




- 1 Program in Molecular Plant Biology, Department of Biology, Colorado State University, Fort Collins, CO, USA
- 2 Department of Computer Science, Colorado State University, Fort Collins, CO, USA
- 3 Centro de Investigação em Biodiversidade e Recursos Genéticos, University of Porto, Vairão, Portugal
- 4 Program in Molecular Plant Biology, Colorado State University, Fort Collins, CO, USA
Extensive alternative splicing (AS) of precursor mRNAs (pre-mRNAs) in multicellular eukaryotes increases the protein-coding capacity of a genome and allows novel ways to regulate gene expression. In flowering plants, up to 48% of intron-containing genes exhibit AS. However, the full extent of AS in plants is not yet known, as only a few high-throughput RNA-Seq studies have been performed. As the cost of obtaining RNA-Seq reads continues to fall, it is anticipated that huge amounts of plant sequence data will accumulate and help in obtaining a more complete picture of AS in plants. Although it is not an onerous task to obtain hundreds of millions of reads using high-throughput sequencing technologies, computational tools to accurately predict and visualize AS are still being developed and refined. This review will discuss the tools to predict and visualize transcriptome-wide AS in plants using short-reads and highlight their limitations. Comparative studies of AS events between plants and animals have revealed that there are major differences in the most prevalent types of AS events, suggesting that plants and animals differ in the way they recognize exons and introns. Extensive studies have been performed in animals to identify cis-elements involved in regulating AS, especially in exon skipping. However, few such studies have been carried out in plants. Here, we review the current state of research on splicing regulatory elements (SREs) and briefly discuss emerging experimental and computational tools to identify cis-elements involved in regulation of AS in plants. The availability of curated alternative splice forms in plants makes it possible to use computational tools to predict SREs involved in AS regulation, which can then be verified experimentally. Such studies will permit identification of plant-specific features involved in AS regulation and contribute to deciphering the splicing code in plants.
1. Introduction
Seminal discoveries in RNA biology in recent years have established a central role for RNAs in gene regulation at the transcriptional, post-transcriptional, and translational level in eukaryotes (reviewed in Chen, 2009; Sharp, 2009; Voinnet, 2009; Licatalosi and Darnell, 2010; Kalsotra and Cooper, 2011; Staiger and Green, 2011). In photosynthetic eukaryotes a vast majority of protein-coding genes (up to 90%) contain non-coding intronic sequences, hence the primary transcripts must undergo splicing to generate mature functional mRNAs (Reddy, 2007; Barbazuk et al., 2008; Labadorf et al., 2010). Pre-mRNA splicing is carried out by the spliceosome, a large ribonucleoprotein complex. In plants, as in animals, there are two types of spliceosomes. The major type is called U2 type, which performs splicing of U2-dependent introns, whereas the minor U12 type is involved in splicing of rare U12-dependent introns (Simpson and Brown, 2008). Both spliceosomes consist of five snRNAs (U1, U2, U4, U5, U6 in the major spliceosome and U11, U12, U4atac, U5, and U6atac in the minor spliceosome). The protein composition of the major spliceosome has been extensively studied in animals, revealing that it contains close to 200 proteins (Wahl et al., 2009; Valadkhan and Jaladat, 2010). Computational analysis has revealed that plants have RNA and protein components of both spliceosomes (Ru et al., 2008; Simpson and Brown, 2008).
Primary transcripts from intron-containing genes can be alternatively spliced by differential selection of splice sites, leading to production of multiple mature mRNAs from a single gene, which is considered a major source for proteome diversity (Black, 2003; Reddy, 2007; Pan et al., 2008; Ru et al., 2008; Kalsotra and Cooper, 2011). Protein isoforms produced by splice variants may have altered functions (Black, 2003; Stamm et al., 2005). In addition, AS plays an important role in gene regulation through regulated production of splice variants with a premature termination codon, which are degraded through nonsense-mediated decay and other RNA surveillance mechanisms (Chang et al., 2007; Kurihara et al., 2009; Barbazuk, 2010; Palusa and Reddy, 2010; Staiger and Green, 2011) or contain target sequences for miRNA so that they are either degraded or not translated (Tan et al., 2007; Chen, 2009). Hence, post-transcriptional regulation of gene expression by pre-mRNA splicing plays a crucial role in generating transcriptome and proteome diversity and provides novel ways to fine-tune gene regulation.
AS in plants was under-appreciated until recently, and it was considered rare as pre-mRNAs of only a few genes were known to undergo AS. For instance, in 2001 pre-mRNAs from only three dozen genes in plants were known to undergo AS (Reddy, 2001). However, the completion of the Arabidopsis genome little over a decade ago, and other plant genomes more recently, as well as the availability of a massive amounts of transcribed sequence data in the form expressed sequence tags (ESTs)/cDNAs and limited RNA sequence data generated with next generation sequencing (NGS) technologies, have allowed the analysis of transcriptome-wide AS in several plants including Arabidopsis, rice, grape, and cucumber (Campbell et al., 2006; Wang and Brendel, 2006; Reddy, 2007; Baek et al., 2008; Barbazuk et al., 2008; Filichkin et al., 2010; Guo et al., 2010; Lu et al., 2010; Sanchez et al., 2010; Zenoni et al., 2010). A variety of splicing-sensitive microarrays such as splice junction arrays and tiling arrays, that are used extensively in animals (Hallegger et al., 2010) have not been widely used in plants to analyze AS globally (Love et al., 2010; Rehrauer et al., 2010; Zenoni et al., 2010). RNA sequencing (RNA-Seq) using NGS platforms has allowed the detection of rare transcripts, precise quantification of transcript levels and global analysis of AS (Pan et al., 2008; Wang et al., 2009a). A recent analysis of AS in plants using RNA-Seq has revealed that over 40% of intron-containing genes in Arabidopsis (Filichkin et al., 2010) and about 48% in rice (Lu et al., 2010) undergo AS, although it is not known how much of AS is due to noise in the splicing process (Melamud and Moult, 2009) and how much is regulated AS with biological consequences. In humans, pre-mRNAs from almost every multi-exon gene are alternatively spliced and misregulation of splicing results in developmental abnormalities and disease (Pan et al., 2008; Ru et al., 2008; Sanford et al., 2009). As more RNA-Seq data from different cell types, tissues, developmental stages and under different biotic and abiotic stresses become available, the known repertoire of AS in plants is likely to increase. In addition, many splice variants are differentially expressed in a tissue- or development-specific manner or in response to developmental cues and stresses (Yoshimura et al., 2002; Iida et al., 2004; Palusa et al., 2007; Reddy, 2007; Schindler et al., 2008; Simpson and Brown, 2008; Filichkin et al., 2010; Simpson et al., 2010; Staiger and Green, 2011).
In addition to AS events that include or delete long sequences in the pre-mRNA, other subtle AS events due to tandem acceptors (NAGNAG, N being any nucleotide) that result in gain or loss of three nucleotides in the spliced mRNA are common in land plants and animals (Iida et al., 2008; Schindler et al., 2008; Sinha et al., 2010). Primary transcripts of miRNAs also undergo AS (Hirsch et al., 2006; Szarzynska et al., 2009). In addition to cis-splicing, occurrence of trans-splicing, which produces chimeric transcripts by joining transcripts derived from two different nuclear protein-coding genes on the same or different chromosomes, has been reported in plants (Kawasaki et al., 1999; He et al., 2008; Guo et al., 2010). In rice, over 200 chimeric transcripts derived from trans-splicing were predicted from short-read sequence data and some of these were verified by RT-PCR (Zhang et al., 2010).
Comparative analysis of prevalence of different types of AS events in plants and animals has revealed that there are some fundamental differences between them. In plants a vast majority of splice variants (up to 56%) are due to intron retention, whereas it is not that prevalent in metazoans (5% in humans; Iida et al., 2004; Ner-Gaon et al., 2004; Wang and Brendel, 2006; Baek et al., 2008; Filichkin et al., 2010; Labadorf et al., 2010). In animals, exon skipping is the most common form of AS (58% in humans) and it is less prevalent in plants (8% in Arabidopsis). The differences in the frequencies of different types of AS events between plants and metazoans are thought to reflect the differences in how plant and animal cells recognize exons and introns. However, it is not known why intron retention is prevalent in plants and what mechanisms contribute to it. Interestingly, transcriptome analysis of 18 accessions of Arabidopsis thaliana has revealed that intron retention events differed between accessions (Gan et al., 2011).
The high rate of occurrence of AS in plants and its regulation by stresses and developmental cues (Kalyna et al., 2006; Palusa et al., 2007; Reddy, 2007; Barbazuk et al., 2008; Simpson et al., 2008, 2010; Barbazuk, 2010; Filichkin et al., 2010; Zenoni et al., 2010; Reddy and Ali, 2011) has sparked a growing interest and led to further studies focused on revealing the full extent of AS in plants by deep sequencing. Such studies will aid in understanding the biological functions of splice variants and the mechanisms by which plant cells regulate AS. This review focuses on computational tools used in predicting AS and splicing regulatory elements (SREs) involved in the regulation of AS. We also briefly discuss some recent experimental approaches that have been used in animals to identify targets of RNA binding proteins (RBPs), which can be applied to plants to discover RNA sequences that bind splicing regulators.
2. Transcriptome-Wide Detection and Visualization of AS
Before the advent of NGS, large-scale studies of AS in plant and mammalian systems were carried out mostly using sequences of Expressed Sequence Tags (ESTs) and full-length cDNAs (Haas et al., 2003; Campbell et al., 2006; Wang and Brendel, 2006; Chen et al., 2007; Gu and Guo, 2007; Ner-Gaon et al., 2007; Wang et al., 2008a; Sablok et al., 2011). These studies have increased dramatically the estimated number of plant genes that exhibit AS, and identified intron retention events as the most common AS event (Reddy, 2007; Wang et al., 2008a). The decline in the cost of sequencing using NGS platforms has made large-scale sequencing readily available, and transcriptome profiling using RNA-Seq has already been carried out in several plant species (Filichkin et al., 2010; Lu et al., 2010; Zenoni et al., 2010; Gan et al., 2011). However, analysis of these massive amounts of sequence data and the short length of sequence reads require novel computational tools, which has become a major bottleneck in mining the data to extract biologically relevant conclusions, especially for accurately predicting AS (Liang et al., 2009; Chodavarapu et al., 2010; Fiume et al., 2010; Marguerat and Bahler, 2010).
2.1. Transcripts vs Splice Graphs
Full-length cDNAs provide the best evidence for a gene’s splice forms – aligning such a sequence to the reference genome provides evidence for the exact exon-intron structure of a transcript. ESTs are shorter, but still usually cover several exons. NGS reads on the other hand, are short (around 100 bp in today’s technology), and only provide local evidence for transcript structure. This makes prediction of splice forms from RNA-Seq difficult, and most of the methods for transcriptome assembly first construct an object called a splice graph (Heber et al., 2002). A splice graph is a compact graphical representation of a gene’s exon-intron structure that captures all the ways in which exons for a given gene may be assembled into a transcript (Heber et al., 2002; Xing et al., 2004; Harrington and Bork, 2008; Sammeth et al., 2008; Bonizzoni et al., 2009; Labadorf et al., 2010; Richardson et al., 2011; Rogers et al., 2012). Figure 1 illustrates the concept. The compact structure allows researchers to visualize a gene’s AS easily (Harrington and Bork, 2008; Rogers et al., 2012), facilitates integration of ESTs into coherent models (Heber et al., 2002; Xing et al., 2004; Bonizzoni et al., 2009), aids statistical analysis of AS across a genome (Labadorf et al., 2010; Rogers et al., 2012), and facilitates comparisons between gene families (Richardson et al., 2011).
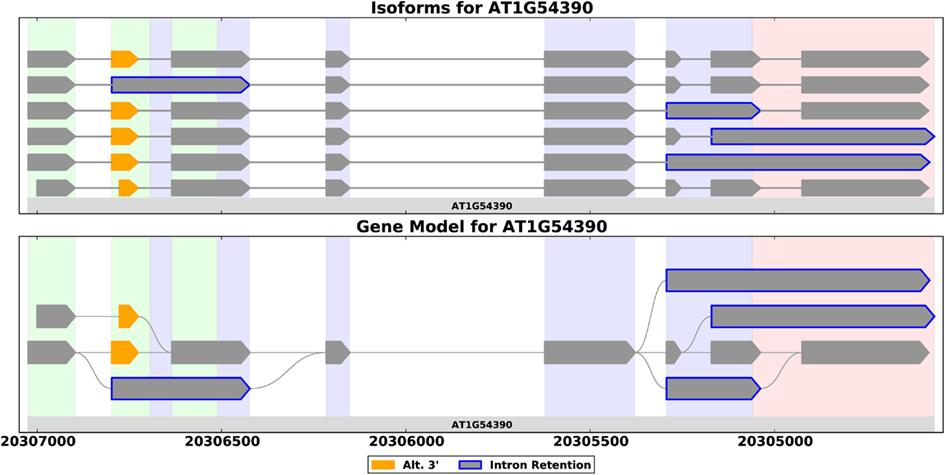
Figure 1. Splice forms for the ING2 gene from A. thaliana shown as a set of transcripts (top) and as a splice graph (bottom). A splice graph is a compact representation that shows all the ways in which a gene’s exons may be combined. These plots, generated by SpliceGrapher, use color coding to highlight AS events.
