 Angel Fernandez i Marti1,2 Blessing Athanson3 Tyson Koepke4
Angel Fernandez i Marti1,2 Blessing Athanson3 Tyson Koepke4 Carolina Font i Forcada5
Carolina Font i Forcada5 Amit Dhingra4
Amit Dhingra4 Nnadozie Oraguzie3*
Nnadozie Oraguzie3*- 1 Departamento Biología Molecular, Parque Científico Tecnológico Aula Dei, Zaragoza, Spain
- 2 Unidad de Fruticultura, Centro de Investigación y Tecnología Agroalimentario de Aragón, Zaragoza, Spain
- 3 Irrigated Agriculture Research and Extension Centre, Washington State University, Pullman, WA, USA
- 4 Department of Horticulture and Landscape Architecture, Washington State University, Pullman, WA, USA
- 5 Departamento de Pomología, Estación Experimental de Aula Dei (CSIC), Zaragoza, Spain
Most previous studies on genetic fingerprinting and cultivar relatedness in sweet cherry were based on isoenzyme, RAPD, and simple sequence repeat (SSR) markers. This study was carried out to assess the utility of single nucleotide polymorphism (SNP) markers generated from 3′ untranslated regions (UTR) for genetic fingerprinting in sweet cherry. A total of 114 sweet cherry germplasm representing advanced selections, commercial cultivars, and old cultivars imported from different parts of the world were screened with seven SSR markers developed from other Prunus species and with 40 SNPs obtained from 3′ UTR sequences of Rainier and Bing sweet cherry cultivars. Both types of marker study had 99 accessions in common. The SSR data was used to validate the SNP results. Results showed that the average number of alleles per locus, mean observed heterozygosity, expected heterozygosity, and polymorphic information content values were higher in SSRs than in SNPs although both set of markers were similar in their grouping of the sweet cherry accessions as shown in the dendrogram. SNPs were able to distinguish sport mutants from their wild type germplasm. For example, “Stella” was separated from “Compact Stella.” This demonstrates the greater power of SNPs for discriminating mutants from their original parents than SSRs. In addition, SNP markers confirmed parentage and also determined relationships of the accessions in a manner consistent with their pedigree relationships. We would recommend the use of 3′ UTR SNPs for genetic fingerprinting, parentage verification, gene mapping, and study of genetic diversity in sweet cherry.
Introduction
Sweet cherry (Prunus avium L.) is an out-breeding, self-incompatible diploid species in the Rosaceae family with a genome of 2n = 16. The species is commonly grown in the temperate climatic zones with cooler temperatures to provide the chilling requirement necessary for flower induction. It is believed that cherries originated in the area between the Black sea and Caspian Sea in Asia Minor (Webster, 1996). Birds may have carried it to Europe prior to human civilization and its cultivation probably began in Roman times and spread to the US in the sixteenth century (Watkins, 1976). Although sexual reproduction in sweet cherry is controlled by a Gametophytic Self-Incompatibility (GSI) system (de Nettancourt, 2001) and open pollinated seedlings are heavily utilized in its traditional culture, the genetic diversity in sweet cherry appears to have been minimized due to repeated use of a few founding clones as parents in breeding programmes (Choi and Kappel, 2004).
The Washington State University (WSU) sweet cherry breeding program came to a stand-still about 25 years ago following the retirement of the then breeder, Dr. Tom Toyama. However, active evaluation of the germplasm continued (although many breeding records were lost) which led to the release of several new sweet cherry cultivars (Oraguzie et al., 2010; Olmstead et al., 2011a,b). The rejuvenated breeding programme has acquired diverse plant materials including advanced selections, commercial varieties, and exotic germplasm which are now fruiting but lack pedigree information. Information on genetic identity and relatedness is necessary to design appropriate conservation and management strategies as well as for selecting diverse individuals with desired fruit quality traits for use as breeding parents. Molecular identification using DNA markers has become the main tool for examination of genetic relationships within and between populations or individuals, mapping of useful genes, construction of genetic linkage maps, marker-assisted selection, and phylogenetic studies in crop species (Arús et al., 2005).
Most published genetic studies in cherry have typically been based on microsatellite or simple sequence repeat (SSR) markers (Dirlewanger et al., 2002; Wünsch and Hormaza, 2002; Schueler et al., 2003; Vaughan and Russell, 2004). SSRs have been recognized as useful genetic markers in plants as well as in animals due to its high degree of polymorphism, abundance in genomes, co-dominance, and suitability for automation (Gupta et al., 1996). Although most of these SSRs have been developed in peach (Cipriani et al., 1999; Sosinski et al., 2000; Testolin et al., 2000), it has been demonstrated in several studies that these markers can also be used in other Prunus species such as cherry, almond, or plum (Schueler et al., 2003; Fernández i Martí et al., 2009; Wünsch, 2009). However, one major limitation of SSR markers is the high cost of the fluorescent labels which renders the assay costly and unaffordable for small scale studies (Agarwal et al., 2008). On the other hand, single nucleotide polymorphism (SNP) markers are gaining popularity as valuable and efficient molecular markers due to their abundance in plant genomes (Rafalski, 2002). They can be used as genetic markers in many applications such as cultivar identification, construction of genetic maps, assessment of genetic diversity, or marker-assisted breeding (Flint-Garcia et al., 2005; Chágne et al., 2008; Wu et al., 2008). Furthermore, the identification of SNPs and INDELs has been simplified by the recent developments in sequencing technology.
Previously, SNP analysis was carried out only in plants with large genome sequence databases including Arabidopsis (Drenkard et al., 2000), barley (Rostoks et al., 2005), maize (Batley et al., 2003), wheat (Qi et al., 2004), grape (Salmaso et al., 2004), etc. But recently, the Rosaceae family including almond (Wu et al., 2008), apple (Chágne et al., 2008), peach, and strawberry (Le Dantec et al., 2010), and Japanese apricot (Fang et al., 2006) joined the bandwagon following the completion of genome sequencing projects for apple, peach, and strawberry. In fact, SNPs from Rosaceae conserved orthologous sequences have been used for comparative analysis of peach (Prunus persicsa), apple (Malus x domestica), and strawberry (Fragaria spp) and recently, for determination of genetic relationships in 30 sweet cherry cultivars and for construction of a SNP-based map (Cabrera et al., 2012).The sweet cherry whole genome sequencing project based on the self-fertile sweet cherry cultivar “Stella” is next in line for completion following the generation of a 10× sequence draft (Dhingra et al., unpublished). The use of next-generation sequencing technologies for SNP discovery and characterization is being demonstrated in large-scale plant studies, which are then used to generate genotyping tools for breeding applications. Such SNPs developed in sweet cherry through 3′ untranslated region (UTR) sequencing on a GS 454 platinum platform (Koepke et al., 2012) have been made available for analyzing genetic variation and relationships in sweet cherry and in the current study it will be validated for use as consistent and reliable genetic marker for establishing genetic identity, parentage verification, study of genetic diversity, and gene mapping.
New methods and technologies are continuously being developed and utilized with the aim to improve the identification and genetic characterization of plant species. For example, high resolution melting (HRM) is a recent advance for the detection of SNPs. This technique measures temperature induced strand separation of short PCR amplicons and is also able to detect variation as small as one base pair difference between samples (Hoffmann et al., 2007). HRM platform is also amenable to visual analysis of the curve profiles to determine variability in case of potential changes in absolute Tm based on reaction conditions (Koepke et al., 2012). In addition, HRM has proven to be an accurate, time-saving, and cost effective approach for SNP detection (Wu et al., 2008).
Our aim in this study is to establish genetic identity, verify parentage, and also determine the relatedness of sweet cherry advanced selections, commercial cultivars, and old cultivars obtained from diverse geographical regions, as well as newly released cultivars from the WSU sweet cherry breeding programme, using SNP markers. We will validate the results by comparison with data generated by screening similar germplasm with SSR markers.
Materials and Methods
Plant Material and DNA Extraction
A total of 110 and 103 sweet cherry cultivars respectively, were used for the SNP experiment and the SSR study (Table 1), while 99 accessions were common to both studies. This germplasm consists of advanced selections, current commercial cultivars, and old cultivars from different parts of the world. These plant materials are grafted on Gisela 6 clonal rootstock and maintained at the Washington State Irrigated Agriculture Research and Extension Center (WSU-IAREC), Prosser, USA. Total genomic DNA was isolated from young unexpanded leaves based on the CTAB method (Doyle and Doyle, 1987), quantified by running on a 2% gel using size standards and diluted to 10 ng/μl prior to PCR amplification. Some cultivars including “Seneca,” “7336-1,” “8008-5,” “Hedelfingen,” “99FL154R4,” “Attika,” “Regina,” “Selah,” “Rainier,” “Santina,” and “JJ” were not available for use in the SSR study while “Sweet September,” “7636-1,” “8014,” and “DD” produced melting curve profiles difficult to interpret and were removed from the SNP study.
