


- 1 INRA, Jean-Pierre Bourgin Institute (IJPB, UMR1318 INRA-AgroParisTech), Laboratory of Excellence “Saclay Plant Sciences” (LabEx SPS); RD10, F-78026 Versailles, France
- 2 AgroParisTech, Chair of Plant Physiology, 16 rue Claude Bernard, F-75231 Paris, France
- 3 CNRS/Bayer CropScience Joint Laboratory (UMR5240), F-69263 Lyon, France
The Seed Proteome Web Portal (SPWP; http://www.seed-proteome.com/) gives access to information both on quantitative seed proteomic data and on seed-related protocols. Firstly, the SPWP provides access to the 475 different Arabidopsis seed proteins annotated from two dimensional electrophoresis (2DE) maps. Quantitative data are available for each protein according to their accumulation profile during the germination process. These proteins can be retrieved either in list format or directly on scanned 2DE maps. These proteomic data reveal that 40% of seed proteins maintain a stable abundance over germination, up to radicle protrusion. During sensu stricto germination (24 h upon imbibition) about 50% of the proteins display quantitative variations, exhibiting an increased abundance (35%) or a decreasing abundance (15%). Moreover, during radicle protrusion (24–48 h upon imbibition), 41% proteins display quantitative variations with an increased (23%) or a decreasing abundance (18%). In addition, an analysis of the seed proteome revealed the importance of protein post-translational modifications as demonstrated by the poor correlation (r2 = 0.29) between the theoretical (predicted from Arabidopsis genome) and the observed protein isoelectric points. Secondly, the SPWP is a relevant technical resource for protocols specifically dedicated to Arabidopsis seed proteome studies. Concerning 2D electrophoresis, the user can find efficient procedures for sample preparation, electrophoresis coupled with gel analysis, and protein identification by mass spectrometry, which we have routinely used during the last 12 years. Particular applications such as the detection of oxidized proteins or de novo synthesized proteins radiolabeled by [35S]-methionine are also given in great details. Future developments of this portal will include proteomic data from studies such as dormancy release and protein turnover through de novo protein synthesis analyses during germination.
Introduction
Biologically, the seed might bethe most critical stage of Angiosperm development. Indeed, the seed number per plant, their size and their ability to germinate are key components of plant fitness (Donohue et al., 2005). Moreover, the seed structure helps plants to survive adverse environmental conditions but also helps to colonize new environments. Concerning seed biology, our comprehension of fundamental biological processes such as dormancy or germination was greatly enhanced by the plant model Arabidopsis thaliana used in combination with global “omics” approaches such as proteomics (North et al., 2010; Rajjou et al., 2012). Thus, modern functional genomics allow the characterization of cellular responses, gene network activation, and metabolic adaptation among a wide range of seed physiological states (Nambara and Nonogaki, 2012). These novel technologies represent powerful tools to accelerate basic and translational seed research. Yet, to our knowledge, there are no publically accessible websites dedicated to the Arabidopsis seed proteome. Thus, we decided to build a “Seed Proteome Web Portal” to give free access to relevant data on the Arabidopsis seed proteome.
Overview of the Seed Proteome Web Portal
The SPWP harbors a presentation of the founding laboratories, proteome data as well as detailed protocols and various links to bioinformatics resources or proteomic journals (please see site map).
The first main part of the SPWP is focused on seed proteome data that can be accessed either from the protein maps or from the protein catalog (Figure 1A). As of today, information on the 475 protein spots identified during Arabidopsis seed germination is available. In the protein map section, the user can retrieve a protein spot on a reference 2D gel obtained from Arabidopsis seeds (Figure 1A). The protein spot can be selected directly on the 2D gel image to open a new page containing protein spot data (Figure 1B). On that new page, the “essential” table gives information on the protein spot class, spot number, protein name, description, gene number (AGI-ID), and Mascot peptide matches are given (Figure 1B). In the “data” and “sequences” table, the user can also find experimental proteomic facts such as the expected/observed molecular weight and isoelectric point together with the peptide sequences that allowed identification of the protein (Figure 1B). Protein spot data can also be accessed via the protein catalog section.

Figure 1. Screenshots of the Arabidopsis seed proteome web portal. Homepage (A), 2D map of protein spots that increase during seed germination (B) and protocols for the detection of carbonylated proteins (C) and detection of de novo synthesized proteins (D) during seed germination.
