, Potsdam-Golm, Germany
- 2 Australian Research Council Centre of Excellence in Plant Energy Biology, The Australian National University, Canberra, ACT, Australia
Although plant metabolomics is largely carried out on Arabidopsis it is essentially genome-independent, and thus potentially applicable to a wide range of species. However, transfer between species, or even between different tissues of the same species, is not facile. This is because the reliability of protocols for harvesting, handling and analysis depends on the biological features and chemical composition of the plant tissue. In parallel with the diversification of model species it is important to establish good handling and analytic practice, in order to augment computational comparisons between tissues and species. Liquid chromatography–mass spectrometry (LC–MS)-based metabolomics is one of the powerful approaches for metabolite profiling. By using a combination of different extraction methods, separation columns, and ion detection, a very wide range of metabolites can be analyzed. However, its application requires careful attention to exclude potential pitfalls, including artifactual changes in metabolite levels during sample preparation under variations of light or temperature and analytic errors due to ion suppression. Here we provide case studies with two different LC–MS-based metabolomics platforms and four species (Arabidopsis thaliana, Chlamydomonas reinhardtii, Solanum lycopersicum, and Oryza sativa) that illustrate how such dangers can be detected and circumvented.
Introduction
Plant metabolomics is a relatively new analytic strategy which provides complementary information to transcriptomic and proteomic studies as well as important information in its own right concerning the regulation of metabolic networks (Hall et al., 2002; Bino et al., 2004). Initial applications of metabolic profiling were largely focused on the model plant Arabidopsis thaliana (von Roepenack-Lahaye et al., 2004; Tohge et al., 2005; Gibon et al., 2006; Trenkamp et al., 2009; Araujo et al., 2010; Kerwin et al., 2011), however, several studies have been carried out on the green algae Chlamydomonas reinhardtii (Giroud et al., 1988; Bolling and Fiehn, 2005; May et al., 2008; Boyle and Morgan, 2009; Renberg et al., 2010) with other successful applications being reported for Catharanthus roseus (Rischer et al., 2006), Fragaria x ananassa (Aharoni et al., 2000, 2002; Hanhineva et al., 2008), Hordeum vulgare (Widodo Patterson et al., 2009), Medicago truncatula (Achnine et al., 2005), Nicotiana tabacum (Goossens et al., 2003), Oryza sativa (Albinsky et al., 2010), Perilla frutescens (Yamazaki et al., 2008), Pisum sativum (Jom et al., 2010), and Solanum lycopersicum (Schauer et al., 2005, 2006; Moco et al., 2006; Fraser et al., 2007) as well as the unicellular prokaryotes Synechocystis sp. (Krall et al., 2009) and the diatom Phaeodactylum tricornutum (Allen et al., 2008).
Initially, the use of metabolic profiling in plants, as indeed in all species, was restricted to diagnostic approaches in which the obtained profiles were used as markers for a range of biological conditions (Sauter et al., 1988; Meyer et al., 2007; Semel et al., 2007; Carmo-Silva et al., 2009; Scherling et al., 2009; Widodo Patterson et al., 2009). Although such studies remain highly important, particularly in medical research (Nicholson and Wilson, 2003; Griffin and Nicholls, 2006), more sophisticated uses of metabolic profiling have recently been developed, including identifying regulated enzymes and exploring the regulatory structure of pathways (Tiessen et al., 2002; Arrivault et al., 2009), searching for unexpected effects of genetic manipulation (Catchpole et al., 2005), screening wild species for beneficial chemical composition (Zhu and Wang, 2000; El-Lithy et al., 2005), gaining a more comprehensive view of metabolic regulation and as part of integrative analyses for the systemic response of environmental genetic perturbations (Hirai et al., 2004, 2005; Fukushima et al., 2009; Sulpice et al., 2009; Trenkamp et al., 2009). In addition to these uses, metabolomics is proving to be a powerful tool for gene functional annotation in plants. There are now several examples of Arabidopsis genes that have been identified with the help of metabolomic approaches including MYB transcription factors (Hirai et al., 2007; Stracke et al., 2007), O-methyltransferase (Tohge et al., 2007), glycosyltransferases (Tohge et al., 2005; Yonekura-Sakakibara et al., 2007, 2008), acyltransferases (Luo et al., 2007), UDP-rhamnose synthase (Yonekura-Sakakibara et al., 2008), and pyrophosphorylase (Okazaki et al., 2009) with the approach being equally effective in other species (Aharoni et al., 2000; Goossens et al., 2003; Achnine et al., 2005; Yamazaki et al., 2008).
One advantage that metabolomics has over transcriptomics [with the exception of next-generation sequencing tools, see Detlef Weigels recent review (Schneeberger and Weigel, 2011)] and proteomics is that it is essentially genome-independent (Stitt and Fernie, 2003) and as such can be applied to a species whose genome has not been sequenced as easily as those whose has. This “democratization” of biology allows in depth functional analyses of many species for which a complete and fully annotated genome is not yet available (Schneeberger and Weigel, 2011). Despite this fact caution needs to be taken when adopting a method set up for one tissue of one species to analyze another tissue of that species or even another species. This is especially so for metabolite profiling. The plant kingdom contains an incredibly rich chemical diversity (St-Pierre and De Luca, 2000). It is obvious that this chemical diversity poses a large challenge and stimulates research in developing new and increasingly powerful approaches to separate, detect, and identify metabolites. However, it also raises important challenges for experimental design, sample handling, and validation of analytic procedures. This is because tissue composition affects the reliability with which a particular metabolite can be reliably extracted and analyzed. This problem is particularly acute when using liquid chromatography–mass spectrometry (LC–MS) due to the so called ion suppression effects wherein the composition of the extract affects the efficiency of ionization of some of its constituent analytes (Fernie et al., 2004). That said, a number of relatively simple control tests, in combination with the growing number of chemoinformatic tools for metabolomics (Tohge and Fernie, 2009; Bais et al., 2010; Carroll et al., 2010; Cottret et al., 2010; Xia and Wishart, 2010), should at least ameliorate this phenomenon and hence facilitate high-quality translational metabolomics.
Driven by the increasing diversification of plant research away from the principle model species A. thaliana we present here case studies in which methods developed for this species are assessed for use in determining metabolite levels either in the unicellular algae C. reinhardtii or in the crop species rice and tomato. For the former we assessed the analysis of primary metabolism using an LC–MS/MS method developed to deliver validated measurements of the levels of Calvin–Benson cycle intermediates, organic acids, nucleotide-sugars, and nucleotides in Arabidopsis rosettes (Arrivault et al., 2009). Given that information documenting the transfer of gas chromatography–mass spectrometry (GC–MS)-based methods of analysis of primary metabolites has already been extensively supplied for potato and tomato (Roessner et al., 2001; Roessner-Tunali et al., 2003), we chose crop species to focus our studies on secondary metabolism. The two LC–MS-based methods applied in this study complement standard and well-established GC–MS methods by greatly increasing the range of metabolites that can be analyzed.
Here some examples of how can be performed using two different LC–MS-based metabolomics platforms, on one algal and two crop species and A. thaliana are shown. The combined results illustrate important experimental controls which should be implemented alongside computation algorithms in order to successfully adapt protocols that have been established for another biological system. This also applies to other LC–MS-based methods (Okazaki et al., 2009; Kanno et al., 2010). We additionally discuss how such studies could be used in conjuncture with novel tools for combined sequence comparison and co-expression analysis (Mutwil et al., 2011, and Ruprecht et al., this issue) in order to improve gene functional predictions from Arabidopsis to crop species.
Materials and Methods
Cell Culture and Extraction Procedures
Chlamydomonas reinhardtii strain CC-1690 wild type mt+ was acquired from the Chlamydomonas Genetics Center (Duke University, Durham, NC, USA). Single colonies were used to inoculate the growth media containing 5 mM Hepes, 1 mM K-phosphates, Beijerinck salts (final concentrations of 7.5 mM NH4Cl, 0.34 mM CaCl2, 0.41 mM MgSO4) and trace salt solution (final concentrations of 184 μM H3BO3, 77 μM ZnSO4, 26 μM MnCl2, 18 μM FeSO4, 7 μM CoCl2, 6 μM CuSO4, 1 μM (NH4)6Mo7O24; Harris, 1989; May et al., 2008; Kempa et al., 2009) at 25°C under constant illumination with 400 μmol photon m−2 s−1 and continuous shaking. The amount of NH4Cl was reduced to 4 mM for the experiment shown in Figure 1 to reduce the impact of ion suppression. Before harvesting, cells were grown to a density of 3 × 106 cells ml−1 and dark-adapted for a minimum of 20 min before transferring 1 ml of cells to a cuvette and exposed them to 660 μmol photon m−2 s−1 under continuous stirring. Before illumination and at different time points after illumination the suspension was quenched by vigorously adding 2 ml of −70°C methanol (70%). The entire mix was then lyophilized to dryness at −80°C and extracted at 4°C by a chloroform:methanol:water (1:2:5 [v/v]) mixture. Water fractions of three subsequent washes were collected, concentrated by lyophilization at −80°C and filtered before metabolite measurement (80 μl of extracted culture in 100 μl sample measured) by ion pair (reverse-phase) chromatography triple quadrupole MS (IPC–MS/MS) detection.
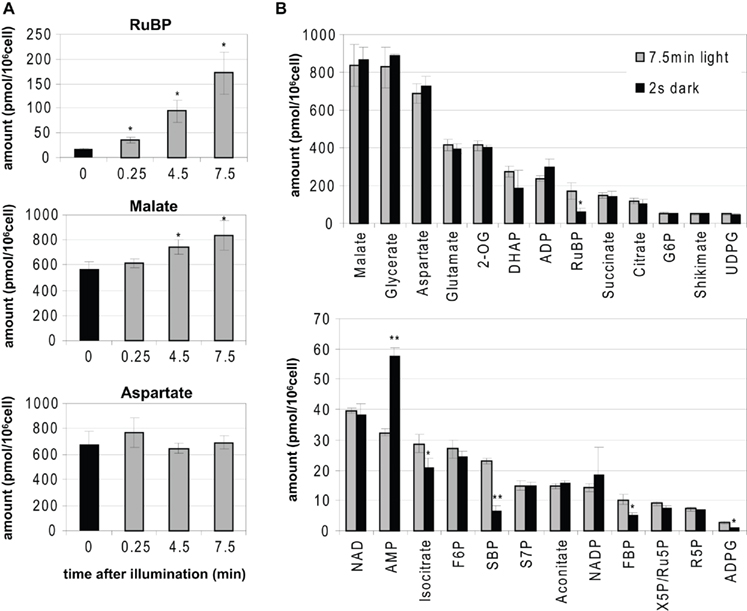
Figure 1. Example of rapid metabolic response by light and darkness. Metabolites in Chlamydomonas reinhardtii CC-1690 were measured after quenching in an excess of cold (−70°C) methanol, lyophilization, and extraction in chloroform-methanol using IPC–MS/MS. (A) Ribulose-1,5-bisphosphate (RuBP), malate, and aspartate levels in Chlamydomonas CC-1690 cells are shown after dark-adaption for 20 min (black bars) and exposure to 660 μmol photon m−2 s−1 for 0.25, 4.5, and 7.5 min (gray bars). Y-axis indicates amount (pmol/106 cell). (B) Chlamydomonas cells were harvested in cuvettes after 20 min dark-adaption and exposure to 660 μmol photon m−2 s−1 for 7.5 min without (gray bars) or with an additional 2 s of darkness (black bars).. Levels of metabolites are presented as absolute values (n = 3, ±SD, two asterisks: Student’s t-test p < 0.01, one asterisk: Student’s t-test p < 0.05). 2-OG, 2-oxoglutarate; DHAP, dihydroxyacetone-phosphate; ADP, adenosine diphosphate; G6P, glucose-6-phosphate; UDPG, UDP-glucose; NAD, nicotinamide adenine dinucleotide; AMP, adenosine monophosphate; F6P, fructose-6-phosphate; SBP, sedoheptulose-1,7-bisphosphate; S7P, sedoheptulose-7-phosphate; NADP, nicotinamide adenine bisnucleotide phosphate; FBP, fructose-1,6-phosphate; X5P, xylulose-5-phosphate; Ru5P, ribulose-5-phosphate; R5P, ribose-5-phosphate; ADPG, ADP-glucose.
