

- Plant Biotechnology, Department of Biology, ETH Zurich, Zurich, Switzerland
The pep2pro database was built to support effective high-throughput proteome data analysis. Its database schema allows the coherent integration of search results from different database-dependent search algorithms and filtering of the data including control for unambiguous assignment of peptides to proteins. The capacity of the pep2pro database has been exploited in data analysis of various Arabidopsis proteome datasets. The diversity of the datasets and the associated scientific questions required thorough querying of the data. This was supported by the relational format structure of the data that links all information on the sample, spectrum, search database, and algorithm to peptide and protein identifications and their post-translational modifications. After publication of datasets they are made available on the pep2pro website at www.pep2pro.ethz.ch. Further, the pep2pro data analysis pipeline also handles data export do the PRIDE database (http://www.ebi.ac.uk/pride) and data retrieval by the MASCP Gator (http://gator.masc-proteomics.org/). The utility of pep2pro will continue to be used for analysis of additional datasets and as a data warehouse. The capacity of the pep2pro database for proteome data analysis has now also been made publicly available through the release of pep2pro4all, which consists of a database schema and a script that will populate the database with mass spectrometry data provided in mzIdentML format.
The pep2pro Public Web Interface
The public web interface of the pep2pro database is available at www.pep2pro.ethz.ch. Here, we provide all information contained in the published Arabidopsis datasets that have been analyzed using the pep2pro data analysis pipeline (Baerenfaller et al., 2008, 2011; Grobei et al., 2009; Reiland et al., 2009, 2011; Bischof et al., 2011; Gfeller et al., 2011). This includes information on all identified proteins, their identified peptides, and whole genome hits, as well as their proteogenomic mapping. The information also includes the number of spectra per peptide together with the scores of peptide spectrum assignments, and the display of the spectra in a Spectrum Viewer. A normalized version of the Spectrum Summary displaying for each protein the number of spectra per organ has been added recently to account for the bias that is introduced based on the different total number of spectra available for the different organs. The data in pep2pro are exported to the PRIDE database1 (Vizcaíno et al., 2010) and the information on spectral counts in the different organs is also provided to MASCP Gator2 (Joshi et al., 2011). In MASCP Gator the information is linked to the pep2pro database. Following this link, all the datasets containing spectra for a given AGI are listed to allow the selection of the dataset that is of interest to the user. Additional functions are now available on the pep2pro website. First, peptides that are detectable in a complex protein sample and that unambiguously identify a protein are displayed. Second, it is now possible to search the database for specific peptide amino acid sequences. Third, all unambiguous theoretical tryptic peptides with minimum length of six amino acids and a molecular mass between 400 and 6000 Da are now provided for a protein of interest. We added this functionality to the database in response to valuable user input. The new functions are useful in the set-up of selected reaction monitoring (SRM) experiments (Lange et al., 2008), in which it is important to select peptides that unambiguously identify a protein. Information on unambiguous theoretical peptide sequences will also help users to assess if reported protein expression evidence for a protein of interest was based on spectrum assignments to unambiguous peptide sequences or whether expression evidence was solely based on a peptide sequence that could also belong to other proteins.
The pep2pro Database
While the different functionalities of the pep2pro website provide useful information in the pep2pro database to the plant community, the pep2pro database system is most powerful for efficient and effective proteome data analysis.
Analysis of High-Throughput Mass Spectrometry Data Using pep2pro
The state of data of a typical mass spectrometry-based proteomics experiment is illustrated in Figure 1. When analyzing and interpreting the results of such an experiment, the data will be queried in different ways depending on the scientific questions of the experiment. For example, in the organ-specific proteome maps of the AtProteome and pep2pro datasets we assessed how many proteins were identified in each plant organ based on the number of peptide spectrum assignments and which proteins were identified only in one organ. We also determined which peptides were identified best in which organ and extract, as sample context was shown to determine peptide detectability (Baerenfaller et al., 2008, 2011). For the characterization of whole genome hits and post-translational modifications we used PepSplice (Roos et al., 2007), for which we determined the optimal search parameters by calculating local false discovery rates based on the number of peptide spectrum assignments against a decoy database (Elias and Gygi, 2007; Baerenfaller et al., 2008). In the Arabidopsis phosphoproteomics datasets, which were obtained following affinity chromatography on IMAC and TiO2, we searched for phosphoproteome differences between end-of-day and end-of-night by integrating the search results from two different search algorithms (Reiland et al., 2009). In wild type and stn8 kinase mutants we identified STN8 kinase substrates (Reiland et al., 2011). To distinguish between changes in phosphorylation state and changes in protein abundance, the proteins from the flow-through fractions were quantified using normalized spectral counting for which all the information was retrieved by querying the pep2pro database. Exact peptide phosphorylation positions were determined for the identified STN8 substrates in each biological replicate. For other proteins their phosphorylation patterns in the stn8 mutant samples revealed that they were independent of STN8 (Reiland et al., 2011). To investigate the extent to which jasmonates control wound-induced re-patterning, we compared the normalized spectral counts in aos mutants to those in wild type leaves after wounding (Gfeller et al., 2011). The identification of proteins that were changed in abundance required statistical tests that are best performed outside of the database in a program such as R (R Development Core Team, 2010) using lists of normalized spectral counts for each biological replicate of each sample that were created as an output of the pep2pro database. The same approach was used to investigate the proteome of Toc159 import receptor mutants, which revealed putative Toc159-dependent and Toc159-independent plastid precursor proteins (Bischof et al., 2011).