The second main part of the SPWP offers a technical goldmine to scientists working on the seed proteome (Figure 1C). The user can find detailed protocols for a complete seed proteome experiment including seed germination, preparation of total protein extracts, 2D electrophoresis, gel staining, analysis of 2D gels, and protein identification by mass spectrometry are all explained in a detailed manner (please also see Rajjou et al., 2011). To our knowledge, there was no publically and freely available technical resource entirely dedicated to the study of the seed proteome. In addition, the SPWP also gives some specific protocols to study seed sub-proteome such as the oxidized or de novo synthesized proteomes (Figure 1D).
The Seed Proteome Illustrates the Developmental Switch Occurring during Arabidopsis Germination
Previous studies on the Arabidopsis seed demonstrated the presence of a high number of long-lived mRNA in the mature dry seed (Nakabayashi et al., 2005) together with the absolute requirement for protein synthesis to germinate (Rajjou et al., 2004). During Arabidopsis seed germination, we found a total of 475 protein spots corresponding unambiguously to 241 non-redundant proteins with a correspondence with a single AGI ID. A total of 18 protein spots were matched to multiple AGI-ID (Table S1 in Supplementary Material). Indeed, because of mRNA processing, protein proteolysis and chemical protein modifications, one gene can produce many different protein isoforms resulting in a wide proteome diversity. Each spot corresponds to a single protein isoform resulting from post-transcriptional or post-translational regulation of gene expression (1 mRNA = X protein isoforms). A total of 250 protein spots showed an increasing abundance during germination sensu stricto (0–24 h) or during radicle protrusion (24–48 h; Figure 2). Otherwise, 143 protein spots displayed a decreasing abundance over the course of seed germination (Figure 2). These results illustrate the major developmental switch that occurs during the germination phase in preparation for seedling establishment. Some of the protein spot abundance variation occurs due to post-translational modifications. Perhaps, the best example comes from 12S globulin subunits. Indeed, while there are only three genes coding for 12S globulin seed storage protein in Arabidopsis (At1g03880, At4g28250, At5g44120), a total of 104 protein spots corresponding to 12S globulin seed storage proteins can be retrieved on a 2D gel of seed proteins (Arc et al., 2011). Spots corresponding to the precursor form of the globulin seed storage proteins can be processed by cleavage and transformed into their mature protein form (Gallardo et al., 2001). Yet, at the seed proteome level, post-translational modifications by cleavage are not the most abundant modification as we found a good correlation between the theoretical and the observed protein molecular weight (Figure 3). In contrast, we found a very poor correlation between the proteins theoretical and the observed isoelectric point (Figure 3; Table S1 in Supplementary Material) illustrating the major impact of post-translational modifications, e.g., phosphorylation (Arc et al., 2011), glycosylation(Vuylsteker et al., 2000), or oxidation (Job et al., 2005) on the seed proteome.
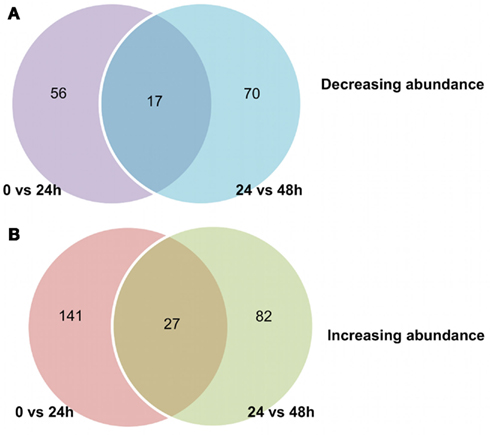
Figure 2. Number of protein spots differentially accumulated during Arabidopsis seed germination. For each time interval, a total number of 475 spots are considered. The normalized volume for each spot was determined in three biological repetitions. The log2 ratio was calculated between 0 and 24 h as well as between 24 and 48 h. When the spot was not detected in one of the time point, a negligible spot abundance value was arbitrary assigned to calculate the log2ratio. (A) Number of protein spots whose abundance is decreasing (log2 ratio < −1) between germination sensu stricto (0–24 h), radicle protrusion (24–48 h) or at both time intervals of seed imbibition. (B) Number of protein spots whose abundance is increasing (log2 ratio >1) between germination sensu stricto (0–24 h), radicle protrusion (24–48 h) or at both time intervals of seed imbibition.