Plant Materials and Extraction Procedures
Arabidopsis thaliana ecotype Col-0 and S. lycopersicum (M82) were grown in soil in a controlled environmental chamber (16 h light/8 h dark photoperiod; 21°C at 145 μmol photon m−2 s−1 and 25°C at 500 μmol photon m−2 s−1, respectively). A. thaliana used for recovery test was grown in general 1/2 MS agar plate in controlled plant growth chamber (16 h light/8 h dark photoperiod; 21°C). Rice (O. sativa, Nipponbare) seeds were pre-germinated in tap water at 28°C for 10 days. Plantlets were transferred to a controlled growth chamber with 12 h day length at 700 μmol photon m−2 s−1. Plant material was frozen in liquid nitrogen, ground into powder and stored at −80°C until use.
For assessment of ion suppression and recovery tests, extraction for secondary metabolite profiling was conducted as described in Tohge and Fernie (2010). Extraction buffer was added to reach 0.2 mg FW μl−1. To evaluate secondary metabolite degradation due to enzymatic activities, four different extraction procedures were carried out using aliquots of frozen powders from a pool of plant materials from at least three plants: (a) extraction was conducted as described in Tohge and Fernie, 2010; extraction buffer was added to frozen material kept at liquid nitrogen temperature); (b) extracts obtained by method (a) were incubated at 37°C for 1 h, (c) extraction buffer was immediately added to frozen material on ice; (d) plant material was incubated at 37°C for 1 h prior addition of extraction buffer. To assess ion suppression in different plant species, an internal standard (IS) mixture containing three standard compounds (isovitexin, CAS: 29702-25-8; saponarin, CAS: 20310-89-3; sinigrin, CAS: 3952-98-5) was prepared at four different concentrations (20, 10, 5, 1 μg ml−1). Identical volume of standard mixture and plant extracts were added, resulting in a final sample containing 0.1 mg FW μl−1 of plant extracts and 10, 5, 2.5, or 0.5 μg ml−1 of standard compounds. Recovery test was carried out with Arabidopsis extracts (0.2 mg FW μl−1) of leaves and roots grown on agar plates for 3 weeks, and flowers harvested from the plants grown on soil for 4 weeks. Extracts from leaves “A” and roots “B” (or flowers) were mixed at different ratios [(A:B), 90:10, 80:20, 50:50, 20:80, 10:90], respectively. The percentage recovery was estimated for evaluation using theoretical concentration of extracts mixture, [(level in leaves × A%) + (level in roots (or flowers) × B%)]/100.
Ion Pair (Reverse-Phase) Chromatography Triple Quadrupole MS (IPC–MS/MS)
Primary metabolite analysis by IPC–MS/MS was carried out on a Dionex HPLC system (Sunnyvale, CA, USA) coupled to a Finnigan TSQ Quantum Discovery MS-Q3 (Thermo Fisher Scientific, Waltham, USA) equipped with an electrospray ionization (ESI) interface. It was operated as described in Arrivault et al. (2009). Chromatographic separation was obtained at 35°C by a multi-step gradient with online-degassed eluent A (10 mM tributylamine aqueous solution, adjusted to pH 4.95 with 15 mM acetic acid) and eluent B (methanol) applied to a Gemini (C18) 150 mm × 2.00 mm inner diameter 5 μm, 110 Å particle column (Phenomenex, Aschaffenburg, Germany). The MS-Q3 device was operated in the negative ion scanning mode with selected reaction monitoring (SRM). The MS-parameters for each compound are documented in the Supplementary Data of Arrivault et al. (2009). Calibration curves using authentic standards were used to calculate absolute amounts of metabolites in algal samples. Data were processed using LC-quan 2.5.6 SP1 software.
Reverse-Phase HPLC–MS Analysis
Secondary metabolite analysis by LC–MS was performed on HPLC system Surveyor (Thermo Finnigan, USA) coupled to Finnigan LTQ-XP system (Thermo Finnigan, USA) as described by Tohge and Fernie (2010). All data were processed using Xcalibur 2.1 software (Thermo Fisher Scientific, Waltham, USA). Metabolite identification and annotation were performed using standard compounds (Nakabayashi et al., 2009) and reference metabolomics databases (Moco et al., 2006; Shinbo et al., 2006; Iijima et al., 2008; Tohge and Fernie, 2009).
Results and Discussion
Harvesting – Obtaining Representative Material and Avoiding Handling-Induced Changes
Expression of genes and activity of enzymes associated with photosynthesis, respiration, and energy metabolism are rapidly affected by changes in environmental conditions. Transcriptional and metabolic regulation by the circadian clock has been defined (Harmer et al., 2000; Gibon et al., 2006; Fukushima et al., 2009; Kerwin et al., 2011). Many metabolites showed marked diurnal changes. Problems related to variation in clock and diurnal rhythms can be circumvented by harvesting plants at the same time in the 24-h cycle. They can also be affected by shorter term fluctuations. The exact timing of harvesting and avoidance of perturbation of metabolism during harvesting, by for instance shading of leaves or changes in the oxygen tension are therefore critical (see Geigenberger et al., 2000). Additionally, rapid and complete quenching of metabolic activity is crucial to ensure faithful measurement of the intracellular metabolite content (discussed in details below).
To highlight the rapid metabolic response following light treatment, metabolite profiling was performed on the model organism C. reinhardtii (Figure 1). Quenching was performed by rapid mixing of the algal suspension in the light with an excess of very cold (−70°C) methanol to instantaneously freeze the cells. Metabolite profiling by IPC–MS/MS analysis of short term light treatment was performed in dark-adapted material and after 0.25, 4.5, and 7.5 min illumination. Ribulose-1,5-bisphosphate (RuBP), which is a major metabolite in the Calvin–Benson cycle, was already significantly elevated by a light treatment of 15 s (Figure 1A). The levels of malate, the late step in TCA cycle, also displayed significant increases upon illumination, whilst the level of the amino acid aspartate was not altered.
Figure 1B illustrates why avoiding perturbations of the conditions by the mean of rapid harvesting is critical for metabolite profiling. Darkening significantly and almost instantaneously influences operation of the photosystems and the delivery of ATP and NADPH. The levels of RuBP, sedoheptulose-1,7-bisphosphate (SBP), fructose-1,6-bisphosphate (FBP), ADP-glucose (ADPG), and isocitrate were significantly decreased, while adenosine 5′-diphosphate (ADP) and adenosine monophosphate (AMP) increased within 2 s of darkening. Thus, harvesting protocols that lead to even very brief decrease or increase in the light intensity preceding quenching or during the quenching process will lead to erroneous estimates for the levels of metabolites. An identical problem arises in higher plants. This is due to the simple fact that the fluxes in the Calvin–Benson cycle are so high that many of the metabolites in the cycle as well as ATP and NADPH have short turnover times of 1 s or less (Arrivault et al., 2009).
This problem is of course especially critical for processes like photosynthesis, where fluxes are very fast and metabolite pools are small and turn over very quickly. However, it illustrates the more general points that (i) all available information about the turnover times of the metabolites-of-interest should be collected, evaluated, and used to design an appropriate harvesting and quenching protocol and (ii) that this protocol should be validated by checking if slowing down or speeding up the harvesting process modifies the levels of metabolites that are found in the harvested material.
Quenching of Enzymatic Activities and Differences between Plant Species and Chemical Properties
Quenching of metabolic activity is not only essential to stop metabolic turnover in the running pathways, but also to inhibit other enzymatic activities that can destroy the metabolite after tissue disruption. An old but still instructive example of this is the precautions needed to determine pyrophosphate levels in plants (Weiner et al., 1987). Pyrophosphatase activity in leaves is so high that it can hydrolyse all the pyrophosphate in a leaf extract in <0.05 s. In an intact tissue, the vast majority of the pyrophosphatase activity is in the plastids whilst the pyrophosphate is in the cytosol. As soon as the tissue is disrupted the pyrophosphatase comes into contact with and destroys the pyrophosphate. To measure pyrophosphate it is therefore essential that enzymatic activity is completely stopped by rapid quenching and remains totally inactive during all subsequent stages in sample handlings as a fraction of a percent would be enough to destroy all the pyrophosphate within few seconds. Such problems can be routinely identified by recovery experiments in which representative amounts of authentic standards are added to the plant material before extraction, and it is checked that the added standard can be quantitatively detected in the final extract (Fernie et al., 2011).
Whilst secondary metabolites do not display such rapid responses to changes in the environment as those observed for primary metabolism (see an example in Kusano et al., 2011), they, like primary metabolites, are highly susceptible to degradation by enzymes that come in contact with them after tissue disruption. For example glucosinolates are converted into isothiocyanates by myrosinase in Arabidopsis (Tierens et al., 2001; Barth and Jander, 2006). Given that degradative enzymes typically remain potent subsequent to freezing in liquid nitrogen when the extract is thawed, particular care must be taken during the extraction procedure. To illustrate this point, three extraction procedures were conducted in addition of our original extraction procedure (extraction a), using frozen powder of plant materials. To test if breakdown enzymes were definitively inactivated during this extraction procedure, extracts were incubated at 37°C for 1 h (extraction b). A pre-incubation of sample material at 37°C for 1 h prior extraction was conducted to test the extent of metabolite degradation upon thawing (extraction d). Secondary metabolite extraction is routinely performed at liquid nitrogen temperature, so to test if another temperature would affect metabolic composition, addition of buffer on frozen sample was performed at ice temperature (extraction c). All extractions were performed using pre-cooled extraction buffer (10°C). Metabolite breakdown was assessed in A. thaliana leaves, O. sativa leaves, and S. lycopersicum fruits by mean of LC–MS. Total ion chromatograms and relative peak areas of selected metabolites are presented in Figures 2 and 3, respectively.