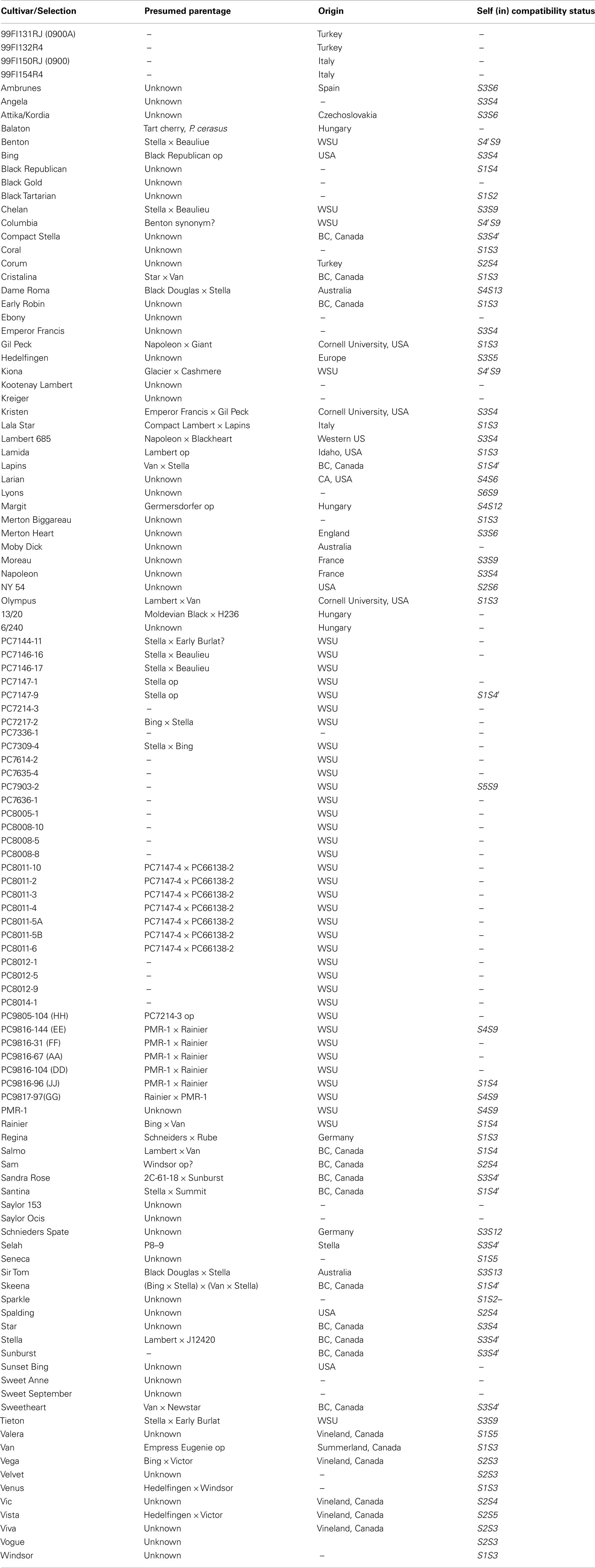
Table 1. Sweet cherry accessions, presumed parentage, origin, and self (in) compatibility status.
SNP Discovery and Primer Design
The methodology for SNP discovery and primer design is described in Koepke et al. (2012). The sequences of the SNP primers used in the study are shown in Table 2.
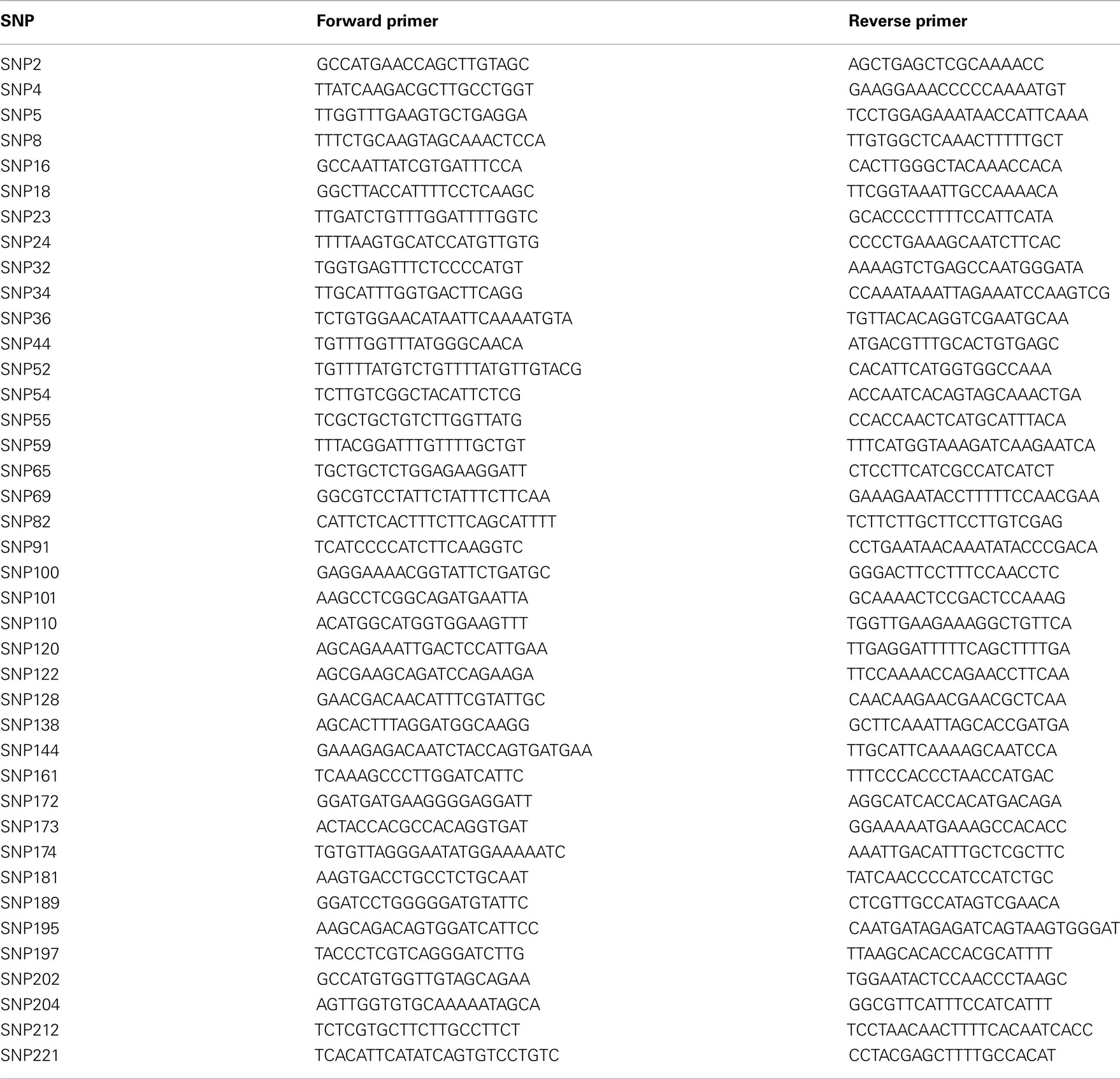
Table 2. SNP primers used and their forward and reverse sequences.
SNP Amplification
PCR amplifications were performed on a LightCycler®480 Real-Time PCR System (Roche, EEUU). A total of 100 SNP primers were used initially to screen a panel of 30 sweet cherry cultivars of which 40 that showed polymorphism were chosen for screening all germplasm (including the initial 30 cultivars and the remaining 80 accessions). Cultivar Dame Roma was also extracted and run twice to check for marker consistency along with the 30 initial sweet cherry cultivars. HRM PCR amplifications were performed in a total volume of 12.5 μl, using the LightCycler®480 HRM Master 1× with 2.5 mM MgCl2, 0.2 μM of each primer, and 2.5 ng of genomic DNA. All the SNP primers were amplified using the same PCR conditions including 10 min of pre-incubation at 95°C; 45 cycles of amplification at 95°C for 10 s, 58°C for 20 s, and 72°C for 30 s; one cycle of HRM at 95°C for 1 min, 40°C for 1 min, 65°C for 1 s, and finally, one cycle of cooling at 40°C for 10 s. Polymorphism was scored based on the melting temperature (Tm) of each accession and visual observation of the melting curves, since the temperature at which a DNA strand separates or melts when heated can vary over a wide range, depending on the sequence, the length of the strand, and the GC/AT ratio (Lehmensiek et al., 2008).