2.2. AS Prediction Using RNA-Seq
Transcriptional activity and AS can be studied using RNA-Seq with and without a reference genome (see Figure 2 for pipeline overview, and Table 1 for a list of tools). We begin our discussion with methods that require a reference genome. In this scenario, the first step is aligning the reads to the genome; we distinguish between two types of short-read mapping methods: those that allow only a limited number of gaps (usually a few bp at the most), and those that are able to map reads across splice junctions. Until a few years ago reads were short (32–36 bp) and most read mapping algorithms such as the Bowtie program (Trapnell and Salzberg, 2009) performed ungapped alignment. As read length continues to increase (100 bp and higher using today’s technology), the number of reads that span splice junctions increases as well, and with it the number of programs that perform spliced alignment (see, e.g., Trapnell et al., 2009; Au et al., 2010; Jean et al., 2010; Wang et al., 2010). Once mapped, read coverage (the distribution of reads that align within a region of interest) then provides evidence for exons and splice junctions recapitulated in the RNA-Seq data (see Figure 3).
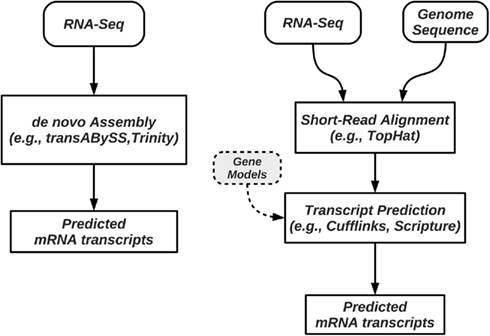
Figure 2. Pipelines for prediction and quantification of splice forms from RNA-Seq data. Methods such as trans-ABySS and Trinity perform de novo prediction, and do not require a reference genome (left); these methods do not rely on alignment programs to map reads to a genome. When a reference genome is available, methods such as Cufflinks first map the reads to the genome, followed by a step of assembly of splice forms or their quantification (right). Some of these methods require, or can use annotated isoforms to guide the process.
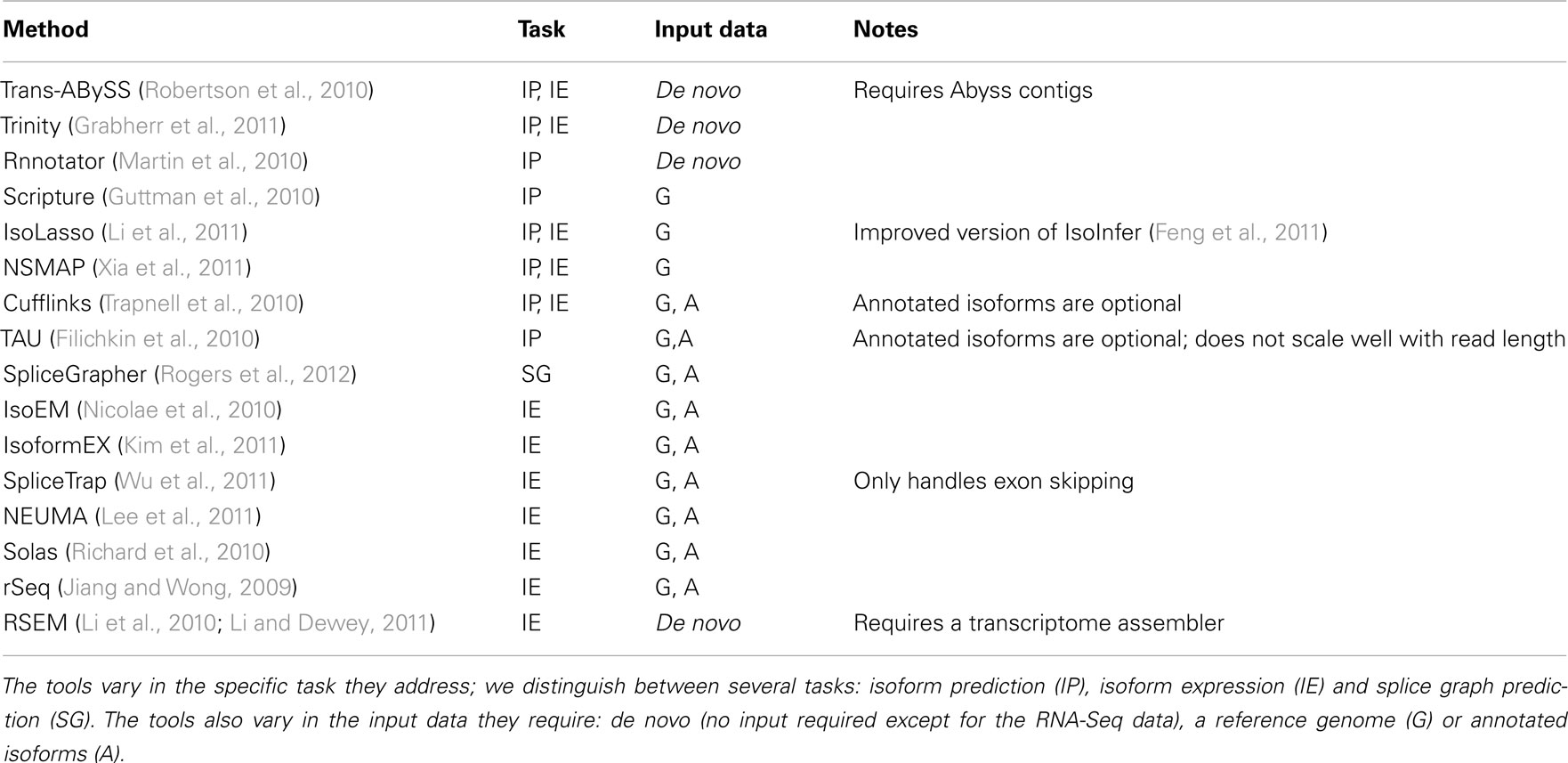
Table 1. Tools for predicting isoforms, their expression, and alternative splicing from RNA-Seq data.
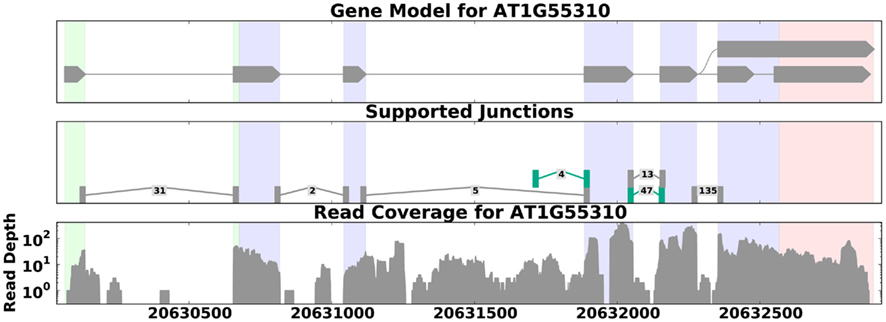
Figure 3. Read coverage for the gene SCL33 in A. thaliana (data from Filichkin et al., 2010). The top panel shows the annotated gene model; the middle panel shows reads that map across splice junctions, with labels showing the number of reads that aligned across each junction and novel splice junctions highlighted in green. The bottom panel shows the distribution of reads across the gene. Here the read depth ranges from 1 to over 300, demonstrating that read coverage can be highly variable even across known exons (shaded regions on the graphs). This variability can make it difficult to distinguish between weakly expressed splice forms and background noise. This figure was generated by SpliceGrapher.
The first studies that predicted AS from RNA-Seq data were performed in mammals and focused on detection of exon skipping, the most common form of AS in these systems (see, e.g., Mortazavi et al., 2008; Pan et al., 2008; Sultan et al., 2008; Wang et al., 2008b; Tang et al., 2009); detection of exon skipping is relatively simple, requiring the detection of reads that span a splice junction that skips a known exon. Several programs are now available for assembling transcripts using ungapped and spliced alignments among them Cufflinks (Trapnell et al., 2009), and Scripture (Guttman et al., 2010). Whereas these programs were designed for mammalian genomes, TAU (Filichkin et al., 2010) was designed to predict transcripts for the model plant A. thaliana. The program predicted an assortment of non-canonical splice junctions associated with splice forms that were later validated via RT-PCR (Filichkin et al., 2010). However, its spliced alignment component has memory requirements that make it prohibitive with increasing read length.
Some methods simultaneously predict transcripts and their abundance, using read depth as information that can be used to untangle transcripts from each other. They are based on the idea that read depth can be expressed as a weighted sum across the transcripts that are represented in a sample. These include IsoLasso (Li et al., 2011) and NSMAP (Xia et al., 2011).
RNA-Seq data alone may not be sufficient to resolve splice forms unambiguously (Lacroix et al., 2008; Rogers et al., 2012). Accordingly, Cufflinks was recently enhanced to incorporate information from gene models into its transcript prediction method (Roberts et al., 2011); the TAU program is also able to leverage gene annotations. We developed the SpliceGrapher tool, which uses gene models to establish a context for interpreting evidence from EST or RNA-Seq alignments, and to predict novel AS events only when the evidence for them is strong (Rogers et al., 2012). Furthermore, our tool leaves the predictions in the form of splice graphs when transcripts cannot be unambiguously resolved. Our work shows the strength of this approach and the tradeoffs associated with using gene annotations to augment TAU or Cufflinks predictions.
Accurate splice junction identification is crucial for making accurate AS predictions, but the short length of NGS reads makes spliced alignment especially challenging. A splice junction may occur anywhere within a read, so the read may have just a few bases on one side of a junction. Therefore, methods that use simple heuristics such as the existence of canonical splice site dimers and acceptable intron lengths can lead to many false-positive splice junctions (Rogers et al., 2012). This highlights the importance of modeling splice junction sequence characteristics as implemented in MapSplice (Wang et al., 2010), for example, or the sophisticated sequence-based splice site models used by SpliceGrapher (Rogers et al., 2012) and PALMapper (Jean et al., 2010).
Not all plants have a reference genome available. In its absence, de novo transcriptome assembly packages can be used. These methods construct transcripts based on overlapping k-mers. One such program is ABySS (Simpson et al., 2009), a de novo genome assembler that has been used to predict AS patterns such as exon skipping, intron retention, and alternative 5′ splice sites (Birol et al., 2009). The recently developed Trinity suite (Grabherr et al., 2011) assembles reads into splice graphs, and predicts splice forms by tracing paths through the graph.
Tiling and exon-junction arrays are an alternative platform for studying AS (Clark et al., 2002; Johnson et al., 2003; Mockler and Ecker, 2005; Cuperlovic-Culf et al., 2006; Hallegger et al., 2010). Tiling arrays provide an unbiased view of a genome using probes evenly spaced across its entire length, thus permitting discovery of novel AS events (Ner-Gaon and Fluhr, 2006; Hazen et al., 2009). Ner-Gaon and Fluhr (2006) found that whole-genome tiling arrays identified as much AS activity in A. thaliana as the most comprehensive EST-based studies. However, microarrays suffer from several limitations such as cross-hybridization and varying probe binding affinities that increase hybridization signal noise, and poor sensitivity for detecting isoforms that differ only in a few nucleotides, and also in cases where minor isoforms are expressed at low levels (Mockler and Ecker, 2005; Hallegger et al., 2010). These factors, combined with the success of RNA-Seq, have impeded adoption of this technology by plant researchers.
Visualization of NGS data is now supported by several genome browsers, including Stein et al. (2002), the Integrated Genome Browser (IGB; Nicol et al., 2009), the Integrative Genomics Viewer (Robinson et al., 2011), and GenomeView (Abeel, 2011), that can display short-read coverage graphs and transcripts. SpliceGrapher (Rogers et al., 2012), designed specifically for AS analysis, includes a plotting tool that displays splice graphs, gene models, and short-read coverage graphs, and highlights AS events. It generates static plots that cannot be manipulated the way a genome browser visualization can, but we have found them to be very useful for viewing of plant transcriptome data, because of the relatively short intron length.
