 Philippe Leroy1*
Philippe Leroy1* Nicolas Guilhot1
Nicolas Guilhot1 Hiroaki Sakai2 Aurélien Bernard1,3
Hiroaki Sakai2 Aurélien Bernard1,3 Frédéric Choulet1
Frédéric Choulet1 Sébastien Theil1 Sébastien Reboux4 Naoki Amano2,5
Sébastien Theil1 Sébastien Reboux4 Naoki Amano2,5 Timothée Flutre4 Céline Pelegrin1 Hajime Ohyanagi6,7
Timothée Flutre4 Céline Pelegrin1 Hajime Ohyanagi6,7 Michael Seidel8 Franck Giacomoni9 Mathieu Reichstadt10
Michael Seidel8 Franck Giacomoni9 Mathieu Reichstadt10 Michael Alaux4 Emmanuelle Gicquello1 Fabrice Legeai11 Lorenzo Cerutti12 Hisataka Numa2
Michael Alaux4 Emmanuelle Gicquello1 Fabrice Legeai11 Lorenzo Cerutti12 Hisataka Numa2 Tsuyoshi Tanaka2 Klaus Mayer8
Tsuyoshi Tanaka2 Klaus Mayer8 Takeshi Itoh2 Hadi Quesneville4
Takeshi Itoh2 Hadi Quesneville4 Catherine Feuillet1*
Catherine Feuillet1*- 1 UMR 1095, Genetics, Diversity and Ecophysiology of Cereals, Institut National de la Recherche Agronomique-Université Blaise Pascal, Clermont-Ferrand, France
- 2 National Institute of Agrobiological Sciences, Tsukuba, Ibaraki, Japan
- 3 ISEM UMR5554, Institut des Sciences de l’Evolution de Montpellier, Montpellier, France
- 4 UR 1164, Unité de Recherche en Génomique Informatique, Institut National de la Recherche Agronomique, Versailles, France
- 5 Center for iPS Cell Research and Application, Kyoto University, Sakyo-ku Kyoto, Japan
- 6 Tsukuba Division, Mitsubishi Space Software Co., Ltd,. Tsukuba, Ibaraki, Japan
- 7 Plant Genetics Laboratory, National Institute of Genetics, Mishima, Shizuoka, Japan
- 8 Institute of Bioinformatics and System Biology/MIPS, Helmholtz Center Munich, Neuherberg, Germany
- 9 UMR1019, Unité de Recherche en Nutrition Humaine, Institut National de la Recherche Agronomique, Saint-Genès-Champanelle, France
- 10 UR1213, Unité de Recherche sur les Herbivores, Institut National de la Recherche Agronomique, Saint-Genès-Champanelle, France
- 11 UMR 1099, Biologie des Organismes et des Populations appliquée à la Protection des Plantes, Institut National de la Recherche Agronomique, Le Rheu, France
- 12 Swiss Institute of Bioinformatics, Geneva, Switzerland
In support of the international effort to obtain a reference sequence of the bread wheat genome and to provide plant communities dealing with large and complex genomes with a versatile, easy-to-use online automated tool for annotation, we have developed the TriAnnot pipeline. Its modular architecture allows for the annotation and masking of transposable elements, the structural, and functional annotation of protein-coding genes with an evidence-based quality indexing, and the identification of conserved non-coding sequences and molecular markers. The TriAnnot pipeline is parallelized on a 712 CPU computing cluster that can run a 1-Gb sequence annotation in less than 5 days. It is accessible through a web interface for small scale analyses or through a server for large scale annotations. The performance of TriAnnot was evaluated in terms of sensitivity, specificity, and general fitness using curated reference sequence sets from rice and wheat. In less than 8 h, TriAnnot was able to predict more than 83% of the 3,748 CDS from rice chromosome 1 with a fitness of 67.4%. On a set of 12 reference Mb-sized contigs from wheat chromosome 3B, TriAnnot predicted and annotated 93.3% of the genes among which 54% were perfectly identified in accordance with the reference annotation. It also allowed the curation of 12 genes based on new biological evidences, increasing the percentage of perfect gene prediction to 63%. TriAnnot systematically showed a higher fitness than other annotation pipelines that are not improved for wheat. As it is easily adaptable to the annotation of other plant genomes, TriAnnot should become a useful resource for the annotation of large and complex genomes in the future.
Introduction
Achieving a robust structural and functional genome sequence annotation is essential to provide the foundation for further relevant biological studies. Genome annotation consists of identifying and attaching biological information to sequence features. It represents one of the most difficult tasks in genome sequencing projects (Elsik et al., 2006), particularly today where the advent of high-throughput next generation sequencing (NGS) technologies enables genome sequences to be produced at a high pace. The reality at present is that new genomes are being sequenced at a faster rate than they are being fully and correctly annotated (Cantarel et al., 2008). It took about 7 years and a large community effort to sequence and fully annotate the Arabidopsis thaliana (The Arabidopsis Genome Initiative, 2000) and rice genomes (International Rice Genome Sequencing Project, 2005) at a quality that none of the other genome sequenced after have reached yet. In the past 5 years, the production of plant genome sequences has grown exponentially (for a review see Feuillet et al., 2011). On August 2011, the NCBI Entrez Genome Project web site1 listed 135 land plant genome sequencing projects including 36 completed or assembled genomes and 101 in progress. Out of the 36 sequenced genomes, 23 have been released in the past 2 years2. Among those, only two genomes larger than 1 Gb, maize (Schnable et al., 2009) and soybean (Schmutz et al., 2010), have been sequenced and annotated.
Genome annotation is generally a long and recursive process, the difficulty of which increases with the size and complexity of the genome. It relies on a successive combination of software, algorithms, and methods, as well as the availability of accurate and updated sequence databanks. To manage the large amount of data generated by >1 Gb genome size sequencing projects, sequence annotation needs to be automated, i.e., performed through a pipeline that combines all different programs and minimizes subsequent manual curation which is long and laborious. Four categories of pipelines are available to support plant genomes annotation, as follows:
(1) Simple commercial software such as Vector NTI3 and DNASTAR4. Usually, these pipelines are not available on the web and they are not free of charge, even for academic research. Most importantly, they cannot be easily customized for specific needs.
(2) Suites of scripts that generate computational evidence for further manual curation. For example, DAWGPAWS5 (Estill and Bennetzen, 2009) – has been developed for annotating wheat BAC contigs and works as a series of command line programs that result in GFF output files. Such a type of pipeline is not available on the web and can only be used by skilled bioinformaticians.
(3) “In-house” pipelines. A number of these have been developed by communities to annotate model plant genomes, e.g., rice (Ouyang and Buell, 2004; International Rice Genome Sequencing Project, 2005) or by major genomic resource centers such as the DOE/JGI6, the MIPS7, Gramene (Liang et al., 2009)8, GenBank9, and EBI (Curwen et al., 2004)10. Although these pipelines are of high quality and are generally based on massive informatics resources, they are not directly accessible to users from outside. In general, these genomic and bioinformatics platforms have their own projects and priorities.
(4) Automated annotation pipelines available on the web. The first pipeline of this kind, RiceGAAS (Sakata et al., 2002) was developed originally for the annotation of the rice genome. Since then a few others have been established such as DNA subway (iPlant, USA)11, FPGP (Amano et al., 2010) and MAKER (Cantarel et al., 2008). They all have web user-friendly interfaces; however, the online access limits the capacity to perform annotation of large genomes within a reasonable time. Thus, until now, none of the publicly available, online pipelines enables a thorough annotation of large genome sequences.
The International Wheat Genome Sequencing Consortium (IWGSC)12 was launched in 2005 with the aim of achieving a reference sequence for the hexaploid (2n = 6× = 42, AABBDD) bread wheat cultivar Chinese Spring genome. The strategy established by the IWGSC follows a chromosome-based approach that relies on the physical mapping and minimal tiling path (MTP) sequencing of each of the 21 individual chromosomes of bread wheat (Feuillet and Eversole, 2007). The first physical map of a wheat chromosome was established in our laboratory in 2008 for the 1-Gb chromosome 3B (Paux et al., 2008). A MTP comprising 8,448 BAC clones and 1,282 contigs has been designed and is used currently to obtain a reference sequence with NGS technologies13. Wheat chromosome sizes range from 600 Mb to 1 Gb (Doležel et al., 2009) and therefore, even with a chromosome-based approach, the annotation of the 17-Gb of the hexaploid wheat genome represents a major bioinformatics challenge. Previous work showed that the wheat genome consists of about 90% of transposable elements (TEs; Flavell et al., 1977; Li et al., 2004; Paux et al., 2006) with less than 10 families representing more than 50% of the TEs (Choulet et al., 2010). TEs are increasingly recognized for their key role in evolutionary changes, regulatory innovation. They are no longer considered “junk DNA,” the annotation of which is not relevant and should simply be “masked” for further gene identification. Therefore, bioinformatics tools, such as REPET (Quesneville et al., 2005), that specifically aim at annotating TEs are needed for TE-rich genomes like wheat. It has also become clear that genes are found all along the wheat chromosomes (Devos et al., 2005; Rustenholz et al., 2010) and are embedded in the form of very small islands of two to three genes on average in the TE matrix (Choulet et al., 2010). Finally, the increasing recognition that small non-coding RNAs (ncRNAs) are key molecules in the regulation of various biological processes in plants (Bonnet et al., 2006; Meyers et al., 2008a,b) has triggered efforts to improve their annotation in genome sequencing projects (Meyers et al., 2008a). Thus, if we want to efficiently and accurately relate genome annotation to biological functions and phenotypes in wheat, genome annotation should not only focus on the prediction and annotation of “genes” and low copy sequences but should also provide an accurate annotation of TEs and other non-protein-coding features.