SSR Amplification
Seven SSR markers previously developed in peach, almond, and sweet cherry (Table 3) were used in this experiment to screen 103 accessions of sweet cherry. These primers were selected because of their polymorphism in Prunus and distribution on the Prunus reference map of “T⊆E” (Joobeur et al., 1998). PCR conditions were the same as described in (Fernández i Martí et al., 2009). PCR products were detected on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems). Forward primers were labeled with the fluorescent dyes NED, PET, VIC, and 6-FAM and the size standard was Genescan 500 Liz (Applied Biosystems). Thirty sweet cherry cultivars (the same as in the SNP study above) along with cultivar Dame Roma were extracted and run twice to check for marker consistency.
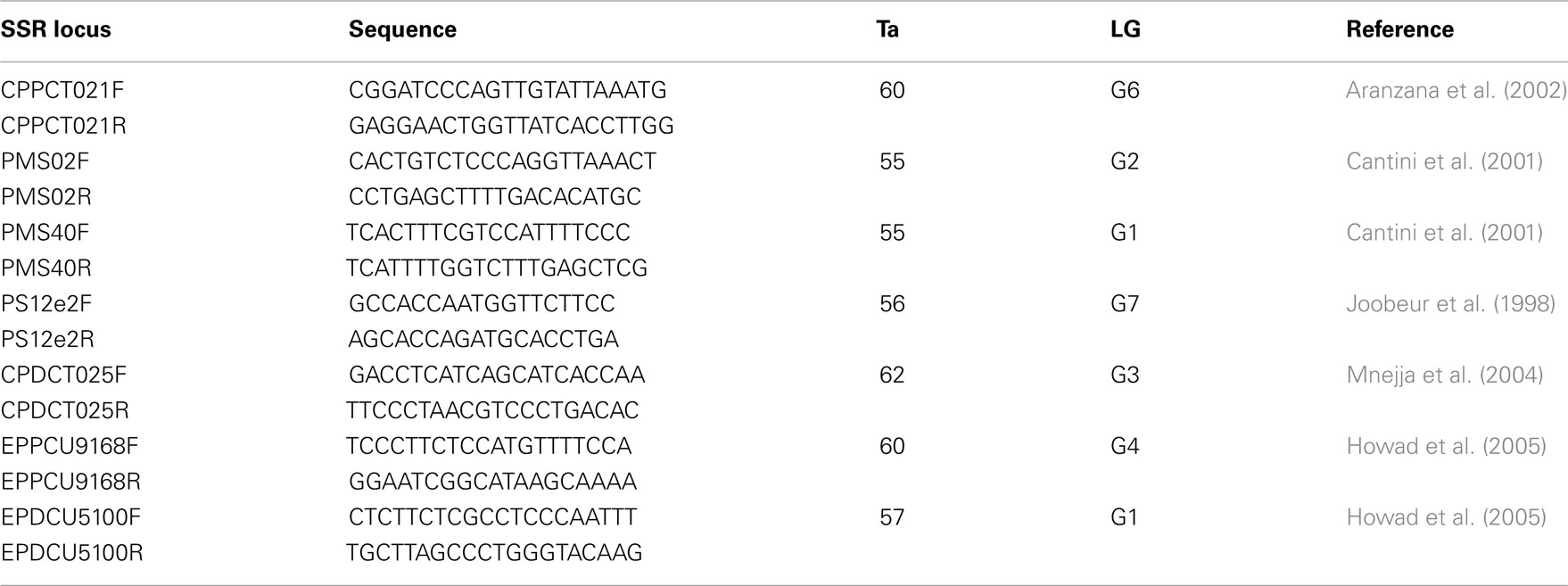
Table 3. SSR primer designation, sequences, annealing temperature (Ta), linkage group (LG), and references.
DATA Analysis for SNP and SSR Markers
Both SNP and SSR data were scored on the basis of presence or absence of marker alleles and this was used to generate a binary matrix. This data was used to estimate genetic similarity between individuals based on Nei and Li (1979) in the NTSYSpc-2.11 software package (Exeter Software Setauket, NY, USA). A dendrogram was generated using the un-weighted pair group method with arithmetic mean (UPGMA) cluster analysis. Genetic parameters were estimated using PopGene 1.31 software (Yeh et al., 1997).
Results
Application of SNP_HRM for Study of Genetic Diversity in Sweet Cherry
Initial screening of a panel of 30 sweet cherry cultivars with 100 SNP markers resulted in the pre-selection of 40 highly polymorphic primers for further amplification of the rest of the germplasm. The allele call was based on the Tm values combined with a thorough analysis of melting peaks. The SNP primer pairs produced a total of 229 alleles in 99 accessions ranging from 2 (including SNP 5, SNP 16, SNP 23, SNP 34) to 9 (SNP 24, SNP 120, SNP 174, SNP 189, or SNP 221, and SNP 100), with a mean of 6 alleles per locus. The observed heterozygosity (Ho) ranged from 0.14 (SNP 59 and SNP 91) to 0.66 (SNP 101). The expected heterozygosity (He) was generally higher and ranged from 0.38 (SNP 174) to 0.73 (SNP 122). The polymorphic information content (PIC) ranged from 0.37 (SNP 174) to 0.73 (SNP 122, Table 4). Based on PIC values, SNP 174 (0.37) appears to be the least informative followed by SNP 120 (0.39), whereas, SNP 122 (0.73) would appear to be the most informative.
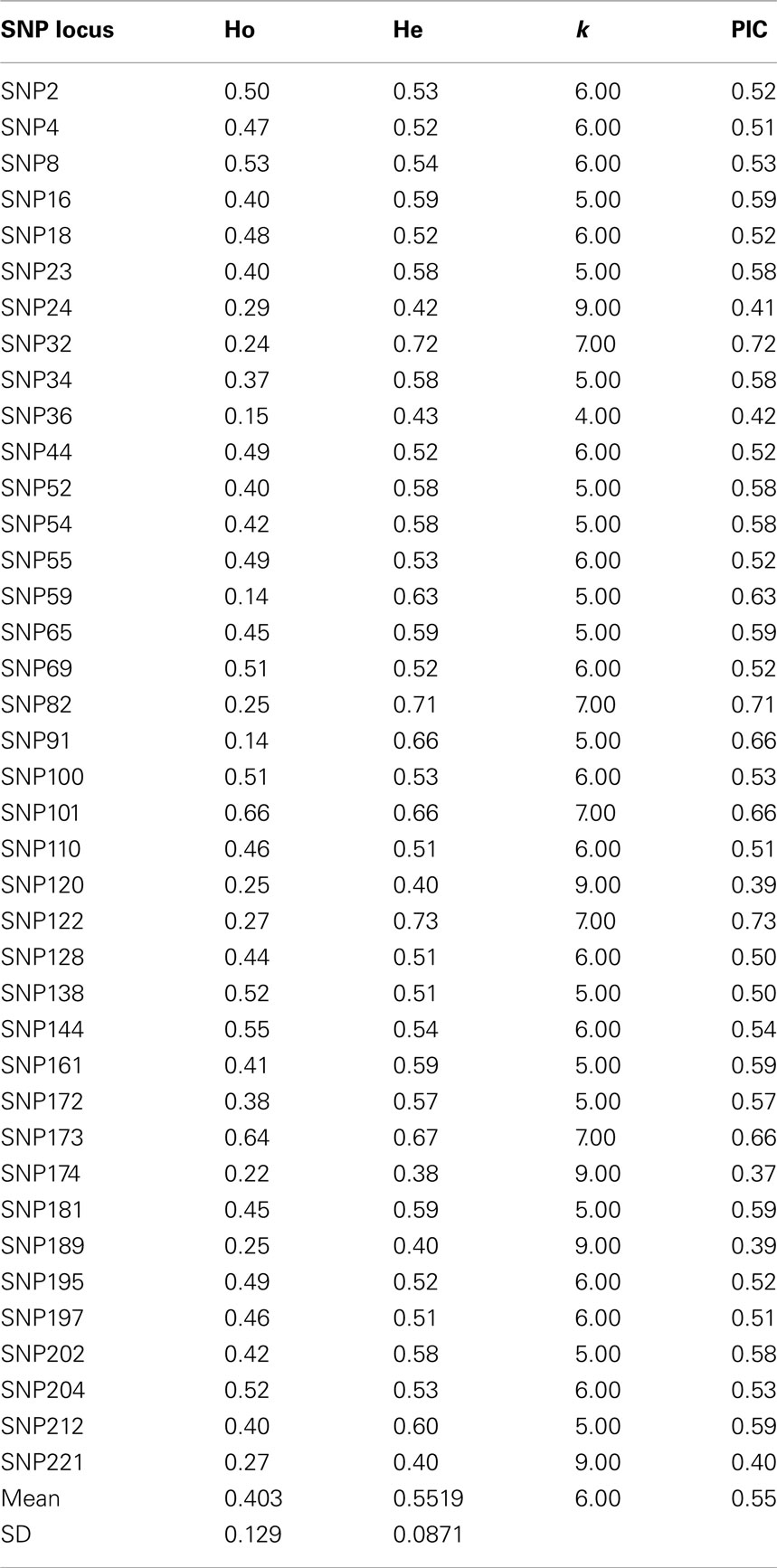
Table 4. SNP locus obtained from the sweet cherry genome analyzed in the sweet cherry cultivars studied, number of alleles obtained (k), observed (Ho), expected heterozygosity (He), and polymorphism information content (PIC).