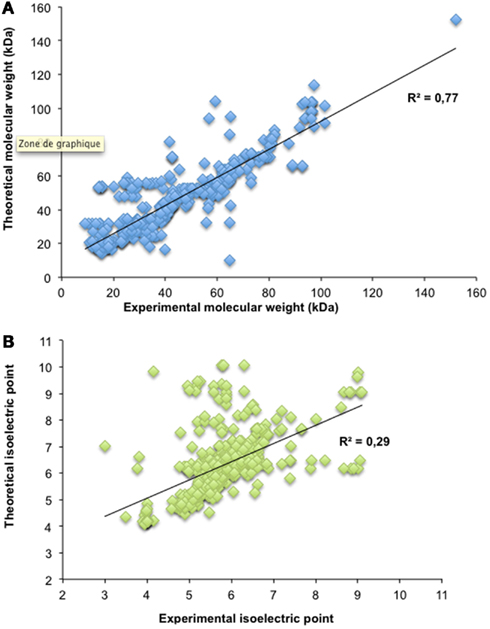
Figure 3. Theoretical and observed molecular weight and isoelectric points. The theoretical and observed molecular weights (A) as well as the theoretical and observed isoelectric point (B) from 457 protein spots with a unique AGI-ID are plotted. For both linear regressions, the correlation coefficient is indicated on each graph.
Originality of the Seed Proteome
We investigated the specificity of the Arabidopsis seed proteome by comparison with the leaf proteome data from the Plant Proteomics Database (Sun et al., 2009) and by the comparison with the entire Arabidopsis proteome (TAIR10 Gene Annotation Data). First, we extracted the unambiguously identified non-redundant AGI-ID from the 475 protein spots of the seed proteome and classified them using the FunCat catalog (Ruepp et al., 2004). We also classified the leaf proteome and the whole genome with the FunCat catalog (Baerenfaller et al., 2008). After classification, we could observe that the “cell rescue, defense and virulence” “energy,” “protein fate,” and “storage protein” categories were over represented in the seed proteome as compared to the leaf proteome or entire proteome datasets (Figure 4, Table S2 in Supplementary Material). The seed proteome “cell rescue” category contains catalase, superoxide dismutase and peroxiredoxin proteins that detoxify reactive oxygen species produced very early during germination (Bailly, 2004). The “energy” category encompasses many proteins from glycolysis (e.g., glyceraldehyde 3-phosphate dehydrogenase, pyruvate carboxylase) as well as mitochondrial proteins from the tricarboxylic acid cycle (e.g., citrate synthase, succinyl-CoA ligase) or the glyoxylate cycle (isocitrate lyase, malate synthase). Thus, the seed proteome illustrates the high-energy demand in the seed upon metabolic resumption and it was recently shown that a majority of a 775 germination-specific gene subset was related to mitochondrial biogenesis (Narsai et al., 2011). The “protein fate” class of the seed proteome is composed of proteins involved in protein degradation (e.g., subunits of the 20S/26S proteasome), protein maturation (e.g., leucine aminopeptidase 1) or protein folding (e.g., heat shock proteins, peptidyl-prolyl cis-trans isomerase 1, protein disulfide isomerase). Finally, the “storage protein” category is overrepresented in the seed proteome due to its developmental specificity and serves both as an amino acid storage pool as well as supplemental roles, since the alpha-subunits are preferentially oxidized during seed germination (Job et al., 2005; Rajjou et al., 2006, 2007). Altogether, contrasting the FunCat classification of the seed proteome, the leaf proteome and the entire Arabidopsis proteome highlights the specificity of the seed proteome particularly concerning the energy demand and the correct protein folding necessary for seed germination.
