

- Max Planck Institute for Molecular Plant Physiology, Potsdam, Germany
As the most studied post-translational modification, protein phosphorylation is analyzed in a growing number of proteomic experiments. These high-throughput approaches generate large datasets, from which specific spectrum-based information can be hard to find. In 2007, the PhosPhAt database was launched to collect and present Arabidopsis phosphorylation sites identified by mass spectrometry from and for the scientific community. At present, PhosPhAt 3.0 consolidates phosphoproteomics data from 19 published proteomic studies. Out of 5460 listed unique phosphoproteins, about 25% have been identified in at least two independent experimental setups. This is especially important when considering issues of false positive and false negative identification rates and data quality (Durek et al., 2010). This valuable data set encompasses over 13205 unique phosphopeptides, with unambiguous mapping to serine (77%), threonine (17%), and tyrosine (6%). Sorting the functional annotations of experimentally found phosphorylated proteins in PhosPhAt using Gene Ontology terms shows an over-representation of proteins in regulatory pathways and signaling processes. A similar distribution is found when the PhosPhAt predictor, trained on experimentally obtained plant phosphorylation sites, is used to predict phosphorylation sites for the Arabidopsis genome. Finally, the possibility to insert a protein sequence into the PhosPhAt predictor allows species independent use of the prediction resource. In practice, PhosPhAt also allows easy exploitation of proteomic data for design of further targeted experiments.
Introduction
Protein post-translational modifications (PTMs) are one of the fastest processes through which plants respond to various stimuli. Thus, they increasingly became the focus of scientific studies. Among the various PTMs, phosphorylation is one of the most studied modifications, due to the large number of affected proteins and its involvement in many cellular processes like signaling, nutrient uptake, and transport. In the mammalian field, the function of particular phosphorylation sites for activation or inactivation of proteins or as docking sites for interaction partners has been particularly well studied (Pawson and Gish, 1992; Chung et al., 1999; Yaffe, 2002; Pawson, 2004). So far, most studies of protein phosphorylation in plant biology have been focused on phosphorylation of specific proteins and protein families (Camoni et al., 2000; Hrabak et al., 2003) and the study of specific signaling pathways (Wang et al., 2005). However, more recently several unbiased large-scale studies of plant protein phosphorylation have been carried out, comparing different physiological states or mutants (Li et al., 2009; Reiland et al., 2009; Nakagami et al., 2010), or by analyzing a time-course after stimulation (Niittyla et al., 2007; Chen et al., 2010; Kline et al., 2010; Engelsberger and Schulze, 2011).
One characteristic of mass spectrometric analyses however is the generation of large datasets, which usually remain difficult to access for the general public. Even if identified peptides are listed, access to spectral data is often very limited. One way of allowing public access is the use of data repositories like Tranche (Smith et al., 2011), but even here much of the data is uploaded in raw and possibly vendor-specific format. This often results in time penalty and high workload when individual verification of only few specific peptides is required. As a consequence, already existing measurements are difficult to re-asses or use for different purposes.
Thus, providing data storage and accessibility to this type of experimental information is of utmost importance. The PhosPhAt database of plant phosphorylation sites including a plant phosphorylation site predictor provides such a resource. It aims to compile predicted and experimental evidence of protein phosphorylation from large-scale proteomic studies with bioinformatics resources. Since its launch in 2007, the database has constantly been updated with new experimental evidence from the growing number of phosphoproteomic experiments (see list at http://phosphat.mpimp-golm.mpg.de/).
Dynamic links to further external resources, such as Aramemnon (Schwacke et al., 2003), eFP browser (Winter et al., 2007), co-expression networks ATTED-II (Obayashi et al., 2009), and subcellular localization (Heazlewood et al., 2008) are implemented in PhosPhAt for each phosphoprotein. Additionally, the phosphorylation prediction function implemented in PhosPhAt allows users to paste any given protein sequence into the prediction query window and obtain prediction of phosphorylation sites. Thus, the support vector machine-based predictor trained on Arabidopsis-specific phosphorylation sites can be used independently of the plant species (Durek et al., 2010). Recently, the PhosPhAt database itself has been integrated into a larger common interface, the GATOR portal (Joshi et al., 2011), which allows concurrent query of various proteomic resources.