Application of SSR for Study of Genetic Diversity in Sweet Cherry
All 7 SSR loci from the different Prunus species were polymorphic and amplified successfully when tested on 103 cultivars and breeding lines of sweet cherry. They produced a total of 50 alleles in 99 accessions (shared in common with the SNP study) ranging from 4 (CPDCT025 and CPPCT021) to 11 (Ps12e2) with a mean of 7 alleles per locus. All primer pairs produced a maximum of two bands per genotype in accordance with the diploid nature of this species. Observed heterozygosity ranged from 0.30 (CPPCT021) to 0.65 (PMS04) while He ranged from 0.49 (CPDCT05) to 0.80 (PMS02). PIC values ranged from 0.48 (CPDCT025) to 0.80 (PMS02). PMS02 is therefore the most informative primer (Table 5). The SSRs developed in other Prunus species such as peach, almond, or Japanese plum can be effectively used for fingerprinting in sweet cherry. Therefore, we confirm the high level of synteny within the Prunus species (Aranzana et al., 2003; Arús et al., 2005).
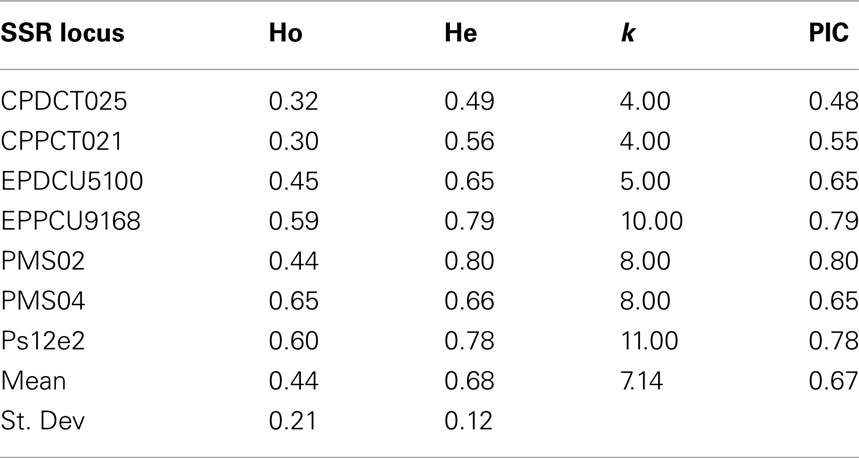
Table 5. SSR loci from different Prunus species analyzed in the sweet cherry cultivars studied, number of alleles obtained (k), observed (Ho), expected (He) heterozygosity, and polymorphism information content (PIC).
Comparison of SSR Dendrogram and SNPs Dendrogram
Although some sweet cherry cultivars have previously been characterized using molecular markers, most selections in this study were genotyped for the first time using SNP and SSR markers. The dendrograms from UPGMA cluster analysis were constructed using 110 and 103 accessions, respectively, for both the SNP and SSR studies. There was no clear pattern of differentiation of the germplasm based on country of origin. However, both “Balaton” (tart cherry) and “NY 54” (wild cherry) used as an out-group grouped together in cluster 1 (SNP dendrogram) and in cluster 3 (SSR dendrogram) were clearly distinguished from sweet cherry (Figures 1 and 2). The lack of separation of numbered selections including “99Fl3131RJ” and “99Fl132R4” from Italy and “99Fl150RJ” and “99Fl154R” from Turkey from one another suggests that both sets of selections are identical and should be treated as one. It should be emphasized that the SNPs allowed the separation of cultivar “Stella” from “Compact Stella.” “Compact Stella” is a smaller more compact tree than “Stella” and the fruit ripens 2 weeks later than “Stella.” This discrimination was not possible with the seven SSR markers although the dendrogram from SSR markers has three main clusters as well. There is about 75% similarity between the dendrogram from both SNPs and SSRs and the genetic distance of the main cultivars are also the same in the two dendrograms. Cultivar Dame Roma was extracted twice to validate the consistency of the SNP markers and this cultivar was identical in the two dendrograms.
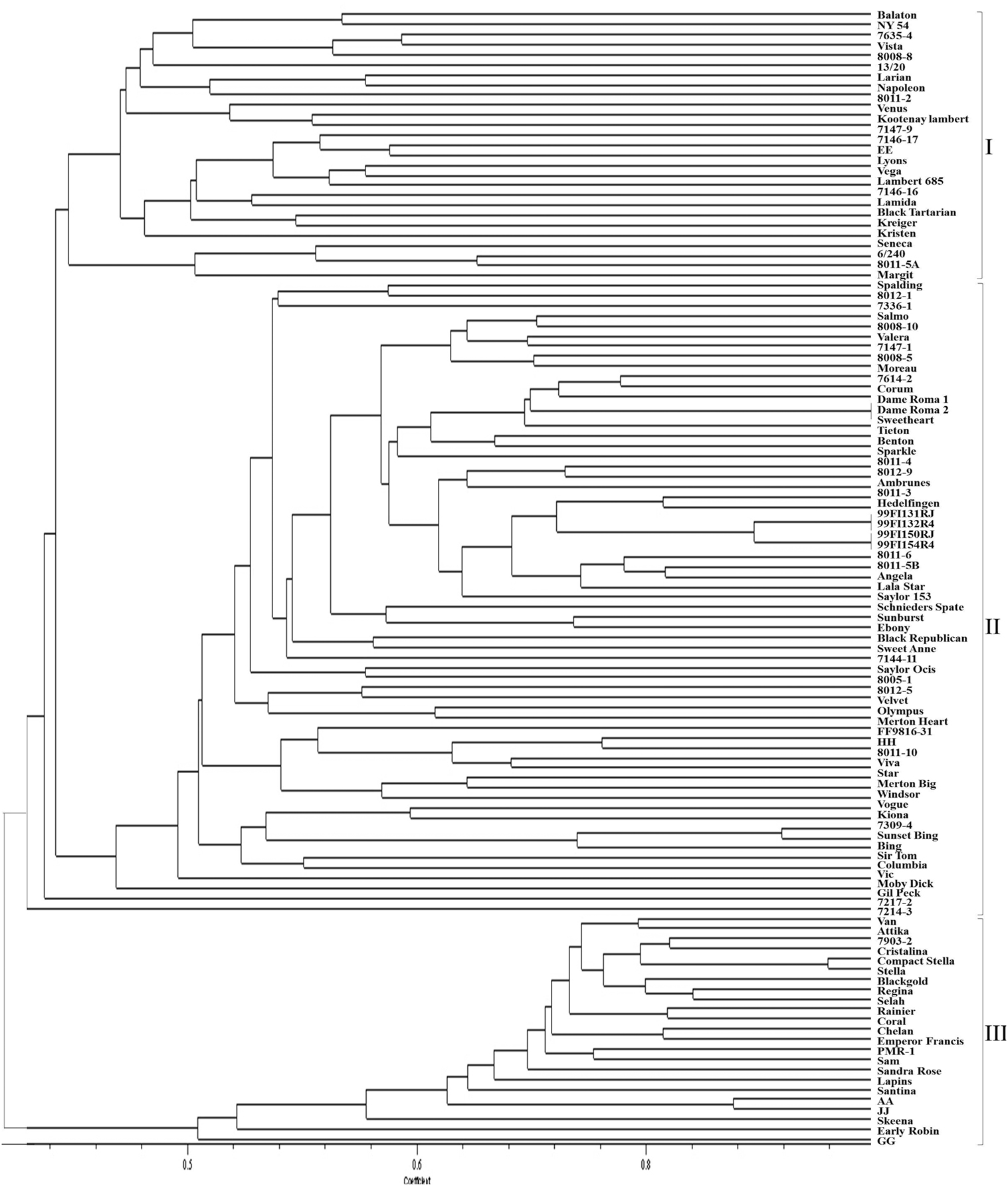
Figure 1. A dendrogram based on SNP data depicting the relationships of the sweet cherry accessions.