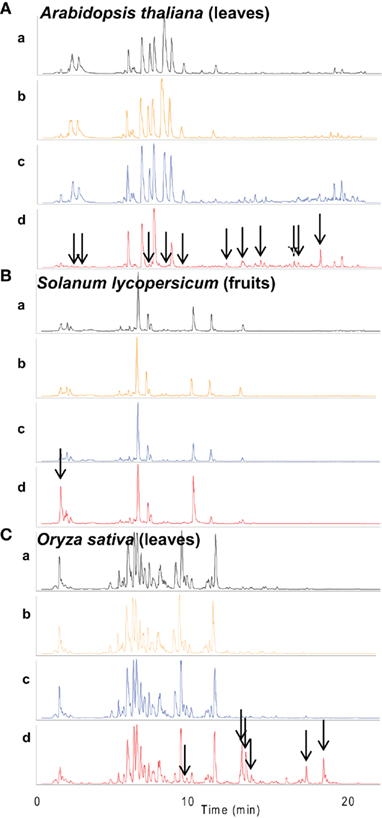
Figure 2. Effect of different extraction methods on secondary metabolite breakdown in Arabidopsis leaves, tomato fruits, and rice leaves. Total ion chromatograms (TIC) monitored by negative ion detection mode of extracts of (A) Arabidopsis leaves, (B) tomato fruits, and (C) rice leaves, are shown. (a–d) indicate different extraction methods. (a) extraction method as described in Tohge and Fernie (2010), (b) extracts obtained by (a) method were incubated at 37°C for 1 h, (c) extraction buffer was immediately added to frozen material on ice, (d) frozen sample was incubated at 37°C for 1 h before extraction. All extractions were performed using pre-cooled extraction buffer (10°C). Arrows show peaks which were newly or not detected in treated samples.
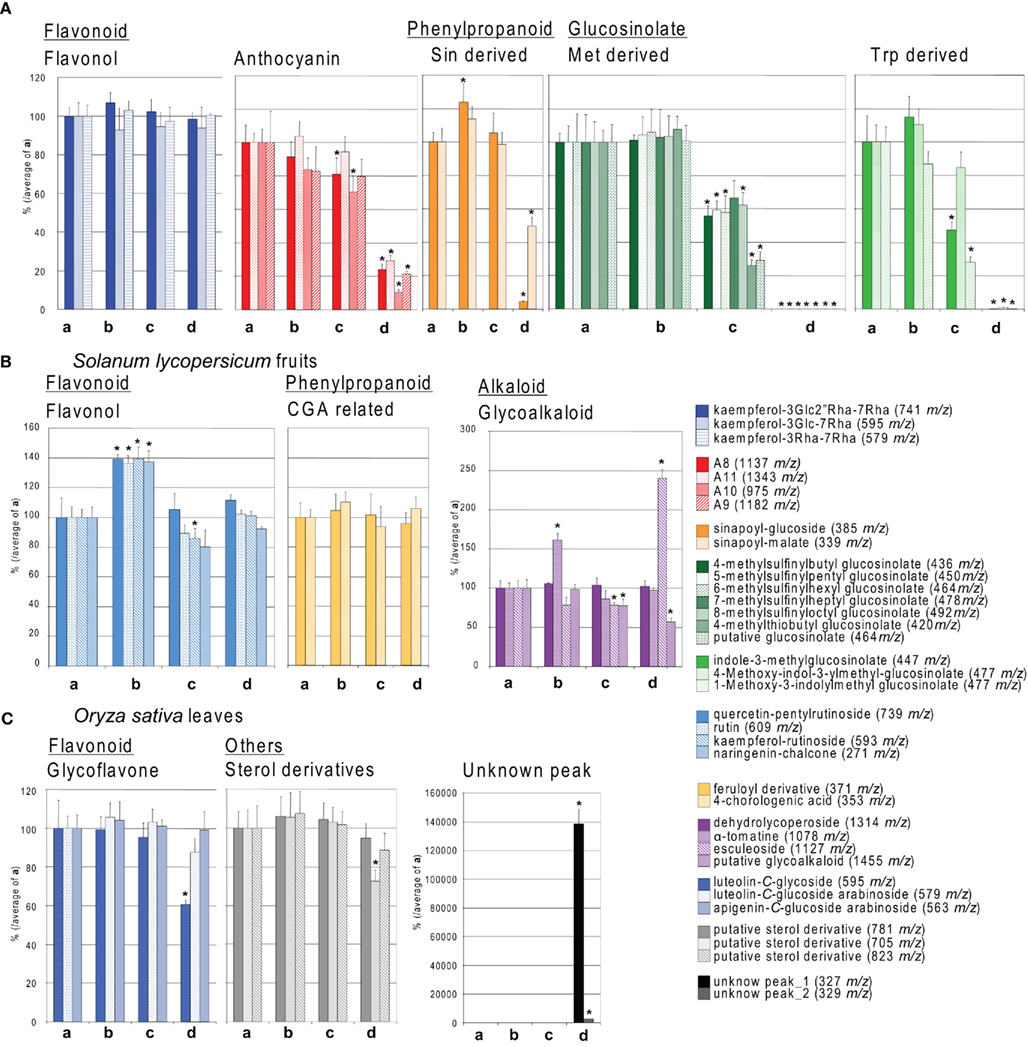
Figure 3. Comparison of secondary metabolite levels between different extraction methods. Relative peak area of the result described in Figure 2 of (A) Arabidopsis leaves, (B) tomato fruits, and (C) rice leaves, are shown. Y-axis indicates percentage calculated by average of peak area of treatment (a). (a–d) indicate different extraction methods as described in Figure 2 and Section “Materials and Methods.” Blue related colors indicate flavonoid related compounds (flavonol and glycoflavone). Red color indicates red-pigmented flavonoid related compounds (anthocyanin). Yellow, phenylpropanoid related compounds (sinapoyl-derivatives and chlorogenate-related compound); green, glucosinolate related compounds in Arabidopsis (methionine-derived aliphatic glucosinolate and tryptophan-related aromatic glucosinolate); purple, indicates glycoalkaloid; gray, putative sterol derivative; black, unknown compounds. The m/z value detected in negative ion detection is indicated between parentheses. Average of three experimental replicates ±SD. Significant differences by t-test (p < 0.05) are marked by asterisks.
For almost all plant species, an incubation of the material after addition of extraction buffer at 37°C for 1 h had no observable consequences on the total ion chromatograms and on the relative peak areas of selected metabolites [Figures 2 and 3, respectively. Comparison between (a) and (b)]. This result indicates that the major secondary metabolites are not broken down at 37°C, provided the tissue has been taken up in the extraction buffer. However, metabolite profiling of samples extracted after 1 h (pre-incubation of the disrupted tissue at 37°C) revealed that samples were significantly changed in some compound species (pointed by arrows in Figure 2). A more detailed analysis revealed that within a plant species and between various plant species the metabolite classes were differently affected [Figure 3, comparison between (a) and (d)]. In general non-pigmented flavonoids such as flavonol glycoside in Arabidopsis leaves and S. lycopersicum fruits, and glycoflavone in O. sativa leaves were stable. By contrast, red-pigmented flavonoids namely anthocyanin derivatives in Arabidopsis were significantly decreased following the 37°C pre-incubation. Furthermore, phenylpropanoids in Arabidopsis such as sinapoyl-derivatives were broken down by pre-incubation. The Brassica species specific secondary metabolites glucosinolates are well-known compounds which can be broken down by myrosinase (Tierens et al., 2001; Barth and Jander, 2006). The glucosinolate levels in extracts with 37°C incubation before extraction were not detected [Figure 3A, comparison between (a) and (d)]. Despite the breakdown of phenylpropanoids by enzyme activity in Arabidopsis, phenylpropanoids in tomato fruit such as chlorogenic acid related compounds were generally unaffected by enzymes. With the exception of esculeoside related compounds, levels of glycoalkaloids which are the major alkaloid in tomato fruits were not significantly changed (Figure 3B). These data show that, whatever the plant species, a pre-incubation of the plant material at 37°C prior to extraction leads generally to various levels of secondary metabolite breakdown due to the presence of active enzymes when plant material is thawed out. Addition of extraction buffer to frozen material not at liquid nitrogen temperature (extraction c) led to a significant decrease in the levels of glucosinolates and anthocyanin derivatives (Figure 3A). This shows that the temperature during addition of the extraction buffer is also an important factor.
These results taken together illustrate that the tissue extraction should be carried out in the proper way with attention being taken to empirically optimize the extraction method for each and every new tissue measured. The effect of 20 min sonication was also evaluated in the same manner, but no differences were observed (data not shown). That said it is important to note that sonication should only be performed if this can be managed without an increase in temperature.
Ion Suppression Effects Caused by Growth Media
Although LC–MS analysis is a highly sensitive technique, ion suppression is a general problem of LC–MS analytical platforms due to altered ESI of a target ion by a contamination (Ikonomou et al., 1990; Kebarle and Tang, 1993; Buhrman et al., 1996; Matuszewski et al., 1998, 2003; King et al., 2000; Fernie et al., 2004). It is actually not a single event but a range of response–reducing phenomena which should be avoided as much as possible. While there is, however, no universal solution to this problem, understanding difference between samples and assessing the effects of ion suppression affords greater confidence in the accuracy of the results.
An example of ion suppression caused by growth conditions of Chlamydomonas is shown in Figure 4; Table 1. This example illustrates how the composition of the growth media can dramatically affect the reliability of metabolite analyses in Chlamydomonas. Following quenching of the algal suspension by mixing with an excess of cold methanol (see above), we took the entire suspension for analysis. This was necessary because some metabolites leak out of the cells into the methanol–water mix and are therefore lost when the quenched cells are harvested by centrifugation (data not shown, see also Krall et al., 2009). This means that metabolites from the cells must be analyzed in a matrix that contains methanol and all the components of the suspension medium. Because the Chlamydomonas cells are quite diluted, components of the growth medium are present in rather large amounts compared to metabolites in the cells. Unfortunately, some components of the growth medium lead to ion suppression.