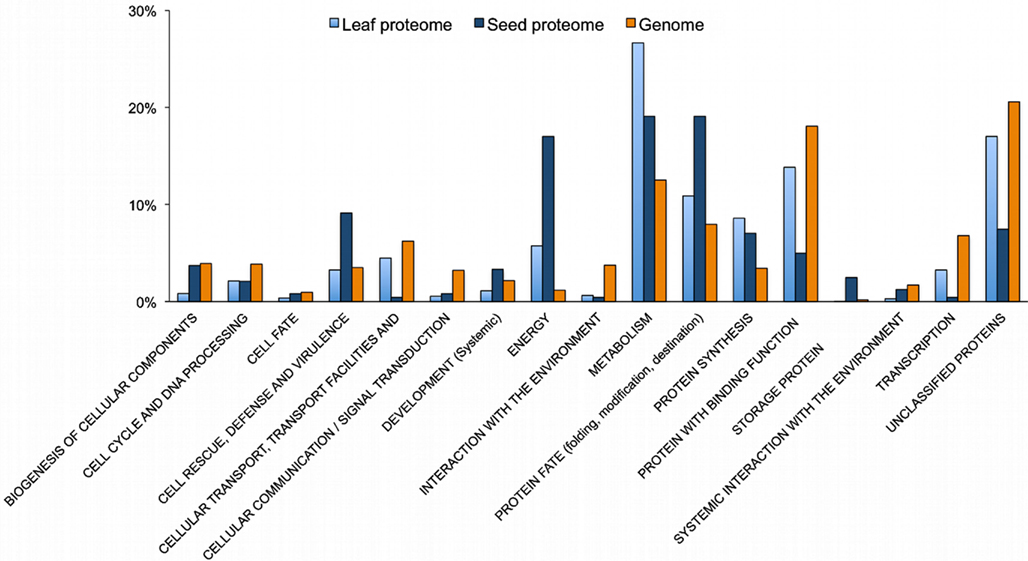
Figure 4. FunCat annotation of the non-redundant proteins detected in the Arabidopsis seed proteome. From the 475 different proteins of the seed proteome, we identified unambiguously 241 non-redundant proteins with a correspondence with a single AGI-ID. A total of 18 protein spots were matched to multiple AGI-ID i.e. grouped under an unique AGI. The FunCat catalog was used to annotate the seed proteome, the leaf proteome (from Baerenfaller et al., 2008) and the entire Arabidopsis proteome TAIR10 Gene Annotation Data; whole genome for comparisons.
In the Arabidopsis Seed, the Transcriptome and Proteome Information Yield Non-Redundant Biological Information
The most highly regulated genes during Arabidopsis seed development tended to be expressed preferentially in seeds compared with other plant organs (Ruuska et al., 2002). Moreover, a recent paper on both transcriptome and proteome during Arabidopsis seed development showed that 56% of 319 protein/transcript pairs had concordant expression patterns (Hajduch et al., 2010). In the Arabidopsis seed dry seed, more than 12000 stored mRNA species were detected (Nakabayashi et al., 2005). This transcriptome is characterized both by a great number of stored mRNA and by the rapid extensive changes occurring a few hours after imbibition (Nakabayashi et al., 2005; Preston et al., 2009; Narsai et al., 2011). Thus, due to the availability of the seed proteome, it was interesting to correlate the transcript and protein abundance variation during germination. Indeed, we wondered if we could expect similar correlations between protein and transcript accumulation profiles during seed development and germination. The 475 protein spots were matched to their corresponding AGI-ID and, we obtained a non-redundant seed proteome of 241 proteins. Then, the normalized abundance of each protein isoform corresponding to the same AGI-ID was summed. Employing the transcriptome data from Nakabayashi et al. (2005), we analyzed the mRNA accumulation corresponding to the 241 genes found in the seed non-redundant proteome. We obtained a probe signal for 218 genes and built a correlation between the transcript change and the protein change between 0 and 24 h after imbibition of non-dormant Arabidopsis seeds (Figure 5, Table S3 in Supplementary Material). It was obvious that, during seed germination, there is no correlation between the transcript and the protein level (r2 = 0.02). This is in accordance with the fact the long-lived stored mRNA are present in the dry seed state and that protein synthesis is required for seed germination while transcription is not (Rajjou et al., 2004; Kimura and Nambara, 2010; Sano et al., 2012). These observations suggest that there are no correlation between mRNA and protein half-lives in Arabidopsis seeds. Therefore, both the transcriptome and the proteome analyses in the Arabidopsis seed result in relevant information about the developmental switch occurring in the dry quiescent state to the metabolically active state.