3. Differential AS
Detection of differentially expressed genes is perhaps the most common analysis task performed on microarray data, and many methods are available (Grant et al., 2007; Durinck, 2008). NGS technology and the availability of microarrays that allow a distinction between splice forms (e.g., exon-junction and tiling arrays) are opening the possibility of extending this idea to detection of differential AS: genes that show differences in the patterns of AS under different conditions or developmental stages. Several studies have revealed regulated AS in plants (Yoshimura et al., 2002; Iida et al., 2004; Palusa et al., 2007; Reddy, 2007; Schindler et al., 2008; Simpson and Brown, 2008; Filichkin et al., 2010; Simpson et al., 2010; Kumar et al., 2011; Staiger and Green, 2011), and detection of differential AS by high-throughput methods will further enhance our understanding of the role of AS, and help in developing a plant condition-dependent splicing code.
A number of studies have used microarrays to estimate isoform expression levels (Mockler and Ecker, 2005; Blencowe et al., 2009; Hallegger et al., 2010), and methods have been proposed to detect splice form expression levels from exon arrays (Purdom et al., 2008; Xing et al., 2008). These methods have been used to study AS in mammals (Xing and Lee, 2008; Warzecha et al., 2009), but have not been widely adopted in plants (Love et al., 2010; Rehrauer et al., 2010). Microarrays have several limitations in this context including a large number of false-positive AS predictions (Gaidatzis et al., 2009).
NGS data address many of the limitations of microarrays – they are more sensitive to weakly expressed isoforms and have a broader dynamic range (Roy et al., 2011) – but they also introduce considerable challenges. For example, it may not be possible to find a unique mapping between reads and the reference genome (Lacroix et al., 2008). Additionally, read coverage can be highly variable due to a variety of factors such as library preparation methods, flow cell characteristics and reads that align to multiple locations (Mortazavi et al., 2008; Fang and Cui, 2011).
Detection of differential AS requires establishing accurate models for the splice forms represented in the data (discussed earlier), quantifying splice form expression in a way that allows detection of weakly expressed splice forms, and performing statistical tests to differentiate between the relative expression of splice forms across samples. Several measures are used to report expression levels based on short-read coverage. A common metric is RPKM (reads per kilobase per million reads), developed in (Mortazavi et al., 2008). Many methods report expression levels in RPKM (Mortazavi et al., 2008; Jiang and Wong, 2009; Guttman et al., 2010; Feng et al., 2011; Kim et al., 2011; Xia et al., 2011) or an equivalent measure such as FPKM (fragments per kilobase per million reads; Nicolae et al., 2010; Trapnell et al., 2010). When comparing expression levels between genes or isoforms, RPKM may bias estimates in favor of longer sequences (Costa et al., 2010), so other measures have been proposed (see, e.g., Lee et al., 2011). Comparisons with microarrays have confirmed that RNA-Seq data can provide at least as much sensitivity as microarrays (Mortazavi et al., 2008), but without the limitations previously mentioned. Several studies have validated RNA-Seq expression estimates using qRT-PCR and these have demonstrated that the two methods produce estimates that are consistent with one another (see, e.g., Wang et al., 2008b; Nicolae et al., 2010; Richard et al., 2010; Lee et al., 2011).
Recently software has been developed to measure differential AS from RNA-Seq. For example, Cuffdiff, an extension of the Cufflinks package, compares transcript expression levels on the basis of read coverage in two experiments (Trapnell et al., 2010). The transcripts may be generated from Cufflinks or may come from annotated gene models. Cuffdiff generates statistical test results for fold changes at either the transcript or gene level. Studies in mammals have used these tools to compare differentially expressed splice forms between different conditions. The authors of the Cufflinks software used it to study transcriptional changes in mouse myoblast cell lines. They identified 70 genes in which the prevalent splice forms changed as cells transitioned from myocyte production to myotube fusion (Trapnell et al., 2010). In another study, Twine et al. (2011) found statistically significant differences in the expression of alternative splice forms between normal and diseased brain cells that yielded insights into the progression of Alzheimer’s disease.
Few plant studies have applied differential AS tools. Differential AS in response to stresses has been reported in A. thaliana (Filichkin et al., 2010). In addition, Jiao and Meyerowitz (2010) investigated flower development in A. thaliana, in part using the program rSeq (Jiang and Wong, 2009) to estimate isoform expression levels. Their analysis revealed differentially expressed splice forms for three genes (APETALA1, APETALA3, and AGAMOUS) between two flowering stages. Analysis of intron retention events showed that they occurred more frequently with the U12 spliceosomes than with the U2 spliceosomes (Jiao and Meyerowitz, 2010).
These results underscore the potential for using RNA-Seq to probe AS under changing conditions such as organism development or stresses. With NGS, researchers are no longer restricted by the cost of ESTs or a dearth of plant-specific microarrays. In addition, a growing number of software tools are helping to automate the analysis of transcript expression from NGS data. The availability of some NGS analysis pipelines through the iPlant cyber infrastructure is going to help plant researchers who do not have the expertise or the required computational infrastructure to analyze their RNA-Seq data (Goff et al., 2011). NGS data analysis is a focus area for iPlant, and several methods are available, including Cufflinks, and several read mapping algorithms; efforts are underway to add the SpliceGrapher package to the iPlant infrastructure. These factors combined, provide unprecedented opportunity for researchers to explore the role of AS across all plant species.
4. Regulation of Splicing
An important question in AS is regulation of splice site choice. The splicing code, i.e., the set of biological rules for determining the splicing outcomes in both constitutive and alternative splicing, is only beginning to be addressed in animals (Barash et al., 2010a) and very little is known about it in plants.
4.1. Gene Architecture and Composition in Pre-mRNA Splicing
Comparative genomics studies on gene structure have revealed major differences in the architecture of plant and animal genes (Reddy, 2007). Plant genes are generally shorter than animal genes with fewer exons and significantly shorter introns. In animals, the average size of exons is about 140 nucleotides, whereas introns are several thousands of nucleotides long (average 3000 nucleotides in humans). In contrast, plant exons and introns are about the same length (in Arabidopsis for example, the average length of exons, and introns is 173 and 172 nucleotides, respectively) or about twice the size of exons in rice (e.g., exons 193 and introns 433 nucleotides; Reddy, 2007; Baek et al., 2008). Based on the smaller size of plant introns relative to animal introns and the high number of intron retention events, it is thought that the primary mode of splicing in plants is accomplished via an intron definition mechanism. However, there are plants whose introns are longer, but still have a high rate of intron retention (e.g., grape, with intron length of around 970, computed using the current genome annotations), and animals with short introns and exon skipping rates characteristic of mammalian systems (e.g., Tetraodon nigroviridis (puffer fish) which has introns of length 600 bp on average).
Plant introns are rich in U and UA nucleotides and exons are G-rich (Goodall and Filipowicz, 1989). Several studies indicate that cis-elements involved in intron recognition in plants are likely to be different from those in yeast and animals (Reviewed in Reddy, 2001; Schuler, 2008). For instance, animal introns are not accurately spliced in plants and vice versa. High U or UA content in introns was found to be important for splice site recognition and efficient splicing of U2 type and U12 type introns (Goodall and Filipowicz, 1989; Lambermon et al., 2000; Lorkovic et al., 2000; Reddy, 2001; Lewandowska et al., 2004; Simpson et al., 2004), suggesting the presence of proteins that interact with U or UA rich elements. In tobacco, three proteins (UBP1, RBP45, RBP47) that bind to intronic sequences have been characterized. UBP1 has been shown to be necessary for efficient splicing of pre-mRNA as well as mRNA accumulation (Goodall and Filipowicz, 1989; Lambermon et al., 2000). An analysis of AS in Medicago, poplar, Arabidopsis and rice, revealed that except for rice, AS was most prevalent for introns with decreased UA content (Baek et al., 2008). In addition to intronic sequences, exonic purine-rich sequences have been shown to influence splice site choice (Egoavil et al., 1997; McCullough and Schuler, 1997; Lewandowska et al., 2004; Schuler, 2008). However, aside from broad surveys of gene architecture, composition, and mutational analysis of splice sites, there has been little work performed to uncover the putative cis-elements that would provide substantial computational evidence for the intron definition model as the primary means by which intron-retaining transcripts are generated.
4.2. Cis-Elements in Pre-mRNAs that Control Splicing
In metazoans, four core sequence elements at the exon/intron boundaries and the intron/exon boundaries are necessary for splice site recognition by the spliceosome. These include (i) a motif at the 5′ splice site (SS) or donor site with a conserved GU dinucleotide, (ii) another motif at the 3′ SS or acceptor site with a conserved AG dinucleotide, (iii) a stretch of pyrimidines (polypyrimidine tract) upstream of the 3′ SS and (iv) a branch point 17–40 nucleotides upstream of the polypyrimidine tract. Components of the spliceosome recognize these core signals. U1 snRNP recognizes the 5′ SS, U2AF35 and U2AF65 recognize the 3′ SS and polypyrimidine tract, respectively and U2 snRNP recognizes the branch point. The 5′ and 3′ SSs are very similar between plants and animals, and the polypyrimidine tract in plants is rich in Us (Reddy, 2007). Several mutants in which splice sites are affected have been isolated and consequences of these mutations have been reviewed extensively (Brown, 1996; Schuler, 2008). Although these core elements are conserved across species, they are very short and they alone, are not sufficient to define exons and introns and recruit the splicing machinery. Additional sequences in exons and introns, which are collectively referred to as splicing regulatory elements (SREs), are important for constitutive splicing as well as AS. The SREs function as either splicing enhancers or suppressors and affect splice site choice. The efficiency by which the spliceosome recognizes exons and introns is in part determined by numerous protein factors that recognize these SREs. Depending on the location of SREs and their effect on splicing, they are grouped into four classes: exonic splicing enhancers (ESEs), exonic splicing silencers (ESSs), intronic splicing enhancers (ISEs), and intronic splicing silencers (ISSs; Black, 2003; Chasin, 2007; Wang and Burge, 2008; Wang et al., 2009b).
4.2.1. Computational studies to predict SREs
In animals systems, many studies have been performed to predict SREs. Most of these studies focused on exon skipping as this is the most prevalent AS event (Fairbrother et al., 2002; Sorek et al., 2004; Dror et al., 2005; Ohler et al., 2005; Ratsch et al., 2005; Yeo et al., 2005; Barash et al., 2010a,b) and some on tandem 3′ splice sites (Akerman and Mandel-Gutfreund, 2006; Xia et al., 2006). Computational tools to predict ESEs such as RESCUE-ESE (Relative Enhancer and Silencer Classification by Unanimous Enrichment) have been used to predict hexameric ESEs in animals (Fairbrother et al., 2002). Several other programs have been developed to identify splicing elements or to simulate the splicing of primary transcripts (Schwartz et al., 2009a). These methods are based on the rationale that alternatively spliced exons may contain sequence elements that are absent in constitutively spliced exons and introns or vice versa. Analysis of sequences that are over or under represented in alternatively spliced exons compared to constitutively spliced exons has led to prediction of sequences that could contribute to AS. Experimental validation of some of these has led to identification of many cis regulatory elements involved in regulating splice site choice either by enhancing or silencing the AS of a particular exon. These studies have shown that cis-elements involved in splicing can exist predominantly in exons and also in introns in some cases (Blencowe, 2000; Chasin, 2007). The theme that emerged from these analyses is that most SREs often share common features: they are short (about 6–10 nucleotides) and loosely conserved between different targets of the same protein, suggesting a flexible nature of the RNA-protein interaction. These motifs, that are individually weak but present in multiple copies, act as binding sites for competing trans factors (Ladd and Cooper, 2002). The most commonly studied elements are ESEs and the SR family of splicing regulators that bind them. It is now clear that many, if not all exons, constitutively or alternatively spliced, harbor ESEs that bind to SR proteins (Black, 2003; Chasin, 2007; Barash et al., 2010a). By contrast, ESS elements are usually bound by hnRNP proteins and can repress interactions across exons and introns that participate in spliceosome assembly (Blencowe, 2006; Chasin, 2007).
A recent compendium of cis-elements was used in the development of a mammalian “splicing code.” It included 171 known motifs and 326 new motifs that were used to predict splicing patterns of exons (Barash et al., 2010a). While most research has focused on discovering these elements within mammalian species, some progress has been made in identifying ESE motifs in Arabidopsis (Pertea et al., 2007). The goal of the study was to improve splice site prediction accuracy in Arabidopsis by incorporating potential ESE motifs into splice site recognition programs. The authors constructed a dataset (ESEAra) comprised of around 4000 high quality Arabidopsis gene models containing close to 17,500 coding exons. They extracted 50 bp regions flanking the boundaries of internal and terminal exons in the ESEAra dataset, established frequency distributions of hexamers near weak and strong splice sites, and used a scoring threshold to identify 84 putative ESE hexamers in these regions. Of these 84 ESE hexamers, 35 (12 at the 5′ end, 6 at the 3′ end, and 17 at both ends) had experimental evidence for ESE activity in Arabidopsis (Pertea et al., 2007). Several of the detected motifs exhibit the GAAGAA hexamer, which is a part of a recognized human ESE.