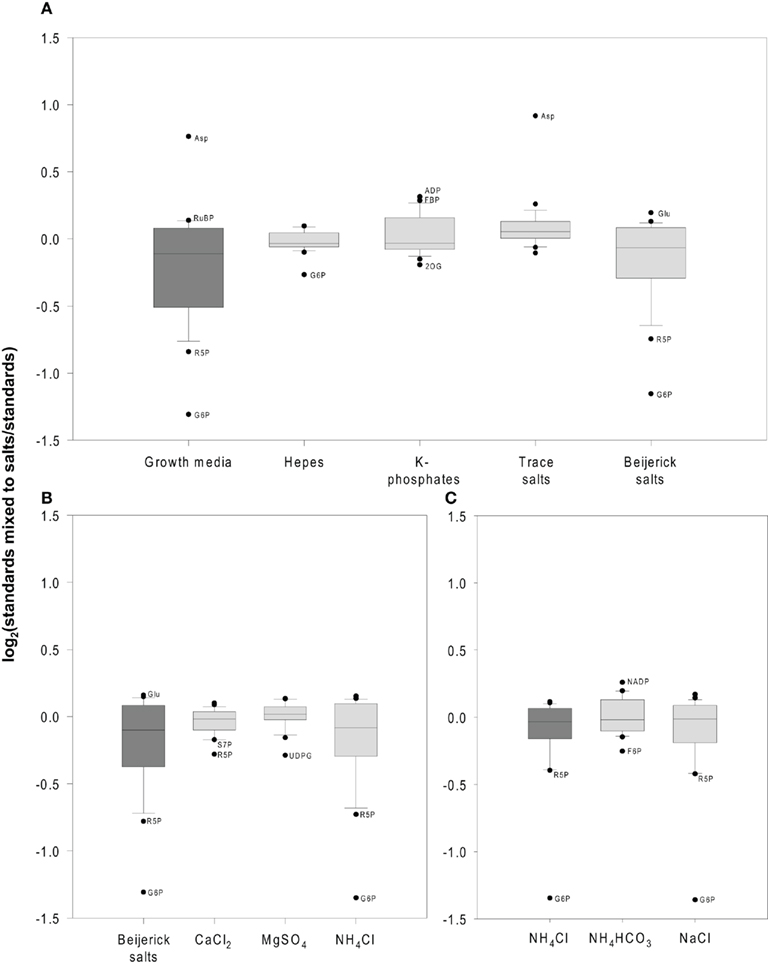
Figure 4. Example of ion suppression caused by growth media. (A–C) A standard mix containing all measured metabolites was mixed with individual components of the growth media to identify which component(s) of the growth media most severely influence ionization during electrospray ionization (ESI). (A) The standard mix was mixed with the whole growth medium (dark gray) and independently with the medium components Hepes, K-phosphates, trace salts, and Beijerinck salts (light gray). (B) In a second experiment, the individual components of the Beijerinck salts were tested for ion suppression and enhancement. Standards were mixed to all Beijerinck salts (dark gray) and independently to its components CaCl2, MgSO4, and NH4Cl (light gray). (C) A third experiment investigated if the anion or the cation was responsible for ion suppression caused by ammonium chloride. Standards were mixed to NH4Cl (dark gray), NH4HCO3, or NaCl (light gray). The data is shown as box plots of the average values (calculated from three technical replicates) for 24 metabolites. Significant outliers (p < 0.05) are identified in the figure panels. For a complete list of percentage of ion suppression and enhancement for all metabolites see Table A1 in Appendix.
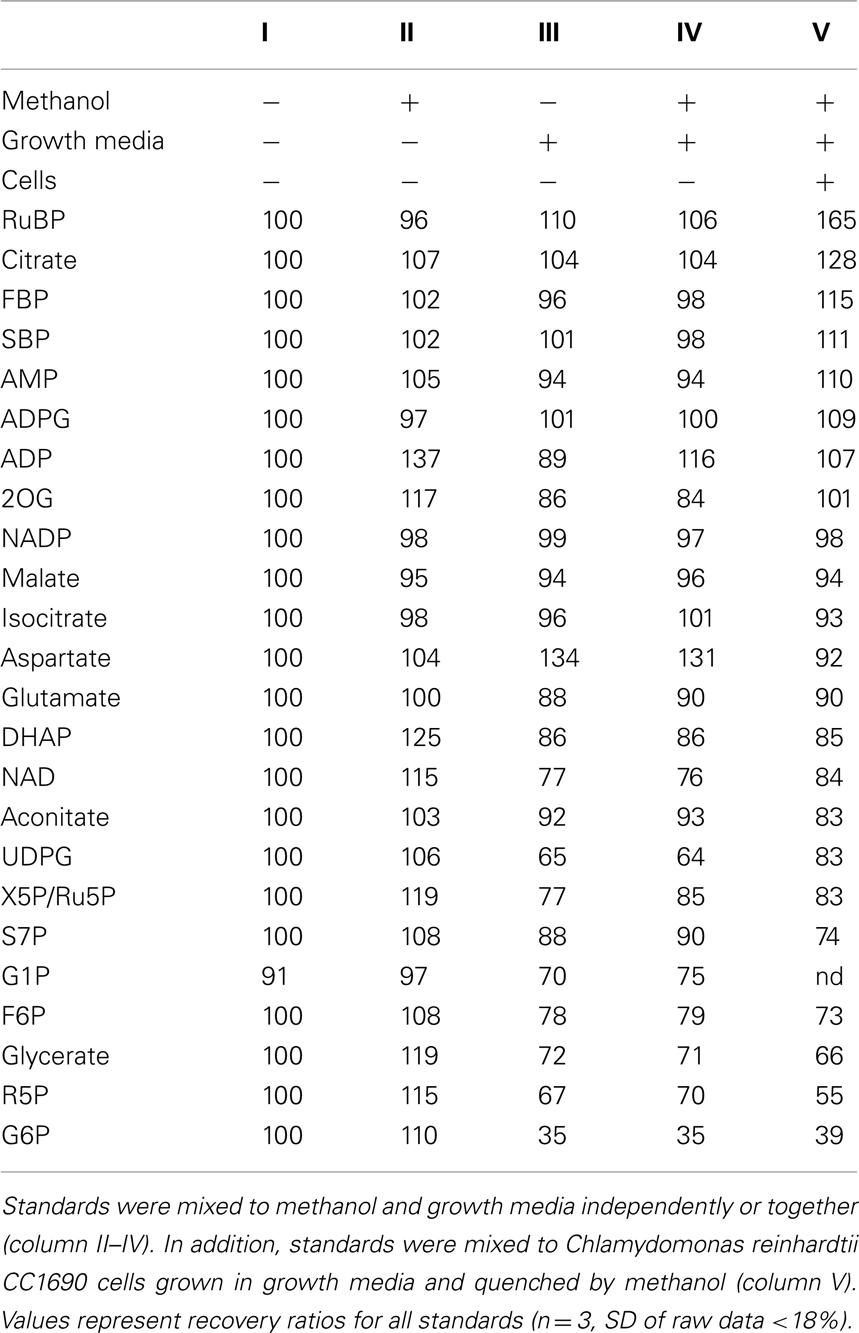
Table 1. Ion suppression mainly caused by growth media.
We were alerted to ion suppression by three routine checks. First, comparison of the spectrum of metabolites with those expected from earlier studies of metabolites during photosynthesis in Arabidopsis showed low levels of several metabolites. Second, we checked whether the signal for each metabolite shows a strictly linear relationship to the amount of extract applied. In the case of ion suppression, the estimated levels of many metabolites decreased when more samples were applied. Third, we checked the recovery of authentic standards added to the extract and found it was very low.
Attempts to analyze high concentrations of sample resulted in an almost complete suppression of all metabolite signals, including those of spiked standards (Figure A1 in Appendix). A five-time dilution allowed an average of 87–93% recovery of the spiked standards (Table 1; Figure 4). However, there was still a residual ion suppression, and this varied from metabolite to metabolite (Table 1). This obviously still prevents reliable and comparable analysis of metabolite levels. We therefore carried out a further series of experiments to identify the major sources of ion suppression, in order to modify the growth medium and circumvent this problem. To show that ion suppression was caused by components of the growth medium and not the biological sample itself or the methanol, we first mixed known amounts of standards with either methanol and/or the growth media compared to the same known amounts of standards mixed with a sample containing Chlamydomonas cells (Table 1). For more than half of the metabolites, <15% of the signal was suppressed by the media, but for many other metabolites including dihydroxyacetone-phosphate (DHAP), fructose-6-phosphate (F6P), glycerate, NAD+, ribose-5-phosphate (R5P), sedoheptulose-7-phosphate (S7P), UDP-glucose (UDPG), and xylulose-5-phosphate/ribulose-5-phosphate (X5P/Ru5P) the signals were reduced by 15–50%. For G6P ion suppression caused a decrease of intensities by >50%. Therefore, for all these metabolites the absolute values have to be treated with extreme caution. Further, small changes in the extent of ion suppression can lead to changes in the relative signals for the various metabolites. It almost goes without saying that mixing ISs of these metabolites to each sample would allow a much more precise determination of their absolute amounts.
To minimize such errors, we systematically investigated which of the salts in the medium could contribute to the loss of signal due to ion suppression or ion enhancement during ionization by ESI. The growth media used in this study consisted of Hepes, K-phosphates, Beijerinck salts, and trace salts (for details see Materials and Methods). A sequence of experiments was performed to unravel the effects of the individual salts from this growth media (Figure 4, for details see Table A1 in Appendix). Hepes, K-phosphate, and the trace salt solution had only minor ion suppression effects (Figure 4A, for details see Table A1 in Appendix). Hepes caused significant ion suppression of G6P but less than the Beijerinck salts. K-phosphates caused weak but significant ion induction of ADP, and FBP, RuBP, SBP, aconitate, and isocitrate. The trace salt solution caused overestimation of aspartate due to ion enhancement. However, most of the ion suppression observed due to the growth media in the sample could be attributed to the Beijerinck salts present in the media (Figure 4A). In a second experiment, ammonium chloride, one component of the Beijerinck salts, was shown to be responsible for the major part of the residual ion suppression whereas MgSO4 and CaCl2 had minor effects (Figure 4B). In a third experiment, the chloride anion was found to be the main reason why ammonium chloride causes ion suppression (Figure 4C). From the 24 metabolites routinely measured with this method, the signal of 10 was significantly suppressed in the presence of the Beijerinck salt (Figure A1 in Appendix). With the exception of UDPG which was found to be suppressed by MgSO4, the chloride anion in the growth media was found to be responsible for ion suppression. Thus, simply replacing the chloride anion with bicarbonate greatly decreased ion suppression (Figure 4C).
For subsequent measurements a growth medium with lower ammonium chloride concentration was used (Figure 1). Alternatively, a medium in which ammonium chloride is replaced with ammonium bicarbonate could be used or, as mentioned earlier ISs for each metabolite could be added to the sample to assure accurate metabolite measurements. This example shows that different degrees of ion suppression, both with respect of individual metabolites and with respect to different samples, can be generated by differences in growth conditions and culture components. These results imply that erroneous results will also be obtained if such changes occur as a result of growing algae in different conditions, or are generated in time as a result of the algae using nutrients.
