 Nora Wehner
Nora Wehner
- Julius-von-Sachs-Institute, University of Würzburg, Würzburg, Germany
In the model plant Arabidopsis thaliana, more than 2000 genes are estimated to encode transcription factors (TFs), which clearly emphasizes the importance of transcriptional control. Although genomic approaches have generated large TF open reading frame (ORF) collections, only a limited number of these genes is functionally characterized, yet. This review evaluates strategies and methods to identify TF functions. In particular, we focus on two recently developed TF screening platforms, which make use of publically available GATEWAY®-compatible ORF collections. (1) The Arabidopsis thaliana TF ORF over-Expression (AtTORF-Ex) library provides pooled collections of transgenic lines over-expressing HA-tagged TF genes, which are suited for screening approaches to define TF functions in stress defense and development. (2) A high-throughput microtiter plate based protoplast trans activation (PTA) system has been established to screen for TFs which are regulating a given promoter:Luciferase construct in planta.
Introduction
DNA-binding transcription factors (TFs) are important transcriptional regulators which either activate or repress transcription of their cognate target genes by binding to regulatory cis-elements in a sequence-specific manner (Ptashne, 2005; Riechmann, 2006). Non-DNA-binding TFs mediate their impact on transcription via protein–protein interaction. In general, TFs are modular in structure and composed of protein domains which facilitate DNA-specific binding, homo- and heterodimerization with other transcriptional regulators and accomplish their transcriptional activation or repression activity (Figure 1A). Roughly 5–10% of all Arabidopsis genes are encoding transcriptional regulators, which clearly emphasizes the importance of transcriptional control (Riechmann et al., 2000; Mitsuda and Ohme-Takagi, 2009). Depending on the bioinformatic approach used, approximately 2000 TF genes have been annotated in Arabidopsis (Riechmann et al., 2000; Guo et al., 2005; Ramirez and Basu, 2009; Perez-Rodriguez et al., 2010; Yilmaz et al., 2011). Based on their evolutionary conserved DNA-binding domains, these TFs are grouped into distinct gene families. In Arabidopsis, more than 60 TF families have been assigned, such as MYBs, MADSs, bHLHs, and AP2/ERFs (Riechmann et al., 2000; Mitsuda and Ohme-Takagi, 2009). Several of these families harbor more than 100 members, which in part share related functions. This redundancy clearly hampers functional TF analysis in plants (Qu and Zhu, 2006; Mitsuda and Ohme-Takagi, 2009). Until now, only a small fraction of the Arabidopsis TFs is functionally well-characterized. Hence, tools are required to assess TF function on a genomic scale (Figure 1B).
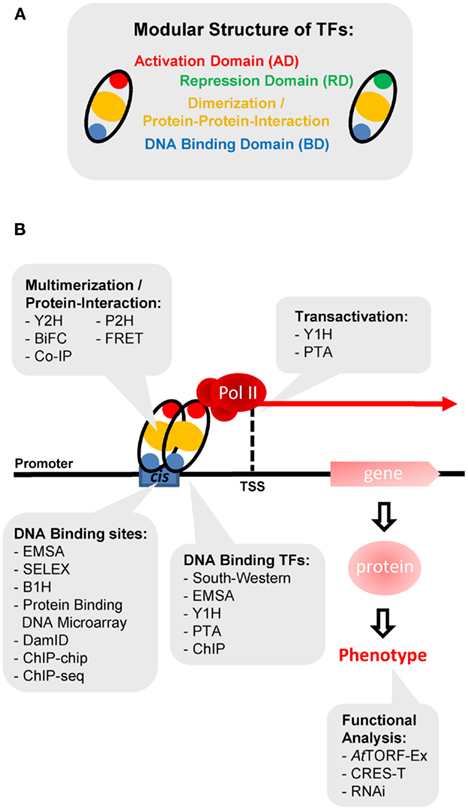
Figure 1. Methods to analyze transcription factor function. (A) The modular structure of TFs. Domains which function in activation, repression, dimerization or protein–protein interaction, and DNA-binding are color coded in red, green, yellow, or blue, respectively. (B) Overview of methods which can be used to elucidate TF functions. For details see text. Y2H, yeast two-hybrid; Y1H, yeast one-hybrid; B1H, bacterial one-hybrid; P2H, protoplast two-hybrid; BiFC, bimolecular fluorescence complementation; FRET, fluorescence resonance energy transfer; Co-IP, co-immuno-precipitation; PTA, protoplast transactivation; EMSA, electrophoretic mobility shift assay; SELEX, systematic evolution of ligands by exponential enrichment; DamID, DNA adenine methylation identification; ChIP, chromatin immuno-precipitation; ChIP-chip, ChIP combined with tilling array technology; ChIP-seq, ChIP combined with sequencing of immunoprecipitated DNA fragments; CRES-T, chimeric repressor gene silencing technology; RNAi, RNA interference; TSS, Transcriptional start site.