Aside from the above study, our catalog of plant cis regulatory elements involved in AS is limited. Over two decades ago, it was shown that AU-rich sequence elements in plant introns are required for plant pre-mRNA splicing (Goodall and Filipowicz, 1989; Filipowicz et al., 1995). Since then, our knowledge of trans factor binding sites has been constrained to putative or few experimentally defined regulatory motifs. Yoshimura et al. (2002) experimentally identified an AU-rich splicing regulatory element (GU[G|C|A]UUGC[C|U]UAUUUGAAUUGCAG) located in an exon/intron boundary responsible for tissue-specific AS of tobacco chloroplast ascorbate peroxidase (chlAPX) pre-mRNA. Four isoforms are produced via AS of the 3′-terminal region of chlAPX pre-mRNA (tAPX-I, sAPX-I, -II, and -III) due to alternative excision of intron 11 and intron 12. Subsequent site directed mutagenesis of the cis regulatory element and RNA gel mobility shift assays confirmed interaction with a particular trans factor expressed in leaves but not roots. The identity of this trans factor remains unknown. Using two 9G8 (one of the mammalian SR proteins) binding sequences in gel shift assays, it was shown that AtRSZ22 also binds to those sequences (Lopato et al., 1999). AtGRP7 (Arabidopsis thaliana glycine-rich RNA binding protein) regulates AS of its own pre-mRNA as well as its paralog, AtGRP8, pre-mRNA by producing a PTC-containing splice variant when its levels increase. Similarly, AtGRP8 autoregulates its AS and AtGRP7 pre-mRNA AS (Schoning et al., 2007, 2008). This regulation involves binding of AtGRP7 to its pre-mRNA and the pre-mRNA of AtGRP8 and vice versa (Schoning et al., 2007, 2008). The sequences in these pre-mRNAs that bind to AtGRP7 and AtGRP8 have been identified (Schoning et al., 2007, 2008). Although there are several other instances of auto- and cross-regulation of AS of several spliceosomal proteins, cis-elements involved in regulation of AS are not known (Kalyna et al., 2003; Reddy, 2007; Barta et al., 2008; Stauffer et al., 2010; Reddy and Ali, 2011).
Because intron retention is so pervasive in plants, uncovering the sequence elements that lead to this splice form would be a valuable breakthrough, as there are fundamental implications for post-transcriptional regulation of gene expression either by NMD/RUST (Lewis et al., 2003), or by generating new target sites for miRNA binding. There is evidence that generation of premature termination codon in plants by AS leads to degradation of mRNAs (Arciga-Reyes et al., 2006; Schoning et al., 2008; Kurihara et al., 2009; Barbazuk, 2010; Palusa and Reddy, 2010; Staiger and Green, 2011). In humans, it has been predicted that retained introns in the 3′ UTR can create and/or increase putative miRNA targets (Tan et al., 2007). Analysis of all known retained introns and their flanking exons can be performed computationally to determine what sequence elements contribute to intron retention. Building a set of potential regulatory sequences would lend predictive power to discern the likelihood that a given gene will produce an intron-retaining transcript.
4.2.2. Validation of computationally predicted SREs
In animals, in vitro and in vivo splicing assays with pre-mRNAs containing wild type and mutated putative SREs have been used to validate predictions. Unfortunately, in vitro methods that employ S100 or nuclear extracts for splicing assays for identifying SREs cannot be applied to plants, as there is no plant-derived in vitro splicing assay system. However, the validity of computationally predicted plant cis-elements in splicing regulation can be tested using two different approaches. In one approach, one can generate mutations in the predicted cis-element and compare the splicing of wild type to mutated gene in a transient or stable expression system, which will allow analysis of splicing in its natural exon/intron context. Alteration of predicted cis-elements can be done in a high-throughput manner using the strategy described in Figure 4. Splicing of the wild type and mutated gene can be first tested in protoplasts by RT-PCR using a forward primer corresponding to the tag and a reverse gene-specific primer. If the candidate gene is not expressed in mesophyll cells then it can be analyzed in transgenic lines. If there is an SRE then one can test the importance of individual bases in that element by site directed mutagenesis. If there are two cis-elements in a gene that are complementary to each other and have the potential to form a stem/stem-loop structure, then a change in the sequence of one of the two elements or both to disrupt base pairing should affect splicing. To confirm that the base pairing in the two cis-elements, not the sequence itself, is necessary, one can mutate both elements in such a way that the sequence is changed but the two elements are complementary to each other. If this still shows regulated splicing then it is likely that the base pairing is involved in regulated splicing.
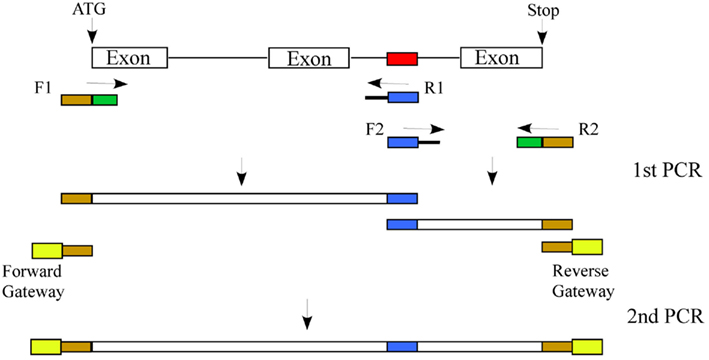
Figure 4. Approach to mutate a predicted cis-element: Generation of a construct in which a predicted cis-element (shown in red) is changed (shown in blue) involves two rounds of PCR. In the first PCR the target gene with two primers sets (F1/R1 and F2/R2). The F1 and R1 primer set amplifies the gene from the initiation codon to the predicted cis-element and F2 and R2 will amplify from the predicted cis-element to the stop codon. Primers F1 and R2, in addition to gene-specific sequence (shown in green), will be tailed with sequences complementary to Gateway vector primers (shown in dark yellow). Similarly, primers R1 and F2, in addition to gene-specific sequence, will be tailed with the changed sequence in the predicted cis-element (shown in blue). In the second PCR, the two gene fragments from the first PCR will be mixed. This overlapping template will be amplified using primers complementary to primers F1 and R2 tailed with the attB1 and attB2 Gateway sequences, which can then be cloned into a Gateway donor vector and into a plant transformation vector with a tag as a fusion to the N-terminus. The wild type gene will be cloned in a similar fashion except that only one PCR will be done with the F1/R2 primer set containing the entire attB1 and attB2 Gateway sequences.
A second approach for validating cis-elements in an intron is to use a reporter gene (GFP) that is interrupted by a test intron that contains predicted cis-elements. By following the gene’s splicing pattern, one can determine if the signals are present exclusively in the inserted part of the gene. A similar approach could be used to study cis-elements in other regions of a gene. Identified SREs can then be further analyzed by using site directed mutagenesis.
In vivo analysis of pre-mRNA splicing by expressing splicing reporters in transient assays (e.g., protoplasts or leaf transfections) have not been used widely (Gniadkowski et al., 1996; Lambermon et al., 2002; Lewandowska et al., 2004; Isshiki et al., 2006; Schuler, 2008; Stauffer et al., 2010). The lack of a plant-derived in vitro splicing system makes transient assay systems very attractive for studying the constitutive splicing and AS of pre-mRNAs for genes that are expressed in leaves.
4.2.3. Experimental approaches for transcriptome-wide identification of SREs
RNA binding proteins (RBPs) with characteristic RNA binding motifs such as the RNA recognition motif (RRM) and the K-homology (KH) domain that interact with specific RNAs, profoundly impact gene expression at various levels (transcription, capping, splicing, polyadenylation, biogenesis of miRNAs and siRNAs, RNA transport, localization and degradation, small RNA regulated gene expression) and have been shown to play important roles in development and disease in animals (Licatalosi and Darnell, 2010). In plants, a large repertoire of RNA binding proteins have been identified using bioinformatic tools. In Arabidopsis there are more than two hundred RNA binding proteins (RBPs), including 200 RRM containing RBPs and 30 KH domain proteins (Lorkovic, 2009). Many plant RBPs are unique to plants, suggesting that they are likely to have novel RNA targets and perform plant-specific functions. A large fraction of RBPs are implicated in pre-mRNA splicing (Reddy, 2007; Simpson et al., 2008; Lorkovic, 2009; Reddy and Ali, 2011; Staiger and Green, 2011). Interestingly, many pre-mRNAs encodings RBPs (e.g., pre-mRNAs of SR and SR-like proteins, glycine-rich RNA binding proteins, and other spliceosomal proteins) are extensively alternatively spliced in plants and there is tight regulation of AS of these genes (Isshiki et al., 2006; Palusa et al., 2007; Barta et al., 2008; Simpson et al., 2010; Stauffer et al., 2010; Staiger and Green, 2011). Protein-RNA interactions are crucial in many aspects of RNA metabolism. These interactions are dependent on RNA sequence elements in RNAs that interact specifically with proteins. Although the in vivo RNA targets of most plant RBPs are unknown, genetic studies with some of the RBPs indicate a crucial role for these in different developmental processes (flowering time, flower development, circadian responses) and various biotic and abiotic stress-signaling pathways (Reddy, 2007; Simpson et al., 2008; Lorkovic, 2009; Staiger and Green, 2011).
RNA sequences that bind to RBPs are generally analyzed using various methods. These include RNA immunoprecipitation (RIP), which involves co-immunoprecipitation of RNAs with RBPs without prior cross-linking to the RNA using an antibody to a specific RBP followed by sequencing of the precipitated RNAs (RIP-seq) or probing microarrays with precipitated RNA (RIP-chip; Brown et al., 2001; Tenenbaum et al., 2002; Barkan, 2009; Wang et al., 2009c). The caveats of this approach include loss of true interactions because the transient and dynamic nature of RNA-protein interactions, thereby precluding co-IP of some RNAs, and at the same time, high noise levels due to the sticky nature of RNAs (Mili and Steitz, 2004). Moreover, the binding RNA sequences represent both direct and indirect targets as most RBPs interact with other proteins and form complexes (Reddy, 2007). Advances in efficient ways to UV crosslink RNAs with RBPs in vivo followed by immunoprecipitation of complexes with an RBP specific antibody (CLIP, Cross-Linking ImmunoPrecipitation) under stringent conditions and sequencing of RNA using high-throughput RNA sequencing (HITS-CLIP) have paved the way to map, in an unbiased manner, transcriptome-wide direct RNA targets of an RBP (Ule et al., 2003, 2005, 2006; Wang et al., 2009b; Darnell, 2010; Licatalosi and Darnell, 2010). In HITS-CLIP, cells/tissues are exposed to ultraviolet (UV) radiation, which generates covalent bonds between RNA and proteins that are in close contact (Ule et al., 2003, 2005). Because of the covalent bonds, RNA-protein complexes can be purified under stringent conditions. A modified version of HITS-CLIP called Photoactivatable-Ribonucleoside-Enhanced Cross-linking and Immunoprecipitation (PAR-CLIP) enhances cross-linking efficiency and allows identification of the locations in the RNA that are involved in interacting with RBP (Hafner et al., 2010a). In PAR-CLIP, a photoreactive ribonucleoside analog, 4-thiouridine, is incorporated into nascent RNAs, which are then cross-linked to RBPs by exposing cells to UV radiation. RNAs that are complexed with a particular RBP are then immunoprecipitated using an antibody to that RBP. RNAs in immunoprecipitated samples are then converted into cDNA and sequenced using NGS platforms. Since the cross-linked RNA region shows T to C mutations in the sequenced cDNA, the binding region in RNA can be precisely identified (Hafner et al., 2010a).
An in-depth discussion on advantages and disadvantages of these methods is available in recent reviews (Barkan, 2009; Darnell, 2010; Hafner et al., 2010b). These methods could be applied to plants to create a transcriptome-wide map of RNA binding sequences that bind to a given RBP. These tools, developed with animal systems, are fairly recent and have the potential to uncover plant SREs, especially those involved in AS events that are prevalent in plants as compared to animals. These procedures can identify the binding site landscape or “RNA map” for plant SR proteins, one of the key regulators of pre-mRNA splicing and other RBPs. Incorporating the results from these studies into computational predictions should lead to a better understanding of the splicing code in plants.