The specific issue with growth medium components does not arise with higher plants. Nevertheless, this example serves as a warning that differential ion accumulation in plant tissues might affect ion suppression. In attempt to circumvent this problem the total ion chromatogram should be carefully checked in areas which appear to be strongly affected. If strong ion suppression is observed, both dilution and recovery tests in which standard compounds are added to the extracts should be performed (see Fernie and Keurentjes, 2011). More generally, such problems can be identified by routine checks that the signal is linear with the amount of applied extract and (where available) that authentic samples can be quantitatively recovered after addition to the extract.
Ion Suppression Effects Caused by Different Tissue Types
Metabolite composition varies between plant species and also between different tissues of the same plant. It is therefore expected to observe different levels of ion suppression within these samples. To evaluate this, an IS mixture (sinigrin, isovitexin, and saponarin) was added to the same volume (ratio 1/1) of extracts from Arabidopsis leaves, tomato fruits, and rice leaves, respectively. This was performed with four different known concentrations of the IS mixture with three experimental replicates of the step of mixing solutions. As control, the 50% diluted original standard mixture was also analyzed without being mixed with plant extract. Peak areas for each IS were determined and are presented in Figure 5. For all IS compounds, the strongest ion suppression was observed when they were added to rice leaves, followed by Arabidopsis leaves, and the lowest was seen for tomato fruit extracts. As expected, the ion suppression in a mixture of Arabidopsis leaf and tomato fruit extracts (ratio 1/1) was intermediate to what was observed in the corresponding independent extracts.
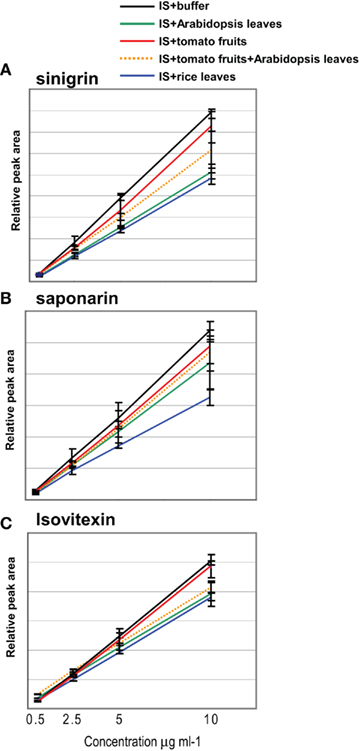
Figure 5. Example of ion suppression caused by plant extracts. Standard solutions (20, 10, 5, 1 μg ml−1) of three standard compounds, (A) sinigrin, (B) saponarin, and (C) isovitexin, were mixed to the same volume of plant extracts (0.2 mg FW μl−1) of Arabidopsis leaves, tomato fruits, rice leaves and mixture of Arabidopsis leaves and tomato fruits (ratio 1/1). Average and ±SD were calculated from three experimental replicates.
Plant material used for metabolic determination is often a mixture of tissues. For example, plant seedling is a mixture of hypocotyl and root, or fruit samples are a mixture of pericarp, seed, and peel. Ion suppression caused by differences between tissue types, in comparative analysis between mutants, transgenic, time course, and stress treatment is relatively minor. But in case of comparison between different plant species (as shown above with Arabidopsis and rice leaves), wild accessions which have important phenotypical differences, or mutants with strong phenotypic differences, such problems due to differential ion suppression could easily arise. For example, seedling samples that differ in the relative amount of shoot/root or hypocotyl/root might be particularly susceptible to differential ion suppression. The same problem is raised in the case of harvesting flower samples with a varying ratio of flower/sepal and/or (flower/pedicel).
To evaluate this problem, recombination analyses (Fernie and Keurentjes, 2011) were evaluated using Arabidopsis samples containing various ratio of leaves and roots or flower extracts, focusing on the general secondary metabolites which were detected in both tissues. The percentage recovery was simply estimated for evaluation using theoretical concentration of extracts mixture, [(level in leaves × A%) + (level in roots (or flowers) × B%)]/100] (Table 2). Mixture of different tissue types results in >100% recovery (i.e., less ion suppression) for some peaks (e.g., IS), presumably because the area which is strongly suppressed differs between leaf and root extracts. Increase of recovery was observed in some peaks (<117%) at an equivalent mixture of leaf and root extracts. On the other hand, recovery ratio in mixture of leaves and flowers showed more significant variance (43 ∼ 122%). This experiment highlights the value of preliminary analyses in order to check for ion suppression. It is furthermore useful to understand the range of variance in instances in which the value of IS is unstable between samples.
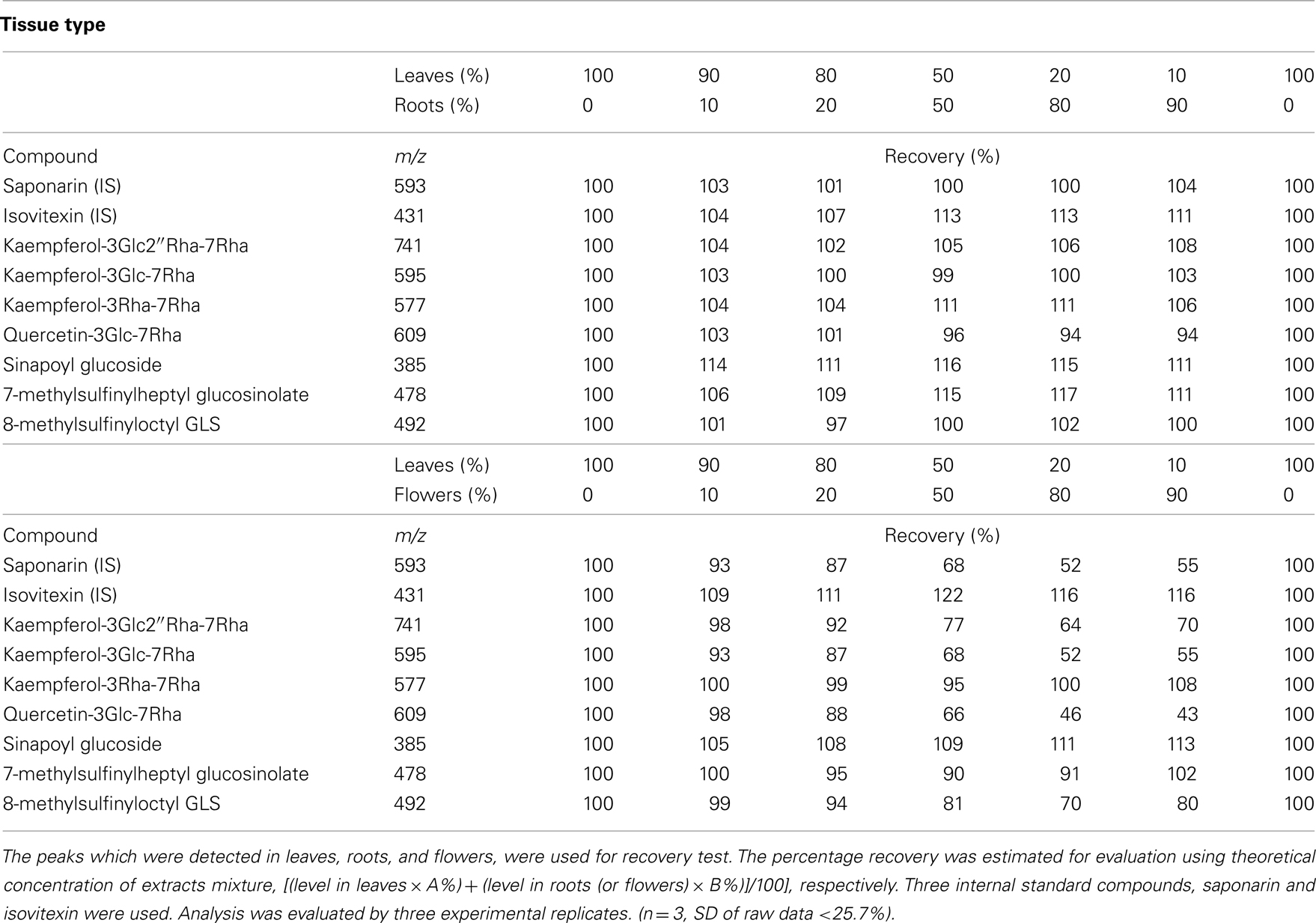
Table 2. Recovery test with mixture of extract of leaves, roots, and flowers.
Chemical Diversity and Peak Annotation Using Cross Species Comparison
Many metabolite databases for LC–MS are available, such as MASSBANK (Horai et al., 2010), METLIN (Smith et al., 2005), MS2T (Matsuda et al., 2009), KNApSAcK (Shinbo et al., 2006), and Flavonoid Viewer (Arita and Suwa, 2008). These greatly aid in the prediction and annotation of detected peaks (Tohge and Fernie, 2010). That said, technical improvement of peak identification and annotation still represents a major hurdle for LC–MS-based metabolite profiling platforms. The identification of secondary metabolites is obstructed by the insufficient availability of standard compounds. It is impossible to comprehensively purchase standard substances since the diversity of their chemical structure is far too large. Moreover, complex compounds are largely unavailable commercially and those that are available are often prohibitively expensive. Furthermore, LC–MS studies are complicated by the fact that the levels of secondary metabolites are highly divergent between different organs, growth conditions, and species (Petersen, 2007; Hanhineva et al., 2008; Matsuda et al., 2010). For these reasons, peak identification is generally performed by the use of combinatorial strategies whereby the literature information is taken alongside available compounds in an attempt to identify specific peaks (see for example Tohge et al., 2005; Giavalisco et al., 2009).
Given the recent explosion of genome information afforded firstly by microarray analyses and more recently by next-generation sequencing (review of (Schneeberger and Weigel, 2011), further tools for translational biology are becoming available. One such example, PlaNet, was described recently by Mutwil et al. (2011). Following this approach gene sequences can be connected between plant species on the basis of BLAST homology searches and then the positions in co-expression networks can be ascertained and finally it is possible to link unknown genes to annotated metabolic genes. As such this approach holds great promise both for gene functional annotation and via use of mutant plants in the annotation of unknown metabolites (Tohge and Fernie, 2010; Mutwil et al., 2011). It demonstrated the utility of this approach by identifying candidate genes of the general and species specific flavonoid pathways. It is likely that integrating metabolomics data on all the species currently in PlaNet will greatly aid this process and is certainly a research avenue that should be pursued in the near future.