ORF Repositories Are a Valuable Source for Functional Genomics of Transcriptional Regulators
In the recent years comprehensive genome sequencing projects have given rise to a vast amount of information about plant genomes and the number of genes they encode. This knowledge enabled the cloning of the Arabidopsis open reading frames (ORFs) to further study this ORFeom (e.g., Arabidopsis Biological Resource Center, ABRC; RIKEN BioResource Center, RBC; Hilson, 2006; Seki and Shinozaki, 2009). Focusing on genes of transcriptional regulators at least five compiled ORF collections have to be highlighted. These are the REGIA collection encompassing approximately 800 TF ORFs (Paz-Ares, 2002) which has been considerably extended to roughly 1200 ORFs (Castrillo et al., 2011), the PKU-Yale collection harboring 1300 ORFs (Gong et al., 2004), and its further enlarged version which consists of ca. 1600 TF clones (Ou et al., 2011). Moreover, the comprehensive TF-only library (Mitsuda et al., 2010) comprising around 1500 TF ORFs has to be considered. Although these collections are highly redundant they corporately cover almost the complete Arabidopsis regulome (Riechmann et al., 2000; Guo et al., 2005; Mitsuda and Ohme-Takagi, 2009). As a common feature, these libraries preserve the cloned TF ORFs in recombinase-compatible vectors (such as the GATEWAY® system), enabling the transfer of single coding-sequences or whole ORF libraries into suitable expression vectors. This immanent transfer-flexibility of these ORF repositories allows the user to apply them for a broad variety of experimental high-throughput screening tools.
High-Throughput Screening Tools for Functional Genomics on Arabidopsis Transcription Factors
Several gain-of-function approaches have been employed to functionally characterize the Arabidopsis ORFeome making use of transgenic plants (Kuromori et al., 2009; Kondou et al., 2010). In this respect, the full-length cDNA over-expressing (FOX) gene hunting system is one of the first described reverse genetic approaches which provides plants expressing a cDNA library of around 10.000 independent Arabidopsis full-length cDNAs (Ichikawa et al., 2006). In order to develop a high-throughput method specifically focusing on functional analysis of TFs, the Arabidopsis thaliana transcription factor ORF over-expression (AtTORF-Ex) seed collection has been established (Weiste et al., 2007). Instead of applying a labor- and cost-intensive one-by-one transformation approach, a parallel batch procedure of pooled collections of GATEWAY®-tagged TF cDNAs has been used to simultaneously recombine ORF libraries into a plant expression vector. This enables the expression of HA-tagged TF-fusion proteins in plants under control of the 35S promoter. As depicted in the scheme in Figure 2, properly recombined vector DNA pools are selected by E. coli transformation. E. coli-derived vector DNA pools are subsequently used for Agrobacterium and Arabidopsis flower-dip transformation. After Basta® selection, a seed library of transgenic lines is obtained which over-expresses HA-tagged TF genes. T2-seeds of these transformants are harvested as seed stocks which can be applied for various screening approaches to identify TF genes involved in plant development or stress response. However, it has to be considered, that the seed stocks hold 25% of wild-type seeds, which enlarge the number of plants which have to enter the screening procedure.
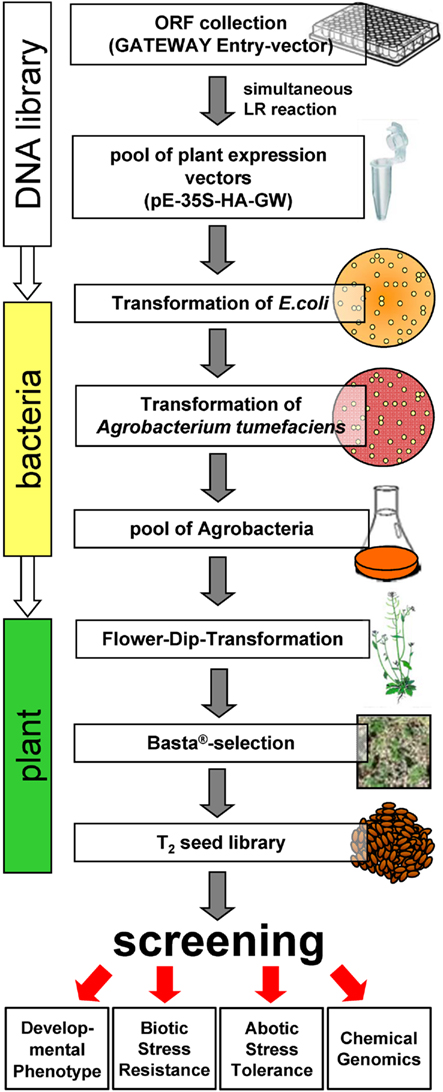
Figure 2. Establishment and application of the Arabidopsis thaliana transcription factor ORF over-expression (At TORF-Ex) seed collection. Pooled GATEWAY®-compatible ORF-TF expression vectors have been transferred from E. coli via Agrobacterium tumefaciens to Arabidopsis. Transgenic Basta®-resistant seeds were collected and stored as seed stocks which can be screened for altered phenotypes. Downstream applications of the AtTORF-Ex collections are indicated. For details see Weiste et al. (2007).