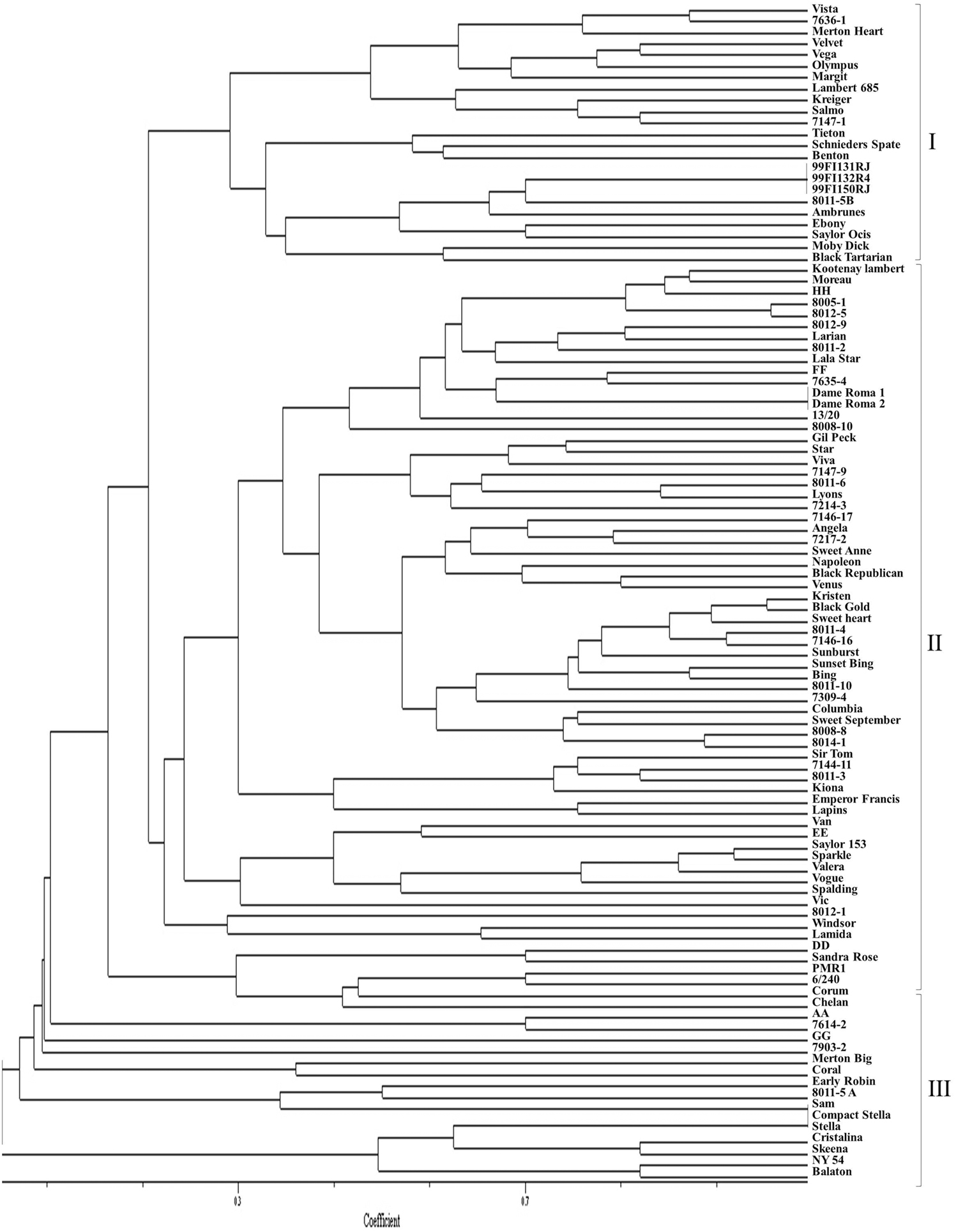
Figure 2. A dendrogram based on SSR data depicting the relationships of the sweet cherry accessions.
Genetic Relatedness of the Accessions and Parentage Verification Based on SNPs
Both SNPs dendrogram and SSR dendrogram showed three main clusters, with Cluster 2 further subdivided into two sub-clusters which include most of the selections and cultivars analyzed (75%). However, in the SSR dendrogram some of the relationships are not consistent with presumed pedigree relationships of the accessions (Figure 2) unlike the SNPs dendrogram (Figure 1). “Larian,” “13/20,” “8011-2,” “Napoleon,” “Lambert,” “Kootenay Lambert,” “Black Tartarian,” “Kreiger,” “Seneca,” “6/240,” “8011-5A,” “Margit” (“Germersdorfer” op), “Vista” (“Hedelfingen” × “Victor”), “Venus” (“Hedelfingen” × “Windsor”), “Kristen” (“Emperor Francis” × “Gil Peck”), and “Lamida” (“Lambert” op) grouped together in Cluster 1 in the SNPs dendrogram. In terms of pedigree relationships, “Lamida” came from open pollinated “Lambert,” while “Lambert” came from “Napoleon” × “BlackHeart” cross, and “Kristen” has “Napoleon” as grandparent. “Vista” and “Venus” both have “Hedelfingen” as the presumed seed parent. The relationship of “Hedelfingen” and “Napoleon” is unclear from anecdotal records however, both cultivars appeared in the same cluster in Wünsch and Hormaza (2002). These authors suggested that their central European origin could be the reason for the close affinity. “Hedelfingen” (S3S5) appeared in the upper part of Cluster 2 in our study and could be related to “Napoleon” (S3S4) through a common parent since both seem to have some alleles in common.
The close relationship of “Dame Roma,” “Tieton,” “Benton,” and “Sweetheart” in the upper part of Cluster 2 is clearly supported with “Stella” being one of the parents of “Dame Roma,” “Tieton,” and “Benton,” and the grand parent of “Sweetheart.” Both “7217-2” (“Bing” × “Stella”) and “7214-3” (with unknown parentage) in the lower part of Cluster 2 are related through both “Bing” and “Stella” as parents based on their inheritance of alleles from both cultivars. Moreover, both came from crosses made in 1972 in the WSU breeding programme. Also, the close affinity of “7309-4” (“Stella” × “Bing”) with “Kiona” (“Glacier” × “Stella” × “Early Burlat”) × “Cashmere” (“Stella” × “Early Burlat”) in the lower part of Cluster 2 confirms their relatedness through “Stella.” Finally, “Lala Star” presumed to be an offspring of “Lambert” and “Lapins” grouped with “8011-6,” “8011-5B,” and “Angela” in the upper part of Cluster 2. Although the pedigree information on these accessions is not available, the close affinity with “Lala Star” is likely to be through “Lapin’s” grandparent, “Stella.” The heavy use of “Stella” in breeding programmes to impart self fertility following introduction in 1968 (Lapins, 1970) is highly evident in these accessions. Self fertility is desired by growers to reduce reliance on bees for pollination, eliminate the need for pollinizers and presumably boast yield. Another interesting finding is the grouping of “Bing” (“Black Republican” op) with “Sir Tom” (“Black Douglas” × “Stella”) in Cluster 2 as well. “Bing” is the most popular sweet cherry variety in the world and controls about 60% of cherry production in the Pacific Northwest region of the US. Identifying the male parent will aid our understanding of inheritance of “Bing” type fruit quality. Perhaps, exploring the connectedness with “Black Douglas” (not available in our collection) could open up possibilities.
Cluster 3 on the other hand, shows some groupings that seem to contradict the pedigree relationships of some accessions. For example, the close grouping of “Black Gold” (with unknown parentage) with “Regina” (“Schneiders” × “Rube”) and “Selah” (“P8-9” × “Stella”). “Selah” is self-fertile (S3S4′) with the S4′ allele inherited from “Stella” while the seed parent “P8-9” is presumed to be a progeny of “Rainier” × “Bing” (Olmstead et al., 2011b). “Regina” (S1S3) shares some alleles with “Schneiders” (S3S12) and the S-genotype of its presumed pollen parent is S1S4 therefore, the true parentage is “Schneiders” × “Rube.” Similarly, “Chelan” presumed to be a progeny of “Stella” × “Beaulieu” grouped closely with “Emperor Francis,” one of the founding clones of sweet cherry (Choi and Kappel, 2004). However, “Emperor Francis” is presumed to be a great grand parent of “Chelan” [Stella = (Lambert × JI2420 (Emperor Francis × Napoleon)] and therefore should transmit 1/8th of its alleles to “Chelan.” We do not have “Beaulieu” in our collection and therefore cannot ascertain that it is the pollen parent of “Chelan” and/or related to “Emperor Francis.” Another interesting relationship is the grouping of “PMR-1” with “Sam.” “PMR-1,” an open pollinated seedling of unknown origin is the original source of powdery mildew resistance found in its progeny including “AA,” “JJ,” and “GG.” This disease appears to be controlled by a single major gene putatively named pmr (Toyama et al., 1993; Olmstead and Lang, 2002). “Chelan,” “Venus,” “Moreau,” and “Hedelfingen” are believed to be resistant however their resistances have not been characterized to dissect the genetic control. Identifying the origin or parentage of “PMR-1” will shed more light on pmr and speed up gene identification and breeding for powdery mildew resistance. “Sam” (S2S4) is susceptible to powdery mildew and is presumed to result from open pollination of “Windsor” but the S-genotype of “Windsor” (S1S3) suggests otherwise. On the other hand, the S-genotype of “PMR-1” is S4S9 and both (i.e., “PMR-1” and “Sam”) have S4 allele in common. Could it be that both have one parent in common? Perhaps more SNP markers may be needed to resolve the relationship. Lastly, “Columbia” is another name for cultivar Benton (Olmstead et al., 2011a). “Benton” is grouped in the upper part of Cluster 2 while “Columbia” is grouped in the lower part with high similarity to “Vic,” suggesting that a tree different from “Columbia” could have been sampled for DNA isolation.