In its current form, the PhosPhAt database contains evidence for 12404 different phosphorylation sites mapping to 5460 different proteins and 94284 high confidence predicted sites mapping to 21764 proteins. We have found a significant overrepresentation of proteins involved in regulatory and signaling processes among the highly confident phosphorylated proteins, while housekeeping and other enzymatic functions are underrepresented (Heazlewood et al., 2008; Figure 1).
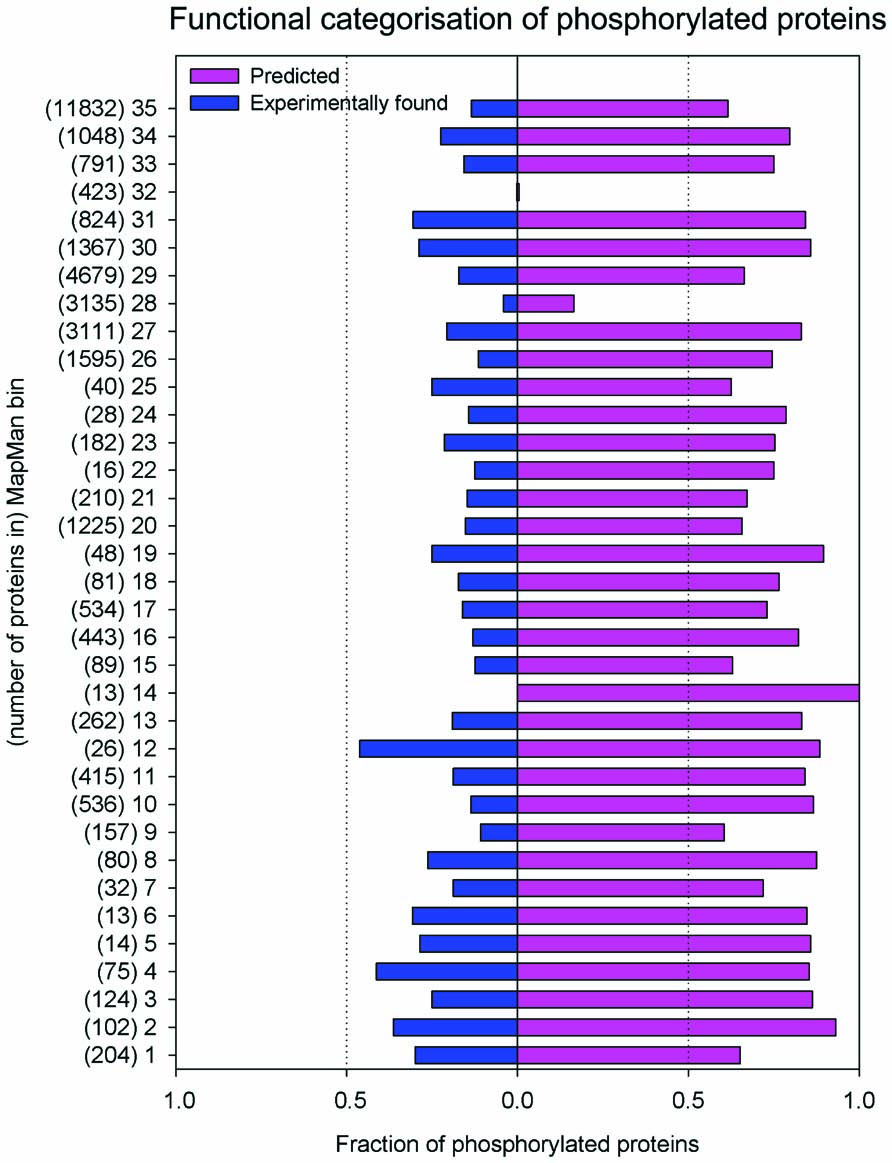
FIGURE 1.Distribution of experimental and predicted phosphorylation sites to functional categories of MapMan. The bins which group encoded proteins in their functional categories are: photosynthesis (PS)-1; major carbohydrate (CHO) metabolism-2; minor CHO metabolism-3; glycolysis-4; fermentation-5; gluconeogenese/glyoxylate cycle-6; OPP-7; TCA/org. transformation- 8; mitochondrial electron transport/ATP synthesis -9; cell wall-10; lipid metabolism-11; N-metabolism-12; amino acid metabolism-13; S-assimilation-14; metal handling-15; secondary metabolism-16; hormone metabolism-17; co-factor and vitamin metabolism-18: tetrapyrrole synthesis-19; stress-20; redox-21; polyamine metabolism-22; nucleotide metabolism-23; biodegradation of xenobiotics-24; C1-metabolism-25; misc-26; RNA-27; DNA-28; protein-29; signaling-30; cell-31; microRNA, natural antisense etc-32; development-33; transport-34; not assigned-35.