The feasibility of the approach has been analyzed using near-complete collections of the AP2/ERF family (Weiste et al., 2007). The cDNAs used as starting material have been equally traced during all steps of the procedure and no significant bias for particular clones and no preference for clone sizes has been observed. The frequency of multiple transformation events has been determined to be relatively low (in the range of 4%). Expression analysis has been performed on RNA and protein level using the HA-tag. Approximately 60 or 30% of the plants show significant transgene expression on RNA or protein level, respectively. In respect to a particular transgene, several independent transgenic lines are present in the AtTORF-Ex collection displaying a high variety in expression. In fact, this finding is important if high expression levels result in lethality. Striking phenotypic alterations have been observed in 4% of the plants. Based on these data and statistical estimations an optimized protocol has been established which ensures a high coverage of TF genes in the library (>99%). Currently, the AtTORF-Ex collection harbors transgenic plants over-expressing 650 TF ORFs which are available as pools consisting of 30–60 TFs (Table 1). Collections which are organized in TF families enable screening approaches which are focused on specific candidates.
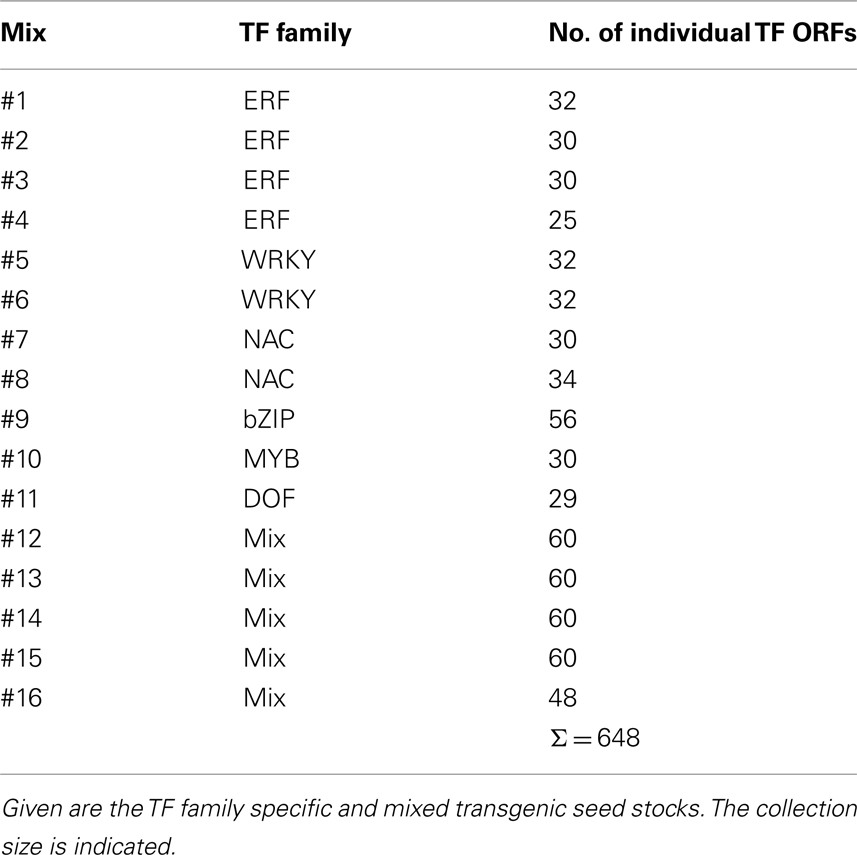
Table 1. Overview of the available AtTORF-Ex screening collections.
In order to define which TF is involved in a particular function, these collections can be used for phenotypical screens. Developmental phenotypes due to ectopic TF expression have successfully been assayed, such as altered leaf shape or early leaf senescence. Resistance to abiotic (e.g., treatment with paraquat, heavy metals, salt) or biotic stresses (e.g., fungal infection) have been accomplished. Beside classical screens based on altered growth or resistance, screens can also be performed by assaying molecular phenotypes, e.g., the production of secondary metabolites.