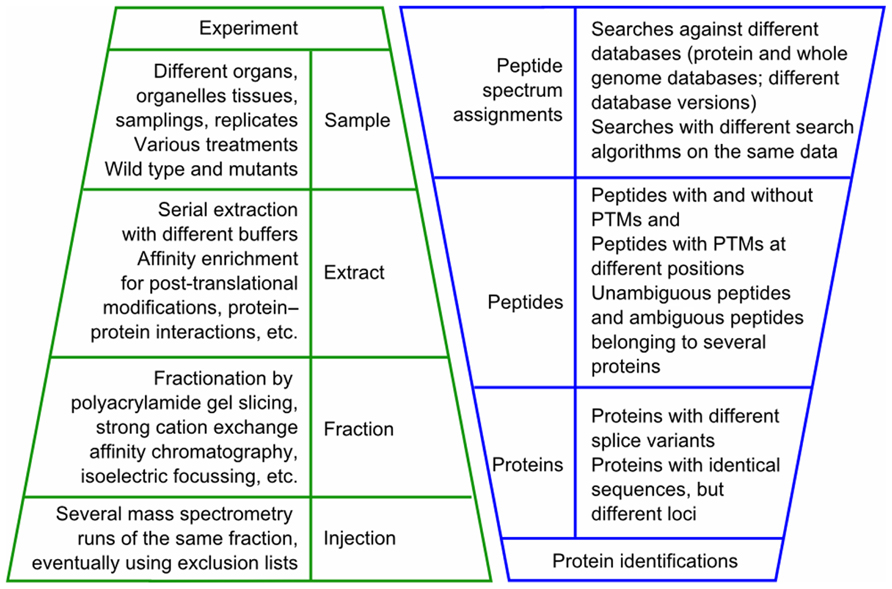
FIGURE 1. State of data in a typical mass spectrometry experiment. In a mass spectrometry-based experiment different samples are analyzed, usually involving different extracts of each sample and different fractions of each extract. When injected several times into the mass spectrometer, this results in a significant multiplication of mass spectrometry runs per experiment. The mass spectrometry data are then subjected to one or several database-dependent peptide assignment algorithms. In the search results one to several spectra are assigned to a peptide sequence and one to several peptides are assigned to a protein sequence. PTM, post-translational modification.
Proteome Data-Specific Challenges Solved in pep2pro
The types of analyses described above required proteome data integration in a relational database. Considering the typical state of proteomics data illustrated in Figure 1, this posed a challenge that we solved in the pep2pro database (Baerenfaller et al., 2011). For example, one challenge was the integration of search results from different search algorithms. Results of the different search algorithms have different formats and quality sores and they can contradict each other. Such contradictions are possible when different search algorithms annotate the same spectrum with different peptide sequences, different post-translational modifications, or with post-translational modifications at different positions. To resolve such contradictions, all contradicting peptide spectrum assignments were flagged during integration of the search results. Inserting a flag for these hits makes them discernible in the database and therefore allows for examining the features of contradicting spectrum assignment and at the same time for exclusion of these hits from the datasets that are subjected to further analyses. The only contradictory peptide spectrum assignments that were not flagged are phosphopeptides with contradicting phosphorylation site assignments. In this case we retained the hit with the attribute that the peptide was phosphorylated but removed the information on the position of the phosphate group.