To support the annotation of the wheat genome as well as to provide other communities coping with large and complex genomes with a useful resource for annotation, we wanted to develop an automated annotation pipeline that: (1) enables rapid and robust structural and functional annotation of genes as well as of TEs and protein non-coding features; (2) is versatile, i.e., is accessible through a user-friendly web interface to allow for the rapid analysis of a few hundred BAC clones/contigs, but can also accommodate large genome scale projects; and (3) provides output files that can be retrieved easily or visualized directly on a web interface. Moreover, to ensure an efficient use of the sequence information, we wanted the annotation to be linked to databases containing genetic and physical maps, markers, genes, and QTL, phenotypes, “omics” data, etc. Since none of the previously mentioned pipelines met all these criteria, we developed a new pipeline called “TriAnnot” with the aim of integrating the best features of different pipelines and linking a versatile system to the integrated wheat databases established at the INRA URGI (GnpIS)14. Here, we provide a detailed description of the features of the TriAnnot V3.5 pipeline15, an evaluation of its performance through the annotation of curated reference sequence sets from wheat and rice, and the comparison of the gene annotation fitness in term of sensitivity (Sn) and specificity (Sp) with other well known annotation pipelines.
Results
General Architecture of the TriAnnot Pipeline
The general architecture is modular and easily customizable using an xml formatted file (step.xml). It consists of four main panels (Figure 1): Panel I for TEs annotation and masking; Panel II for structural and functional annotation of protein-coding genes; Panel III for the identification of ncRNA genes and conserved non-coding sequences; and, Panel IV for molecular markers development. Each panel is divided into different modules or steps that correspond to a bioinformatics program (see Table S1 in Supplementary Material for a description of each module).
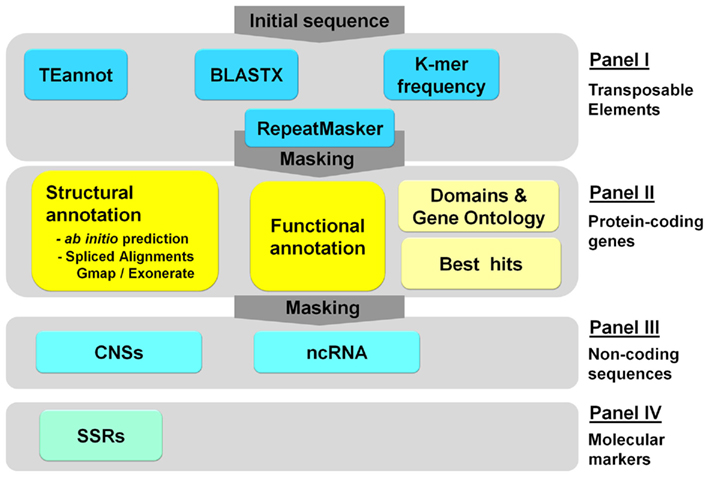
Figure 1. An overview of the workflow supported by the TriAnnot pipeline V3.5. The four main panels are displayed. Each panel contains modules and each module can use one or more bioinformatics programs and databanks. The detailed description of each panel and module is provided in the text. CNSs, conserved non-coding sequences; ncRNA, non-coding RNA; SSRs, simple sequence repeats or microsatellites; TEannot, pipeline for transposable elements annotation (REPET package – Quesneville et al., 2005).
Panel I – transposable elements
Three strategies are followed to annotate the TEs. First, TriAnnot uses a sophisticated approach based on TEannot which is part of the REPET package developed by Quesneville et al. (2005). The main utility of TEannot is that it links segmental portions of TEs that are fragmented into several pieces through the insertions of other elements, thereby allowing the analysis of the nested pattern of TEs in wheat (Flutre et al., 2011). TriAnnot follows the guideline and the three-letters code of Wicker et al. (2007) for the classification of TEs. The second approach is based on a classical similarity search performed by RepeatMasker (Smit, 1993) against the TREP databank (Wicker et al., 2002) and “in-house” annotated TEs (Choulet et al., 2010). Seven other repeat databanks are also available for more exhaustive analyses (Tables S1 and S2 in Supplementary Material). Subsequently, TriAnnot performs a similarity search at the protein level using BLASTX against TREPprot16. In a third complementary approach, TriAnnot uses the k-mer composition to mask repeated regions using an Mathematically Defined Repeats index of 17-mer frequency that was computed with Tallymer (Kurtz et al., 2008) on an Illumina reads sample representing 2× coverage of sorted chromosome 3B (Choulet et al., 2010). With this index, TriAnnot masks highly repeated 17-mers within a query sequence. Eventually, Panel I produces soft and hard-masked sequences that are further analyzed in Panel II and, a graph of the k-mer frequency along the sequence that can be displayed under the graphical viewer ARTEMIS (Carver et al., 2008).
Panel II – structural and functional annotation of protein-coding genes
Structural annotation. Exon–intron structures and the protein-coding sequences (CDS) can be predicted ab initio, by sequence similarity, or through a combination of the two approaches. TriAnnot follows these three strategies. For ab initio gene prediction, TriAnnot uses four programs: FGeneSH17, GeneID (Guigo et al., 1992), GeneMarkHMM (Lukashin and Borodovsky, 1998; Lomsadze et al., 2005), and augustus (Stanke and Waack, 2003). Because of the lack of training dataset, none of these predictors has been trained specifically for wheat. Only, FGeneSH has been trained for monocotyledons. The TriAnnot pipeline can launch each of these programs either on the initial sequence or on the TE-masked sequence obtained after Panel I analysis. Currently, augustus is emphasized within the TriAnnot pipeline as it gives the best specificity/sensitivity ratio (see evaluation section below). Similarity approaches, based on BLAST (Altschul et al., 1997), can also be performed on the initial sequence or on the TE-masked sequence following a two-step methodology. First, BLASTN and BLASTX are used to find significant similarities within transcript and protein databanks, respectively. TriAnnot currently uses 73 databanks (Table S2 in Supplementary Material) that are updated twice a year. Then, BLAST hit sequences are retrieved and aligned against the sequence using exonerate (Slater and Birney, 2005) for proteins and transcripts or Gmap (Wu and Watanabe, 2005) for transcripts only. These two programs compute spliced alignments to identify exon/intron junctions precisely.
The outputs of the ab initio and similarity search analyses are then used to perform gene modeling following two strategies. The first one relies on SIMsearch, a gene modeling program based on FPGP (Amano et al., 2010) that was developed specifically for the TriAnnot pipeline. SIMsearch follows five main steps to build a gene model:
• Step 1: BLASTN (≥80% nucleotide identity and ≥80% nucleotide coverage) is performed against a databank (SIMnuc) comprising plant FL-cDNAs and CDSs from grass genomes.
• Step 2: BLASTN hit sequences are retrieved and a spliced alignment against the sequence is produced with est2genome (Mott, 1997).
• Step 3: BLASTX is performed against the SIMprot databank which is composed of refSeqPlantProt (from NCBI), proteins derived from the annotation of Oryza sativa (IRGSP) and Brachypodium distachyon as well as proteomes of Hordeum and Triticum species. The best hit is used by SIMsearch to define an Open Reading Frame (ORF). If start and/or stop codons cannot be found within the aligned region, the ORF is extended in both 5′ and 3′ directions as described by Amano et al. (2010). If no protein hit is found, then SIMsearch can use a relevant ab initio prediction to predict the ORF. Homologous hits without initiation and/or termination codon or for which no ab initio prediction can be found are discarded.
• Step 4: The best gene model is defined using a priority list (gene coverage, gene identity, category of source transcript, mapped region of the transcript, number of exon, CDS length, and amino acid identity). NB: The present version of TriAnnot does not display yet alternative spliced transcripts variants.
The second strategy uses the gene combiner EuGene (Schiex et al., 2001). In the current version of TriAnnot, EuGene combines augustus predictions (with a wheat matrix) with spliced alignments of wheat-ESTs, SIMnuc, and SIMprot generated by exonerate.
Six categories of gene models have been defined to reflect the reliability of the predictions and provide a quality index to the annotator. Categories 0–3 correspond to similarity search with SIMsearch based on the following biological evidence:
○ Cat0: mRNA of gene manually curated from previous wheat genome annotation,
○ Cat1: Triticum and Aegilops Full-length cDNAs,
○ Cat2: Poaceae Full-length cDNAs,
○ Cat3: CDS from O. sativa (IRGSP) and B. distachyon genomes annotation.
The gene models predicted by EuGene belong to Category 4 (Cat4) whereas ab initio predictions fall into Category 5 (Cat5).
In a final step, the gene models predicted by the ab initio program, SIMsearch and EuGene are merged using a “Merge” program in a stepwise manner which retains EuGene models that do not overlap with SIMsearch models and ab initio models that do not overlap with either SIMsearch or EuGene models. “Merge” also prioritizes the different categories of prediction obtained in the previous steps with the following order: Cat0 > Cat1 > Cat2 > Cat3 > Cat4 > Cat5. If a gene is identified in two categories, e.g., Cat1 and Cat4, then the Cat1 gene prediction is kept and the Cat4 that relies on less solid biological evidence is discarded. To provide users with a representation of the quality index for the gene prediction, TriAnnot displays a color coded system in which each of the above mentioned six categories is symbolized with a specific color (Figure 2). The gene models are soft-masked for further analysis in Panel III.
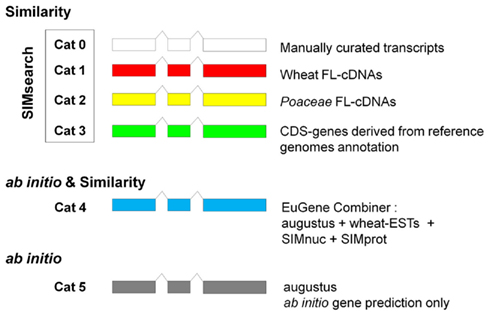
Figure 2. Color coded system established to provide a quality index for the gene annotation in TriAnnot. Six categories (Cat0–Cat5) have been defined depending on the approach and the biological evidences used for the analysis. FL-cDNAs, full-length cDNAs. The SMInuc and SIMprot databanks are described in more details Table S2 in Supplementary Material.