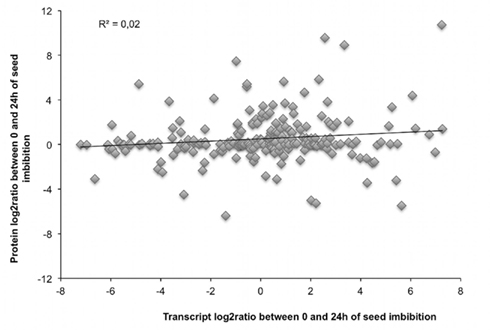
Figure 5. The transcript and protein abundance variation during seed germination are poorly correlated. From the 475 different proteins of the seed proteome, we identified 241 non-redundant proteins (241 AGI). The transcript normalized abundance of 218 of these proteins was retrieved in the data from Nakabayashi et al. (2005). For these 218 proteins, the log2 ratio between 0 and 24 h of seed germination was calculated for both transcript and protein. The corresponding values were then plotted and a correlation coefficient (indicated on the graph) was calculated from the linear regression.
In a complementary approach, we took advantage of two independent studies designed to identify tissue-specific genes in Arabidopsis by genome-wide transcriptome (Schmid et al., 2005) or proteome (Baerenfaller et al., 2008) approaches. To compare both transcriptome and proteome studies, we restricted our analysis to the “flower,” “leaves,” “root,” and “seed” tissues as they were common in the two studies. Altogether, 151 tissue-specific genes could be identified in these four tissues by transcriptomics while 469 could be identified by proteomics (Figure 6A, Table S4 in Supplementary Material). Surprisingly, we found that only 29 tissue-specific genes were commonly identified by the two approaches (Figure 6A). On closer examination, we found that 20 seed-specific genes were identified by both approaches out of a total of 190 seed-specific genes, i.e., 57 seed-specific genes identified by transcriptomics plus 133 seed-specific genes identified by proteomics (Figure 6B, Table S4 in Supplementary Material). This is in accordance with the poor correlation between RNA and protein levels that we found for the 241 non-redundant proteins of the seed proteome (Figure 5). Finally, in a study on 319 protein/transcript pairs during Arabidopsis seed filling, the observed correlation was equal to 56% (Hajduch et al., 2010). It suggests that the correlation between RNA and protein levels is strongly reduced during seed germination, a key transition in a plants life. Altogether, these analyses show that the seed transcriptome and proteome are not redundant and that each technique yields complementary biological information that reflects the importance of post-transcriptional as well as post-translational modifications in the seed.
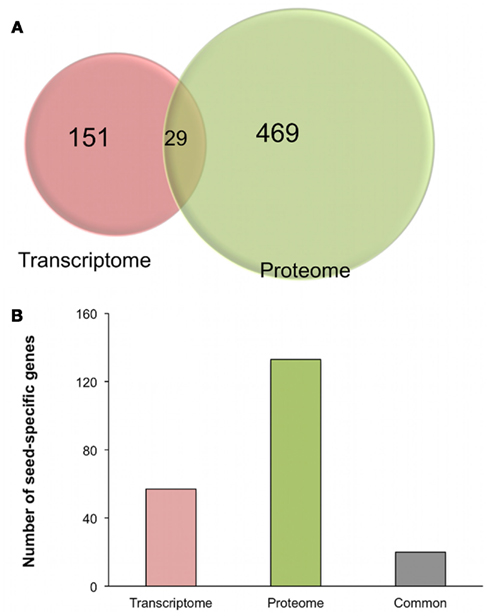
Figure 6. Comparison of Arabidopsis tissue-specific genes as defined by transcriptomic or proteomic approaches. (A) Number of distinct and common tissue-specific genes defined by transcriptome (151 genes, Schmid et al., 2005) or by proteome (469 genes, Baerenfaller et al., 2008) studies in the “leaf,” “root,” “flower,” and “seed” samples. Only samples commonly used in the two studies (i.e., flower, leaves, root, and seed) were compared. (B) Number of seed-specific genes identified by transcriptome (Schmid et al., 2005), proteome (Baerenfaller et al., 2008) or both approaches.