Search algorithms also use different definitions of a non-tryptic end or a missed cleavage site, which will create inconsistencies in a database if not properly addressed. To solve this problem we pre-defined true tryptic peptides in pep2pro by digesting the TAIR Arabidopsis protein sequence database in silico. The results are stored in a look-up table and the peptide is flagged with the attribute whether or not it is a true tryptic peptide during uploading of the data. Another important issue to solve was the protein inference problem because a same peptide sequence can occur in several proteins (Nesvizhskii and Aebersold, 2005). In the TAIR10 protein database release (Lamesch et al., 2012) 15.2% of all theoretical tryptic peptide sequences and 5.1% of the theoretical tryptic peptides with a minimum length of six amino acids and a molecular mass between 400 and 6000 Da are contained in more than one protein, while 138 protein sequences are assigned to several loci. We therefore developed a peptide ambiguity filter, which only integrates peptide sequences into pep2pro that are unambiguously assigned to one protein only. This does not apply to different splice variants of the same locus that most of the times cannot be distinguished by the identified peptides anyway or to different genes with exactly the same amino acid protein sequence. Another important aspect during construction of pep2pro was the integration of the decoy database hits. For this we first needed to create a look-up table containing those peptide sequences that both occur in the forward and the decoy database (553 peptide sequences in the TAIR10 database). Upon import of the decoy hits into the pep2pro database it was first checked, whether their sequences were contained in the look-up table, and if not, they were flagged so that they can be distinguished from the forward hits. Integration and flagging of decoy database hits allows the assessment of local false discovery rates in the datasets after data upload and filtering and the exclusion of the decoy hits from the final datasets that can then be used for subsequent analyses and biological interpretations (Baerenfaller et al., 2011).
Together, the upload and filtering algorithms implemented and integrated in the pep2pro data analysis pipeline address most issues related to proteome data. The relational database schema in which the proteome data reside allows users to perform flexible and detailed data analysis by querying pep2pro for different information.
pep2pro4all: Making the pep2pro Database System Broadly Available
While the data in pep2pro are public, until now the pep2pro database system was available only to users who provided their mass spectrometry raw files, which we then analyzed using our data analysis pipeline. Alternatively, users could re-build the pep2pro analysis pipeline in their own environment based on the published database schema and the detailed explanations of the different building blocks (Baerenfaller et al., 2011). To make the analysis pipeline more broadly available to users we now built the pep2pro4all system3.
The pep2pro4all Database Schema
The pep2pro4all database schema enables users to locally build a light version of the pep2pro data analysis pipeline and integrate their mass spectrometry search results in mzIdentML format into the pep2pro4all database schema (Figure 2). For this, users can download all necessary files and data from www.pep2pro4all.ethz.ch, install the empty pep2pro4all database schema and use the p2p4a script for populating the database. To deal with unambiguous peptides and forward peptide sequences in the decoy database, information on loci, splice variants, and protein sequences from the TAIR10 Arabidopsis search database is provided and used by the p2p4a script for data processing. This assures that only unambiguous peptides are uploaded for analysis and that only peptide sequences that exclusively occur in the decoy database are flagged as decoy hits. Further, upon population of the pep2pro4all database, the sequence-specific protein sequences and protein and locus information are stored in the corresponding database tables. In the pep2pro4all version provided here the sequence-specific information is based on the TAIR10 release of the Arabidopsis protein search database. However, this can be extended to other search databases, plant species, and organisms upon request.
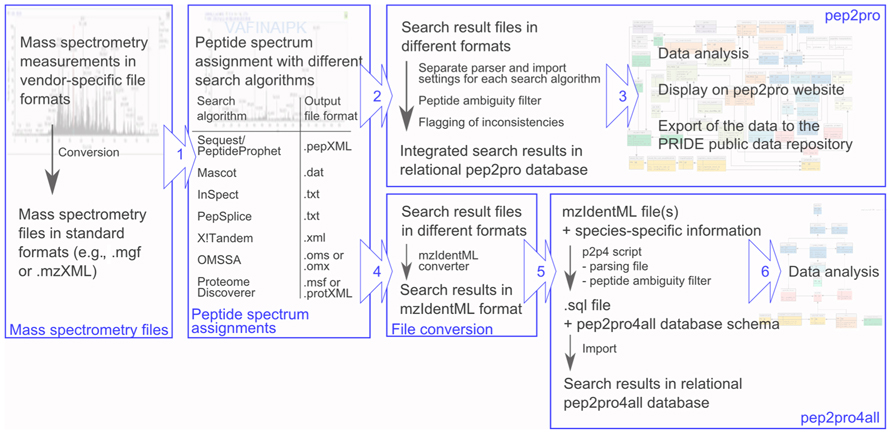
FIGURE 2. Workflow for the integration of mass spectrometry result files into the pep2pro and pep2pro4all relational databases. Mass spectrometry files are first converted to standard formats. (1) They are then searched with different peptide spectrum assignment algorithms, which create search results files in different formats. (2) For integration of the mass spectrometry result files into pep2pro, a separate parser reads out the different formats and applies different import settings. Before integration into the relational database, the data are filtered with a peptide ambiguity filter and inconsistencies in the results are flagged. (3) After data integration, the data are ready for data analysis, are displayed on the pep2pro website and can be exported to the PRIDE database. (4) For import of mass spectrometry result files into pep2pro4all, they are first converted into mzIdentML standard format using a mzIdentML converter. (5) The p2p4a script then parses the data, performs a peptide ambiguity check using the provided sequence-specific information from the search protein database, assembles the dataset, and finally writes the data and all necessary SQL commands into a .sql file. For creating the pep2pro4all database populated with mass spectrometry result data, first the empty pep2pro4all database schema is imported into a local MySQL server, and afterward the .sql output file containing all the data is imported into the database. (6) The mass spectrometry result files now reside in relational format in the pep2pro4all database and can be analyzed by data querying.