Functional annotation. Putative function for the gene models are assigned via a combination of similarity search (BLASTP) against several protein databanks and against the Pfam (Sammut et al., 2008; Finn et al., 2010) protein domain collection with HMMER 3.018. TriAnnot follows a nomenclature based on the guideline established in 2006 by the IWGSC annotation working group19:
• “known function”: when >80% identity over >80% of the protein length is found with a known protein in UniProtKB/Swiss-Prot. This category reflects the highest quality for functional annotation.
• “putative function”: when >45% similarity over >50% of the protein length is found with a known protein in UniProtKB/Swiss-Prot and UniProtKB/TrEMBL.
• “domain-containing-protein”: when there is no significant BLASTP hit with a known or putative function in the previous steps, but one or more Pfam domains (Sammut et al., 2008; Finn et al., 2010) are identified.
• “expressed sequence”: based on TBLASTN against plant EST databanks with >45% identity and >50% coverage.
• “conserved-unknown function”: when no expressed sequence is found, and when >45% similarity over >50% of the protein length is found only with an unknown function (i.e., a protein annotated as “putative” or “hypothetical”) in UniProtKB/Swiss-Prot and UniProtKB/TrEMBL.
• “hypothetical protein”: when no similarity is found, either in UniProtKB/Swiss-Prot or UniProtKB/TrEMBL, or Pfam domain or ESTs.
In addition, TriAnnot provides Gene Ontology (GO) terms20 for each gene model and protein domain predictions based on InterProScan (Zdobnov and Apweiler, 2001) search against Pfam (Sammut et al., 2008; Finn et al., 2010), Prosite (Sigrist et al., 2010), and SMART (Letunic et al., 2009).
Identification of homologous proteins in other plant species. Comparative sequence analysis of genomic regions from related species can greatly support gene identification in the annotation process. For all gene models, TriAnnot searches for the best BLASTP hit with plant proteomes including A. thaliana, O. sativa (IRSGP annotations), Zea mays, Sorghum bicolor, B. distachyon, and Saccharum officinarum as well as with the NCBI non-redundant protein databank (nr; Table S2 in Supplementary Material). In addition, the alignment with the best hit is parsed in order to check for the presence of gaps (>9 amino acids) that can reveal missing or additional exons in the gene model compared to its homolog.
Panel III – identification of non-coding RNA genes and conserved non-coding sequences
ncRNAs. TriAnnot allows for the identification of other sequence features based on specific bioinformatics programs such as tRNAscan (Lowe and Eddy, 1997). This module will be completed by programs for the identification of small non-coding RNAs (siRNA, miRNA) by rnaspace21 in the next version of TriAnnot.
Conserved non-coding sequences. TriAnnot is also seeking for other sequence features based on comparative genomics using BLASTN/BLASTX search similarities against major plant genomes (Arabidopsis, Oryza, Zea, Sorghum, Brachypodium). This similarity search is performed on un-annotated portions of the query sequence (hard-masked for TEs and gene models).This module also allows identifying pseudogenes using BLASTX against public protein databanks and searches against the plastids and mitochondrial genomes (Table S2 in Supplementary Material) to identify fragment of such sequences integrated into the nuclear genomes.
Panel IV – marker design
Simple sequence repeats (SSR) or microsatellites have been extensively used for molecular marker design in plants (Paux and Sourdille, 2009). In wheat, their density was estimated to one SSR every 13.1 kb (Choulet et al., 2010). TriAnnot uses the TRF program (Tandem Repeats Finder; Benson, 1999) with specific parameters to enhance the finding of such repeats (Table S1 in Supplementary Material). This will be complemented with other marker type detection modules.
TriAnnot Runs on a Parallel Computing Environment
To deal with the annotation of Gb-sized sequences, such as the 1-Gb wheat chromosome 3B, and thereafter the annotation of the remaining 20 wheat chromosomes under the umbrella of IWGSC22, the architecture of the pipeline is oriented toward parallel computing. To speed up the annotation process, the pipeline executes parallel tasks taking into consideration task dependencies so that the pipeline can manage a logical data flow. It reads a Tasks list XML file that defines the list of tasks to be executed and enters the main loop until each task is completed (Figure 3). For job monitoring, the master program (MP) relies on the REPET Application Programming Interface which uses a MySQL database to exchange status information between jobs and the MP. When all dependencies are satisfied for a given task, the MP submits a Program Launcher Job to the cluster. When the Program Launcher results are available, the MP submits a Parser Launcher Job to the cluster which generates GFF and EMBL files. Both Program and Parser launcher jobs update their status in the MySQL database and generate an XML Result file. This file gives detailed information about the task execution status (e.g., CPU and memory usage, created files, execution/parsing results…). Along the process, the MP constantly checks the status of each submitted job (waiting for execution in the cluster queue, running on a computing node, finished or failed). In case of failure, since errors are reported in the MySQL database or in the Result file, it becomes possible to resume the pipeline at the exact step where it failed instead of launching the entire analysis again. This contributes to the quality and efficiency of the pipeline. At present, the TriAnnot pipeline runs on a high-throughput cluster composed of 712 CPU representing 8.5 Tflops that enabled the annotation of 96 fragments of 200 Kb of the wheat genome (145 genes; Choulet et al., 2010) in less than 5 h with a default analysis step.xml file (Table S3 in Supplementary Material) and in less than 7 h with a full analysis (Table S1 in Supplementary Material). With this, the automatic structural and functional annotation of the whole 3B chromosome, representing 1 Gb scattered into ~16,000 scaffolds, has been performed in less than 5 days.
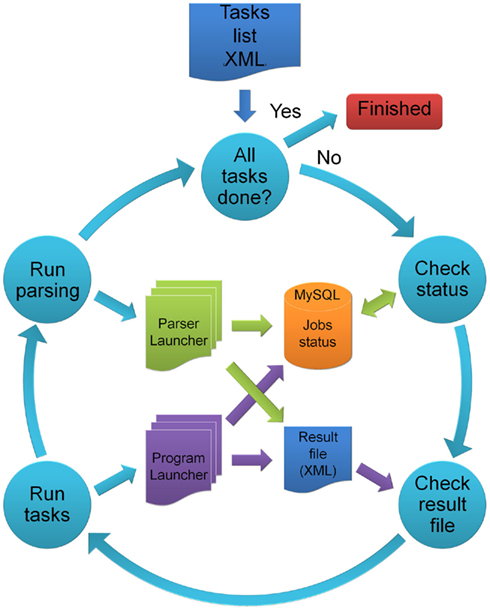
Figure 3. Schematic representation of the master program (MP). “Tasks list”: list of tasks to be executed and their parameters (XML file). Each task may depend on the results produced by a preceding task and this information is also specified in the XML file. When all the dependencies are satisfied for a given task, it is submitted to the computing cluster by running a “Program Launcher” job (Run tasks). When the “Program Launcher” is completed, a “Parser Launcher” job is submitted (Run parsing) to generate GFF and EMBL files from the program output. These scripts update their status in a MySQL database and write XML files to summarize the execution result. The main program checks both the database (Check Status) and the result files (Check result files) to monitor running jobs. When all tasks are completed, the master program ends the pipeline (Finished).
TriAnnot Can be Used for Small and Large Scale Analyses
The TriAnnot pipeline can be accessed at http://www.clermont.inra.fr/triannot/with a login and password that is provided, for server security reasons, after the signature of an “Agreement and Access Rights” document23.
In principle, the pipeline can be used to annotate full genomes. However for technical reasons and parallelization purposes, the upper limit for submitting a sequence at once is set to 3 Mb in the current version. Annotating several Mb or Gb of sequence this way would be cumbersome and therefore, the online access is more adapted to small scale analyses (i.e., BAC or small BAC contigs) in which the user can submit its sequence directly on the webpage (copy/paste or download) and start the analysis with a single click. In this configuration, TriAnnot can deliver a BAC annotation in less than 1 h.
Large datasets (>10 Mb) can be uploaded, upon request todHJpYW5ub3Qtc3VwcG9ydEBjbGVybW9udC5pbnJhLmZyLA== in a specific repository on the cluster at URGI (Figure 4). A simple program launcher is then used to launch the TriAnnot pipeline on the parallelized environment. In this case, pending that all nodes are available, 1 Gb of sequence can be analyzed in less than 5 days.
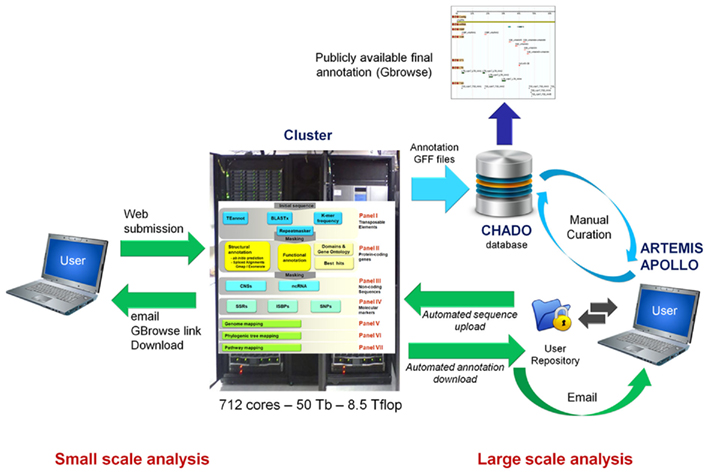
Figure 4. Schematic representation of the different options to access and use TriAnnot for genome sequence annotation. The process for small scale analyses (individual BACs or a few BAC contigs) that are performed directly on the web is represented on the left hand side. The process that enables large scale analysis (several thousand of sequences) through the automated download and annotation with direct manual curation in a CHADO database is described on the right hand side. The curation can be performed either with ARTEMIS, GenomeView or APOLLO graphical editors. Curated annotation can then be displayed with a GBrowse graphical viewer through internet. The future architecture of the pipeline with seven panels is represented on the Cluster.