Conclusion
Whilst applying a method established for another species is likely not to be overly problematic for screening purposes and for a first insight into the metabolome of an organism, the examples presented here demonstrate that when more precise information is required considerable effort should be put into establishing both the qualitative and quantitative reliability of any LC–MS-based metabolic profiling method. As evidenced by the ion suppression (and ion enhancement) examples particular care must be taken with this issue as well as in ensuring that the extraction procedure is appropriate for the tissue under study. Once these important controls have been adhered to a wide array of computational resources are available (Tohge and Fernie, 2009), which will greatly aid in translational research. Given that the trend in plant science research is to move away from the single model species of A. thaliana, such tools will become increasingly important. However, it is prudent to note that uncritical use of such tools without adequate controls of the type demonstrated here may well result in inaccurate representations of the metabolome. The best way to approach a new tissue, species, or even a dramatic mutant/transgenic line is to adopt both experimental and computational approaches to ensure the highest possible data quality.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Yozo Okazaki in RIKEN PSC for useful discussion, and Dr. Mark-Aurel Schöttler at the Max-Planck-Institute of Molecular Plant Physiology (MPIMP) for kindly providing instrumentation for Chlamydomonas illumination and harvesting. We thank Prof. Dr. Martin Steup in University of Potsdam and Dr. Wagner L. Araújo at MPIMP for expert comments. This work is partially funded by the German Federal Ministry of Education and Research by the FORSYS BMBF grant (GoFORSYS) and the Max-Planck Society (MPG). Research activity of Takayuki Tohge was supported by the Alexander von Humboldt Foundation. Tabea Mettler was supported by the International Max-Planck Research School (IMPRS). Adam James Carroll was supported through a grant to the Australian Research Council Centre of Excellence in Plant Energy Biology.
References
Achnine, L., Huhman, D. V., Farag, M. A., Sumner, L. W., Blount, J. W., and Dixon, R. A. (2005). Genomics-based selection and functional characterization of triterpene glycosyltransferases from the model legume Medicago truncatula. Plant J. 41, 875–887.
Aharoni, A., Keizer, L. C. P., Bouwmeester, H. J., Sun, Z. K., Alvarez-Huerta, M., Verhoeven, H. A., Blaas, J., van Houwelingen, A., De Vos, R. C. H., van der Voet, H., Jansen, R. C., Guis, M., Mol, J., Davis, R. W., Schena, M., van Tunen, A. J., and O’Connell, A. P. (2000). Identification of the SAAT gene involved in strawberry flavor biogenesis by use of DNA microarrays. Plant Cell 12, 647–661.
Aharoni, A., Ric de Vos, C. H., Verhoeven, H. A., Maliepaard, C. A., Kruppa, G., Bino, R. J., and Goodenowe, D. B. (2002). Nontargeted metabolome analysis by use of Fourier transform ion cyclotron mass spectrometry. OMICS 6, 217–234.
Albinsky, D., Kusano, M., Higuchi, M., Hayashi, N., Kobayashi, M., Fukushima, A., Mori, M., Ichikawa, T., Matsui, K., Kuroda, H., Horii, Y., Tsumoto, Y., Sakakibara, H., Hirochika, H., Matsui, M., and Saito, K. (2010). Metabolomic screening applied to rice FOX Arabidopsis lines leads to the identification of a gene-changing nitrogen metabolism. Mol. Plant 3, 125–142.
Allen, A. E., LaRoche, J., Maheswari, U., Lommer, M., Schauer, N., Lopez, P. J., Finazzi, G., Fernie, A. R., and Bowler, C. (2008). Whole-cell response of the pennate diatom Phaeodactylum tricornutum to iron starvation. Proc. Natl. Acad. Sci. U.S.A. 105, 10438–10443.
Araujo, W. L., Ishizaki, K., Nunes-Nesi, A., Larson, T. R., Tohge, T., Krahnert, I., Witt, S., Obata, T., Schauer, N., Graham, I. A., Leaver, C. J., and Fernie, A. R. (2010). Identification of the 2-hydroxyglutarate and isovaleryl-CoA dehydrogenases as alternative electron donors linking lysine catabolism to the electron transport chain of Arabidopsis mitochondria. Plant Cell 22, 1549–1563.
Arita, M., and Suwa, K. (2008). Search extension transforms Wiki into a relational system: a case for flavonoid metabolite database. BioData Min. 7, 8.
Arrivault, S., Guenther, M., Ivakov, A., Feil, R., Vosloh, D., van Dongen, J. T., Sulpice, R., and Stitt, M. (2009). Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 59, 824–839.
Bais, P., Moon, S. M., He, K., Leitao, R., Dreher, K., Walk, T., Sucaet, Y., Barkan, L., Wohlgemuth, G., Roth, M. R., Wurtele, E. S., Dixon, P., Fiehn, O., Lange, B. M., Shulaev, V., Sumner, L. W., Welti, R., Nikolau, B. J., Rhee, S. Y., and Dickerson, J. A. (2010). Plant metabolomics. org: a web portal for plant metabolomics experiments. Plant Physiol. 152, 1807–1816.
Barth, C., and Jander, G. (2006). Arabidopsis myrosinases TGG1 and TGG2 have redundant function in glucosinolate breakdown and insect defence. Plant J. 46, 549–562.
Bino, R. J., Hall, R. D., Fiehn, O., Kopka, J., Saito, K., Draper, J., Nikolau, B. J., Mendes, P., Roessner-Tunali, U., Beale, M. H., Trethewey, R. N., Lange, B. M., Wurtele, E. S., and Sumner, L. W. (2004). Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9, 418–425.
Bolling, C., and Fiehn, O. (2005). Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 139, 1995–2005.
Boyle, N. R., and Morgan, J. A. (2009). Flux balance analysis of primary metabolism in Chlamydomonas reinhardtii. BMC Syst. Biol. 3, 4.
Buhrman, D. L., Price, P. I., and Rudewicz, P. J. (1996). Quantitation of SR 27417 in human plasma using electrospray liquid chromatography tandem mass spectrometry: a study of ion suppression. J. Am. Soc. Mass Spectrom. 7, 1099–1105.
Carmo-Silva, A. E., Keys, A. J., Beale, M. H., Ward, J. L., Baker, J. M., Hawkins, N. D., Arrabaca, M. C., and Parry, M. A. J. (2009). Drought stress increases the production of 5-hydroxynorvaline in two C-4 grasses. Phytochemistry 70, 664–671.
Carroll, A. J., Badger, M. R., and Harvey Millar, A. (2010). The MetabolomeExpress Project: enabling web-based processing, analysis and transparent dissemination of GC/MS metabolomics datasets. BMC Bioinformatics 11, 376.
Catchpole, G. S., Beckmann, M., Enot, D. P., Mondhe, M., Zywicki, B., Taylor, J., Hardy, N., Smith, A., King, R. D., Kell, D. B., Fiehn, O., and Draper, J. (2005). Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proc. Natl. Acad. Sci. U.S.A. 102, 14458–14462.
Cottret, L., Wildridge, D., Vinson, F., Barrett, M. P., Charles, H., Sagot, M. F., and Jourdan, F. (2010). MetExplore: a web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res. 38, W132–W137.
El-Lithy, M. E., Rodrigues, G. C., van Rensen, J. J. S., Snel, J. F. H., Dassen, H., Koornneef, M., Jansen, M. A. K., Aarts, M. G. M., and Vreugdenhil, D. (2005). Altered photosynthetic performance of a natural Arabidopsis accession is associated with atrazine resistance. J. Exp. Bot. 56, 1625–1634.
Fernie, A. R., Aharoni, A., Willmitzer, L., Stitt, M., Tohge, T., Kopka, J., Carroll, A. J., Saito, K., Fraser, P. D., and DeLuca, V. (2011). Recommendations for reporting metabolite data. Plant Cell 23, 2477–2482.
Fernie, A. R., and Keurentjes, J. J. B. (2011). Genetics, genomics and metabolomics. Annu. Plant Rev. 43, 219–246.
Fernie, A. R., Trethewey, R. N., Krotzky, A. J., and Willmitzer, L. (2004). Innovation – metabolite profiling: from diagnostics to systems biology. Nat. Rev. Mol. Cell Biol. 5, 763–769.
Fraser, P. D., Enfissi, E. M. A., Goodfellow, M., Eguchi, T., and Bramley, P. M. (2007). Metabolite profiling of plant carotenoids using the matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Plant J. 49, 552–564.
Fukushima, A., Kusano, M., Nakamichi, N., Kobayashi, M., Hayashi, N., Sakakibara, H., Mizuno, T., and Saito, K. (2009). Impact of clock-associated Arabidopsis pseudo-response regulators in metabolic coordination. Proc. Natl. Acad. Sci. U.S.A. 106, 7251–7256.
Geigenberger, P., Fernie, A. R., Gibon, Y., Christ, M., and Stitt, M. (2000). Metabolic activity decreases as an adaptive response to low internal oxygen in growing potato tubers. Biol. Chem. 381, 723–740.
Giavalisco, P., Kohl, K., Hummel, J., Seiwert, B., and Willmitzer, L. (2009). C-13 isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Anal. Chem. 81, 6546–6551.
Gibon, Y., Usadel, B., Blaesing, O. E., Kamlage, B., Hoehne, M., Trethewey, R., and Stitt, M. (2006). Integration of metabolite with transcript and enzyme activity profiling during diurnal cycles in Arabidopsis rosettes. Genome Biol. 7, R76.
Giroud, C., Gerber, A., and Eichenberger, W. (1988). Lipids of Chlamydomonas reinhardtii – analysis of molecular-species and intracellular site(s) of biosynthesis. Plant Cell Physiol. 29, 587–595.
Goossens, A., Hakkinen, S. T., Laakso, I., Seppanen-Laakso, T., Biondi, S., De Sutter, V., Lammertyn, F., Nuutila, A. M., Soderlund, H., Zabeau, M., Inze, D., and Oksman-Caldentey, K. M. (2003). A functional genomics approach toward the understanding of secondary metabolism in plant cells. Proc. Natl. Acad. Sci. U.S.A. 100, 8595–8600.
Griffin, J. L., and Nicholls, A. W. (2006). Metabolomics as a functional genomic tool for understanding lipid dysfunction in diabetes, obesity and related disorders. Pharmacogenomics 7, 1095–1107.
Hall, R., Beale, M., Fiehn, O., Hardy, N., Sumner, L., and Bino, R. (2002). Plant metabolomics: the missing link in functional genomics strategies. Plant Cell 14, 1437–1440.
Hanhineva, K., Rogachev, I., Kokko, H., Mintz-Oron, S., Venger, I., Karenlampi, S., and Aharoni, A. (2008). Non-targeted analysis of spatial metabolite composition in strawberry (Fragaria x ananassa) flowers. Phytochemistry 69, 2463–2481.
Harmer, S. L., Hogenesch, L. B., Straume, M., Chang, H. S., Han, B., Zhu, T., Wang, X., Kreps, J. A., and Kay, S. A. (2000). Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290, 2110–2113.
Hirai, M. Y., Klein, M., Fujikawa, Y., Yano, M., Goodenowe, D. B., Yamazaki, Y., Kanaya, S., Nakamura, Y., Kitayama, M., Suzuki, H., Sakurai, N., Shibata, D., Tokuhisa, J., Reichelt, M., Gershenzon, J., Papenbrock, J., and Saito, K. (2005). Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 280, 25590–25595.
Hirai, M. Y., Sugiyama, K., Sawada, Y., Tohge, T., Obayashi, T., Suzuki, A., Araki, R., Sakurai, N., Suzuki, H., Aoki, K., Goda, H., Nishizawa, O. I., Shibata, D., and Saito, K. (2007). Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 6478–6483.
