
 Christiana Staudinger
Christiana Staudinger

- Department of Molecular Systems Biology, University of Vienna, Vienna, Austria
The ProMEX database is one of the main collection of annotated tryptic peptides in plant proteomics. The main objective of the ProMEX database is to provide experimental MS/MS-based information for cell type-specific or sub-cellular proteomes in Arabidopsis thaliana, Medicago truncatula, Chlamydomonas reinhardtii, Lotus japonicus, Lotus corniculatus, Phaseolus vulgaris, Lycopersicon esculentum, Solanum tuberosum, Nicotiana tabacum, Glycine max, Zea mays, Bradyrhizobium japonicum, and Sinorhizobium meliloti. Direct links at the protein level to the most relevant databases are present in ProMEX. Furthermore, the spectral sequence information are linked to their respective pathways and can be viewed in pathway maps.
Introduction
Mass spectrometry (MS)-based proteomics is the most widely used high-throughput technology for identification, quantification, and characterization of the proteins that constitute a proteome (Bensimon et al., 2012). Currently, there is a large amount of sequence information for several plant species that has been generated from MS-based proteomics projects (Cao et al., 2011). As a further result important meta-information has been obtained, such as sub-cellular localization and semi-quantitative protein levels, but data interpretation of complex experiments is still a challenge. Thus sharing empirical data is becoming increasingly important and database development will be an ongoing process.
The ProMEX library (http://ProMEX.pph.univie.ac.at/ProMEX/) launched in 2007 (Hummel et al., 2007), enables spectral identification of unknown samples. Moreover, the database provides access to information of previously identified modifications as well as metadata of the spectra originating experiments. At present, the library contains 30,483 spectra of 12,119 different Proteins and 25,311 different peptides to date. The spectral information of Arabidopsis thaliana is also connected to the MASCP Gator (Joshi et al., 2011).
Mass Spectral Information
Previously identified high quality spectra provide information about sequence coverage as well as missing sequence information. The sequence coverage may give semi-quantitative information about the abundance of a protein or peptide while missing peptide sequences could provide important information about unknown modifications. In addition, the most common post-translational modifications such as oxidation and phosphorylation are indicated, if present. Moreover, the peptide fragment ions indicated by a, b, c, x, y, or z can be visualized to get an impression of the quality of the displayed spectra (Figure 1).
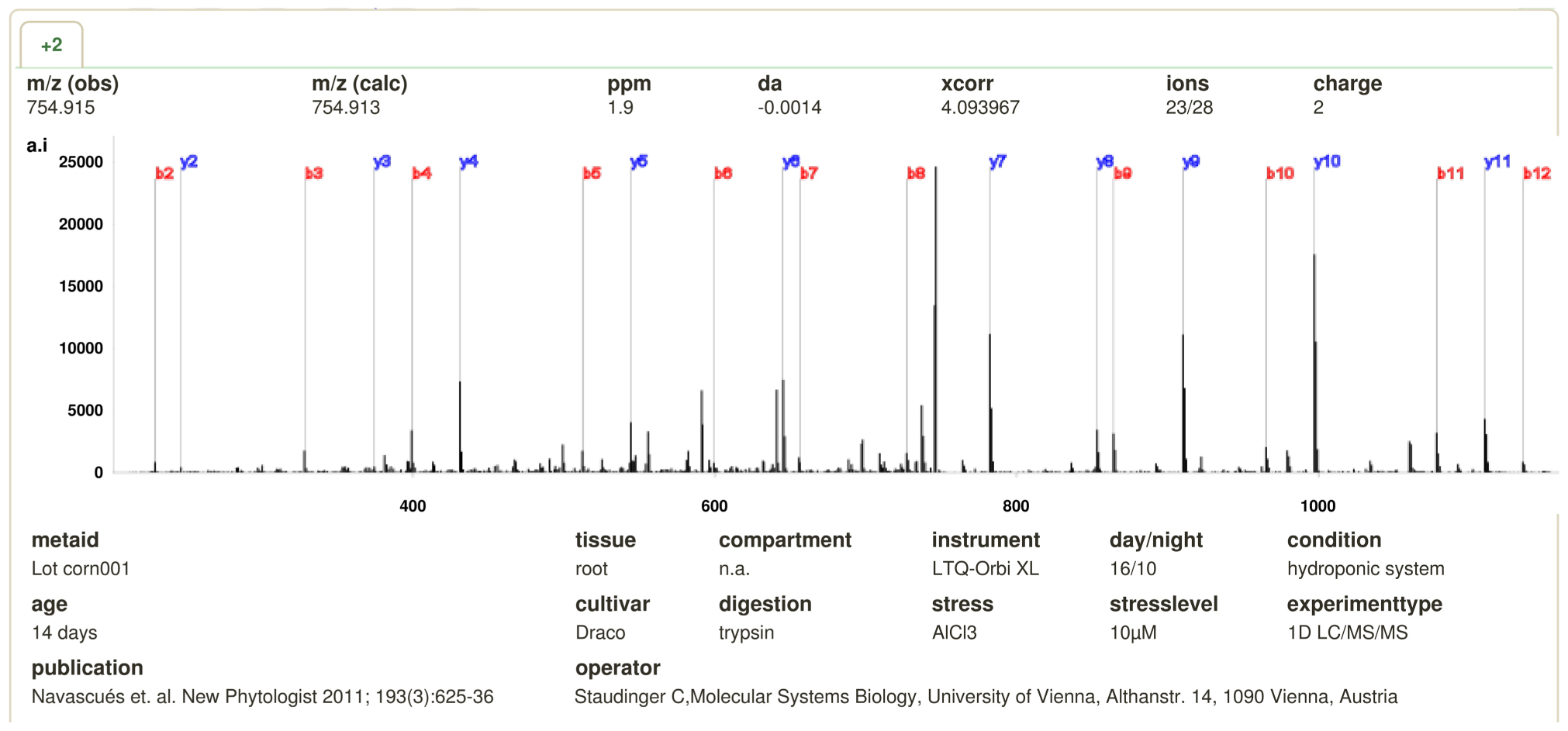
FIGURE 1. MS/MS spectrum of peptide DLVALSGAHTIGQAR with precursor mass at 754.915. The dominant fragments ions (b, y) are labeled. The experimental conditions are shown below the spectrum.