As a proof-of-principle, Weiste et al. (2007) have demonstrated that over-expression of At3g23220 (ERF95; Nakano et al., 2006) leads to resistance to oxidative stress when seedlings were grown on MS media supplemented with paraquat. The GATEWAY®-tagged TF genes can easily be recovered from the selected plants by PCR and sequencing using att-site specific primers. A recurring correlation between phenotype and PCR-amplified transgenes discloses likely TF candidates encoding the function of interest. This was the case in 50% of the identified paraquat resistant plants. However, the observed phenotype might not necessarily be linked to TF expression, e.g., if the T-DNA insertion leads to dominant mutations due to truncated gene products. Alternatively, the phenotype might be due to co-suppression and not caused by over-expression. In conclusion, promising candidates should be obtained several times during the screen and carefully evaluated by molecular means.
As a general drawback of a gain-of-function method, one has to consider that by ectopic expression, both hypermorphs (altered phenotype due to high expression levels) and neomorphs (new function caused by inappropriate tissue or developmental stage dependent expression) might appear (Qu and Zhu, 2006). Therefore, loss-of-function approaches should be used to further validate the findings. With respect to the mentioned example, ERF95 loss-of-function plants show wt resistance to paraquat, probably due to functional redundancy of closely related TF family members (Nakano et al., 2006). Redundancy within large gene families can easily be addressed by AtTORF-Ex screening, as homologous TFs involved in related functions are frequently identified during exhaustive screens. In summary, this high-throughput procedure for TF ORFeome analysis can efficiently be used in unbiased screening approaches to unravel the TF phenome.
Future Perspectives of Screens to Decipher Transcription Factor Function
As constitutive expression might result in severe phenotypical alterations, inducible expression collections would clearly be useful and can be generated both by one-by-one or batch procedures. To complement the ectopic expression experiments, loss-of-function approaches would also provide straight-forward screening tools. However, comprehensive RNAi- or amiRNA-collections for studying TF function are not published, yet. As a loss-of-function approach, the chimeric repressor gene silencing technology (CRES-T) procedure is currently applied for studying TFs on a genome-wide basis (http://www.cres-t.org/fiore/public_db/index.shtml). This resource uses a one-by-one transformation procedure expressing TF-fusions with an ERF-associated amphiphilic repression (EAR) domain (Hiratsu et al., 2003). However, it has to be mentioned that fusion of a repressor domain to a TF which functions as a negative regulator does not necessarily phenocopy the loss-of-function mutant. Thus, careful molecular and phenotypical analyses are needed to validate the results.
As it has been described for the AtTORF-Ex collection, the CRES-T approach can also be performed as a pooled expression system. As a major advantage, these artificial repressors block DNA-binding sites of target promoters and therefore, are particularly useful when redundant TFs are studied. This Arabidopsis thaliana transcription factor ORF repression (AtTORF-Rep) collection is currently under investigation. Although single phenotypes described for knock-out plants have been reproduced, the high-throughput analysis of these phenotypes are somewhat difficult, as suppression is partial and might also lead to target gene activation if a TF-specific activation domain competes with the EAR repression domain. Hence, molecular interpretation of phenotypes might not always be easy to perform.
Screening Tools to Identify the Cognate Transcription Factor Regulating a Promoter of Interest
Defining TFs which bind and/or regulate a promoter of choice is a standard experimental approach. Classically, in vitro DNA-binding of phage-expressed cDNA-derived proteins (“South-Western” screening) has been used (Singh et al., 1988; Figure 1B). However, in vitro DNA-binding might not reflect the situation in a cell (Heinekamp et al., 2002). Yeast one-hybrid (Y1H) screenings (Fields and Song, 1989) are favored methods to rapidly detect protein–DNA interactions in vivo. Traditional Y1H screenings are based on the interaction of complete cDNA expression libraries with a promoter sequence driving a reporter gene. In order to identify TF–DNA interactions, libraries prepared from total mRNA might not be appropriate as TF genes are frequently expressed at low levels and are thus under-represented. In addition, regulatory proteins with high affinity to un-specific DNA regions may frequently lead to false positives.
Recently, several Y1H approaches have been published, making use of normalized collections composed of up to 1500 TF cDNAs (Mitsuda et al., 2010; Castrillo et al., 2011; Ou et al., 2011). In particular, TF collections arrayed in 96-well plates which can be independently transferred in a high-throughput mating-type set-up, appear to be promising tools (Castrillo et al., 2011). Hence, it is assured that each cDNA clone of the collection is assayed. Although TF-specific libraries enhance the screening efficiency, yeast systems still have the disadvantage that the conditions inside the yeast nucleus might differ from those in plant cells (Ehlert et al., 2006). In comparison to plants, yeast promoters are shorter in size and therefore, additional transcriptional start events might compete when promoters are assayed which exceeds the size of approximately 300 bps (Dobi and Winston, 2007; Mitsuda et al., 2010). Hence, Y1H screens are limited to short promoter fragments or multimerized cis-elements.