Hirai, M. Y., Yano, M., Goodenowe, D. B., Kanaya, S., Kimura, T., Awazuhara, M., Arita, M., Fujiwara, T., and Saito, K. (2004). Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 101, 10205–10210.
Horai, H., Arita, M., Kanaya, S., Nihei, Y., Ikeda, T., Suwa, K., Ojima, Y., Tanaka, K., Tanaka, S., Aoshima, K., Oda, Y., Kakazu, Y., Kusano, M., Tohge, T., Matsuda, F., Sawada, Y., Hirai, M. Y., Nakanishi, H., Ikeda, K., Akimoto, N., Maoka, T., Takahashi, H., Ara, T., Sakurai, N., Suzuki, H., Shibata, D., Neumann, S., Iida, T., Tanaka, K., Funatsu, K., Matsuura, F., Soga, T., Taguchi, R., Saito, K., and Nishioka, T. (2010). MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 45, 703–714.
Iijima, Y., Nakamura, Y., Ogata, Y., Tanaka, K., Sakurai, N., Suda, K., Suzuki, T., Suzuki, H., Okazaki, K., Kitayama, M., Kanaya, S., Aoki, K., and Shibata, D. (2008). Metabolite annotations based on the integration of mass spectral information. Plant J. 54, 949–962.
Ikonomou, M. G., Blades, A. T., and Kebarle, P. (1990). Investigations of the electrospray interface for liquid-chromatography mass-spectrometry. Anal. Chem. 62, 957–967.
Jom, K. N., Frank, T., and Engel, K. H. (2010). A metabolite profiling approach to follow the sprouting process of mung beans (Vigna radiata). Metabolomics 7, 102–117.
Kanno, Y., Jikumaru, Y., Hanada, A., Nambara, E., Abrams, S. R., Kamiya, Y., and Seo, M. (2010). Comprehensive hormone profiling in developing Arabidopsis seeds: examination of the site of ABA biosynthesis, ABA transport and hormone interactions. Plant Cell Physiol. 51, 1988–2001.
Kebarle, P., and Tang, L. (1993). From ions in solution to ions in the gas-phase – the mechanism of electrospray mass-spectrometry. Anal. Chem. 65, A972–A986.
Kempa, S., Hummel, J., Schwemmer, T., Pietzke, M., Strehmel, N., Wienkoop, S., Kopka, J., and Weckwerth, W. (2009). An automated GCxGC-TOF-MS protocol for batch-wise extraction and alignment of mass isotopomer matrixes from differential C-13-labelling experiments: a case study for photoautotrophic-mixotrophic grown Chlamydomonas reinhardtii cells. J. Basic Microbiol. 49, 82–91.
Kerwin, R. E., Jimenez-Gomez, J. M., Fulop, D., Harmer, S. L., Maloof, J. N., and Kliebenstein, D. J. (2011). Network quantitative trait loci mapping of circadian clock outputs identifies metabolic pathway-to-clock linkages in Arabidopsis. Plant Cell 23, 471–485.
King, R., Bonfiglio, R., Fernandez-Metzler, C., Miller-Stein, C., and Olah, T. (2000). Mechanistic investigation of ionization suppression in electrospray ionization. J. Am. Soc. Mass Spectrom. 11, 942–950.
Krall, L., Huege, J., Catchpole, G., Steinhauser, D., and Willmitzer, L. (2009). Assessment of sampling strategies for gas chromatography-mass spectrometry (GC-MS) based metabolomics of cyanobacteria. J. Chromatogr. 877, 2952–2960.
Kusano, M., Tohge, T., Fukushima, A., Kobayashi, M., Hayashi, N., Otsuki, H., Kondou, Y., Goto, H., Kawashima, M., Matsuda, F., Niida, R., Matsui, M., Saito, K., and Fernie, A. R. (2011). Metabolomics reveals comprehensive reprogramming involving two independent metabolic responses of Arabidopsis to ultraviolet-B light. Plant J. 67, 354–369.
Luo, J., Nishiyama, Y., Fuell, C., Taguchi, G., Elliott, K., Hill, L., Tanaka, Y., Kitayama, M., Yamazaki, M., Bailey, P., Parr, A., Michael, A. J., Saito, K., and Martin, C. (2007). Convergent evolution in the BAHD family of acyltransferases: identification and characterization of anthocyanin acyltransferases from Arabidopsis thaliana. Plant J. 50, 678–695.
Matsuda, F., Hirai, M. Y., Sasaki, E., Akiyama, K., Yonekura-Sakakibara, K., Provart, N. J., Sakurai, T., Shimada, Y., and Saito, K. (2010). AtMetExpress development: a phytochemical atlas of Arabidopsis development. Plant Physiol. 152, 566–578.
Matsuda, F., Yonekura-Sakakibara, K., Niida, R., Kuromori, T., Shinozaki, K., and Saito, K. (2009). MS/MS spectral tag-based annotation of non-targeted profile of plant secondary metabolites. Plant J. 57, 555–577.
Matuszewski, B. K., Constanzer, M. L., and Chavez-Eng, C. M. (1998). Matrix effect in quantitative LC/MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 70, 882–889.
Matuszewski, B. K., Constanzer, M. L., and Chavez-Eng, C. M. (2003). Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 75, 3019–3030.
May, P., Wienkoop, S., Kempa, S., Usadel, B., Christian, N., Rupprecht, J., Weiss, J., Recuenco-Munoz, L., Ebenhoh, O., Weckwerth, W., and Walther, D. (2008). Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics 179, 157–166.
Meyer, R. C., Steinfath, M., Lisec, J., Becher, M., Witucka-Wall, H., Torjek, O., Fiehn, O., Eckardt, A., Willmitzer, L., Selbig, J., and Altmann, T. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 104, 4759–4764.
Moco, S., Bino, R. J., Vorst, O., Verhoeven, H. A., de Groot, J., van Beek, T. A., Vervoort, J., and de Vos, C. H. R. (2006). A liquid chromatography-mass spectrometry-based metabolome database for tomato. Plant Physiol. 141, 1205–1218.
Mutwil, M., Klie, S., Tohge, T., Giorgi, F. M., Wilkins, O., Campbell, M. M., Fernie, A. R., Usadel, B., Nikoloski, Z., and Persson, S. (2011). PlaNet: combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell 23, 895–910.
Nakabayashi, R., Kusano, M., Kobayashi, M., Tohge, T., Yonekura-Sakakibara, K., Kogure, N., Yamazaki, M., Kitajima, M., Saito, K., and Takayama, H. (2009). Metabolomics-oriented isolation and structure elucidation of 37 compounds including two anthocyanins from Arabidopsis thaliana. Phytochemistry 70, 1017–1029.
Nicholson, J. K., and Wilson, I. D. (2003). Understanding “global” systems biology: metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2, 668–676.
Okazaki, Y., Shimojima, M., Sawada, Y., Toyooka, K., Narisawa, T., Mochida, K., Tanaka, H., Matsuda, F., Hirai, A., Hirai, M. Y., Ohta, H., and Saito, K. (2009). A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell 21, 892–909.
Renberg, L., Johansson, A. I., Shutova, T., Stenlund, H., Aksmann, A., Raven, J. A., Gardestrom, P., Moritz, T., and Samuelsson, G. (2010). A metabolomic approach to study major metabolite changes during acclimation to limiting CO2 in Chlamydomonas reinhardtii. Plant Physiol. 154, 187–196.
Rischer, H., Oresic, M., Seppanen-Laakso, T., Katajamaa, M., Lammertyn, F., Ardiles-Diaz, W., Van Montagu, M. C. E., Inze, D., Oksman-Caldentey, K. M., and Goossens, A. (2006). Gene-to-metabolite networks for terpenoid indole alkaloid biosynthesis in Catharanthus roseus cells. Proc. Natl. Acad. Sci. U.S.A. 103, 5614–5619.
Roessner, U., Willmitzer, L., and Fernie, A. R. (2001). High-resolution metabolic phenotyping of genetically and environmentally diverse potato tuber systems. Identification of phenocopies. Plant Physiol. 127, 749–764.
Roessner-Tunali, U., Hegemann, B., Lytovchenko, A., Carrari, F., Bruedigam, C., Granot, D., and Fernie, A. R. (2003). Metabolic profiling of transgenic tomato plants overexpressing hexokinase reveals that the influence of hexose phosphorylation diminishes during fruit development. Plant Physiol. 133, 84–99.
Sauter, H., Lauer, M., and Fritsch, H. (1988). Metabolite profiling of plants – a new diagnostic technique. Abstr. Pap. Am. Chem. Soc. 195, 129.
Schauer, N., Semel, Y., Roessner, U., Gur, A., Balbo, I., Carrari, F., Pleban, T., Perez-Melis, A., Bruedigam, C., Kopka, J., Willmitzer, L., Zamir, D., and Fernie, A. R. (2006). Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 24, 447–454.
Schauer, N., Zamir, D., and Fernie, A. R. (2005). Metabolic profiling of leaves and fruit of wild species tomato: a survey of the Solanum lycopersicum complex. J. Exp. Bot. 56, 297–307.
Scherling, C., Ulrich, K., Ewald, D., and Weckwerth, W. (2009). A metabolic signature of the beneficial interaction of the endophyte paenibacillus sp isolate and in vitro-grown poplar plants revealed by metabolomics. Mol. Plant Microbe Interact. 22, 1032–1037.
Schneeberger, K., and Weigel, D. (2011). Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci. 16, 282–288.
Semel, Y., Schauer, N., Roessner, U., Zamir, D., and Fernie, A. (2007). Metabolite analysis for the comparison of irrigated and non-irrigated field grown tomato of varying genotype. Metabolomics 3, 289–295.
Shinbo, Y., Nakamura, Y., Altaf-Ul-Amin, M., Asahi, H., Kurokawa, K., Arita, M., Saito, K., Ohta, D., Shibata, D., and Kanaya, S. (2006). KNApSAcK: a comprehensive species-metabolite relationship database. Plant Metabolomics 165–181.
Smith, C. A., O’Maille, G., Want, E. J., Qin, C., Trauger, S. A., Brandon, T. R., Custodio, D. E., Abagyan, R., and Siuzdak, G. (2005). METLIN – a metabolite mass spectral database. Ther. Drug Monit. 27, 747–751.
Stitt, M., and Fernie, A. R. (2003). From measurements of metabolites to metabolomics: an “on the fly” perspective illustrated by recent studies of carbon-nitrogen interactions. Curr. Opin. Biotechnol. 14, 136–144.
St-Pierre, B., and De Luca, V. (2000). “Evolution of acyltransferase genes: origin and diversification of the BAHD superfamily of acyltransferases involved in secondary metabolism,” in Evolution of Metabolic Pathways, ed J. T. Romeo (Amsterdam: Elsevier Science), 285–315.