Discussion
Origin and General Characteristics of the SNPs Ued in This Study
Molecular markers have been successfully used for the study of genetic relationship in sweet cherry. These studies have been carried out using isoenzyme markers (Ducci and Santi, 1997), RAPDs (Gerlach and Stosser, 1998; Hormaza, 1999, AFLP (Zhu et al., 2003), and finally SSRs (Dirlewanger et al., 2002; Wünsch and Hormaza, 2002; Schueler et al., 2003). However, the introduction of new sequencing technologies has changed the landscape for detecting genome-wide polymorphism (Craig et al., 2008). The availability and stability of SNPs in comparison to SSRs provide better prospects for cultivar identification and assessment of genetic diversity. In addition, the cost of analysis and development of SNPs is rapidly coming down. These SNPs were discovered from 3′ UTR sequencing of “Rainier” and “Bing” sweet cherry from developing floral buds using the Roche 454 GS platform (Koepke et al., 2012). The 3′ UTR of messenger RNAs (mRNAs) contain several regulatory elements that play a role in plant development, differentiation, and cell cycle (Ohta et al., 2005; Kertesz et al., 2006; Merritt et al., 2008). It has also been reported that 3′ UTR has a high frequency of SNP polymorphism (Bhattramakki et al., 2002). Zhu et al. (2003) reported that one-third of the 3′ UTR ends of soybean genes show SNP polymorphism. Also, Wu et al. (2008) reported that SNP frequencies were found to be lowest in the coding region (1:157), moderate in the intron (1:130), and highest in the UTR (1:50) in almond.
Simple single nucleotides loci are generally bi-allelic and it is supposed to have occurred only once in evolutionary time. As a consequence SNPs are generally polymorphic only for a particular species or genetic material. Thus, contrary to SSRs, these markers are being created to evaluate variation within a specific species, which is in this study Prunus avium L., and are not readily transferable to other Prunus species such as peach, apricot, or almond.
Assessment of Genetic Diversity
The Ho obtained with SNPs and SSRs were similar; 0.40 vs. 0.44. Furthermore, the Ho is very close to those reported in other sweet cherries studies such as Ohta et al. (2005; Ho 0.46), Wünsch and Hormaza(2002; Ho 0.49), Lisek and Rozpara(2009; Ho 0.38), and Cabrera et al. (2012; Ho 0.45). The number of alleles per locus in our SNP study ranged from 9 (SNP 24, SNP 120, and SNP 174) to 4 (SNP 36), with a mean of 6 alleles per locus. With SSR markers, the primer pair Ps12e2 showed the highest number of alleles (11) while primer pairs CPDCT025 and CPPCT021, amplified in both cases only four alleles per locus. The average number of alleles per locus obtained here (7.14) was very similar to that reported by Schueler et al. (2003) (6) but higher than that reported by Wünsch and Hormaza (2002) (3.3). The low level of heterozygosity observed in this study as well as in other sweet cherry studies (Wünsch and Hormaza, 2002; Ohta et al., 2005), compared with other Prunus species (Xie et al., 2006; Fernández i Martí et al., 2009) may be due to limited gene flow among accessions. The heterozygote deficiency could be due to limited number of founder cultivars used as parents in breeding programmes (Choi et al., 2000).
Although sweet cherry is characterized by GSI, the level of heterozygosity observed here is much lower than in other Prunus species such as almond, where a heterozygosity value of 0.75 has been reported (Xie et al., 2006; Fernández i Martí et al., 2009). No differences between the heterozygosity levels in self-compatible and self-incompatible cultivars have been observed in this study however, it is expected that self-compatible cultivars would be less heterozygous than self-incompatible cultivars (Socias i Company, 1990).
Assessment of genetic diversity of cultivated sweet cherries may aid in crop improvement strategies. Molecular markers such as SNPs may be applied to assess diversity at the DNA level and thus provide an effective tool for decision making regarding the choice of parental genotypes for use in crosses and for germplasm conservation.
Determination of Genetic Relationships and Parentage Verification
In our study, a dendrogram using SSR markers was also generated in order to be compared with the one generated using SNPs. We observed in both cases, that the cultivars were grouped into three main clusters, with the second cluster subdivided into additional subgroups. As expected, both dendrograms are not identical, but share at least 75% of the clusters. The cultivar “Compact Stella,” which is a mutation of “Stella,” could not be differentiated from “Stella” using SSR markers but this was possible with SNPs. This is probably due to the intrinsic nature of SSRs and its inability to differentiate mutants that differ from the original genotype in one gene. It also suggests that SNPs may be able to distinguish somatic mutants from the mother plants.
The history of the pedigree relationships of sweet cherry is fraught with challenges including complications with selfing, outcrossing, and presence of synonyms. Of a total of 243 seedlings from 22 populations screened with PCR-based S-locus markers, Haldar et al. (2010) reported that 22% were selfs, 11% were unintended outcrosses, and 59% had the intended parentage. This highlights the importance of parentage verification with molecular markers before carrying out genetic studies in sweet cherry. In this study, SNPs were better able to determine relationships and verify parentage of accessions than SSRs. This suggests that more SSRs may be needed to resolve the relationships of the accessions. In fact, Falush et al. (2003) suggested that the number of genome-wide SSRs used should at least equal the number of linkage groups (LG) in any species in order to successfully identify population structure. In our own case, there are eight LG in sweet cherry but only six SSRs were used in the study. The heavy use of “Stella” as a parent in breeding programmes was very notable with many accessions having “Stella” in their pedigree. It would be worthwhile to explore other sources of self fertility in sweet cherry to minimize inbreeding and increase genetic diversity. “Cristobalina” a landrace cultivar from Spain appears to be a source of non-S gametophytic self fertility in sweet cherry (Cachi and Wünsch, 2011) that could be exploited. The relationship between “PMR-1” and “Sam” needs to be further investigated to identify the genetic origin of “PMR-1” and facilitate identification of the gene that underpins powdery mildew resistance in “PMR-1” and its progeny. “Windsor,” widely touted as the seed parent of “Sam” was found not to be true since both cultivars have a similarity coefficient of 0.3 and also do not share any S-alleles in common (S2S4 for “Sam” and S1S3 for “Windsor”). Further research may be necessary to resolve the relationship of other important cultivars including “Napoleon” and “Hedelfingen,” and “Selah” and “Regina.”
Although the 40 SNPs used in this study were able to identify duplicates, confirm most parentages and also determine relatedness of the accessions, more SNPs may be necessary for a better resolution of the relationships.
HRM Analysis and Tm Calling
Although contrary to conventional approaches, we analyzed melting peaks and scored Tm values in metric units. Graham et al. (2005)reported that the meaning of Tm is ambiguous and less useful as a metric unit than curve shape. However, in breeding and population genetics studies, scoring allelic forms, and heterozygosity numerically is necessary for calculation of population parameters. Since the relationships generated with SNPs data is supported by our SSR data, we would argue that calling Tm values as alleles is appropriate and valid. Thus, with the strategy used in this study and with the results obtained we confirm the efficiency of SNP markers for fingerprinting purposes in sweet cherry. In fact, HRM has been proven to be an accurate, time-saving, and cost-efficient approach for SNP genotyping and when this technique was compared with SSR markers, we were able to realize both savings in terms of time and cost.