The use of plant cell screening systems prevents most of these conceivable disadvantages. High-throughput microtiter plate based protoplast transfection systems have been recently described (De Sutter et al., 2005; Wehner et al., 2011) which can be used in combination with arrayed TF expression collections to identify TFs which regulate a promoter of choice (protoplast trans activation System, PTA; Wehner et al., 2011). Applying this procedure, the transactivation properties of 96 TFs can simultaneously be defined (Figure 3).
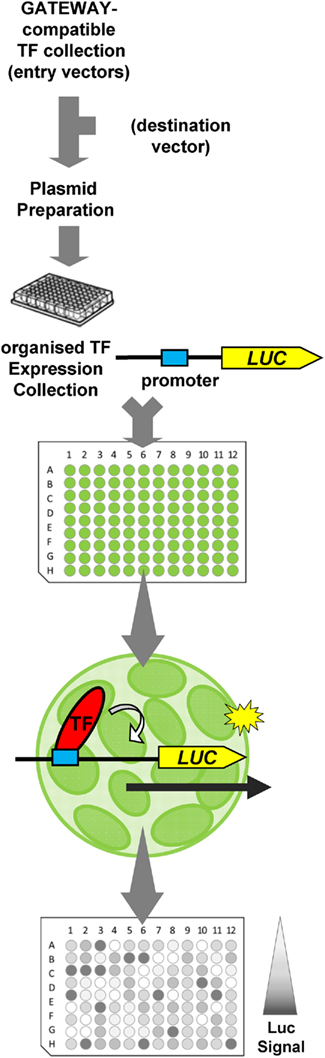
Figure 3. Schematic overview of the protoplast trans activation (PTA) system to identify TFs regulating a Promoter:LUC reporter. For details see Wehner et al. (2011).
Using the GATEWAY® technology, collections of TF ORFs can easily be mobilized into a plant expression vector to enable the expression of HA-tagged TF-fusion proteins in Arabidopsis protoplasts. Screens for operating TFs can be performed by co-transfection of promoter:LUCIFERASE reporters and assaying the TF’s activation or repression potential by luciferase imaging. The measurement is performed in vivo and does not need any cell extraction. Thus this system is suited for automatization or liquid handling by using multichannel pipettes or robotic systems (De Sutter et al., 2005). Currently a screening collection of roughly 850 TF expression vectors is available, which is arrayed in microtiter plates. In contrast to classical Y1H screens no time consuming DNA re-isolation steps are necessary.
As a proof-of-principle several full-length Arabidopsis promoters (up to 1500 bps) have been analyzed in the PTA system (Wehner et al., 2011). Importantly, those TFs which have been described to regulate these promoters could be re-isolated but moreover, several closely related family members, which appear to show redundant transactivation properties are likewise identified. Furthermore, promoters with both, low and high background activities can be used in this screening system.
The PTA system offers a wide range of applications. For instance, it has been shown that activators as well as repressors can be studied (Wehner et al., 2011). Moreover, in contrast to yeast cells, protoplasts provide the necessary perception and signaling system to study signal-induced plant processes. Several stress or hormone treatments such as salt, abscisic acid, auxin, jasmonic acid, or photosynthetic inhibitors have already been successfully applied (Wehner et al., 2011). Finally, by using protoplasts from different sources (e.g., leaves, roots, suspension culture) tissue specific conditions can be taken into account.
Future Perspectives of the PTA System: Identification of Signaling Compounds Which Functionally Interact with Transcription Factors
Transcription factor function requires the interaction with other proteins, e.g., transcriptional regulation often depends on the formation of heterodimers. Protoplast two-hybrid (P2H) approaches have already been successfully established to define in vivo protein–protein interactions (Ehlert et al., 2006; Weltmeier et al., 2006; Böttner et al., 2009). This approach can also be used in the protoplast high-throughput system. Moreover, the involvement of TFs in signaling cascades can be assayed. For instance, TF activity is frequently modulated by phosphorylation (Schütze et al., 2008). Making use of the PTA system, the operating kinases can be identified by co-transformation of a collection of kinase expression vectors. As a proof-of-principle, the functional interplay of the SnRK1 kinase KIN10 (Baena-Gonzalez et al., 2007) and bZIP TFs has already been addressed in the PTA system (Wehner et al., 2011). Furthermore, Y1H assays have been used to screen for TFs interacting with proteins such as co-regulators (Ou et al., 2011). These experiments can also be performed in the PTA system, but in a homologous cellular background. In order to rapidly model plant signaling cascades, TFs and candidate signaling effectors can be co-expressed in specific mutant backgrounds if these plants are used as a source for protoplast preparation. Finally, the high-throughput system might also be applied as a loss-of-function approach. RNAi-based silencing has been demonstrated to work efficiently in protoplasts (Zhai et al., 2009). Hence, GATEWAY® TF–RNAi collections are needed to evaluate the potential of this approach.