Future Developments
As outlined here, the SPWP gives precise information for seed biologists. These data are complementary and non-redundant in comparison to seed transcriptome data in particular during Arabidopsis seed germination. In addition, the great number of protein isoforms revealed by the 2D analysis highlights the seed proteome diversity currently underestimated due to the difficulty to detect low-abundant proteins. Future developments of the SPWP will first include proteins regulated by dormancy release in Arabidopsis (Chibani et al., 2006) as well as protein turnover (L. Rajjou, unpublished data). Moreover, we will include proteomic studies on sugar beet (Beta vulgaris) and rice (Oryza sativa) germination. In addition, LC-MS/MS data on specific protein modifications such as carbonylation or phosphorylation during seed germination will be included to highlight the extent of post-translational modifications at this key development stage. Finally, the recent progress in laser-assisted microdissection combined with shotgun proteomics by LC-MS/MS could be applied to describe the metabolic compartmentalization in the Arabidopsis seed as it was done on other species (Gallardo et al., 2007; Finnie and Svensson, 2009).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Plant_Proteomics/10.3389/fpls.2012.00098/abstract
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arc, E., Galland, M., Cueff, G., Godin, B., Lounifi, I., Job, D., and Rajjou, L. (2011). Reboot the system thanks to protein post-translational modifications and proteome diversity: how quiescent seeds restart their metabolism to prepare seedling establishment. Proteomics 11, 1606–1618.
Baerenfaller, K., Grossmann, J., Grobei, M. A., Hull, R., Hirsch-Hoffmann, M., Yalovsky, S., Zimmermann, P., Grossniklaus, U., Gruissem, W., and Baginsky, S. (2008). Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320, 938–941.
Bailly, C. (2004). Active oxygen species and antioxidants in seed biology. Seed Sci. Res. 14, 93–107.
Chibani, K., Ali-Rachedi, S., Job, C., Job, D., Jullien, M., and Grappin, P. (2006). Proteomic analysis of seed dormancy in Arabidopsis. Plant Physiol. 142, 1493–1510.
Donohue, K., Dorn, L., Griffith, C., Kim, E., Aguilera, A., Polisetty, C. R., and Schmitt, J. (2005). The evolutionary ecology of seed germination of Arabidopsis thaliana: variable natural selection on germination timing. Evolution 59, 758–770.
Finnie, C., and Svensson, B. (2009). Barley seed proteomics from spots to structures. J. Proteomics 72, 315–234.
Gallardo, K., Firnhaber, C., Zuber, H., Héricher, D., Belghazi, M., Henry, C., Küster, H., and Thompson, R. (2007). A combined proteome and transcriptome analysis of developing Medicago truncatula seeds: evidence for metabolic specialization of maternal and filial tissues. Mol. Cell. Proteomics 6, 2165–2179.
Gallardo, K., Job, C., Groot, S. P., Puype, M., Demol, H., Vandekerckhove, J., and Job, D. (2001). Proteomic analysis of Arabidopsis seed germination and priming. Plant Physiol. 126, 835–848.
Hajduch, M., Hearne, L. B., Miernyk, J. A., Casteel, J. E., Joshi, T., Agrawal, G. K., Song, Z., Zhou, M., Xu, D., and Thelen, J. J. (2010). Systems analysis of seed filling in Arabidopsis: using general linear modeling to assess concordance of transcript and protein expression. Plant Physiol. 152, 2078–2087.
Job, C., Rajjou, L., Lovigny, Y., Belghazi, M., and Job, D. (2005). Patterns of protein oxidation in Arabidopsis seeds and during germination. Plant Physiol. 138, 790–802.
Kimura, M., and Nambara, E. (2010). Stored and neosynthesized mRNA in Arabidopsis seeds: effects of cycloheximide and controlled deterioration treatment on the resumption of transcription during imbibition. Plant Mol. Biol. 73, 119–129.
Nakabayashi, K., Okamoto, M., Koshiba, T., Kamiya, Y., and Nambara, E. (2005). Genome-wide profiling of stored mRNA in Arabidopsis thaliana seed germination: epigenetic and genetic regulation of transcription in seed. Plant J. 41, 697–709.
Nambara, E., and Nonogaki, H. (2012). Seed biology in the 21st century: perspectives and new directions. Plant Cell Physiol. 53, 1–4.
Narsai, R., Law, S. R., Carrie, C., Xu, L., and Whelan, J. (2011). In-depth temporal transcriptome profiling reveals a crucial developmental switch with roles for RNA processing and organelle metabolism that are essential for germination in Arabidopsis. Plant Physiol. 157, 1342–1362.
North, H., Baud, S., Debeaujon, I., Dubos, C., Dubreucq, B., Grappin, P., Jullien, M., Lepiniec, L., Marion-Poll, A., Miquel, M., Rajjou, L., Routaboul, J. M., and Caboche, M. (2010). Arabidopsis seed secrets unravelled after a decade of genetic and omics-driven research. Plant J. 61, 971–981.