There is also the possibility to perform an advanced text search, which allows for the extraction of information about target proteins such as protein accession numbers, peptide sequences, organism – also experimental data like publications, tissues, compartments, and stress factors can be searched. In addition, multiple terms can be combined together with the Boolean operators to form a more complex query.
Peptide/Protein Identification
ProMEX allows for the identification of proteins/peptides by matching spectra of unknown shotgun analyses against the reference spectra of the library “spectrum-to-spectrum search.” The advantages of spectrum-to-spectrum-searching are on the one hand a more simple visual discrimination of false positive identifications and on the other hand search time reduction due to smaller number of peptide sequence information.
Database Structure
ProMEX was designed in a Linux/Apache environment using Php5, JavaScript, Python, and C++ for dynamic generation of HTML pages, core database generation, and the implementation of the search algorithms. The database structure can be divided into three areas: Spectra Information, Protein Annotation Information, and Experimental Information. Although these sections are linked together, they are independent enough to allow for the update of each without affecting the others.
Experimental Metadata
In ProMEX the spectral information is linked to experimental metadata viz. localizations like species, tissues, or treatment conditions like “drought stress” and MS instrumentation such as Orbitrap, LTQ, and TSQ.
Case Example
ProMEX provides a web-based view on the underlying reference database. The main screen can be divided into four sub windows. If you wish to see data from a specific species you can simply click on the corresponding image link (window bottom right) and all available annotation data for that species will be retrieved. The records are listed in a table on the left side of the window. Selecting a link from the first column opens a window above the current table. The new window contains, among other things, the peptide- and protein sequence information of the selected entry. Clicking on a peptide sequence will open another window, showing both the corresponding spectra and the corresponding experimental data below each spectra (see also Figure 1).
Future Directions
We will keep working on the program interface and usability improvement. The ProMEX database is continuously updated with new in-house generated data. In this context, a next step will be the implementation of an interface for loading external data into ProMEX.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the University of Vienna and the Faculty of Life Sciences for their great support.
References
Bensimon, A., Heck, A. J., and Aebersold, R. (2012). Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem. 81, 18.1–18.27.
Cao, J., Schneeberger, K., Ossowski, S., Günther, T., Bender, S., Fitz, J., Koenig, D., Lanz, C., Stegle, O., Lippert, C., Wang, X., Ott, F., Müller, J., Alonso-Blanco, C., Borgwardt, K., Schmid, K. J., and Weigel, D. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963.
Hummel, J., Niemann, M., Wienkoop, S., Schulze, W., Steinhauser, D., Selbig, J., Walther, D., and Weckwerth, W. (2007). ProMEX: a mass spectral reference database for proteins and protein phosphorylation sites. BMC Bioinformatics 8, 216. doi: 10.1186/1471-2105-8-216
Joshi, H. J., Hirsch-Hoffmann, M., Baerenfaller, K., Gruissem, W., Baginsky, S., Schmidt, R., Schulze, W. X., Sun, Q., van Wijk, K. J., Egelhofer, V., Wienkoop, S., Weckwerth, W., Bruley, C., Rolland, N., Toyoda, T., Nakagami, H., Jones, A. M., Briggs, S. P., Castleden, I., Tanz, S. K., Millar, A. H., and Heazlewood, J. L. (2011). MASCP Gator: an aggregation portal for the visualization of Arabidopsis proteomics data. Plant Physiol. 155, 259–270.
Keywords: proteomics, mass spectrometry, bioinformatics, functional databases, plant biology, system biology
Citation: Wienkoop S, Staudinger C, Hoehenwarter W, Weckwerth W and Egelhofer V (2012) ProMEX – a mass spectral reference database for plant proteomics. Front. Plant Sci. 3:125. doi:10.3389/fpls.2012.00125
Received: 31 March 2012; Accepted: 25 May 2012;
Published online: 06 June 2012.
Edited by:
Joshua L. Heazlewood, Lawrence Berkeley National Laboratory, USAReviewed by:
Chris Petzold, Lawrence Berkeley National Laboratory, USAMartin Hajduch, Slovak Academy of Sciences, Slovakia
Copyright: © 2012 Wienkoop, Staudinger, Hoehenwarter, Weckwerth and Egelhofer. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Volker Egelhofer, Department of Molecular Systems Biology, University of Vienna, Althanstr. 14, Vienna 1090, Austria. e-mail:dm9sa2VyLmVnZWxob2ZlckB1bml2aWUuYWMuYXQ=