4.2.4. RNA secondary structure and other properties that affect AS
In addition to the presence of SREs, there are several properties of pre-mRNA transcripts that were shown to affect AS. These include intron length, GC-content, splice site strength, and pre-mRNA secondary structure (Ladd and Cooper, 2002; Chasin, 2007; Shepard and Hertel, 2008; Schwartz et al., 2009b; Labadorf et al., 2010). A comparison of retained and constitutively spliced introns identified using Arabidopsis tiling arrays revealed that retained introns tended to be shorter and to have higher GC-content than constitutively spliced introns (Ner-Gaon and Fluhr, 2006). Similar characteristics were also observed for introns in rice (Zhang et al., 2010).
Sequence signals surrounding the 5′ and 3′ splice site junctions, polypyrimidine tract (PPT), and branch sites also impact splice site selection. For instance, in humans and plants it has been shown that splice sites flanking retained introns contain weaker signals than those flanking constitutively spliced introns (Kurmangaliyev and Gelfand, 2008; Labadorf et al., 2010). Studies on PPT and branch sites (Brown, 1996; Tolstrup et al., 1997) in plants suggest that their metazoan equivalents are more pronounced. However, their importance in correctly splicing pre-mRNA was shown by Simpson et al. (2002). This evidence suggests a need to generate a catalog for all AS variants centered at these signal-rich regions.
Cis regulatory elements in pre-mRNA secondary structure are known to regulate AS by sometimes affecting the recruitment of SR proteins (Buratti and Baralle, 2004; Buratti et al., 2004). Formation of secondary structure between complementary repeat-pair sequences in two introns flanking a cassette exon is proposed to mask splice sites and cause exon skipping events (Lian and Garner, 2005). A similar mechanism where complementary sequences in two exons can base pair across an intron may lead to intron retention. There are only a few examples in plants where RNA secondary structure has been shown to regulate splicing. In photosynthetic eukaryotes, AS of pre-mRNAs from genes encoding proteins involved in thiamine metabolism (THIC and THI4 in Chlamydomonas and THIC in Arabidopsis) is regulated by a thiamin pyrophosphate (TPP) binding riboswitch (Bocobza et al., 2007; Croft et al., 2007; Wachter et al., 2007). High levels of TPP result in its binding to an RNA aptamer in the pre-mRNA, which leads to production of splice variants with a PTC or long 3′UTR that are unstable (Bocobza and Aharoni, 2008; Wachter, 2010). In plants, the transcription factor IIIA (TFIIIA), which is necessary for transcription of 5S RNA, is alternatively spliced by inclusion or exclusion of its third exon; the splice variant with the included exon contains a PTC and is a target of NMD (Barbazuk, 2010). This exon is thought to be derived by exonization of 5S RNA and is conserved from moss to flowering plants, but not in green algae and metazoans (Fu et al., 2009; Hammond et al., 2009). The 5S RNA-like element assumes a 5S RNA structure, which regulates AS of its own pre-mRNA. The 5S RNA-like structure binds ribosomal protein L5, which in turn promotes exon skipping and generation of a functional transcript, whereas in the absence of L5, this exon is included, and results in the production of a PTC-containing unstable transcript (Hammond et al., 2009), suggesting a tight regulation of functional TFIIIA mRNA by the L5 protein.
5. Regulation of Alternative Splicing by Chromatin Organization
Apart from the features mentioned above, several recent studies point to epigenetic regulation of AS. Access to DNA may be affected by chromatin organization or methylation, which could impact the rate at which a gene is transcribed thereby affecting AS (Luco et al., 2011). In animals, it is now well established that the recruitment of splicing factors to pre-mRNAs occurs co-transcriptionally by the RNA Pol II carboxy-terminal domain, but the completion of splicing may occur co-transcriptionally or post-transcriptionally (Pandya-Jones and Black, 2009; Luco et al., 2011). Furthermore, the rate of elongation of transcription regulates AS, where rapid elongation favors recruitment of the splicing machinery to strong splice sites, thereby including exons with strong splice sites; whereas slow elongation allows recruitment of spliceosome to weak sites (Luco et al., 2011).
Some studies implicate a more direct role for chromatin remodeling enzymes. For instance, histone deacetylases in yeast and humans interact directly with U2 snRNP (Gunderson and Johnson, 2009) and the histone methyltransferase with U1 snRNP-specific protein U1C (Ohkura et al., 2005) and alter splicing patterns. Furthermore, chromatin remodelers SWI/SNF in yeast and humans (e.g., Braham) also regulate AS by interacting with spliceosomal proteins and recruiting snRNPs (Batsche et al., 2006; Tyagi et al., 2009). In Arabidopsis, mutations in protein arginine methyl transferase 5 (PRMT5), which methylates arginine residues in histones and Sm spliceosomal proteins, impair the circadian rhythm (Sanchez et al., 2010). It has been shown that AS of core-clock regulated genes (e.g., pseudo response regulator 9) and other pre-mRNAs is altered in the prmt5 mutants (Sanchez et al., 2010). Whether the observed effects in prmt5 mutants are due to altered methylation of Sm proteins and/or due to epigenetic defects remains to be studied.
Genome-wide nucleosome positioning and methylation studies in Arabidopsis and humans revealed that DNA associated with nucleosomes is more highly methylated than the flanking DNA (Nahkuri et al., 2009; Chodavarapu et al., 2010). Furthermore, nucleosomes are enriched in exons, particularly at exon-intron and intron-exon boundaries. Included exons and exons with weak splice sites are highly enriched in nucleosomes (Schwartz et al., 2009c; Spies et al., 2009; Tilgner et al., 2009). These studies suggest a role for nucleosome positioning in defining exons as well as regulating splicing (Nahkuri et al., 2009; Schwartz et al., 2009c; Spies et al., 2009; Tilgner et al., 2009; Chodavarapu et al., 2010). Small interfering RNAs (siRNAs) trigger transcriptional gene silencing by inducing heterochromatin formation in a sequence specific manner. Targeting of siRNA to intronic or exonic sequences that are close to an alternative exon has been shown to regulate the splicing of that exon (Allo et al., 2009), suggesting a role for siRNAs in AS.
Conclusion
Elucidation of multiple layers of gene regulation is critical to understand how plants grow, differentiate and respond appropriately to their environment. Regulated pre-mRNA splicing is emerging as an important layer in gene regulation. Extensive studies aimed at identifying features that control splicing in animal cells suggest that the combination of multiple characteristics in pre-mRNAs, including loosely conserved cis-elements and/or secondary structure(s) in transcripts, chromatin modification, and the rate of transcription regulate splicing (Barash et al., 2010a; Schor et al., 2010; Luco et al., 2011). Thus far, our knowledge of AS regulation in plants is limited to a few experimentally determined ESE motifs in plants. A prerequisite for elucidating the splicing code for plants is to identify candidate SREs. New technological advances such as deep sequencing of RNA, together with in vivo RNA-RBP cross-linking methods (HITS-CLIP, PAR-CLIP) have paved the way to find SREs globally in animals. Although these methods are applicable to plants, they have yet to be employed. Determining the splicing code in plants requires analysis of entire transcriptomes across tissues and conditions, and relating the observed patterns of AS to a variety of sequence signals, including SREs, nucleosome positioning signals, and possibly other as-yet unknown factors that affect splice site choice. Addressing these challenges and integrating the results to formulate the plant splicing code is a daunting task that will require a significant effort from the plant research community.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Pre-mRNA research in our laboratories is funded by a grant from the National Science Foundation. We thank Julie Thomas for her comments on the manuscript.
References
Abeel, T. (2011). Genomeview: visualizing the next-generation of data. J. Biomol. Tech. 22(Suppl.), :S19.
Akerman, M., and Mandel-Gutfreund, Y. (2006). Alternative splicing regulation at tandem 3’ splice sites. Nucleic Acids Res. 34, 23–31.
Allo, M., Buggiano, V., Fededa, J. P., Petrillo, E., Schor, I., de la Mata, M., Agirre, E., Plass, M., Eyras, E., Elela, S. A., Klinck, R., Chabot, B., and Kornblihtt, A. R. (2009). Control of alternative splicing through sirna-mediated transcriptional gene silencing. Nat. Struct. Mol. Biol. 16, 717–724.
Arciga-Reyes, L., Wootton, L., Kieffer, M., and Davies, B. (2006). UPF1 is required for nonsense-mediated mRNA decay (NMD) and RNAi in Arabidopsis. Plant J. 47, 480–489.
Au, K. F., Jiang, H., Lin, L., Xing, Y., and Wong, W. H. (2010). Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 38, 4570.
Baek, J. M., Han, P., Iandolino, A., and Cook, D. R. (2008). Characterization and comparison of intron structure and alternative splicing between Medicago truncatula, Populus trichocarpa, Arabidopsis and rice. Plant Mol. Biol. 67, 499–510.
Barash, Y., Calarco, J. A., Gao, W., Pan, Q., Wang, X., Shai, O., Blencowe, B. J., and Frey, B. J. (2010a). Deciphering the splicing code. Nature 465, 53–59.
Barash, Y., Blencowe, B. J., and Frey, B. J. (2010b). Model-based detection of alternative splicing signals. Bioinformatics 26, i325–i33.
Barbazuk, W. B. (2010). A conserved alternative splicing event in plants reveals an ancient exonization of 5s rRNA that regulates TFIIIA. RNA Biol. 7, 397–402.
Barbazuk, W. B., Fu, Y., and McGinnis, K. M. (2008). Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res. 18, 1381–1392.
Barkan, A. (2009). Genome-wide analysis of RNA-protein interactions in plants. Methods Mol. Biol. 553, 13–37.
Barta, A., Kalyna, M., and Lorkovic, Z. J. (2008). Plant SR proteins and their functions. Curr. Top. Microbiol. Immunol. 326, 83–102.
Batsche, E., Yaniv, M., and Muchardt, C. (2006). The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat. Struct. Mol. Biol. 13, 22–29.
Birol, I., Jackman, S. D., Nielsen, C. B., Qian, J. Q., Varhol, R., Stazyk, G., Morin, R. D., Zhao, Y., Hirst, M., Schein, J. E., Horsman, D. E., Connors, J. M., Gascoyne, R. D., Marra, M. A., and Jones, S. J. (2009). De novo transcriptome assembly with ABySS. Bioinformatics 25, 2872.
Black, D. L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336.
Blencowe, B. J. (2000). Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci. 25, 106–110.
Blencowe, B. J., Ahmad, S., and Lee, L. J. (2009). Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev. 23, 1379.
Bocobza, S., Adato, A., Mandel, T., Shapira, M., Nudler, E., and Aharoni, A. (2007). Riboswitch-dependent gene regulation and its evolution in the plant kingdom. Genes Dev. 21, 2874–2879.
Bocobza, S. E., and Aharoni, A. (2008). Switching the light on plant riboswitches. Trends Plant Sci. 13, 526–533.
Bonizzoni, P., Mauri, G., Pesole, G., Picardi, E., Pirola, Y., and Rizzi, R. (2009). Detecting alternative gene structures from spliced ESTs: a computational approach. J. Comput. Biol. 16, 43–66.
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., Jin, X., Feng, Y., Wilkinson, K. D., Keene, J. D., Darnell, R. B., and Warren, S. T. (2001). Microarray identification of fmrp-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487.
Buratti, E., and Baralle, F. E. (2004). Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell Biol. 24, 10505–10514.
Buratti, E., Muro, A. F., Giombi, M., Gherbassi, D., Iaconcig, A., and Baralle, F. E. (2004). RNA folding affects the recruitment of SR proteins by mouse and human polypurinic enhancer elements in the fibronectin EDA exon. Mol. Cell Biol. 24, 1387–1400.
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M., and Buell, C. R. (2006). Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7, 327. doi:10.1186/1471-2164-7-327
Chang, Y. F., Imam, J. S., and Wilkinson, M. F. (2007). The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 76, 51–74.
Chen, W. H., Lv, G., Lv, C., Zeng, C., and Hu, S. (2007). Systematic analysis of alternative first exons in plant genomes. BMC Plant Biol. 7, 55. doi:10.1186/1471-2164-7-327
Chen, X. (2009). Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 25, 21–44.
Chodavarapu, R. K., Feng, S., Bernatavichute, Y. V., Chen, P. Y., Stroud, H., Yu, Y., Hetzel, J. A., Kuo, F., Kim, J., Cokus, S. J., Casero, D., Bernal, M., Huijser, P., Clark, A. T., Kramer, U., Merchant, S. S., Zhang, X., Jacobsen, S. E., and Pellegrini, M. (2010). Relationship between nucleosome positioning and DNA methylation. Nature 466, 388–392.