Once the analysis is completed, an email containing links to download all output files (EMBL and GFF files, masked sequences, best hit alignments, gene model and translated sequences) and visualize the annotation in GBrowse is sent to the user (Figure 4). Finally, a log file summarizing the entire pipeline process is provided for traceability. The GFF files are in a format suitable for further integration into a CHADO database (Zhou et al., 2006; Figure 4). The first line of each GFF file contains information about the databanks and software versions used during the analysis. The EMBL files are suitable for manual curation under ARTEMIS (Carver et al., 2008) and GenomeView24. The GBrowse has been configured to display nine tracks based on the default analysis (Figure 5):
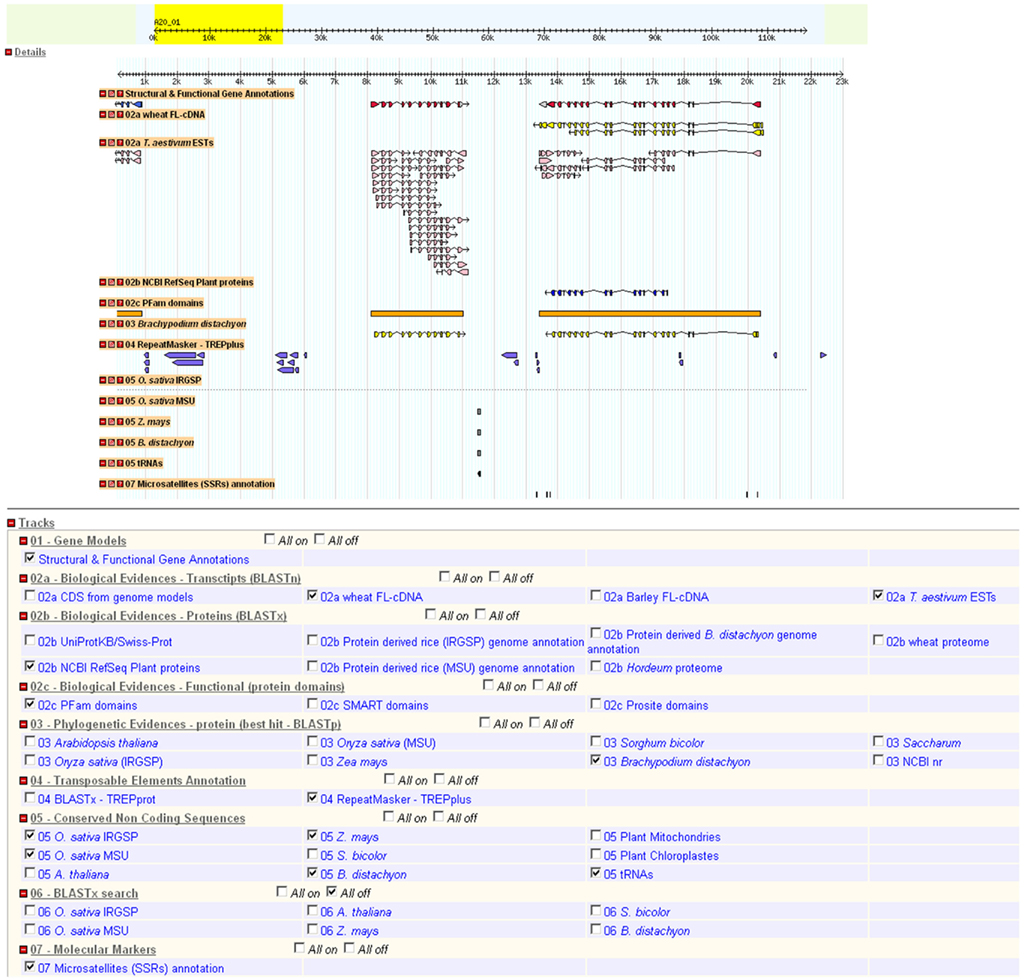
Figure 5. GBrowse graphical display of a 117-kb sequence scaffold from the wheat chromosome 3B. The upper part shows the sequence and the window corresponding to the region for which annotation features are displayed in the central part. The bottom part presents the different databases that are used for the annotation. The ticked boxes indicate the databases that were used for the annotation of this sequence. The “Structural and Functional Gene Annotations” track represents the final gene models with the six color index categories described in Figure 2. All other tracks are biological or ab initio evidences. The GBrowse display is available only for a default analysis.
– 1. Gene models (with the confidence color code),
– 2a,b,c. Biological evidences,
– 3. Best hits in related species,
– 4. TEs,
– 5. Conserved non-coding sequences, tRNAs and organelle-like sequences,
– 6. BLASTX search,
– 7. Molecular markers.
Gbrowse allows the user to retrieve individual features such as gene, mRNA, CDS, or protein sequences for further analyses. The results are available online for 15 days.
The code of TriAnnot (Perl and Python) is available upon request and groups can choose to install the program in-house instead of running the analysis on the URGI server. However, such installation may require extensive skills in informatics and bioinformatics. INRA will not be able to provide technical support for the installation except in the framework of formal collaborations.
Evaluation of TriAnnot Performances
Evaluation of TriAnnot using a wheat curated dataset
A reference dataset of 145 manually curated genes, carried by 96 fragments of 200 Kb belonging to 12 contigs of the wheat chromosome 3B (Choulet et al., 2010), was used to evaluate the accuracy of the TriAnnot gene predictions. The CDS coordinates were checked with the Eval software (Keibler and Brent, 2003) that estimates the specificity (Sp) and the sensitivity (Sn) of the gene predictions. They are defined as: Sp = TP/(TP + FP) and Sn = TP/(TP + FN) where TP are true positives (a reference gene which is predicted with exact CDS coordinates), FP are false positives (a predicted gene the CDS coordinates of which are not exact or a predicted gene that does not correspond to a reference gene), and FN are false negatives (a reference gene which is not predicted or a predicted gene that does not correspond to a reference gene). This mode of calculation ensures that the Sn and Sp values never exceed 100%. Sp and Sn are calculated systematically for genes (SnG, SpG) and exons (SnE, SpE). Both then are considered to calculate a fitness value defined as Ft = (SnG × SpG × SnE × SpE)0.25.
In a first analysis, we wanted to evaluate the accuracy of TriAnnot, i.e., the capacity to identify correctly the 145 manually predicted genes. All additional predictions (FPs) were not considered. The results reveal that 80 genes (∼55%) were annotated correctly by TriAnnot (TP genes). Among them, 47 (58.7%) belong to Cat1; 19 (23.7%) to Cat2; 6 (7.5%) to Cat3; 2 (0.02%) to Cat4, and 6 (7.5%) to Cat5. In addition, 55 genes (∼38%) were predicted but with inconsistencies in their structure compared to the reference annotation. They were considered as FP and FN. Finally, 10 genes (∼7%) were missing in the TriAnnot predictions and were considered as FN. With 80 TP, 55 FP, and 65 FN, the sensitivity (Sn) and the specificity (Sp) at the gene level were of 55 and 59%, respectively. New biological evidence enabled us to modify the manual reference annotation for 12 genes among the 55 FPs and consider them as TP genes. Taking these into account, the number of TP genes is 92 (∼63.0%) leading to Sn and Sp values, at the gene level, of 63 and 68% respectively. Thus, in total, more than 93% of the 145 reference genes were identified by the TriAnnot pipeline including ∼30% that showed discrepancies (ATG, intron/exon junction, number of exon) with the reference annotation. These results made us confident that the TriAnnot pipeline delivers a robust automated annotation.
In a second analysis, we evaluated the performance of TriAnnot compared to that of three other pipelines (MIPS, RiceGAAS and FPGP) that were used for the annotation of other plant species (rice, Brachypodium…) and therefore, were not optimized for wheat. For this analysis, all FP genes were taken into account to enable the assessment of specificity. RiceGAAS predicted the highest number of genes (848) and the lowest fitness (22.9%) of all (Table 1). This is because this pipeline relies mostly on ab initio predictions obtained with gene predictors that are not trained for wheat but rice. The TriAnnot SIMsearch module was derived from FPGP (Amano et al., 2010) and adapted to wheat. The results show that SIMsearch has a higher specificity resulting in a higher fitness (63.7 versus 45.8%) than FPGP demonstrating that it is well adapted to wheat. Finally, comparisons between TriAnnot and the MIPS pipeline that also combines ab initio gene predictions and similarity searches, showed that the TriAnnot annotation results in a higher fitness (49.5 versus 40.3%; Table 1). The main difference is likely the result of the higher sensitivity and specificity at the gene and exon levels provided by the SIMsearch module which is specifically adapted to wheat. In all cases, TriAnnot found more true positives than the other pipelines (Table 1). Thus, we conclude that by using an optimized pipeline with trained algorithms and adapted sequence resources, TriAnnot is a powerful and robust pipeline for the automated annotation of the wheat genome sequence with potential application to other genomes.