Conclusion
The study has demonstrated the utility of SNPs markers for genetic fingerprinting in sweet cherry. Although SSRs had a higher mean number of alleles per locus as well as higher heterozygosity and PIC values than SNPs, both markers showed similar groupings for the sweet cherry accessions as shown in the dendrogram. In fact, SNPs were able to distinguish between mutants and their wild type germplasm thus making SNPs a more valuable tool for cultivar fingerprinting and identification than SSRs. Further, SNPs confirmed the parentage of some accessions and determined relationships consistent with pedigree relationships. We recommend the use of SNPs for genetic fingerprinting, parentage verification, gene mapping, and study of genetic diversity in sweet cherry and believe it will prove useful in most other related species within Amygdaloidea as well.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sarah O’Neill, a High School intern in Dr. Oraguzie’s lab, for carrying out the initial DNA extraction and HRM analysis. Nnadozie Oraguzie, Amit Dhingra, Tyson Koepke, Angel Fernandez i Marti, and BlessingAthanson designed the study. Amit Dhingra and Tyson Koepke performed transcriptomic sequencing and identified SNPs while Nnadozie Oraguzie and Amit Dhingra designed the primers. BlessingAthanson and Angel Fernandez i Marti collected leaf samples and performed melting curve analysis on the LightCycler 480. Angel Fernandez i Marti performed SSR analyses and along with CF carried out data analyses while Nnadozie Oraguzie supervised the research and guided data interpretation. Angel Fernandez i Marti and Nnadozie Oraguzie wrote the paper. All authors read and approvedthe final manuscript. Angel Fernandez i Marti was supported by a post-doctoral fellowship (CA-1-10) from the CAI/government of Aragon/(Spain). This work was funded in part with Royalty funds from the Sweet cherry breeding program, Washington State University, Irrigated Agriculture and Extension Centre, Prosser, WA, USA. We thank Drs. Chuck Brown and Kate Evans for their discussion and critical review of the manuscript.
References
Agarwal, M., Shrivastava, N., and Padh, H. (2008). Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep. 27, 617–631.
Aranzana, M. J., Garcia-MAS, J., Carbo, J., and Arus, P. (2002). Development and variability analysis of microsatellite markers in peach. Plant Breeding 121, 87–92.
Aranzana, M. J., Pineda, A., Cosson, P., Dirlewanger, E., Ascasibar, J., Cipriani, G., Ryder, C., Testolin, R., Abbott, A., King, G., Iezzoni, A., and Arús, P. (2003). A set of simple-sequence repeat (SSR) markers covering the Prunus genome. Theor. Appl. Genet. 106, 819–825.
Arús, P., Yamamoto, T., Dirlewanger, E., and Abbott, A. G. (2005). Synteny in the Rosaceae. Plant Breed. Rev. 27, 175–211.
Batley, J., Barker, G., O’Sullivan, H., and Edwards, K. J. (2003). Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data. Plant Physiol. 132, 84–91.
Bhattramakki, D., Dolan, M., Hanafey, M., Wineland, R., Vaske, D., Register, J. C., Tingey, S. V., and Rafalski, A. (2002). Insertion-deletion polymorphisms in 3′ regions of maize genes occur frequently and can be used as highly informative genetic markers. Plant Mol. Biol. 48, 539–547.
Cabrera, A., Umesh, R., De Franceschi, P., Sebolt, A., Sooriyapathirana, S., Dirlewanger, E., Quero-Garcia, J., Schuster, M., Iezzoni, A. F., and van der Knaap, E. (2012). Rosaceae conserved orthologous sequences marker polymorphism in sweet cherry germplasm and construction of a SNP-based map. Tree Genet. Genomes 8, 237–247.
Cachi, A. M., and Wünsch, A. (2011). Characterization and mapping of non-S gametophytic self-incompatibility in sweet cherry (Prunus avium L.) J. Exp. Bot. 62, 1847–1865.
Cantini, C., Iezzoni, A. F., Lamboy, W. F., Boritzki, M., and Struss, D. (2001). DNA fingerprinting of tetraploid cherry germplasm using SSR. J. Am. Soc. Hortic. Sci. 126, 205–209.
Chágne, D., Gasic, K., Crowhurst, R. N., Han, Y., Bassett, H. C., Bowatte, D. R., Lawrence, T. J., Rikkerink, E. H. A., Gardiner, S. E., and Korban, S. S. (2008). Development of a set of SNP markers present in expressed genes of the apple. Genomics 92, 353–358.
Choi, C., and Kappel, F. (2004). Inbreeding, coancestry and founding clones of sweet cherries from North America. J. Am. Soc. Hortic. Sci. 129, 535–543.
Choi, C., Livermore, K., and Andersen, R. L. (2000). Sweet cherry pollination: recommendation based on compatibility groups and bloom time. J. Am. Pomol. Soc. 54, 148–152.
Cipriani, G., Lot, G., Huang, H. G., Marrazzo, M. T., Peterlunger, E., and Testolin, R. (1999). AC/GT and AG/CT microsatellite repeats in peach (Prunus persica (L.) Batsch): isolation, characterization and cross-species amplification in Prunus. Theor. Appl. Gen. 99, 65–72.
Craig, D. W., Pearson, J. V., Szelinger, S., Sekar, A., Redman, M., Corneveaux, J. J., Laub, T., Nunn, G., Stephan, D. A., and Homer, N. (2008). Identification of genetic variants using bar-coded multiplexed sequencing. Nat. Methods 5, 887–893.
de Nettancourt, D. (2001). Incompatibility and Incongruity in Wild and Cultivated Plants, 2nd Edn. Berlin: Springer.
Dirlewanger, E., Cosson, P., Travaud, M., Aranzana, M. J., Poizat, C., Zanetto, A., Arús, P., and Laigret, F. (2002). Development of microsatellite markers in peach [Prunus persica (L.)Batsch] and their use in genetic diversity analysis in peach and sweet cherry (Prunus avium L.). Theor. Appl. Genet. 105, 127–138.33.
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Drenkard, E., Richter, B. G., Rozen, S., Stutius, L. M., Angell, N. A., Mindrinos, M., Cho, R. J., Oefner, P. J., Davis, R. W., and Ausubel, F. M. (2000). A simple procedure for the analysis of single nucleotide polymorphisms facilitates map-based cloning in Arabidopsis. Plant Physiol. 124, 1483–1492.
Ducci, F., and Santi, F. (1997). The distribution of clones in managed and unmanaged populations of wild cherry (Prunu savium). Can. J. Forest res. 27, 1998–2004
Falush, D., Stephens, M., and Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587.
Fang, J., Tao, J., and Chao, C. T. (2006). Genetic diversity in fruiting-mei, apricot, plum and peach revealed by AFLP analysis. J. Hortic. Sci. Biotechnol. 81, 898–920.
Fernández i Martí, A., Alonso, J. M., Espiau, M. T., Rubio-Cabetas, M. J., and Socias i Company, R. (2009). Genetic diversity in Spanish and foreign almond germplasm assessed by molecular characterization with SSRs. J. Am. Soc. Hortic. Sci. 134, 535–542.
Flint-Garcia, S. A., Thuillet, A.-C., Yu, J. M., Pressoir, G., Romero, S. M., Mitchell, S. E., Doebley, J., Kresovich, S., Goodman, M. M., and Buckler, E. S. (2005). Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J. 44, 1054–1064.
Gerlach, H. K., and Stosser, R. (1998). Sweet cherry cultivar identification using RAPD derived DNA fingerprints. Acta Hortic. 468, 63–69.
Graham, R., Liew, M., Meadows, C., Lyon, E., and Wittwer, C. T. (2005). Distinguishing different DNA heterozygotes by high-resolution melting. Clin. Chem. 51, 1295–1298.
Gupta, P. K., Balyan, H. S., Sharma, P. C., and Ramesh, B. (1996). Microsatellites in plants: a new class of molecular markers. Curr. Sci. 70, 45–54.
Haldar, S., Haedinges, S., Edge-Garza, D., Oraguzie, N., Olmstead, J., Iezzoni, A., and Peace, C. (2010). Applying genetic markers for seed fertility in the WSU sweet cherry breeding programme. Acta Hortic. 859, 375–380.
Hoffmann, M., Hurlebaus, J., and Weilke, C. (2007). High resolution melting curve analysis on the LightCycler (R) 480 PCR system. Nat. Methods (Suppl. S), AN17–AN18.
Hormaza, J. I. (1999). Early selection in cherry combining RAPDs with embryo culture. Sci. Hortic. 79, 121–126.
Howad, W., Yamamoto, T., Dirlewanger, E., Testolin, R., Cosson, P., Cipriani, G., Monforte, A. J., Georgi, L., Abbott, A. G., and Arús, P. (2005). Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics 171, 1305–1309.
Joobeur, T., Viruel, M. A., Vicente, M. C., de Jáuregui, B., Ballester, J., Dettori, M. T., Verde, I., Truco, M. J., Messeguer, R., Batlle, I., Quarta, R., Dirlewanger, E., and Arús, P. (1998). Construction of a saturated linkage map for Prunus using an almond x peach F2 progeny. Theor. Appl. Genet. 97, 1034–1041.