Clark, T. A., Sugnet, C. W., and Ares, M. (2002). Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 296, 907.
Costa, V., Angelini, C., De Feis, I., and Ciccodicola, A. (2010). Uncovering the complexity of transcriptomes with RNA-seq. J. Biomed. Biotechnol. 2010, 1–19.
Croft, M. T., Moulin, M., Webb, M. E., and Smith, A. G. (2007). Thiamine biosynthesis in algae is regulated by riboswitches. Proc. Natl. Acad. Sci. U.S.A. 104, 20770–20775.
Cuperlovic-Culf, M., Belacel, N., Culf, A. S., and Ouellette, R. J. (2006). Data analysis of alternative splicing microarrays. Drug Discov. Today 11, 983–990.
Darnell, R. B. (2010). HITS-CLIP: panoramice views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 1, 266–286.
Dror, G., Sorek, R., and Shamir, R. (2005). Accurate identification of alternatively spliced exons using support vector machine. Bioinformatics 21, 897–901.
Durinck, S. (2008). Pre-processing of microarray data and analysis of differential expression. Methods Mol. Biol. 452, 89.
Egoavil, C., Marton, H. A., Baynton, C. E., McCullough, A. J., and Schuler, M. A. (1997). Structural analysis of elements contributing to 5’ splice site selection in plant pre-mRNA transcripts. Plant J. 12, 971–980.
Fairbrother, W. G., Yeh, R. F., Sharp, P. A., and Burge, C. B. (2002). Predictive identification of exonic splicing enhancers in human genes. Science 297, 1007–1013.
Fang, Z., and Cui, X. (2011). Design and validation issues in RNA-seq experiments. Brief. Bioinformatics 12, 280.
Feng, J., Li, W., and Jiang, T. (2011). Inference of isoforms from short sequence reads. J. Comput. Biol. 18, 305–321.
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R., Bryant, D. W., Fox, S. E., Wong, W. K., and Mockler, T. C. (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 20, 45–58.
Filipowicz, W., Gniadkowski, M., Klahre, U., and Liu, H.-X. (1995). Pre-mRNA splicing in plants. Trends Plant Sci. 4, 65–77.
Fiume, M., Williams, V., Brook, A., and Brudno, M. (2010). Savant: genome browser for high-throughput sequencing data. Bioinformatics 26, 1938–1944.
Fu, Y., Bannach, O., Chen, H., Teune, J. H., Schmitz, A., Steger, G., Xiong, L., and Barbazuk, W. B. (2009). Alternative splicing of anciently exonized 5S rRNA regulates plant transcription factor TFIIIA. Genome Res. 19, 913–921.
Gaidatzis, D., Jacobeit, K., Oakeley, E. J., and Stadler, M. B. (2009). Overestimation of alternative splicing caused by variable probe characteristics in exon arrays. Nucleic Acids Res. 37, e107.
Gan, X., Stegle, O., Behr, J., Steffen, J. G., Drewe, P., Hildebrand, K. L., Lyngsoe, R., Schultheiss, S. J., Osborne, E. J., Sreedharan, V. T., Kahles, A., Bohnert, R., Jean, G., Derwent, P., Kersey, P., Belfield, E. J., Harberd, N. P., Kemen, E., Toomajian, C., Kover, P. X., Clark, R. M., Ratsch, G., and Mott, R. (2011). Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423.
Gniadkowski, M., Hemmings-Mieszczak, M., Klahre, U., Liu, H. X., and Filipowicz, W. (1996). Characterization of intronic uridine-rich sequence elements acting as possible targets for nuclear proteins during pre-mRNA splicing in Nicotiana plumbaginifolia. Nucleic Acids Res. 24, 619–627.
Goff, S. A., Vaughn, M., McKay, S., Lyons, E., Stapleton, A. E., Gessler, D., Matasci, N., Wang, L., Hanlon, M., Lenards, A., Muir, A., Merchant, N., Lowry, S., Mock, S., Helmke, M., Kubach, A., Narro, M., Hopkins, N., Micklos, D., Hilgert, U., Gonzales, M., Jordan, C., Skidmore, E., Dooley, R., Cazes, J., McLay, R., Lu, Z., Pasternak, S., Koesterke, L., Piel, W. H., Grene, R., Noutsos, C., Gendler, K., Feng, X., Tang, C., Lent, M., Kim, S. -J., Kvilekval, K., Manjunath, B. S., Tannen, V., Stamatakis, A., Sanderson, M., Welch, S. M., Cranston, K. A., Soltis, P., Soltis, D., O’Meara, B., Ane, C., Brutnell, T., Kleibenstein, D. J., White, J. W., Leebens-Mack, J., Donoghue, M. J., Spalding, E. P., Vision, T. J., Myers, C. R., Lowenthal, D., Enquist, B. J., Boyle, B., Akoglu, A., Andrews, G., Ram, S., Ware, D., Stein, L., and Stanzione, D. (2011). The iPlant collaborative: cyberinfrastructure for plant biology. Front. Plant Sci. 2:34. doi:10.3389/fpls.2011.00034
Goodall, G. J., and Filipowicz, W. (1989). The au-rich sequences present in the introns of plant nuclear pre-mRNAs are required for splicing. Cell 58, 473–483.
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., Adiconis, X., Fan, L., Raychowdhury, R., Zeng, Q., Chen, Z., Mauceli, E., Hacohen, N., Gnirke, A., Rhind, N., di Palma, F., Birren, B. W., Nusbaum, C., Lindblad-Toh, K., Friedman, N., and Regev, A. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652.
Grant, G. R., Manduchi, E., and Stoeckert, C. J. Jr. (2007). Analysis and management of microarray gene expression data. Curr. Protoc. Mol. Biol. 77, 19.6.1–19.6.30.
Gu, L., and Guo, R. (2007). Genome-wide detection and analysis of alternative splicing for nucleotide binding site-leucine-rich repeats sequences in rice. J. Genet. Genomics 34, 247–257.
Gunderson, F. Q., and Johnson, T. L. (2009). Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 5, e1000682. doi:10.1371/journal.pgen.1000682
Guo, S., Zheng, Y., Joung, J. G., Liu, S., Zhang, Z., Crasta, O. R., Sobral, B. W., Xu, Y., Huang, S., and Fei, Z. (2010). Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types. BMC Genomics 11, 384. doi:10.1186/1471-2164-11-384
Guttman, M., Garber, M., Levin, J. Z., Donaghey, J., Robinson, J., Adiconis, X., Fan, L., Koziol, M. J., Gnirke, A., Nusbaum, C., Rinn, J. L., Lander, E. S., and Regev, A. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 28, 503–510.
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K. Jr., Hannick, L. I., Maiti, R., Ronning, C. M., Rusch, D. B., Town, C. D., Salzberg, S. L., and White, O. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654.
Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser, J., Berninger, P., Rothballer, A. Jr., Ascano, M., Jungkamp, A. C., Munschauer, M., Ulrich, A., Wardle, G. S., Dewell, S., Zavolan, M., and Tuschl, T. (2010a). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141.
Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser, J., Berninger, P., Rothballer, A., Ascano, M., Jungkamp, A. C., Munschauer, M., Ulrich, A., Wardle, G. S., Dewell, S., Zavolan, M., and Tuschl, T. (2010b). PAR-CliP–a method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. doi:10.3791/2034.
Hallegger, M., Llorian, M., and Smith, C. W. (2010). Alternative splicing: global insights. FEBS J. 277, 856–866.
Hammond, M. C., Wachter, A., and Breaker, R. R. (2009). A plant 5S ribosomal RNA mimic regulates alternative splicing of transcription factor IIIA pre-mRNAs. Nat. Struct. Mol. Biol. 16, 541–549.
Harrington, E. D., and Bork, P. (2008). Sircah: a tool for the detection and visualization of alternative transcripts. Bioinformatics 24, 1959.
Hazen, S. P., Naef, F., Quisel, T., Gendron, J. M., Chen, H., Ecker, J. R., Borevitz, J. O., and Kay, S. A. (2009). Exploring the transcriptional landscape of plant circadian rhythms using genome tiling arrays. Genome Biol. 10, R17.
He, Z. S., Zou, H. S., Wang, Y. Z., Zhu, J. B., and Yu, G. Q. (2008). Maturation of the nodule-specific transcript MsHSF1c in Medicago sativa may involve interallelic trans-splicing. Genomics 92, 115–121.
Heber, S., Alekseyev, M., Sze, S. H., Tang, H., and Pevzner, P. A. (2002). Splicing graphs and EST assembly problem. Bioinformatics 18, 181–188.
Hirsch, J., Lefort, V., Vankersschaver, M., Boualem, A., Lucas, A., Thermes, C., D’Aubenton Carafa, Y., and Crespi, M. (2006). Characterization of 43 non-protein-coding mRNA genes in Arabidopsis, including the MIR162a-derived transcripts. Plant Physiol. 140, 1192–1204.
Iida, K., Seki, M., Sakurai, T., Satou, M., Akiyama, K., Toyoda, T., Konagaya, A., and Shinozaki, K. (2004). Genome-wide analysis of alternative pre-mRNA splicing in Arabidopsis thaliana based on full-length cDNA sequences. Nucleic Acids Res. 32, 5096–5103.
Iida, K., Shionyu, M., and Suso, Y. (2008). Alternative splicing at NAGNAG acceptor sites shares common properties in land plants and mammals. Mol. Biol. Evol. 25, 709–718.
Isshiki, M., Tsumoto, A., and Shimamoto, K. (2006). The serine/arginine-rich protein family in rice plays important roles in constitutive and alternative splicing of pre-mRNA. Plant Cell 18, 146–158.
Jean, G., Kahles, A., Sreedharan, V. T., Bona, F. D., and Rätsch, G. (2010). RNA-seq read alignments with PALMapper. Curr. Protoc. Bioinformatics 32, 11.6.1–11.6.37.
Jiang, H., and Wong, W. H. (2009). Statistical inferences for isoform expression in RNA-seq. Bioinformatics 25, 1026.
Jiao, Y., and Meyerowitz, E. M. (2010). Cell-type specific analysis of translating RNAs in developing flowers reveals new levels of control. Mol. Syst. Biol. 6, 419.
Johnson, J. M., Castle, J., Garrett-Engele, P., Kan, Z., Loerch, P. M., Armour, C. D., Santos, R., Schadt, E. E., Stoughton, R., and Shoemaker, D. D. (2003). Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302, 2141.
Kalsotra, A., and Cooper, T. A. (2011). Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715–729.
Kalyna, M., Lopato, S., and Barta, A. (2003). Ectopic expression of atRSZ33 reveals its function in splicing and causes pleiotropic changes in development. Mol. Biol. Cell 14, 3565–3577.
Kalyna, M., Lopato, S., Voronin, V., and Barta, A. (2006). Evolutionary conservation and regulation of particular alternative splicing events in plant SR proteins. Nucleic Acids Res. 34, 4395–4405.
Kawasaki, T., Okumura, S., Kishimoto, N., Shimada, H., Higo, K., and Ichikawa, N. (1999). RNA maturation of the rice SPK gene may involve trans-splicing. Plant J. 18, 625–632.
Kim, H., Bi, Y., Pal, S., Gupta, R., and Davuluri, R. V. (2011). IsoformEx: isoform level gene expression estimation using weighted non-negative least squares from mRNA-seq data. BMC Bioinformatics 12, 305. doi:10.1186/1471-2105-12-305
Kumar, S., Asif, M. H., Chakrabarty, D., Tripathi, R. D., and Trivedi, P. K. (2011). Differential expression and alternative splicing of rice sulphate transporter family members regulate sulphur status during plant growth, development and stress conditions. Funct. Integr. Genomics 11, 259–273.
Kurihara, Y., Matsui, A., Hanada, K., Kawashima, M., Ishida, J., Morosawa, T., Tanaka, M., Kaminuma, E., Mochizuki, Y., Matsushima, A., Toyoda, T., Shinozaki, K., and Seki, M. (2009). Genome-wide suppression of aberrant mRNA-like noncoding RNAs by NMD in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 106, 2453–2458.
Kurmangaliyev, Y. Z., and Gelfand, M. S. (2008). Computational analysis of splicing errors and mutations in human transcripts. BMC Genomics 9, 13. :10.1186/1471-2164-9-13
Labadorf, A., Link, A., Rogers, M. F., Thomas, J., Reddy, A. S., and Ben-Hur, A. (2010). Genome-wide analysis of alternative splicing in Chlamydomonas reinhardtii. BMC Genomics 11, 114. doi:10.1186/1471-2164-11-114
Lacroix, V., Sammeth, M., Guigo, R., and Bergeron, A. (2008). “Exact transcriptome reconstruction from short sequence reads,” in Proceedings of the 8th International Workshop on Algorithms in Bioinformatics (Karlsruhe: Springer), 63.