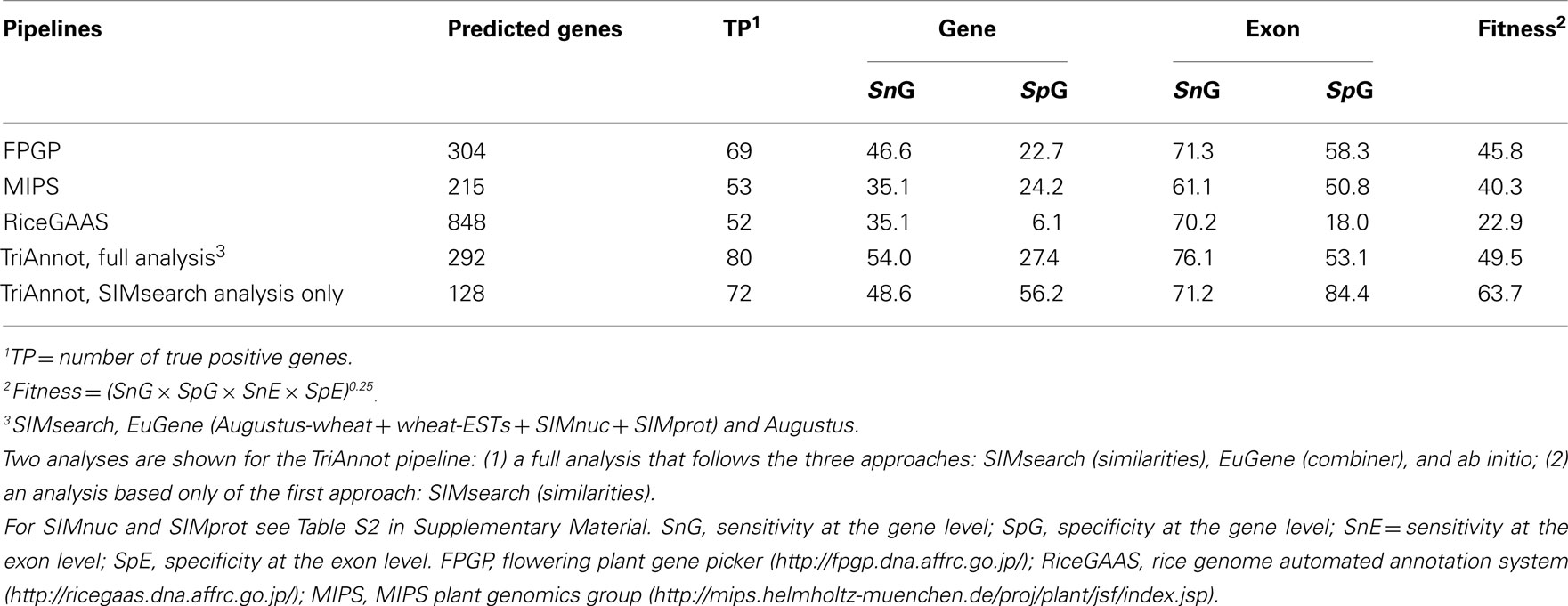
Table 1. Comparisons of the fitness of TriAnnot with other well known annotation pipelines based on a reference dataset containing 145 genes (17.9 Mb of wheat chromosome 3B).
Re-annotation of rice chromosome 1 using TriAnnot
To confirm the robustness of TriAnnot and demonstrate its potential for application to other plant genomes, we wanted to evaluate the performance of the pipeline on a reference genome sequence. For this analysis, we selected rice chromosome 1 (∼45 Mb) and used the IRGSP/RAP build5 as a reference sequence (released on December 2009, last updated on August 2010). The comparison was performed using the 4,848 “representative” gene models (RAP3_locus_chr01.gff3) that correspond to evidence-based models. The 1,138 “predicted” gene models (predicted_orf_chrom01.fna) that correspond only to ab initio predictions were excluded (masked). The IRGSP/RAP build5 dataset gives several spliced predicted variants for a given gene (837 genes have more than one mRNA) and here, the longest mRNA was selected as a reference. In addition, we observed that 207 “genes” had no CDS (annotated as non-protein-coding gene or transcript) while 9 genes contained at least one exon corresponding to a single nucleotide. These genes were removed resulting in 4,632 “representative” gene models that were used as a reference for the TriAnnot analyses. A first analysis was performed in optimal conditions, i.e., with all rice databanks including the reference annotation. Similarity search (SIMsearch module) was performed with the rice and Poaceae FL-cDNA, annotated CDS from genome annotation of rice (IRGSP and MSU) and Brachypodium, NCBI RefSeq protein databank and, the rice proteome and proteins derived from the IRGSP and MSU annotations. ab initio gene prediction was performed using augustus with a maize matrix (no rice matrix available). Finally, the combined analysis was performed with EuGene using the above mentioned databanks and rice ESTs. A second analysis was performed without the IRGSP build5 (i.e., without CDS and protein derived from rice IRGSP and MSU genome annotations). Sensitivity (Sn) and Specificity (Sp) of the two analyses were evaluated using Eval as described previously for the wheat data (Table 1).
Out of the 4,632 representative gene models, TriAnnot predicted 3,885 and 3,387 genes in analysis 1 and 2, respectively (Table 2). As expected, less genes (∼500) were predicted with analysis 2 compared to analysis 1, resulting in less true positive genes: 2,050 in analysis 2 versus 2,368 in analysis 1. Interestingly, the main impact concerned the sensitivity, the specificity remaining almost the same in both analyses (Table 2). The fitness was of 66.2% for analysis 1 and 62.3% for analysis 2 (Table 2). To determine the origin of the discrepancy between the results obtained by TriAnnot in analysis 1 and the IRGSP/RAP build5 dataset, we re-examined the 4,632 “representative” rice gene models. Among those, 862 derived-proteins showed inconsistencies: 50 had no start and stop codons, 86 had a start codon but no stop codon, and 726 had a stop codon without a start codon and likely correspond to pseudogenes. Because TriAnnot does not annotate pseudogenes automatically, the pipeline could not predict these 862 genes. In addition, 22 genes appeared to correspond to TEs. After removal of these 884 “genes,” the rice dataset comprised 3,748 genes of which 3,121 (83.3%) were predicted by TriAnnot. 2,017 (53.8%) of them were predicted with perfect coordinates. It is not possible to determine the exact number of not perfectly predicted genes since Eval does not distinguish them from missing or additional genes. It is likely that this number is close to the ∼40% observed in the wheat analysis. All together, these results demonstrate that TriAnnot can be used efficiently to annotate and curate genome sequence from other plant species.

Table 2. Evaluation of the TriAnnot fitness for the annotation of rice chromosome 1 using the IRGSP/RAP build5 dataset.
Discussion and Perspectives
TriAnnot Provides a Versatile Resource for Large Genome Sequence Annotation
The TriAnnot project aimed at developing an annotation pipeline with architectural and computing capacities that enable the efficient automated annotation of large and complex genomes and that could be adapted to different scales of analysis. The largest plant genome sequenced and annotated to date is the 2.5-Gb maize genome (Schnable et al., 2009). In this case, the annotation was not performed using a single automated pipeline but through a large series of individual programs dedicated to specific features. For example, the TEs fraction that represents the majority of the maize sequence was annotated either by iterative BLAST searches to identify and mask highly represented families, or through searches with individual programs for specific elements (Helitrons, LINES, MULES, or LTR retrotransposons). The sequences were masked using the MIPs REdat v4.3 library and used to predict genes with a combination of the Gramene evidence-based gene build pipeline and/or FGeneSH ab initio predictions (Schnable et al., 2009). With the ongoing revolution in sequencing technologies, it is now feasible to sequence de novo >3 Gb genomes at reasonable costs. While whole genome approaches remain problematic for large and complex genomes, reference sequences can be obtained using BAC pools of a MTP thereby reducing cost without losing essential information (Rounsley et al., 2009). Thus, it is very likely that in the next few years, de novo sequencing of large genomes will become more popular and will be performed by groups that may not be as large as the consortia which sequenced the rice and maize genomes. Even in cases where large sequencing centers produce the sequence, international collaborative projects in which individual groups want to perform and monitor the annotation personally will take place. This is already underway for the wheat genome sequencing project in which individual laboratories are in charge of individual chromosomes25. Annotation remains a challenge for wheat chromosomes that are each two to three times larger than any model plant genome sequenced thus far and the cluster-based version of TriAnnot with its capacity to analyze 1 Gb of sequence in less than a week will greatly support the annotation of the wheat genome. Already, this version of TriAnnot is being utilized to annotate the chromosome 3B sequence and is available for other groups worldwide.
TriAnnot is not only limited to annotating the wheat genome. As demonstrated with the re-annotation of rice chromosome 1, the TriAnnot pipeline can be used for other species with good performances. First, it can be used to quickly re-annotate reference sequences taking the advantage of new biological evidence that are present in the databanks used by TriAnnot (updated regularly) and were not available to the communities at the time of the reference annotation release. Second, and most importantly, TriAnnot can be adapted for the de novo annotation of new genomes. In this case, optimal annotation will be obtained if predictors can be trained with the specific datasets and the related databanks are fed into TriAnnot.
TriAnnot V4: Getting Better, Broader, Deeper, and Faster
Improving annotation
TEannot is one of the unique features of TriAnnot compared to other pipelines for TEs annotation. To date, it is performing well on the Drosophila and Arabidopsis model genomes (Flutre et al., 2011) but it needs to be further improved to cope with the complexity of the nested TE organization in the wheat genome. TEannot belongs to the REPET package (Quesneville et al., 2005) together with TEdenovo, another pipeline that identifies new TEs families (Flutre et al., 2011). TEdenovo will be used on the wheat chromosome 3B sequence to implement a dedicated databank (TREPcons) that will be utilized to improve the accuracy of TEannot for TEs annotation and masking in wheat.