The mzidentML Data Standard
The second version (v1.1) of the mzIdentML file format was released recently and represents the stable and recommended exchange format for peptide and protein identification data (Jones et al., 2012). It was developed by the Proteomics Standards Initiative (PSI) in collaboration with instrument and software vendors, and the developers of major open-source proteomics projects. Software implementations have been developed to enable conversion into mzIdentML format from various search algorithm output formats and the major public proteomics data repositories will soon support the format. Additionally, the various methods used by mzIdentML to reference external spectra are supported by a common interface implemented by parsers of the jmzReader library that parse the most commonly used mass spectrometry data formats (Griss et al., 2012). Therefore we decided to develop the p2p4a script based on the mzIdentML standard file format to facilitate the exchange of MS proteomics data irrespective of search engine and analysis software.
From Search Results to an Integrated Dataset in Relational Format
The p2p4a script using the mzIdentML format was developed based on the examples provided with the release of mzIdentML v1.1 (Jones et al., 2012) and on a mzIdentML file converted with the id convert algorithm provided by the openMS platform (Sturm et al., 2008) from a pep.xml Sequest/PeptideProphet (Eng et al., 1994; Keller et al., 2002) search output of Arabidopsis proteome measurements. The p2p4a script parses the mzIdentML file(s), performs several filtering steps, assembles the final dataset, and writes the data and all necessary SQL commands into a file with which users can then populate the previously prepared database. During parsing the p2p4a script checks for ambiguity of the peptide by determining if it was assigned to several proteins with different amino acid sequences but excluding splice variants. The peptide will be considered only when it is unambiguous. After population of the pep2pro4all database with the mass spectrometry search results, the data now reside in relational format, which enables flexible and detailed data analysis by querying the database. To ease querying we have also provided the entity-relationship diagram of the pep2pro4all database schema and a file with canned typical queries on the www.pep2pro4all.ethz.ch website.
Availability of the pep2pro4all System
All the files of the pep2pro4all system are available from our website at www.pep2pro4all.ethz.ch. The p2p4a script is provided as perl program and detailed instructions can be found in the readme.txt file. In addition, we provide the Arabidopsis TAIR10 protein database in FASTA format and example input and output files.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by the AGRON-OMICS integrated project funded in the Sixth European Framework Programme (LSHG-CT-2006-037704).
Footnotes
References
Baerenfaller, K., Grossmann, J., Grobei, M. A., Hull, R., Hirsch-Hoffmann, M., Yalovsky, S., Zimmermann, P., Grossniklaus, U., Gruissem, W., and Baginsky, S. (2008). Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320, 938–941.
Baerenfaller, K., Hirsch-Hoffmann, M., Svozil, J., Hull, R., Russenberger, D., Bischof, S., Lu, Q., Gruissem, W., and Baginsky, S. (2011). pep2pro: a new tool for comprehensive proteome data analysis to reveal information about organ-specific proteomes in Arabidopsis thaliana. Integr. Biol. (Camb) 3, 225–237.
Bischof, S., Baerenfaller, K., Wildhaber, T., Troesch, R., Vidi, P. A., Roschitzki, B., Hirsch-Hoffmann, M., Hennig, L., Kessler, F., Gruissem, W., and Baginsky, S. (2011). Plastid proteome assembly without Toc159: photosynthetic protein import and accumulation of N-acetylated plastid precursor proteins. Plant Cell 23, 3911–3928.
Elias, J. E., and Gygi, S. P. (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214.
Eng, J., Mccormack, A., and Yates, J. (1994). An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989.
Gfeller, A., Baerenfaller, K., Loscos, J., Chételat, A., Baginsky, S., and Farmer, E. E. (2011). Jasmonate controls polypeptide patterning in undamaged tissue in wounded Arabidopsis leaves. Plant Physiol. 156, 1797–1807.