Ladd, A. N., and Cooper, T. A. (2002). Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol. 3, reviews0008.
Lambermon, M. H., Fu, Y., Kirk, D. A., Dupasquier, M., Filipowicz, W., and Lorkovic, Z. J. (2002). UBA1 and UBA2, two proteins that interact with UBP1, a multifunctional effector of pre-mRNA maturation in plants. Mol. Cell Biol. 22, 4346–4357.
Lambermon, M. H., Simpson, G. G., Wieczorek, D., Kirk, A., Hemmings-Mieszczak, M., Klahre, U., and Filipowicz, W. (2000). UBP1, a novel hnRNP-like protein that functions at multiple steps of higher plant nuclear pre-mRNA maturation. EMBO J. 19, 1638–1649.
Lee, S., Seo, C. H., Lim, B., Yang, J. O., Oh, J., Kim, M., Lee, S., Lee, B., Kang, C., and Lee, S. (2011). Accurate quantification of transcriptome from RNA-seq data by effective length normalization. Nucleic Acids Res. 39, e9.
Lewandowska, D., Simpson, C. G., Clark, G. P., Jennings, N. S., Barciszewska-Pacak, M., Lin, C. F., Makalowski, W., Brown, J. W., and Jarmolowski, A. (2004). Determinants of plant u12-dependent intron splicing efficiency. Plant Cell 16, 1340–1352.
Lewis, B. P., Green, R. E., and Brenner, S. E. (2003). Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. U.S.A. 100, 189–192.
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. doi:10.1186/1471-2105-12-323
Li, B., Ruotti, V., Stewart, R. M., Thomson, J. A., and Dewey, C. N. (2010). RNA-seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493.
Li, W., Feng, J., and Jiang, T. (2011). “Isolasso: a lasso regression approach to RNA-seq based transcriptome assembly,” in Research in Computational Molecular Biology eds V. Bafna and S. Sahinalp (Berlin: Springer), 168–188.
Lian, Y., and Garner, H. R. (2005). Evidence for the regulation of alternative splicing via complementary DNA sequence repeats. Bioinformatics 21, 1358–1364.
Liang, C., Mao, L., Ware, D., and Stein, L. (2009). Evidence-based gene predictions in plant genomes. Genome Res. 19, 1912–1923.
Licatalosi, D. D., and Darnell, R. B. (2010). RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 11, 75–87.
Lopato, S., Gattoni, R., Fabini, G., Stevenin, J., and Barta, A. (1999). A novel family of plant splicing factors with a zn knuckle motif: examination of RNA binding and splicing activities. Plant Mol. Biol. 39, 761–773.
Lorkovic, Z. J. (2009). Role of plant RNA-binding proteins in development, stress response and genome organization. Trends Plant Sci. 14, 229–236.
Lorkovic, Z. J., Wieczorek, D., Kirk, A., Lambermon, M. H., and Filipowicz, W. (2000). Pre-mRNA splicing in higher plants. Trends Plant Sci. 5, 160–167.
Love, C. G., Graham, N. S., Lochlainn, S. O., Bowen, H. C., May, S. T., White, P. J., Broadley, M. R., Hammond, J. P., and King, G. J. (2010). A Brassica exon array for whole-transcript gene expression profiling. PLoS ONE 5, e12812. doi:10.1371/journal.pone.0012812
Lu, T., Lu, G., Fan, D., Zhu, C., Li, W., Zhao, Q., Feng, Q., Zhao, Y., Guo, Y., Huang, X., and Han, B. (2010). Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 20, 1238–1249.
Luco, R. F., Allo, M., Schor, I. E., Kornblihtt, A. R., and Misteli, T. (2011). Epigenetics in alternative pre-mRNA splicing. Cell 144, 16–26.
Marguerat, S., and Bahler, J. (2010). RNA-seq: from technology to biology. Cell. Mol. Life Sci. 67, 569–579.
Martin, J., Bruno, V. M., Fang, Z., Meng, X., Blow, M., Zhang, T., Sherlock, G., Snyder, M., and Wang, Z. (2010). Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-seq reads. BMC Genomics 11, 663. doi:10.1186/1471-2164-11-663
McCullough, A. J., and Schuler, M. A. (1997). Intronic and exonic sequences modulate 5’ splice site selection in plant nuclei. Nucleic Acids Res. 25, 1071–1077.
Melamud, E., and Moult, J. (2009). Stochastic noise in splicing machinery. Nucleic Acids Res. 37, 4873–4886.
Mili, S., and Steitz, J. A. (2004). Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10, 1692–1694.
Mockler, T. C., and Ecker, J. R. (2005). Applications of DNA tiling arrays for whole-genome analysis. Genomics 85, 1–15.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 5, 621–628.
Nahkuri, S., Taft, R. J., and Mattick, J. S. (2009). Nucleosomes are preferentially positioned at exons in somatic and sperm cells. Cell Cycle 8, 3420–3424.
Ner-Gaon, H., and Fluhr, R. (2006). Whole-genome microarray in Arabidopsis facilitates global analysis of retained introns. DNA Res. 13, 111.
Ner-Gaon, H., Halachmi, R., Savaldi-Goldstein, S., Rubin, E., Ophir, R., and Fluhr, R. (2004). Intron retention is a major phenomenon in alternative splicing in Arabidopsis. Plant J. 39, 877–885.
Ner-Gaon, H., Leviatan, N., Rubin, E., and Fluhr, R. (2007). Comparative cross-species alternative splicing in plants. Plant Physiol. 144, 1632.
Nicol, J. W., Helt, G. A., Blanchard, S. G. Jr., Raja, A., and Loraine, A. E. (2009). The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25, 2730–2731.
Nicolae, M., Mangul, S., Mandoiu, I., and Zelikovsky, A. (2010). Estimation of alternative splicing isoform frequencies from RNA-seq data. Algorithm. Bioinformat. 6293, 202–214.
Ohkura, N., Takahashi, M., Yaguchi, H., Nagamura, Y., and Tsukada, T. (2005). Coactivator-associated arginine methyltransferase 1, CARM1, affects pre-mRNA splicing in an isoform-specific manner. J. Biol. Chem. 280, 28927–28935.
Ohler, U., Shomron, N., and Burge, C. B. (2005). Recognition of unknown conserved alternatively spliced exons. PLoS Comput. Biol. 1, e15. doi:10.1371/journal.pcbi.0010015
Palusa, S. G., Ali, G. S., and Reddy, A. S. (2007). Alternative splicing of pre-mRNAs of Arabidopsis serine/arginine-rich proteins: regulation by hormones and stresses. Plant J. 49, 1091–1107.
Palusa, S. G., and Reddy, A. S. (2010). Extensive coupling of alternative splicing of pre-mRNAs of serine/arginine (SR) genes with nonsense-mediated decay. New Phytol. 185, 83–89.
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415.
Pandya-Jones, A., and Black, D. L. (2009). Co-transcriptional splicing of constitutive and alternative exons. RNA 15, 1896–1908.
Pertea, M., Mount, S. M., and Salzberg, S. L. (2007). A computational survey of candidate exonic splicing enhancer motifs in the model plant Arabidopsis thaliana. BMC Bioinformatics 8, 159. doi:10.1186/1471-2105-8-159
Purdom, E., Simpson, K. M., Robinson, M. D., Conboy, J. G., Lapuk, A. V., and Speed, T. P. (2008). FIRMA: a method for detection of alternative splicing from exon array data. Bioinformatics 24, 1707.
Ratsch, G., Sonnenburg, S., and Scholkopf, B. (2005). RASE: recognition of alternatively spliced exons in C. elegans. Bioinformatics 21(Suppl. 1), i369–i377.
Reddy, A. S. N. (2007). Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 58, 267–294.
Reddy, A. S. N., and Ali, G. S. (2011). Plant SR proteins: roles in pre-mRNA splicing, plant development and stress responses. Wiley Interdiscip. Rev. RNA 2, 875–889.
Rehrauer, H., Aquino, C., Gruissem, W., Henz, S. R., Hilson, P., Laubinger, S., Naouar, N., Patrignani, A., Rombauts, S., Shu, H., Van de Peer, Y., Vuylsteke, M., Weigel, D., Zeller, G., and Hennig, L. (2010). Agronomics1: a new resource for Arabidopsis transcriptome profiling. Plant Physiol. 152, 487–499.
Richard, H., Schulz, M. H., Sultan, M., Nürnberger, A., Schrinner, S., Balzereit, D., Dagand, E., Rasche, A., Lehrach, H., Vingron, M., Haas, S. A., and Yaspo, M. L. (2010). Prediction of alternative isoforms from exon expression levels in RNA-seq experiments. Nucleic Acids Res. 38, e112.
Richardson, D. N., Rogers, M. F., Labadorf, A., Ben-Hur, A., Guo, H., Paterson, A. H., and Reddy, A. S. N. (2011). Comparative analysis of serine/arginine-rich proteins across 27 eukaryotes: insights into sub-family classification and extent of alternative splicing. PLoS ONE 6, e24542. doi:10.1371/journal.pone.0024542
Roberts, A., Pimentel, H., Trapnell, C., and Pachter, L. (2011). Identification of novel transcripts in annotated genomes using RNA-seq. Bioinformatics. 27, 2325–2329.
Robertson, G., Schein, J., Chiu, R., Corbett, R., Field, M., Jackman, S. D., Mungall, K., Lee, S., Okada, H. M., Qian, J. Q., Griffith, M., Raymond, A., Thiessen, N., Cezard, T., Butterfield, Y. S., Newsome, R., Chan, S. K., She, R., Varhol, R., Kamoh, B., Prabhu, A. L., Tam, A., Zhao, Y., Moore, R. A., Hirst, M., Marra, M. A., Jones, S. J., and Hoodless, P. A. (2010). De novo assembly and analysis of RNA-seq data. Nat. Methods 7, 909–912.
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., and Mesirov, J. P. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26.
Rogers, M. F., Reddy, A. S. N., and Ben-Hur, A. (2012). SpliceGrapher: detecting patterns of alternative splicing from RNA-seq data in the context of gene models and EST data. Genome Biol. 13, R4.
Roy, N. C., Altermann, E., Park, Z. A., and McNabb, W. C. (2011). A comparison of analog and next-generation transcriptomic tools for mammalian studies. Brief. Funct. Genomics 10, 135–150.
Ru, Y., Wang, B. B., and Brendel, V. (2008). Spliceosomal proteins in plants. Curr. Top. Microbiol. Immunol. 326, 1–15.
Sablok, G., Gupta, P. K., Baek, J. M., Vazquez, F., and Min, X. J. (2011). Genome-wide survey of alternative splicing in the grass Brachypodium distachyon: a emerging model biosystem for plant functional genomics. Biotechnol. Lett. 33, 629–636.
Sammeth, M., Valiente, G., and Guigo, R. (2008). Bubbles: alternative splicing events of arbitrary dimension in splicing graphs. Lect. Notes Comput. Sci. 4955, 372.
Sanchez, S. E., Petrillo, E., Beckwith, E. J., Zhang, X., Rugnone, M. L., Hernando, C. E., Cuevas, J. C., Godoy, M., Herz, A., Depetris-Chauvin, A., Simpson, C. G., Brown, J. W., Cerdan, P. D., Borevitz, J. O., Mas, P., Ceriani, M. F., Kornblihtt, A. R., and Yanovsky, M. J. (2010). A methyl transferase links the circadian clock to the regulation of alternative splicing. Nature 468, 112–116.
Sanford, J. R., Wang, X., Mort, M., Vanduyn, N., Cooper, D. N., Mooney, S. D., Edenberg, H. J., and Liu, Y. (2009). Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 19, 381–394.
Schindler, S., Szafranski, K., Hiller, M., Ali, G. S., Palusa, S. G., Backofen, R., Platzer, M., and Reddy, A. S. (2008). Alternative splicing at NAGNAG acceptors in Arabidopsis thaliana SR and SR-related protein-coding genes. BMC Genomics 9, 159. doi:10.1186/1471-2164-9-159
Schoning, J. C., Streitner, C., Meyer, I. M., Gao, Y., and Staiger, D. (2008). Reciprocal regulation of glycine-rich RNA-binding proteins via an interlocked feedback loop coupling alternative splicing to nonsense-mediated decay in Arabidopsis. Nucleic Acids Res. 36, 6977–6987.
Schoning, J. C., Streitner, C., Page, D. R., Hennig, S., Uchida, K., Wolf, E., Furuya, M., and Staiger, D. (2007). Auto-regulation of the circadian slave oscillator component AtGRP7 and regulation of its targets is impaired by a single RNA recognition motif point mutation. Plant J. 52, 1119–1130.