TriAnnot uses homology-based methods, gene prediction and a combination of the two to provide a single gene model with a priority given to homology searches against biological evidences. This and the quality index that is attached with the annotation to provide the biologists with information about the type of evidence which supports the gene models are other unique features of TriAnnot compared to existing pipelines. Although the evaluation results indicate that TriAnnot is providing a robust automated annotation, it can still be improved to increase the sensitivity and, most importantly, boost the specificity by reducing the amount of FPs. The main improvement will come from enhanced training of the ab initio predictors augustus and EuGene. EuGene is a powerful ab initio predictor that efficiently combines biological evidences. It has been used for the annotation of A. thaliana (Moskal et al., 2007), Medicago truncatula26, Theobroma cacao (Argout et al., 2011), the brown algae Ectocarpus (Cock et al., 2010), and Ostreococcus tauri (Derelle et al., 2006). Augustus (Stanke and Waack, 2003) also combines ab initio predictions and biological evidences and it has been used to annotate genomes such as Aspergillus sojae (Sato et al., 2011) and Schistosoma japonicum (Brejova et al., 2009). As few wheat genomic sequences were available in the public databases until now, a relevant training dataset, e.g., with more than 300 representative genes, could not be established and the performances of these two predictors have been limited. Currently, augustus is used only as an ab initio gene prediction program and EuGene only as a combiner. With the genomic sequences that will soon be available from the chromosome 3B (1 Gb) sequencing project27 and the transcript sequences that are available already for wheat (17,525 FL-cDNA (NCBI/EBI and Riken) + 1,067,223 EST), training sets will be created and a “EuGene-wheat” and an “augustus-wheat” will be established. After training and evaluation, the best combiner will be selected eventually and used as the main program in future versions of TriAnnot. While TriAnnot V3.5 has been optimized for wheat sequence annotation with default parameters (Table S3 in Supplementary Material), a customized interface will be available in the near future to allow each user to define or import his own procedure via the upload of the “step.xml” file.
Accuracy of the annotation depends also on the capacity to identify unknown TEs and pseudo genes. Ab initio prediction programs often annotate these features as genes thereby increasing the number of FPs and decreasing the specificity of the annotation. PPFINDER (Van Baren and Brent, 2006) may help to remove fragments of processed pseudo genes from predictions (Brent, 2008) and it will be implemented and tested in future versions of the TriAnnot pipeline.
With the advent of the NGS platforms, functional analyses are increasingly performed through RNA-Seq experiments (Wang et al., 2009). These data are also of great value to support structural annotation and we will integrate new programs in TriAnnot to take advantage of the RNA-Seq data that are currently under production for wheat in different projects worldwide. New versions of EuGene (Schiex, personal communication) and augustus28 that will integrate RNA-Seq data analysis are currently under development.
Synteny-based annotation will also be improved. To date, TriAnnot only identifies the best hit between a gene model and other plant genomes. In the near future, all possible orthologs/paralogs will be displayed in a new “Genome mapping” panel (Panel V, Figure 4). Two other panels dedicated to phylogenetic analysis (Panel VI) and “metabolic pathway” (Panel VII) mapping will also be developed (Figure 4). In Panel VI, gene models will be mapped on pre-calculated phylogenetic trees to enable the rapid identification of putative orthologous and paralogous relationships for the gene models. Panel VII will map gene models on pre-calculated metabolic pathways, such as RiceCyc and SorghumCyc29, to provide hypotheses about the potential biological function of the gene models.
Finally, in the past decade, various groups of ncRNAs (Ren, 2010) have been identified as genome features that are essential for the regulation of gene expression. TriAnnot will integrate a new package, “rnaspace30,” to support the identification of non-protein-coding RNA (ncRNA). This will enable, in particular, the identification and mapping of microRNAs (miRNAs) that have been shown to regulate gene expression in plants (Jones-Rhoades et al., 2006; Meyers et al., 2008b) and to play a major role in plant development (Chitwood and Timmermans, 2010).
Enhancing genetic marker design
The vision of the TriAnnot project is to provide tools that help scientists and breeders rapidly mine genome sequence information for marker development and accelerate marker-assisted selection programs. Sequencing pilot projects showed the potential of the wheat genome sequence for high-throughput marker design. For example Choulet et al. (2010) indicated a density of about one SSR every 13.1 kb. To date, TriAnnot identifies SSR motives in Panel IV but the automated design of primers is not implemented yet. This will be done in the near future with the addition of the in-house developed SSRdesign program that produces a tabulated output file which can be used easily to order primers. Additionally, a new type of marker based on the identification of junctions between TEs has been developed recently (Paux et al., 2006). A program, ISBPfinder, dedicated to the automated design of ISBPs has been developed and preliminary experiments show that it can define one ISBP marker per 3.8 kb on average (Paux et al., 2010). ISBPfinder will also be integrated in TriAnnot Panel IV.
Improving query length size and on line expertise
With a computing cluster comprising 712 CPU units and 50 TB of disk storage, TriAnnot can run on a fully parallelized system and launch analysis of ∼100 BACs, contigs, or scaffolds at the same time. At present, the maximal query sequence length than can be annotated by TriAnnot is 3 Mb, and to be annotated, large sequences are split in fragments of 1–3 Mb (depending of cluster power and parallelization optimization). This approach has been followed to re-annotate the 45-Mb of the rice chromosome 1 in this study. In future versions of TriAnnot we intend to implement a “sliding window” system that should enable the annotation of much larger size sequences, perhaps as much as the 1-Gb wheat chromosome 3B pseudomolecule at once.
Another essential feature of an easy-to-use annotation pipeline is that its output formats enable efficient manual curation of the data. This task has been simplified by the Generic Model Organism Database (GMOD) project31 which provides a generic genome database scheme and genome visualization tools. Therefore, a common thread of each TriAnnot module is that computational evidence is translated from the native annotation program output into the standard general feature format GFF32 and, in turn, the GFF files are formatted for loading the annotation results into relational databases (e.g., CHADO) that enable online manual curation through ARTEMIS or APOLLO33 graphical editors. This system, however, will rapidly become limiting with the exponential growth of sequence data. Further, integrated environments, such as the “Bioinformatics Online Genome Annotation System” (BOGAS) developed at the VIB Institute in Gent, Belgium34, will need to be taken into consideration to maintain manual curation efficiency.
Conclusion
Genome annotation is a continuous process (e.g., five versions of the rice genome have been released so far) and TriAnnot which is hosted by a sustainable bioinformatics platform at the INRA URGI will also enable ongoing community annotation. The preliminary phase of the TriAnnot project, to provide the international wheat community with an efficient, user-friendly, online pipeline for the annotation of the sequence of the 21 bread wheat chromosomes under the umbrella of the IWGSC, has been accomplished. Even though improvement is still needed for training the predictors with wheat data, TriAnnot is operational already and in use for the 3BSEQ project35 which will serve as the proof of concept and will assist in the continuing improvement of TriAnnot pipeline for additional wheat chromosomes and plant genome annotation projects. As demonstrated here, TriAnnot can be easily adapted to other plant species with minor modifications.
TriAnnot Accessibility
Project Name: TriAnnot.
Login/password request: http://urgi.versailles.inra.fr/Species/Wheat/Triannot-Pipeline/Help.
Project Home Page: http://www.clermont.inra.fr/triannot/ with a full and precise description of the TriAnnot pipeline architecture, regularly updated.
The source code is available upon request todHJpYW5ub3Qtc3VwcG9ydEBjbGVybW9udC5pbnJhLmZy.
Programming language: Perl and Python.
Dependencies: bioinformatics programs (see Table S1 in Supplementary Material); Databanks (see Table S2 in Supplementary Material); Oracle Grid Engine; MySQL database.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the “Programme Régional d’Actions Innovatrices” (PRAI) e-nnovergne LifeGrid through a European project “Actions innovatrices du FEDER” (2006–2008) coordinated by the “Région Auvergne” (http://www.lifegrid.fr/fr.html), the European Community’s Seventh Framework Programme Triticeae Genome (grant agreement no FP7-212019; http://www.triticeaegenome.eu/), the ANR Blanche (EXEGESE-BLE; ANR-05-BLANC-0258-01/2005-2008), the ANR(09-GENM-025) – FranceAgriMer (201006-015-104) project 3BSEQ, the competitiveness cluster “Céréales Vallée” (http://www.cereales-vallee.org/default_gb.cfm) and the Ministry of Agriculture, Forestry, and Fisheries of Japan (Genomics for Agricultural Innovation, GIR-1001). The authors are grateful to Dr. S. Rombault (VIB, Gent, Belgium) and Dr. E. Paux (INRA, Clermont-Ferrand, France) for helpful discussions and to Kellye Eversole for critical editing of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/plant_genetics_and_genomics/10.3389/fpls.2012.00005/abstract
Footnotes
- ^http://www.ncbi.nlm.nih.gov/genomes/
- ^http://www.ncbi.nlm.nih.gov/genomes/leuks.cgi
- ^http://www.invitrogen.com/
- ^http://www.gatc-biotech.com/en/bioinformatics/dnastar-software.html
- ^http://dawgpaws.sourceforge.net/
- ^http://www.phytozome.net/
- ^http://mips.helmholtz-muenchen.de/plant/genomes.jsp
- ^http://www.gramene.org/info/docs/genebuild/index.html
- ^http://www.ncbi.nlm.nih.gov/genome/guide/build.shtml
- ^http://www.ensembl.org/info/docs/genebuild/index.html
- ^http://dnasubway.iplantcollaborative.org/
- ^http://www.wheatgenome.org
- ^http://urgi.versailles.inra.fr/Projects/3BSeq
- ^http://urgi.versailles.inra.fr/gnpis/
- ^http://www.clermont.inra.fr/triannot
- ^http://wheat.pw.usda.gov/ITMI/Repeats/index.shtml
- ^http://linux1.softberry.com/berry.phtml
- ^http://hmmer.janelia.org/software
- ^http://www.wheatgenome.org/tools.php
- ^http://www.geneontology.org/
- ^http://rnaspace.org
- ^http://www.wheatgenome.org/
- ^http://urgi.versailles.inra.fr/Species/Wheat/Triannot-Pipeline/Help
- ^http://genomeview.org/
- ^http://www.wheatgenome.org/Projects/IWGSC-Bread-Wheat-Projects/Sequencing/Whole-Chromosome-Reference-Sequencing-Projects
- ^http://medicago.jcvi.org/cgi-bin/medicago/overview.cgi
- ^http://urgi.versailles.inra.fr/Projects/3BSeq
- ^http://bioinf.uni-greifswald.de/augustus/binaries/readme.rnaseq.html
- ^http://www.gramene.org/pathway/
- ^http://www.rnaspace.org
- ^http://www.gmod.org
- ^http://www.sanger.ac.uk/resources/software/gff/spec.html
- ^http://apollo.berkeleybop.org/current/index.html
- ^http://bioinformatics.psb.ugent.be/webtools/bogas/
- ^http://urgi.versailles.inra.fr/Projects/3BSeq
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Amano, N., Tanaka, T., Numa, H., Sakai, H., and Itoh, T. (2010). Efficient plant gene identification based on interspecies mapping of full-length cDNAs. DNA Res. 17, 271–279.