Screening Tools to Identify TF Binding Sites
The identification of TF target sites is crucial for deciphering their biological function. Several in vitro selection methods have been proposed to identify a DNA–binding-site for a given recombinant TF protein (Figure 1B). Basically, systematic evolution of ligands by exponential enrichment (SELEX)-type methods (Tuerk and Gold, 1990) have been proposed (Xue, 2005). Recently, Godoy et al. (2011) described a protein-binding microarray (PBM11) containing a saturated collection of double-stranded 11-mers for determining DNA-binding TFs. Although these methods are clearly limited as they assay in vitro interactions, biological relevant TF–DNA interactions have been reproduced. An alternative in vivo bacterial one-hybrid approach using a library of cloned random DNA-binding sites has been successfully applied in the animal field (Meng et al., 2005). However, this tool has not been described for plant TFs, yet.
Several methods are used to identify in vivo DNA-binding sites of TFs, such as the DNA adenine methylation identification (DamID) approach (Germann and Gaudin, 2011). Expression of a TF-fusion with the prokaryotic DNA adenine methyltransferase (Dam) enzyme leads to methylation fingerprints on the DNA which are located in close vicinity to the TF binding-site. These fingerprints can be disclosed by methylation-sensitive restriction enzymes. Chromatin immuno-precipitation (ChIP) combined with tilling array technology (ChIP-chip) or sequencing of immunoprecipitated DNA fragments (ChIP-seq) appear to be straight-forward methods to define TF binding sites in vivo (Fode and Gatz, 2009; Mitsuda and Ohme-Takagi, 2009; Kaufmann et al., 2010; Muino et al., 2011). These methods have successfully been applied in plants, however they are demanding with respect to technical expertise and bioinformatic analysis. Using this approach, highly expressed tagged TFs are easier to study and do not require TF-specific antibodies. However, miss-expression and protein fusions require careful additional analysis to validate the obtained results.
Conclusion
Understanding the transcriptional networks is the final goal of plant TF research. In order to define distinct or partly redundant functions of highly related TF family members, the described screening approaches are valuable additions to the molecular biology tool-box which will significantly speed-up functional analysis of plant TFs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the DFG grant DR273/12-1 within the framework of the Arabidopsis Functional Genomics Network (AFGN) and the University of Würzburg in the funding program “Open Access Publishing.”
References
Baena-Gonzalez, E., Rolland, F., Thevelein, J. M., and Sheen, J. (2007). A central integrator of transcription networks in plant stress and energy signalling. Nature 448, 938–942.
Böttner, S., Iven, T., Carsjens, C. S., and Dröge-Laser, W. (2009). Nuclear accumulation of the ankyrin repeat protein ANK1 enhances the auxin-mediated transcription accomplished by the bZIP transcription factors BZI-1 and BZI-2. Plant J. 58, 914–926.
Castrillo, G., Turck, F., Leveugle, M., Lecharny, A., Carbonero, P., Coupland, G., Paz-Ares, J., and Oñate-Sánchez, L. (2011). Speeding cis-trans regulation discovery by phylogenomic analyses coupled with screenings of an arrayed library of Arabidopsis transcription factors. PLoS ONE 6, e21524. doi:10.1371/journal.pone.0021524
De Sutter, V., Vanderhaeghen, R., Tilleman, S., Lammertyn, F., Vanhoutte, I., Karimi, M., Inze, D., Goossens, A., and Hilson, P. (2005). Exploration of jasmonate signalling via automated and standardized transient expression assays in tobacco cells. Plant J. 44, 1065–1076.
Dobi, K. C., and Winston, F. (2007). Analysis of transcriptional activation at a distance in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 5575–5586.
Ehlert, A., Weltmeier, F., Wang, X., Mayer, C. S., Smeekens, S., Vicente-Carbajosa, J., and Dröge-Laser, W. (2006). Two-hybrid protein-protein interaction analysis in Arabidopsis protoplasts: establishment of a heterodimerization map of group C and group S bZIP transcription factors. Plant J. 46, 890–900.
Fields, S., and Song, O. (1989). A novel genetic system to detect protein-protein interactions. Nature 340, 245–246.