Preston, J., Tatematsu, K., Kanno, Y., Hobo, T., Kimura, M., Jikumaru, Y., Yano, R., Kamiya, Y., and Nambara, E. (2009). Temporal expression patterns of hormone metabolism genes during imbibition of Arabidopsis thaliana seeds: a comparative study on dormant and non-dormant accessions. Plant Cell Physiol. 50, 1786–1800.
Rajjou, L., Belghazi, M., Catusse, J., Ogé, L., Arc, E., Godin, B., Chibani, K., Ali-Rachidi, S., Collet, B., Grappin, P., Jullien, M., Gallardo, K., Job, C., and Job, D. (2011). Proteomics and posttranslational proteomics of seed dormancy and germination. Methods Mol. Biol. 773, 215–236.
Rajjou, L., Belghazi, M., Huguet, R., Robin, C., Moreau, A., Job, C., and Job, D. (2006). Proteomic investigation of the effect of salicylic acid on Arabidopsis seed germination and establishment of early defense mechanisms. Plant Physiol. 141, 910–923.
Rajjou, L., Duval, M., Gallardo, K., Catusse, J., Bally, J., Job, C., and Job, D. (2012). Seed germination and vigor. Annu. Rev. Plant Biol. 63, 507–533.
Rajjou, L., Gallardo, K., Debeaujon, I., Vandekerckhove, J., Job, C., and Job, D. (2004). The effect of alpha-amanitin on the Arabidopsis seed proteome highlights the distinct roles of stored and neosynthesized mRNAs during germination. Plant Physiol. 134, 1598–1613.
Rajjou, L., Lovigny, Y., Job, C., Belghazi, M., Groot, S., and Job, D. (2007). “Seed quality and germination,” in Seeds: Biology, Development and Ecology, eds S. Navie, S. Adkins, and S. Ashmore (Cambridge: CAB International Publishing), 324–332.
Ruepp, A., Zollner, A., Maier, D., Albermann, K., Hani, J., Mokrejs, M., Tetko, I., Güldener, U., Mannhaupt, G., Münsterkötter, M., and Mewes, H. W. (2004). The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 32, 5539–5545.
Ruuska, S. A., Girke, T., Benning, C., and Ohlrogge, J. B. (2002). Contrapuntal networks of gene expression during Arabidopsis seed filling. Plant Cell 14, 1191–1206.
Sano, N., Permana, H., Kumada, R., Shinozaki, Y., Tanabata, T., Yamada, T., Hirasawa, T., and Kanekatsu, M. (2012). Proteomic analysis of embryonic proteins synthesized from long-lived mRNAs during germination of rice seeds. Plant Cell Physiol. 53, 687–698.
Schmid, M., Davison, T. S., Henz, S. R., Pape, U. J., Demar, M., Vingron, M., Schölkopf, B., Weigel, D., and Lohmann, J. U. (2005). A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37, 501–506.
Sun, Q., Zybailov, B., Majeran, W., Friso, G., Olinares, P. D., and van Wijk, K. J. (2009). PPDB, the Plant Proteomics Database at Cornell. Nucleic Acids Res. 37, D969–D974.
Keywords: seed, proteome, website, Arabidopsis, germination, dormancy, longevity, plant
Citation: Galland M, Job D and Rajjou L (2012) The seed proteome web portal. Front. Plant Sci. 3:98. doi: 10.3389/fpls.2012.00098
Received: 23 March 2012; Accepted: 26 April 2012;
Published online: 11 June 2012.
Edited by:
Joshua L. Heazlewood, Lawrence Berkeley National Laboratory, USAReviewed by:
Natalia V. Bykova, Memorial University of Newfoundland, CanadaJesus V. Jorrin Novo, University of Cordoba, Spain
Copyright: © 2012 Galland, Job and Rajjou. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marc Galland and Loïc Rajjou, Laboratory of Excellence “Saclay Plant Sciences”, Institut National de la Recherche Agronomique, Jean-Pierre Bourgin Institute (UMR1318 INRA-AgroParisTech), RD10, F-78026 Versailles, France. e-mail:bWFyYy5nYWxsYW5kQHZlcnNhaWxsZXMuaW5yYS5mcg==;bG9pYy5yYWpqb3VAYWdyb3BhcmlzdGVjaC5mcg==