Argout, X., Salse, J., Aury, J. M., Guiltinan, M. J., Droc, G., Gouzy, J., Allegre, M., Chaparro, C., Legavre, T., Maximova, S. N., Abrouk, M., Murat, F., Fouet, O., Poulain, J., Ruiz, M., Roguet, Y., Rodier-Goud, M., Barbosa-Neto, J. F., Sabot, F., Kudrna, D., Ammiraju, J. S., Schuster, S. C., Carlson, J. E., Sallet, E., Schiex, T., Dievart, A., Kramer, M., Gelley, L., Shi, Z., Berard, A., Viot, C., Boccara, M., Risterucci, A. M., Guignon, V., Sabau, X., Axtell, M. J., Ma, Z., Zhang, Y., Brown, S., Bourge, M., Golser, W., Song, X., Clement, D., Rivallan, R., Tahi, M., Akaza, J. M., Pitollat, B., Gramacho, K., D’Hont, A., Brunel, D., Infante, D., Kebe, I., Costet, P., Wing, R., Mccombie, W. R., Guiderdoni, E., Quetier, F., Panaud, O., Wincker, P., Bocs, S., and Lanaud, C. (2011). The genome of Theobroma cacao. Nat. Genet. 43, 101–108.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580.
Bonnet, E., Van De Peer, Y., and Rouze, P. (2006). The small RNA world of plants. New Phytol. 171, 451–468.
Brejova, B., Vinar, T., Chen, Y., Wang, S., Zhao, G., Brown, D. G., Li, M., and Zhou, Y. (2009). Finding genes in Schistosoma japonicum: annotating novel genomes with help of extrinsic evidence. Nucleic Acids Res. 37, e52.
Brent, M. R. (2008). Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat. Rev. Genet. 9, 62–73.
Cantarel, B. L., Korf, I., Robb, S. M. C., Parra, G., Ross, E., Moore, B., Holt, C., Sánchez Alvarado, A., and Yandell, M. (2008). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196.
Carver, T., Berriman, M., Tivey, A., Patel, C., Bohme, U., Barrell, B. G., Parkhill, J., and Rajandream, M. A. (2008). Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24, 2672–2676.
Choulet, F., Wicker, T., Rustenholz, C., Paux, E., Salse, J., Leroy, P., Schlub, S., Le Paslier, M. C., Magdelenat, G., Gonthier, C., Couloux, A., Budak, H., Breen, J., Pumphrey, M., Liu, S., Kong, X., Jia, J., Gut, M., Brunel, D., Anderson, J. A., Gill, B. S., Appels, R., Keller, B., and Feuillet, C. (2010). Megabase level sequencing reveals contrasted organization and evolution patterns of the wheat gene and transposable element spaces. Plant Cell 22, 1686–1701.
Cock, J. M., Sterck, L., Rouze, P., Scornet, D., Allen, A. E., Amoutzias, G., Anthouard, V., Artiguenave, F., Aury, J. M., Badger, J. H., Beszteri, B., Billiau, K., Bonnet, E., Bothwell, J. H., Bowler, C., Boyen, C., Brownlee, C., Carrano, C. J., Charrier, B., Cho, G. Y., Coelho, S. M., Collen, J., Corre, E., Da Silva, C., Delage, L., Delaroque, N., Dittami, S. M., Doulbeau, S., Elias, M., Farnham, G., Gachon, C. M., Gschloessl, B., Heesch, S., Jabbari, K., Jubin, C., Kawai, H., Kimura, K., Kloareg, B., Kupper, F. C., Lang, D., Le Bail, A., Leblanc, C., Lerouge, P., Lohr, M., Lopez, P. J., Martens, C., Maumus, F., Michel, G., Miranda-Saavedra, D., Morales, J., Moreau, H., Motomura, T., Nagasato, C., Napoli, C. A., Nelson, D. R., Nyvall-Collen, P., Peters, A. F., Pommier, C., Potin, P., Poulain, J., Quesneville, H., Read, B., Rensing, S. A., Ritter, A., Rousvoal, S., Samanta, M., Samson, G., Schroeder, D. C., Segurens, B., Strittmatter, M., Tonon, T., Tregear, J. W., Valentin, K., Von Dassow, P., Yamagishi, T., Van De Peer, Y., and Wincker, P. (2010). The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465, 617–621.
Curwen, V., Eyras, E., Andrews, T. D., Clarke, L., Mongin, E., Searle, S. M., and Clamp, M. (2004). The Ensembl automatic gene annotation system. Genome Res. 14, 942–950.
Derelle, E., Ferraz, C., Rombauts, S., Rouze, P., Worden, A. Z., Robbens, S., Partensky, F., Degroeve, S., Echeynie, S., Cooke, R., Saeys, Y., Wuyts, J., Jabbari, K., Bowler, C., Panaud, O., Piegu, B., Ball, S. G., Ral, J.-P., Bouget, F.-Y., Piganeau, G., De Baets, B., Picard, A., Delseny, M., Demaille, J., Van De Peer, Y., and Moreau, H. (2006). Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc. Natl. Acad. Sci. U.S.A. 103, 11647–11652.
Devos, K. M., Ma, J., Pontaroli, A. C., Pratt, L. H., and Bennetzen, J. L. (2005). Analysis and mapping of randomly chosen bacterial artificial chromosome clones from hexaploid bread wheat. Proc. Natl. Acad. Sci. U.S.A. 102, 19243–19248.
Doležel, J., Šimková, H., Kubaláková, M., Šafár, J., Suchánková, P., Cíhalíková, J., Bartoš, J., and Valárik, M. (2009). “Chromosome genomics in the Triticeae,” in Genetics and Genomics of the Triticeae, eds G. J. Muehlbauer, and C. Feuillet (New York: Springer), 285–316.
Elsik, C. G., Worley, K. C., Zhang, L., Milshina, N. V., Jiang, H., Reese, J. T., Childs, K. L., Venkatraman, A., Dickens, C. M., Weinstock, G. M., and Gibbs, R. A. (2006). Community annotation: procedures, protocols, and supporting tools. Genome Res. 16, 1329–1333.
Estill, J. C., and Bennetzen, J. L. (2009). The DAWGPAWS pipeline for the annotation of genes and transposable elements in plant genomes. Plant Methods 5, 8.
Feuillet, C., and Eversole, K. (2007). Physical mapping of the wheat genome: a coordinated effort to lay the foundation for genome sequencing and develop tools for breeders. Isr. J. Plant Sci. 55, 307–313.
Feuillet, C., Leach, J. E., Rogers, J., Schnable, P. S., and Eversole, K. (2011). Crop genome sequencing: lessons and rationales. Trends Plant Sci. 16, 77–88.
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R., and Bateman, A. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222.
Flavell, R. B., Rimpau, J., and Smith, D. B. (1977). Repeated sequence DNA relationship in four cereals genomes. Chromosoma 63, 205–222.
Flutre, T., Duprat, E., Feuillet, C., and Quesneville, H. (2011). Considering transposable element diversification in de novo annotation approaches. PLoS ONE 6, e16526. doi: 10.1371/journal.pone.0016526
Guigo, R., Knudsen, S., Drake, N., and Smith, T. (1992). Prediction of gene structure. J. Mol. Biol. 226, 141–157.
International Rice Genome Sequencing Project. (2005). The map-based sequence of the rice genome. Nature 436, 793–800.
Jones-Rhoades, M. W., Bartel, D. P., and Bartel, B. (2006). MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 57, 19–53.
Keibler, E., and Brent, M. R. (2003). Eval: a software package for analysis of genome annotations. BMC Bioinformatics 4, 50. doi: 10.1186/1471-2105-4-50
Kurtz, S., Narechania, A., Stein, J. C., and Ware, D. (2008). A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics 9, 517. doi: 10.1186/1471-2164-9-517
Letunic, I., Doerks, T., and Bork, P. (2009). SMART 6: recent updates and new developments. Nucleic Acids Res. 37, D229–D232.
Li, W., Zhang, P., Fellers, J. P., Friebe, B., and Gill, B. S. (2004). Sequence composition, organization, and evolution of the core Triticeae genome. Plant J. 40, 500–511.
Liang, C., Mao, L., Ware, D., and Stein, L. (2009). Evidence-based gene predictions in plant genomes. Genome Res. 19, 1912–1923.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506.
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964.
Lukashin, A. V., and Borodovsky, M. (1998). GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115.
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., Cao, X., Carrington, J. C., Chen, X., Green, P. J., Griffiths-Jones, S., Jacobsen, S. E., Mallory, A. C., Martienssen, R. A., Poethig, R. S., Qi, Y., Vaucheret, H., Voinnet, O., Watanabe, Y., Weigel, D., and Zhu, J.-K. (2008a). Criteria for annotation of plant microRNAs. Plant Cell 20, 3186–3190.
Meyers, B. C., Matzke, M., and Sundaresan, V. (2008b). The RNA world is alive and well. Trends Plant Sci. 13, 311–313.
Moskal, W. A. Jr., Wu, H. C., Underwood, B. A., Wang, W., Town, C. D., and Xiao, Y. (2007). Experimental validation of novel genes predicted in the un-annotated regions of the Arabidopsis genome. BMC Genomics 8, 18. doi: 10.1186/1471-2164-8-18
Mott, R. (1997). EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 13, 477–478.