Fode, B., and Gatz, C. (2009). Chromatin immunoprecipitation experiments to investigate in vivo binding of Arabidopsis transcription factors to target sequences. Methods Mol. Biol. 479, 261–272.
Germann, S., and Gaudin, V. (2011). Mapping in vivo protein-DNA interactions in plants by DamID, a DNA adenine methylation-based method. Methods Mol. Biol. 754, 307–321.
Godoy, M., Franco-Zorrilla, J. M., Perez-Perez, J., Oliveros, J. C., Lorenzo, O., and Solano, R. (2011). Improved protein-binding microarrays for the identification of DNA-binding specificities of transcription factors. Plant J. 66, 700–711.
Gong, W., Shen, Y. P., Ma, L. G., Pan, Y., Du, Y. L., Wang, D. H., Yang, J. Y., Hu, L. D., Liu, X. F., Dong, C. X., Ma, L., Chen, Y. H., Yang, X. Y., Gao, Y., Zhu, D., Tan, X., Mu, J. Y., Zhang, D. B., Liu, Y. L., Dinesh-Kumar, S. P., Li, Y., Wang, X. P., Gu, H. Y., Qu, L. J., Bai, S. N., Lu, Y. T., Li, J. Y., Zhao, J. D., Zuo, J., Huang, H., Deng, X. W., and Zhu, Y. X. (2004). Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes. Plant Physiol. 135, 773–782.
Guo, A., He, K., Liu, D., Bai, S., Gu, X., Wei, L., and Luo, J. (2005). DATF: a database of Arabidopsis transcription factors. Bioinformatics 21, 2568–2569.
Heinekamp, T., Kuhlmann, M., Lenk, A., Strathmann, A., and Dröge-Laser, W. (2002). The tobacco bZIP transcription factor BZI-1 binds to G-box elements in the promoters of phenylpropanoid pathway genes in vitro, but it is not involved in their regulation in vivo. Mol. Genet. Genomics 267, 16–26.
Hilson, P. (2006). Cloned sequence repertoires for small- and large-scale biology. Trends Plant Sci. 11, 133–141.
Hiratsu, K., Matsui, K., Koyama, T., and Ohme-Takagi, M. (2003). Dominant repression of target genes by chimeric repressors that include the EAR motif, a repression domain, in Arabidopsis. Plant J. 34, 733–739.
Ichikawa, T., Nakazawa, M., Kawashima, M., Iizumi, H., Kuroda, H., Kondou, Y., Tsuhara, Y., Suzuki, K., Ishikawa, A., Seki, M., Fujita, M., Motohashi, R., Nagata, N., Takagi, T., Shinozaki, K., and Matsui, M. (2006). The FOX hunting system: an alternative gain-of-function gene hunting technique. Plant J. 48, 974–985.
Kaufmann, K., Muino, J. M., Osteras, M., Farinelli, L., Krajewski, P., and Angenent, G. C. (2010). Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat. Protoc. 5, 457–472.
Kondou, Y., Higuchi, M., and Matsui, M. (2010). High-throughput characterization of plant gene functions by using gain-of-function technology. Annu. Rev. Plant Biol. 61, 373–393.
Kuromori, T., Takahashi, S., Kondou, Y., Shinozaki, K., and Matsui, M. (2009). Phenome analysis in plant species using loss-of-function and gain-of-function mutants. Plant Cell Physiol. 50, 1215–1231.
Meng, X., Brodsky, M. H., and Wolfe, S. A. (2005). A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat. Biotechnol. 23, 988–994.
Mitsuda, N., Ikeda, M., Takada, S., Takiguchi, Y., Kondou, Y., Yoshizumi, T., Fujita, M., Shinozaki, K., Matsui, M., and Ohme-Takagi, M. (2010). Efficient yeast one-/two-hybrid screening using a library composed only of transcription factors. Plant Cell Physiol. 2145–2151.
Mitsuda, N., and Ohme-Takagi, M. (2009). Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiol. 50, 1232–1248.
Muino, J. M., Kaufmann, K., Van Ham, R. C., Angenent, G. C., and Krajewski, P. (2011). ChIP-seq analysis in R (CSAR): an R package for the statistical detection of protein-bound genomic regions. Plant Methods 7, 11.
Nakano, T., Suzuki, K., Fujimura, T., and Shinshi, H. (2006). Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 140, 411–432.
Ou, B., Yin, K. Q., Liu, S. N., Yang, Y., Gu, T., Wing Hui, J. M., Zhang, L., Miao, J., Kondou, Y., Matsui, M., Gu, H. Y., and Qu, L. J. (2011). A high-throughput screening system for Arabidopsis transcription factors and its application to med25-dependent transcriptional regulation. Mol. Plant 4, 546–555.