Stracke, R., Ishihara, H., Barsch, G. H. A., Mehrtens, F., Niehaus, K., and Weisshaar, B. (2007). Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 50, 660–677.
Sulpice, R., Pyl, E. T., Ishihara, H., Trenkamp, S., Steinfath, M., Witucka-Wall, H., Gibon, Y., Usadel, B., Poree, F., Piques, M. C., Von Korff, M., Steinhauser, M. C., Keurentjes, J. J. B., Guenther, M., Hoehne, M., Selbig, J., Fernie, A. R., Altmann, T., and Stitt, M. (2009). Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. U.S.A. 106, 10348–10353.
Tierens, K., Thomma, B. P. H., Brouwer, M., Schmidt, J., Kistner, K., Porzel, A., Mauch-Mani, B., Cammue, B. P. A., and Broekaert, W. F. (2001). Study of the role of antimicrobial glucosinolate-derived isothiocyanates in resistance of Arabidopsis to microbial pathogens. Plant Physiol. 125, 1688–1699.
Tiessen, A., Hendriks, J. H. M., Stitt, M., Branscheid, A., Gibon, Y., Farre, E. M., and Geigenberger, P. (2002). Starch synthesis in potato tubers is regulated by post-translational redox modification of ADP-glucose pyrophosphorylase: a novel regulatory mechanism linking starch synthesis to the sucrose supply. Plant Cell 14, 2191–2213.
Tohge, T., and Fernie, A. R. (2009). Web-based resources for mass-spectrometry-based metabolomics: a user’s guide. Phytochemistry 70, 450–456.
Tohge, T., and Fernie, A. R. (2010). Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nat. Protoc. 5, 1210–1227.
Tohge, T., Nishiyama, Y., Hirai, M. Y., Yano, M., Nakajima, J., Awazuhara, M., Inoue, E., Takahashi, H., Goodenowe, D. B., Kitayama, M., Noji, M., Yamazaki, M., and Saito, K. (2005). Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 42, 218–235.
Tohge, T., Yonekura-Sakakibara, K., Niida, R., Watanabe-Takahashi, A., and Saito, K. (2007). Phytochemical genomics in Arabidopsis thaliana: a case study for functional identification of flavonoid biosynthesis genes. Pure Appl. Chem. 79, 811–823.
Trenkamp, S., Eckes, P., Busch, M., and Fernie, A. R. (2009). Temporally resolved GC-MS-based metabolic profiling of herbicide treated plants treated reveals that changes in polar primary metabolites alone can distinguish herbicides of differing mode of action. Metabolomics 5, 277–291.
von Roepenack-Lahaye, E., Degenkolb, T., Zerjeski, M., Franz, M., Roth, U., Wessjohann, L., Schmidt, J., Scheel, D., and Clemens, S. (2004). Profiling of Arabidopsis secondary metabolites by capillary liquid chromatography coupled to electrospray ionization quadrupole time-of-flight mass spectrometry. Plant Physiol. 134, 548–559.
Weiner, H., Stitt, M., and Heldt, H. W. (1987). Subcellular compartmentation of pyrophosphate and alkaline pyrophosphatase in leaves. Biochim. Biophys. Acta 893, 13–21.
Widodo Patterson, J. H., Newbigin, E., Tester, M., Bacic, A., and Roessner, U. (2009). Metabolic responses to salt stress of barley (Hordeum vulgare L.) cultivars, Sahara and Clipper, which differ in salinity tolerance. J. Exp. Bot. 60, 4089–4103.
Xia, J., and Wishart, D. S. (2010). MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 38, W71–W77.
Yamazaki, M., Shibata, M., Nishiyama, Y., Springob, K., Kitayama, M., Shimada, N., Aoki, T., Ayabe, S. I., and Saito, K. (2008). Differential gene expression profiles of red and green forms of Perilla frutescens leading to comprehensive identification of anthocyanin biosynthetic genes. FEBS J. 275, 3494–3502.
Yonekura-Sakakibara, K., Tohge, T., Matsuda, F., Nakabayashi, R., Takayama, H., Niida, R., Watanabe-Takahashi, A., Inoue, E., and Saito, K. (2008). Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell 20, 2160–2176.
Yonekura-Sakakibara, K., Tohge, T., Niida, R., and Saito, K. (2007). Identification of a flavonol 7-O-rhamnosyltransferase gene determining flavonoid pattern in Arabidopsis by transcriptome coexpression analysis and reverse genetics. J. Biol. Chem. 282, 14932–14941.
Zhu, T., and Wang, X. (2000). Large-scale profiling of the Arabidopsis transcriptome. Plant Physiol. 124, 1472–1476.
Appendix
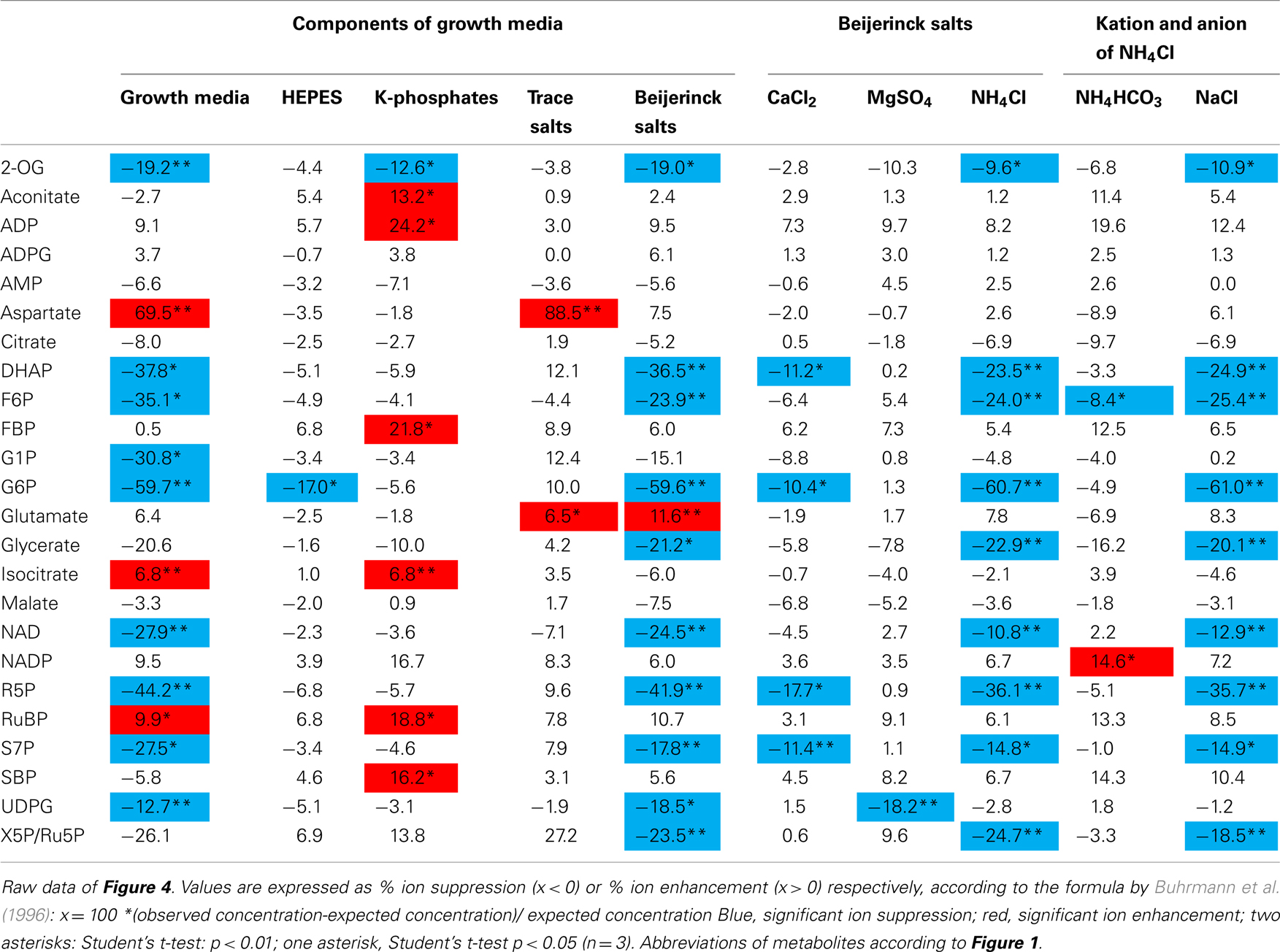
Table A1. Ion suppression of growth media mainly caused by ammonium chloride.
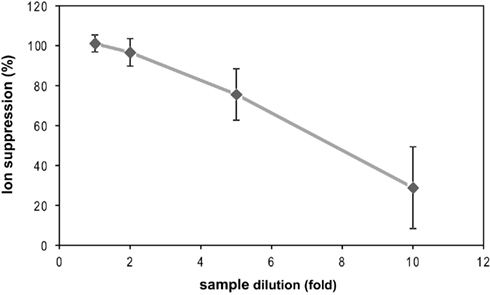
Figure A1. Sample dilution led to decreased ion suppression. Illuminated Chlamydomonas reinhardtii CC-1690 suspension was quenched, harvested, and extracted as in Figure 1. Four different dilutions of the extract (diluted with water) were then spiked with a reference standard mix and compared to the standard mix on its own to assess the extent of ion suppression. The fivefold dilution was selected for experiments shown in Figures 1 and 4; Table 1 as for several metabolites the 10-fold dilution was below detection limit. Note that the average of ion suppression was 77% for the fivefold dilution in this experiment. For experiment shown in Figures 1 and 4; Table 1, the average ion suppression for the fivefold dilution was <13%. This improvement was vey likely achieved by using a quadrupole (Finning TSQ Quantum Ultra) with a larger ion transfer tube diameter. The plot shows the mean ion suppression for 24 metabolites, each measured with two technical replicates, ±SD.
Keywords: plant metabolomics, sample preparation, ion suppression, chemical diversity, translational biology, LC–MS
Citation: Tohge T, Mettler T, Arrivault S, Carroll AJ, Stitt M and Fernie AR (2011) From models to crop species: caveats and solutions for translational metabolomics. Front. Plant Sci. 2:61. doi: 10.3389/fpls.2011.00061
Received: 04 May 2011; Accepted: 13 September 2011;
Published online: 03 October 2011.
Edited by:
Wolf B. Frommer, Carnegie Institution for Science, USAReviewed by:
Jin Chen, Michigan State University, USAWoei-Jiun Guo, National Cheng Kung University, Taiwan
Copyright: © 2011 Tohge, Mettler, Arrivault, Carroll, Stitt and Fernie. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Takayuki Tohge, Max-Planck-Institute for Molecular Plant Physiology, Am Muehlenberg 1, 14476 Potsdam-Golm, Germany. e-mail:dG9oZ2VAbXBpbXAtZ29sbS5tcGcuZGU=