Ouyang, S., and Buell, C. R. (2004). The TIGR plant repeat databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res. 32, D360–D363.
Paux, E., Faure, S., Choulet, F., Roger, D., Gauthier, V., Martinant, J. P., Sourdille, P., Balfourier, F., Le Paslier, M. C., Chauveau, A., Cakir, M., Gandon, B., and Feuillet, C. (2010). Insertion site-based polymorphism markers open new perspectives for genome saturation and marker-assisted selection in wheat. Plant Biotechnol. J. 8, 196–210.
Paux, E., Legeai, F., Guilhot, N., Adam-Blondon, A. F., Alaux, M., Salse, J., Sourdille, P., Leroy, P., and Feuillet, C. (2008). Physical mapping in large genomes: accelerating anchoring of BAC contigs to genetic maps through in silico analysis. Funct. Integr. Genomics 8, 29–32.
Paux, E., Roger, D., Badaeva, E., Gay, G., Bernard, M., Sourdille, P., and Feuillet, C. (2006). Characterizing the composition and evolution of homoeologous genomes in hexaploid wheat through BAC-end sequencing on chromosome 3B. Plant J. 48, 463–474.
Paux, E., and Sourdille, P. (2009). “A toolbox for Triticeae genomics,” in Genetics and Genomics of the Triticeae, eds C. Feuillet, and J. G. Muehlbauer (New York: Springer), 255–284.
Quesneville, H., Bergman, C. M., Andrieu, O., Autard, D., Nouaud, D., Ashburner, M., and Anxolabehere, D. (2005). Combined evidence annotation of transposable elements in genome sequences. PLoS Comput. Biol. 1, e22. doi: 10.1371/journal.pcbi.0010022
Rounsley, S., Marri, P., Yu, Y., He, R., Sisneros, N., Goicoechea, J., Lee, S., Angelova, A., Kudrna, D., Luo, M., Affourtit, J., Desany, B., Knight, J., Niazi, F., Egholm, M., and Wing, R. (2009). De novo next generation sequencing of plant genomes. Rice 2, 35–43.
Rustenholz, C., Hedley, P., Morris, J., Choulet, F., Feuillet, C., Waugh, R., and Paux, E. (2010). Specific patterns of gene space organisation revealed in wheat by using the combination of barley and wheat genomic resources. BMC Genomics 11, 714. doi: 10.1186/1471-2164-11-714
Sakata, K., Nagamura, Y., Numa, H., Antonio, B. A., Nagasaki, H., Idonuma, A., Watanabe, W., Shimizu, Y., Horiuchi, I., Matsumoto, T., Sasaki, T., and Higo, K. (2002). RiceGAAS: an automated annotation system and database for rice genome sequence. Nucleic Acids Res. 30, 98–102.
Sammut, S. J., Finn, R. D., and Bateman, A. (2008). Pfam 10 years on: 10,000 families and still growing. Brief. Bioinformatics 9, 210–219.
Sato, A., Oshima, K., Noguchi, H., Ogawa, M., Takahashi, T., Oguma, T., Koyama, Y., Itoh, T., Hattori, M., and Hanya, Y. (2011). Draft genome sequencing and comparative analysis of Aspergillus sojae NBRC4239. DNA Res. 18, 165–176.
Schiex, T., Moisan, A., and Rouzé, P. (2001). “EuGene: an eucaryotic gene finder that combines several sources of evidence,” in Computational Biology, eds O. Gascuel, and M.-F. Sagot (France: Springer Verlag), 111–125.
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., Hyten, D. L., Song, Q., Thelen, J. J., Cheng, J., Xu, D., Hellsten, U., May, G. D., Yu, Y., Sakurai, T., Umezawa, T., Bhattacharyya, M. K., Sandhu, D., Valliyodan, B., Lindquist, E., Peto, M., Grant, D., Shu, S., Goodstein, D., Barry, K., Futrell-Griggs, M., Abernathy, B., Du, J., Tian, Z., Zhu, L., Gill, N., Joshi, T., Libault, M., Sethuraman, A., Zhang, X. C., Shinozaki, K., Nguyen, H. T., Wing, R. A., Cregan, P., Specht, J., Grimwood, J., Rokhsar, D., Stacey, G., Shoemaker, R. C., and Jackson, S. A. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183.
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., Liang, C., Zhang, J., Fulton, L., Graves, T. A., Minx, P., Reily, A. D., Courtney, L., Kruchowski, S. S., Tomlinson, C., Strong, C., Delehaunty, K., Fronick, C., Courtney, B., Rock, S. M., Belter, E., Du, F., Kim, K., Abbott, R. M., Cotton, M., Levy, A., Marchetto, P., Ochoa, K., Jackson, S. M., Gillam, B., Chen, W., Yan, L., Higginbotham, J., Cardenas, M., Waligorski, J., Applebaum, E., Phelps, L., Falcone, J., Kanchi, K., Thane, T., Scimone, A., Thane, N., Henke, J., Wang, T., Ruppert, J., Shah, N., Rotter, K., Hodges, J., Ingenthron, E., Cordes, M., Kohlberg, S., Sgro, J., Delgado, B., Mead, K., Chinwalla, A., Leonard, S., Crouse, K., Collura, K., Kudrna, D., Currie, J., He, R., Angelova, A., Rajasekar, S., Mueller, T., Lomeli, R., Scara, G., Ko, A., Delaney, K., Wissotski, M., Lopez, G., Campos, D., Braidotti, M., Ashley, E., Golser, W., Kim, H., Lee, S., Lin, J., Dujmic, Z., Kim, W., Talag, J., Zuccolo, A., Fan, C., Sebastian, A., Kramer, M., Spiegel, L., Nascimento, L., Zutavern, T., Miller, B., Ambroise, C., Muller, S., Spooner, W., Narechania, A., Ren, L., Wei, S., Kumari, S., Faga, B., Levy, M. J., Mcmahan, L., Van Buren, P., Vaughn, M. W., Ying, K., Yeh, C. T., Emrich, S. J., Jia, Y., Kalyanaraman, A., Hsia, A. P., Barbazuk, W. B., Baucom, R. S., Brutnell, T. P., Carpita, N. C., Chaparro, C., Chia, J. M., Deragon, J. M., Estill, J. C., Fu, Y., Jeddeloh, J. A., Han, Y., Lee, H., Li, P., Lisch, D. R., Liu, S., Liu, Z., Nagel, D. H., McCann, M. C., SanMiguel, P., Myers, A. M., Nettleton, D., Nguyen, J., Penning, B. W., Ponnala, L., Schneider, K. L., Schwartz, D. C., Sharma, A., Soderlund, C., Springer, N. M., Sun, Q., Wang, H., Waterman, M., Westerman, R., Wolfgruber, T. K., Yang, L., Yu, Y., Zhang, L., Zhou, S., Zhu, Q., Bennetzen, J. L., Dawe, R. K., Jiang, J., Jiang, N., Presting, G. G., Wessler, S. R., Aluru, S., Martienssen, R. A., Clifton, S. W., McCombie, W. R., Wing, R. A., and Wilson, R. K. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115.
Sigrist, C. J., Cerutti, L., De Castro, E., Langendijk-Genevaux, P. S., Bulliard, V., Bairoch, A., and Hulo, N. (2010). PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 38, D161–D166.
Slater, G. S., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31. doi: 10.1186/1471-2105-6-31
Smit, A. F. (1993). Identification of a new, abundant superfamily of mammalian LTR-transposons. Nucleic Acids Res. 21, 1863–1872.
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl. 2), ii215–ii225.
The Arabidopsis Genome Initiative. (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–814.
Van Baren, M. J., and Brent, M. R. (2006). Iterative gene prediction and pseudogene removal improves genome annotation. Genome Res. 16, 678–685.
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63.
Wicker, T., Matthews, D. E., and Keller, B. (2002). TREP: a database for Triticeae repetitive elements. Trends Plant Sci. 7, 561–562.
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., Flavell, A., Leroy, P., Morgante, M., Panaud, O., Paux, E., Sanmiguel, P., and Schulman, A. H. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982.
Wu, T. D., and Watanabe, C. K. (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875.
Keywords: cluster, gene models, pipeline, plant genome, structural and functional annotation, transposable elements, wheat
Citation: Leroy P, Guilhot N, Sakai H, Bernard A, Choulet F, Theil S, Reboux S, Amano N, Flutre T, Pelegrin C, Ohyanagi H, Seidel M, Giacomoni F, Reichstadt M, Alaux M, Gicquello E, Legeai F, Cerutti L, Numa H, Tanaka T, Mayer K, Itoh T, Quesneville H and Feuillet C (2012) TriAnnot: a versatile and high performance pipeline for the automated annotation of plant genomes. Front. Plant Sci. 3:5. doi: 10.3389/fpls.2012.00005
Received: 02 October 2011; Accepted: 04 January 2012;
Published online: 31 January 2012.
Edited by:
Takuji Sasaki, National Institute of Agrobiological Sciences, JapanCopyright: © 2012 Leroy, Guilhot, Sakai, Bernard, Choulet, Theil, Reboux, Amano, Flutre, Pelegrin, Ohyanagi, Seidel, Giacomoni, Reichstadt, Alaux, Gicquello, Legeai, Cerutti, Numa, Tanaka, Mayer, Itoh, Quesneville and Feuillet. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Philippe Leroy and Catherine Feuillet, UMR 1095, Genetics, Diversity and Ecophysiology of Cereals, Institut National de la Recherche Agronomique-Université Blaise Pascal, 234 Avenue du Brézet, Domaine de Crouel, F-63000 Clermont-Ferrand, France. e-mail:bGVyb3lAY2xlcm1vbnQuaW5yYS5mcg==;Y2F0aGVyaW5lLmZldWlsbGV0QGNsZXJtb250LmlucmEuZnI=