Paz-Ares, J. (2002). REGIA, an EU project on functional genomics of transcription factors from Arabidopsis thaliana. Comp. Funct. Genomics 3, 102–108.
Perez-Rodriguez, P., Riano-Pachon, D. M., Correa, L. G., Rensing, S. A., Kersten, B., and Mueller-Roeber, B. (2010). PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, D822–D827.
Ptashne, M. (2005). Regulation of transcription: from lambda to eukaryotes. Trends Biochem. Sci. 30, 275–279.
Qu, L. J., and Zhu, Y. X. (2006). Transcription factor families in Arabidopsis: major progress and outstanding issues for future research. Curr. Opin. Plant Biol. 9, 544–549.
Ramirez, S. R., and Basu, C. (2009). Comparative analyses of plant transcription factor databases. Curr. Genomics 10, 10–17.
Riechmann, J. L. (2006). “Transcription factors of Arabidopsis and rice: a genomic perspective” in Regulation of Transcription in Plants, ed. K. D. Grasser (Oxford, UK: Blackwell Publishing Ltd), 28–53.
Riechmann, J. L., Heard, J., Martin, G., Reuber, L., Jiang, C., Keddie, J., Adam, L., Pineda, O., Ratcliffe, O. J., Samaha, R. R., Creelman, R., Pilgrim, M., Broun, P., Zhang, J. Z., Ghandehari, D., Sherman, B. K., and Yu, G. (2000). Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290, 2105–2110.
Schütze, K., Harter, K., and Chaban, C. (2008). Post-translational regulation of plant bZIP factors. Trends Plant Sci. 13, 247–255.
Seki, M., and Shinozaki, K. (2009). Functional genomics using RIKEN Arabidopsis thaliana full-length cDNAs. J. Plant Res. 122, 355–366.
Singh, H., Lebowitz, J. H., Baldwin, A. S. Jr., and Sharp, P. A. (1988). Molecular cloning of an enhancer binding protein: isolation by screening of an expression library with a recognition site DNA. Cell 52, 415–423.
Tuerk, C., and Gold, L. (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510.
Wehner, N., Hartmann, L., Ehlert, A., Böttner, S., Onate-Sanchez, L., and Dröge-Laser, W. (2011). High-throughput protoplast trans activation (PTA) system for the analysis of Arabidopsis transcription factor function. Plant J. doi:10.1111/j.1365-1313X.2011.04704.x
Weiste, C., Iven, T., Fischer, U., Onate-Sanchez, L., and Dröge-Laser, W. (2007). In planta ORFeome analysis by large-scale over-expression of GATEWAY-compatible cDNA clones: screening of ERF transcription factors involved in abiotic stress defense. Plant J. 52, 382–390.
Weltmeier, F., Ehlert, A., Mayer, C. S., Dietrich, K., Wang, X., Schütze, K., Alonso, R., Harter, K., Vicente-Carbajosa, J., and Dröge-Laser, W. (2006). Combinatorial control of Arabidopsis proline dehydrogenase transcription by specific heterodimerisation of bZIP transcription factors. EMBO J. 25, 3133–3143.
Xue, G. P. (2005). A CELD-fusion method for rapid determination of the DNA-binding sequence specificity of novel plant DNA-binding proteins. Plant J. 41, 638–649.
Yilmaz, A., Mejia-Guerra, M. K., Kurz, K., Liang, X., Welch, L., and Grotewold, E. (2011). AGRIS: the Arabidopsis gene regulatory information server, an update. Nucleic Acids Res. 39, D1118–D1122.
Keywords: Arabidopsis thaliana, transcription factor function, screening tools
Citation: Wehner N, Weiste C and Dröge-Laser W (2011) Molecular screening tools to study Arabidopsis transcription factors. Front. Plant Sci. 2:68. doi: 10.3389/fpls.2011.00068
Received: 02 September 2011; Paper pending published: 22 September 2011;
Accepted: 08 October 2011; Published online: 04 November 2011.
Edited by:
Andreas P. M. Weber, University of Duesseldorf, GermanyReviewed by:
Kian Hematy, Institut National de la Recherche Agronomique, FranceImre E. Somssich, Max-Planck Institute for Plant Breeding, Germany
Copyright: © 2011 Wehner, Weiste and Dröge-Laser. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Wolfgang Dröge-Laser, Julius-von-Sachs-Institute, University of Würzburg, Julius-von-Sachs-Platz 2, D-97082 Würzburg, Germany. e-mail:d29sZmdhbmcuZHJvZWdlLWxhc2VyQHVuaS13dWVyemJ1cmcuZGU=