Kertesz, S., Kerenyi, Z., Merai, Z., Bartos, I., Palfy, T., Barta, E., and Silhavy, D. (2006). Bothintronsandlong3′-UTRs operate as cis-acting elements to trigger nonsense-mediated decay in plants. Nucleic Acids Res. 34, 6147–6157.
Koepke, T., Schaeffer, S., Krishnan, V., Jiwan, D., Whiting, M., Oraguzie, N., and Dhingra, A. (2012). Rapid SNP and haplotype development in non-model crops using 3′UTR sequencing. BMC Genomics 12, 13–18. doi:10.1186/1471-2164-13-18
Le Dantec, L., Cardinet, G., Bonet, J., Fouche, M., Boudehri, K., Monfort, A., Poessel, J. L., Moing, A., and Dirlewanger, E. (2010). Development and mapping of peach candidate genes involved in fruit quality and their transferability and potential use in other Rosaceae species. Tree Genet. Genome 6, 995–1012.
Lehmensiek, A., Mark, W., Sutherland, R., and McNamara, B. (2008). The use of high resolution melting (HRM) to map single nucleotide polymorphism markers linked to a covered smut resistance gene in barley. Theor. Appl. Genet. 117, 721–728.
Lisek, A., and Rozpara, E. (2009). Identification and genetic diversity assessment of cherry cultivars and rootstocks using the ISSR-PCR technique. J. Fruit Ornamental Plant Res. 17, 95–106.
Merritt, C., Rasoloson, D., Ko, D., and Seydoux, G. (2008) 0.3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr. Biol. 18, 1476–1482.
Mnejja, M., Garcia-Mas, M., Howad, W., Badenes, M. L., and Arús, P. (2004). Simple sequence repeat (SSR) markers of Japanese plum (Prunus salicina Lindl.) are highly polymorphic and transferable to peach and almond. Mol. Ecol. Notes 4, 163–166.
Nei, M., and Li, W. H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273.
Ohta, S., Katsuki, T., Tanaka, T., Hayashi, T., Sato, Y. I., and Yamamoto, T. (2005). Genetic variation in flowering cherries (Prunus subgenus Cerasus) characterized by SSR markers. Breed. Sci. 55, 415–424.
Olmstead, J., Whiting, M., Ophardt, D., and Oraguzie, N. (2011a). PC7146-8 (Benton®) sweet cherry. Hortscience 46, 121–122.
Olmstead, J., Whiting, M., Ophardt, D., and Oraguzie, N. (2011b). PC7064-3 (Selah®) sweet cherry. Hortscience 46, 123–124.
Olmstead, J. W., and Lang, G. A. (2002). pmr1, a gene for resistance to powdery mildew in sweet cherry. HortScience 37, 1098–1099.
Oraguzie, N. C., Ophardt, D., Whiting, M. D., Lang, G. A., and Long, L. E. (2010). Kiona™ sweet cherry. HortScience 45, 1906–1907.
Qi, L. L., Echalier, B., Chao, S., Lazo, G. R., Butler, G. E., Anderson, O. D., Akhunov, E. D., Dvorak, J., Linkiewicz, A. M., Ratnasiri, A., Dubcovsky, J., Bermudez- Kandianis, C. E., Greene, R. A., Kantety, R., La Rota, C. M., Munkvold, J. D., Sorrells, S. F., Sorrells, M. E., Dilbirligi, M., Sidhu, D., Erayman, M., Randhawa, H. S., Sandhu, D., Bondareva, S. N., Gill, K. S., Mahmoud, A. A., Ma, X., Miftahudin, F., Gustafson, J. P., Conley, E. J., Nduati, V., Gonzalez-Hernandez, J. L., Anderson, J. A., Peng, J. H., Lapitan, N. L. V., Hossain, K. G., Kalavacharla, V., Kianian, S. F., Pathan, M. S., Zhang, D. S., Nguyen, H. T., Choi, D. W., Fenton, R. D., Close, T. J., McGuire, P. E., Qualset, C. O., and Gill, B. S. (2004). A chromosome bin map of 16,000 expressed sequence tag loci and distribution of genes among the three genomes of polyploid wheat. Genetics 168, 701–712.
Rafalski, A. (2002). Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant Biol. 5, 94–100.
Rostoks, N., Mudie, S., Cardle, L., Russell, J., Ramsay, L., Booth, A., Svensson, J. T., Wanamaker, S. I., Walia, H., Rodriguez, E. M., Hedley, P. E., Liu, H., Morris, J., Close, T. J., Marshall, D. F., and Waugh, R. (2005). Genome-wide SNP discovery and linkage analysis in barley based on genes responsive to abiotic stress. Mol. Genet. Genomics 274, 515–527.
Salmaso, M., Faes, G., Segala, C., and Stefanini, M. (2004). Genome diversity and gene haplotypes in the grapevine (Vitis vinifera L.), as revealed by single nucleotide polymorphisms. Mol. Breed. 14, 385–395.
Schueler, S., Tusch, A., Schuster, M., and Ziegenhagen, B. (2003). Characterization of microsatellites in wild and sweet cherry (Prunu savium L.) markers for individual identification and reproductive processes. Genome 46, 95–102.
Sosinski, B., Gannavarapu, M., Hager, L. D., Beck, L. E., King, G. J., Ryder, C. D., Rajapakse, S., Baird, W. V., Ballard, R. E., and Abbott, A. G. (2000). Characterization of microsatellite markers in peach (Prunu spersica (L.)Batsch). Theor. Appl. Genet. 101, 421–428.
Testolin, R., Marrazzo, T., Cipriani, G., Quarta, R., Verde, I., Dettori, T., Pancaldi, M., and Sansavini, S. (2000). Microsatellite DNA in peach (Prunu spersica (L.)Batsch) and its use in fingerprinting and testing the genetic origin of cultivars. Genome 43, 512–520.
Toyama, T. K., Ophardt, D. R., Howell, W. E., and Grove, G. G. (1993). New powdery mildew resistant sweet cherry. Fruit Var. J. 47, 234–235.
Vaughan, S. P., and Russell, K. (2004). Characterization of novel microsatellites and development of multiplex PCR for large-scale population studies in wild cherry, Prunu savium. Mol. Ecol. Notes 4, 429–431.
Watkins, R. (1976). “Cherry, plum, peach, apricot, and almond,”in Evolution of Crop Plants, ed. N. W. Simmonds (New York: Longman), 242–247.
Webster, A. D. (1996). “The taxonomic classification of sweet and sour cherries and a brief history of their cultivation,” in Cherries: Crop Physiology, Production and Uses, eds A. D. Webster, and N. E. Looney (Wallingford, Oxon: CAB International), 3–24.
Wu, S. H., Wirthensohn, M., Hunt, P., Gibson, J., and Sedgley, M. (2008). High resolution melting analysis of almond SNPs derived from ESTs. Theor. Appl. Genet. 118, 1–14.
Wünsch, A. (2009). Cross-transferable polymorphic SSR loci in Prunus species. Sci. Hortic. 120, 348–352.
Wünsch, A., and Hormaza, J. I. (2002). Molecular characterisation of sweet cherry (Prunus avium L.) genotypes using peach [Prunus persica (L.) Batsch] SSR sequences. Heredity 89, 56–63.
Xie, H., Sui, Y., Chang, F. Q., Xu, Y., and Ma, R. C. (2006). SSR allelic variation in almond (Prunus dulcis Miller). Theor. Appl. Genet. 112, 366–372.
Yeh, F. C., Yang, R.-C., and Boyle, T. (1997). POPGENE, the User-Friendly Shareware for Population Genetic Analysis. Molecular Biology and Biotechnology Centre. Edmonton: University of Alberta
Keywords: Prunus avium L., 3′ UTR sequencing, high resolution melting analysis, molecular markers, genetic parameters, parentage verification
Citation: Fernandez i Marti A, Athanson B, Koepke T, Font i Forcada C, Dhingra A and Oraguzie N (2012) Genetic diversity and relatedness of sweet cherry (Prunus Avium L.) cultivars based on single nucleotide polymorphic markers. Front. Plant Sci. 3:116. doi: 10.3389/fpls.2012.00116
Received: 27 April 2012; Paper pending published: 15 May 2012;
Accepted: 15 May 2012; Published online: 25 June 2012.
Edited by:
Sun Hee Woo, Chungbuk National University, South KoreaReviewed by:
Md. Shafikur Rahman, Patuakhali Science and Technology University, BangladeshAbu Hena Mostafa Kamal, Korea Research Institute of Bioscience and Biotechnology, South Korea
Copyright: © 2012 Fernandez i Marti, Athanson, Koepke, Font i Forcada, Dhingra and Oraguzie. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nnadozie Oraguzie, Irrigated Agriculture Research and Extension Centre, Washington State University, 24106 North Bunn Road, Pullman, WA 99350, USA. e-mail:bm9yYWd1emllQHdzdS5lZHU=