Griss, J., Reisinger, F., Hermjakob, H., and Vizcaino, J. A. (2012). jmzReader: a Java parser library to process and visualize multiple text and XML-based mass spectrometry data formats. Proteomics 12, 795–798.
Grobei, M. A., Qeli, E., Brunner, E., Rehrauer, H., Zhang, R., Roschitzki, B., Basler, K., Ahrens, C. H., and Grossniklaus, U. (2009). Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res. 19, 1786–1800.
Jones, A. R., Eisenacher, M., Mayer, G., Kohlbacher, O., Siepen, J., Hubbard, S., Selley, J., Searle, B., Shofstahl, J., Seymour, S., Julian, R., Binz, P. A., Deutsch, E. W., Hermjakob, H., Reisinger, F., Griss, J., Vizcaino, J. A., Chambers, M., Pizarro, A., and Creasy, D. (2012). The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics. doi: 10.1074/mcp.M111.014381 [Epub ahead of print].
Joshi, H. J., Hirsch-Hoffmann, M., Baerenfaller, K., Gruissem, W., Baginsky, S., Schmidt, R., Schulze, W. X., Sun, Q., van Wijk, K. J., Egelhofer, V., Wienkoop, S., Weckwerth, W., Bruley, C., Rolland, N., Toyoda, T., Nakagami, H., Jones, A. M., Briggs, S. P., Castleden, I., Tanz, S. K., Millar, A. H., and Heazlewood, J. L. (2011). MASCP Gator: an aggregation portal for the visualization of Arabidopsis proteomics data. Plant Physiol. 155, 259–270.
Keller, A., Nesvizhskii, A. I., Kolker, E., and Aebersold, R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392.
Lamesch, P., Berardini, T. Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R., Muller, R., Dreher, K., Alexander, D. L., Garcia-Hernandez, M., Karthikeyan, A. S., Lee, C. H., Nelson, W. D., Ploetz, L., Singh, S., Wensel, A., and Huala, E. (2012). The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210.
Lange, V., Picotti, P., Domon, B., and Aebersold, R. (2008). Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222.
Nesvizhskii, A. I., and Aebersold, R. (2005). Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440.
R Development Core Team. (2010). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Reiland, S., Finazzi, G., Endler, A., Willig, A., Baerenfaller, K., Grossmann, J., Gerrits, B., Rutishauser, D., Gruissem, W., Rochaix, J. D., and Baginsky, S. (2011). Comparative phosphoproteome profiling reveals a function of the STN8 kinase in fine-tuning of cyclic electron flow (CEF). Proc. Natl. Acad. Sci. U.S.A. 108, 12955–12960.
Reiland, S., Messerli, G., Baerenfaller, K., Gerrits, B., Endler, A., Grossmann, J., Gruissem, W., and Baginsky, S. (2009). Large-scale Arabidopsis phosphoproteome profiling reveals novel chloroplast kinase substrates and phosphorylation networks. Plant Physiol. 150, 889–903.
Roos, F. F., Jacob, R., Grossmann, J., Fischer, B., Buhmann, J. M., Gruissem, W., Baginsky, S., and Widmayer, P. (2007). PepSplice: cache-efficient search algorithms for comprehensive identification of tandem mass spectra. Bioinformatics 23, 3016– 3023.
Sturm, M., Bertsch, A., Gröpl, C., Hildebrandt, A., Hussong, R., Lange, E., Pfeifer, N., Schulz-Trieglaff, O., Zerck, A., Reinert, K., and Kohlbacher, O. (2008). OpenMS – an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. doi: 10.1186/1471-2105-9-163
Keywords: database, mzIdentML, pep2pro, plant proteomics, standard format
Citation: Hirsch-Hoffmann M, Gruissem W and Baerenfaller K (2012) pep2pro: the high-throughput proteomics data processing, analysis, and visualization tool. Front. Plant Sci. 3:123. doi: 10.3389/fpls.2012.00123
Received: 30 April 2012; Accepted: 21 May 2012;
Published online: 11 June 2012.
Edited by:
Joshua L. Heazlewood, Lawrence Berkeley National Laboratory, USAReviewed by:
Martin Hajduch, Slovak Academy of Sciences, SlovakiaHolger Eubel, Leibniz Universität Hannover, Germany
Copyright: © 2012 Hirsch-Hoffmann, Gruissem and Baerenfaller. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Katja Baerenfaller, Plant Biotechnology, Department of Biology, ETH Zurich, Universitaetsstrasse 2, 8092 Zurich, Switzerland. e-mail:a2JhZXJlbmZhbGxlckBldGh6LmNo