Schor, I. E., Allo, M., and Kornblihtt, A. R. (2010). Intragenic chromatin modifications: a new layer in alternative splicing regulation. Epigenetics 5, 174–179.
Schuler, M. A. (2008). Splice site requirements and switches in plants. Curr. Top. Microbiol. Immunol. 326, 39–59.
Schwartz, S., Hall, E., and Ast, G. (2009a). SROOGLE: webserver for integrative, user-friendly visualization of splicing signals. Nucleic Acids Res. 37, W189–W192.
Schwartz, S., Gal-Mark, N., Kfir, N., Oren, R., Kim, E., and Ast, G. (2009b). Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput. Biol. 5, e1000300. doi:10.1371/journal.pcbi.1000300
Schwartz, S., Meshorer, E., and Ast, G. (2009c). Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 16, 990–995.
Shepard, P. J., and Hertel, K. J. (2008). Conserved RNA secondary structures promote alternative splicing. RNA 14, 1463–1469.
Simpson, C. G., and Brown, J. W. (2008). U12-dependent intron splicing in plants. Curr. Top. Microbiol. Immunol. 326, 61–82.
Simpson, C. G., Fuller, J., Maronova, M., Kalyna, M., Davidson, D., McNicol, J., Barta, A., and Brown, J. W. (2008). Monitoring changes in alternative precursor messenger RNA splicing in multiple gene transcripts. Plant J. 53, 1035–1048.
Simpson, C. G., Jennings, S. N., Clark, G. P., Thow, G., and Brown, J. W. (2004). Dual functionality of a plant u-rich intronic sequence element. Plant J. 37, 82–91.
Simpson, C. G., Manthri, S., Raczynska, K. D., Kalyna, M., Lewandowska, D., Kusenda, B., Maronova, M., Szweykowska-Kulinska, Z., Jarmolowski, A., Barta, A., and Brown, J. W. (2010). Regulation of plant gene expression by alternative splicing. Biochem. Soc. Trans. 38, 667–671.
Simpson, C. G., Thow, G., Clark, G. P., Jennings, S. N., Watters, J. A., and Brown, J. W. (2002). Mutational analysis of a plant branchpoint and polypyrimidine tract required for constitutive splicing of a mini-exon. RNA 8, 47–56.
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J. M., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117.
Sinha, R., Zimmer, A. D., Bolte, K., Lang, D., Reski, R., Platzer, M., Rensing, S. A., and Backofen, R. (2010). Identification and characterization of NAGNAG alternative splicing in the moss Physcomitrella patens. BMC Plant Biol. 10, 76. doi:10.1186/1471-2229-10-76
Sorek, R., Shemesh, R., Cohen, Y., Basechess, O., Ast, G., and Shamir, R. (2004). A non-EST-based method for exon-skipping prediction. Genome Res. 14, 1617–1623.
Spies, N., Nielsen, C. B., Padgett, R. A., and Burge, C. B. (2009). Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell 36, 245–254.
Staiger, D., and Green, R. (2011). RNA-based regulation in the plant circadian clock. Trends Plant Sci. 16, 517–523.
Stamm, S., Ben-Ari, S., Rafalska, I., Tang, Y., Zhang, Z., Toiber, D., Thanaraj, T. A., and Soreq, H. (2005). Function of alternative splicing. Gene 344, 1–20.
Stauffer, E., Westermann, A., Wagner, G., and Wachter, A. (2010). Polypyrimidine tract-binding protein homologues from Arabidopsis underlie regulatory circuits based on alternative splicing and downstream control. Plant J. 64, 243–255.
Stein, L. D., Mungall, C., Shu, S. Q., Caudy, M., Mangone, M., Day, A., Nickerson, E., Stajich, J. E., Harris, T. W., Arva, A., and Lewis, S. (2002). The generic genome browser: a building block for a model organism system database. Genome Res. 12, 1599–1610.
Sultan, M., Schulz, M. H., Richard, H., Magen, A., Klingenhoff, A., Scherf, M., Seifert, M., Borodina, T., Soldatov, A., Parkhomchuk, D., Schmidt, D., O’Keeffe, S., Haas, S., Vingron, M., Lehrach, H., and Yaspo, M. L. (2008). A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321, 956–959.
Szarzynska, B., Sobkowiak, L., Pant, B. D., Balazadeh, S., Scheible, W. R., Mueller-Roeber, B., Jarmolowski, A., and Szweykowska-Kulinska, Z. (2009). Gene structures and processing of Arabidopsis thaliana HYL1-dependent pri-miRNAs. Nucleic Acids Res. 37, 3083–3093.
Tan, S., Guo, J., Huang, Q., Chen, X., Li-Ling, J., Li, Q., and Ma, F. (2007). Retained introns increase putative microRNA targets within 3’ UTRs of human mRNA. FEBS Lett. 581, 1081–1086.
Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., Wang, X., Bodeau, J., Tuch, B. B., Siddiqui, A., Lao, K., and Surani, M. A. (2009). mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382.
Tenenbaum, S. A., Lager, P. J., Carson, C. C., and Keene, J. D. (2002). Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods 26, 191–198.
Tilgner, H., Nikolaou, C., Althammer, S., Sammeth, M., Beato, M., Valcarcel, J., and Guigo, R. (2009). Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 16, 996–1001.
Tolstrup, N., Rouze, P., and Brunak, S. (1997). A branch point consensus from Arabidopsis found by non-circular analysis allows for better prediction of acceptor sites. Nucleic Acids Res. 25, 3159–3163.
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 25, 1105–1111.
Trapnell, C., and Salzberg, S. L. (2009). How to map billions of short reads onto genomes. Nat. Biotechnol. 27, 455–457.
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J., and Pachter, L. (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515.
Twine, N. A., Janitz, K., Wilkins, M. R., and Janitz, M. (2011). Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS ONE 6, e16266. doi:10.1371/journal.pone.0016266
Tyagi, A., Ryme, J., Brodin, D., Ostlund Farrants, A. K., and Visa, N. (2009). SWI/SNF associates with nascent pre-mRNPs and regulates alternative pre-mRNA processing. PLoS Genet. 5, e1000470. doi:10.1371/journal.pgen.1000470
Ule, J., Jensen, K., Mele, A., and Darnell, R. B. (2005). CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods 37, 376–386.
Ule, J., Jensen, K. B., Ruggiu, M., Mele, A., Ule, A., and Darnell, R. B. (2003). CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215.
Ule, J., Stefani, G., Mele, A., Ruggiu, M., Wang, X., Taneri, B., Gaasterland, T., Blencowe, B. J., and Darnell, R. B. (2006). An RNA map predicting nova-dependent splicing regulation. Nature 444, 580–586.
Valadkhan, S., and Jaladat, Y. (2010). The spliceosomal proteome: at the heart of the largest cellular ribonucleoprotein machine. Proteomics. 10, 4128–4141.
Wachter, A. (2010). Riboswitch-mediated control of gene expression in eukaryotes. RNA Biol. 7, 67–76.
Wachter, A., Tunc-Ozdemir, M., Grove, B. C., Green, P. J., Shintani, D. K., and Breaker, R. R. (2007). Riboswitch control of gene expression in plants by splicing and alternative 3’ end processing of mRNAs. Plant Cell 19, 3437–3450.
Wahl, M. C., Will, C. L., and Luhrmann, R. (2009). The spliceosome: design principles of a dynamic rnp machine. Cell 136, 701–718.
Wang, B. B., and Brendel, V. (2006). Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. U.S.A. 103, 7175–7180.
Wang, B. B., O’Toole, M., Brendel, V., and Young, N. D. (2008a). Cross-species EST alignments reveal novel and conserved alternative splicing events in legumes. BMC Plant Biol. 8, 17. doi:10.1186/1471-2229-8-17
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P., and Burge, C. B. (2008b). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
Wang, K., Singh, D., Zeng, Z., Coleman, S. J., Huang, Y., Savich, G. L., He, X., Mieczkowski, P., Grimm, S. A., Perou, C. M., MacLeod, J. N., Chiang, D. Y., Prins, J. F., and Liu, J. (2010). MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178.
Wang, Z., and Burge, C. B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813.
Wang, Z., Gerstein, M., and Snyder, M. (2009a). RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63.
Wang, X., Wang, K., Radovich, M., Wang, Y., Wang, G., Feng, W., Sanford, J. R., and Liu, Y. (2009b). Genome-wide prediction of cis-acting RNA elements regulating tissue-specific pre-mRNA alternative splicing. BMC Genomics 10(Suppl. 1), S4. doi:10.1186/1471-2164-10-S1-S4
Wang, Z., Tollervey, J., Briese, M., Turner, D., and Ule, J. (2009c). CLIP: construction of cDNA libraries for high-throughput sequencing from RNAs cross-linked to proteins in vivo. Methods 48, 287–293.
Warzecha, C. C., Shen, S., Xing, Y., and Carstens, R. P. (2009). The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 6, 546.
Wu, J., Akerman, M., Sun, S., McCombie, W. R., Krainer, A. R., and Zhang, M. Q. (2011). SpliceTrap: a method to quantify alternative splicing under single cellular conditions. Bioinformatics. 27, 3010–3016.
Xia, H., Bi, J., and Li, Y. (2006). Identification of alternative 5’/3’splice sites based on the mechanism of splice site competition. Nucleic Acids Res. 34, 6305–6313.
Xia, Z., Wen, J., Chang, C., and Zhou, X. (2011). NSMAP: a method for spliced isoforms identification and quantification from RNA-seq. BMC Bioinformatics 12, 162. doi:10.1186/1471-2105-12-162
Xing, Y., and Lee, C. (2008). Reconstruction of full-length isoforms from splice graphs. Methods Mol. Biol. 452, 199.
Xing, Y., Resch, A., and Lee, C. (2004). The multiassembly problem: reconstructing multiple transcript isoforms from EST fragment mixtures. Genome Res. 14, 426.
Xing, Y., Stoilov, P., Kapur, K., Han, A., Jiang, H., Shen, S., Black, D. L., and Wong, W. H. (2008). MADS: a new and improved method for analysis of differential alternative splicing by exon-tiling microarrays. RNA 14, 1470.
Yeo, G. W., Van Nostrand, E., Holste, D., Poggio, T., and Burge, C. B. (2005). Identification and analysis of alternative splicing events conserved in human and mouse. Proc. Natl. Acad. Sci. U.S.A. 102, 2850–2855.
Yoshimura, K., Yabuta, Y., Ishikawa, T., and Shigeoka, S. (2002). Identification of acid element for tissue-specific alternative splicing of chloroplast ascorbate peroxidase pre-mRNA in higher plants. J. Biol. Chem. 277, 40623–40632.
Zenoni, S., Ferrarini, A., Giacomelli, E., Xumerle, L., Fasoli, M., Malerba, G., Bellin, D., Pezzotti, M., and Delledonne, M. (2010). Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-seq. Plant Physiol. 152, 1787–1795.
Zhang, G., Guo, G., Hu, X., Zhang, Y., Li, Q., Li, R., Zhuang, R., Lu, Z., He, Z., Fang, X., Chen, L., Tian, W., Tao, Y., Kristiansen, K., Zhang, X., Li, S., Yang, H., Wang, J., and Wang, J. (2010). Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 20, 646–654.
Keywords: pre-mRNA splicing, alternative splicing, splicing regulators, splicing regulatory elements, plants, Arabidopsis, splicing code, RNA-Seq
Citation: Reddy ASN, Rogers MF, Richardson DN, Hamilton M and Ben-Hur A (2012) Deciphering the plant splicing code: experimental and computational approaches for predicting alternative splicing and splicing regulatory elements. Front. Plant Sci. 3:18. doi: 10.3389/fpls.2012.00018
Received: 27 November 2011;
Accepted: 18 January 2012;
Published online: 07 February 2012.
Edited by:
Richard A. Jorgensen, Laboratorio Nacional de Genómica para la Biodiversidad, MexicoReviewed by:
William Brad Barbazuk, University of Florida, USASteve M. Mount, University of Maryland, USA
Copyright: © 2012 Reddy, Rogers, Richardson, Hamilton and Ben-Hur. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Anireddy S. N. Reddy, Program in Molecular Plant Biology, Department of Biology, Colorado State University, Fort Collins, CO 80523-1878, USA. e-mail:cmVkZHlAY29sb3N0YXRlLmVkdQ==; Asa Ben-Hur, Department of Computer Science, Colorado State University, Fort Collins, CO 80523, USA. e-mail:YXNhQGNzLmNvbG9zdGF0ZS5lZHU=
†Anireddy S. N. Reddy and Mark F. Rogers are co-first authors.