The proteome-wide magnitude of protein phosphorylation becomes apparent when looking at the high confidence prediction of protein phosphorylation: mapping these predicted phosphorylation sites to the number of proteins that are affected by phosphorylation, about 64% of the proteins listed in TAIR9 (January 2010; Lamesch et al., 2012) are predicted to be phosphorylated with high confidence (score >1). However, until now only about one-quarter of these predicted sites have been experimentally confirmed using mass spectrometry (Figure 1). Probably due to the focus of various proteomic studies on particular cellular compartments (i.e., plasma membrane, chloroplasts), larger numbers of experimentally confirmed phosphorylated proteins have been found for particular functional categories (MapMan bins; Thimm et al., 2004). Examples of these include proteins with functions in photosynthesis (bin 1), glycolysis (bin 4), N-metabolism (bin 12), C1 metabolism (bin 25), as well as microRNA and natural antisense-related proteins (bin 32). In other functional categories, the fraction of proteins with only predicted phosphorylation is very high, while often only one-third of the phosphorylated proteins has been identified experimentally (Figure 1). These include signaling functions (bin 30), cytoskeleton and vesicle trafficking (bin 31), as well as major carbohydrate metabolism (bin 2). This points not only to the versatility of processes in which phosphorylation plays a role, but also from experimental point of view indicates remaining work to confirm predicted phosphorylations.
The most challenging part, however, lies in the precise molecular characterization of the identified phosphorylation sites with regards to their effect on protein function. In this regards, current knowledge is still very limited. To this end, it is extremely valuable to study these experimentally determined phosphorylation sites and their role in specific physiological conditions, tissue types, or in the whole organism context.
Thus, besides global analysis of protein phosphorylation and discovery of new phosphorylation sites, precise targeted studies of particular proteins of interest are necessary to finely elucidate the role of phosphorylation sites in particular proteins. This becomes especially important as protein phosphorylation functionally interacts with other protein modifications such as methionine oxidation (Hardin et al., 2009), lysine-acetylation (van Noort et al., 2012), and ubiquitination (Hunter, 2007; Thomas et al., 2009).
Therefore, in this mini review, we aim at providing a detailed overview of the PhosPhAt features through a specific example for further utilization of the PhosPhAt resources in new experimental design of targeted phosphoprotein analysis.
Functions of PhosPhAt Resource
The PhosPhAt web resource allows the user to search for experimental and predicted phosphorylation sites in a given protein (see phosphat.mpimp-golm.mpg.de). Queries can be run based on Arabidopsis gene identifiers (AGI coded) or based on peptide sequences or protein annotation text queries. The advanced search possibilities allow users to include meta-information from experimental context (tissue type, experimental treatment, etc.). Both, for queries of experimental sites as well as for queries of phosphorylation site predictions, multiple AGI codes can be submitted (see phosphat.mpimp-golm.mpg.de). Query results will then be displayed on a multipage result window, sorted by gene identifiers.
Upon selecting one of the protein identifiers, followed by a peptide, the protein prediction tab becomes activated and upon clicking displays a detailed protein view tab. The top right corner of this protein tab contains links to various other resources: SUBA, TAIR, ATTED, Aramemnon, and GabiPD. Below the protein ID, its functional description and the MapMan bin classification, the middle part of the protein tab is allocated to the phosphorylation site predictor. Here the amino acids from experimentally identified peptides are underlined, and predicted phosphorylated amino acids are marked with a green background. Amino acids that were experimentally confirmed to be phosphorylated are shown in bold, and hovering with the mouse over one of those will display the details for this identification or prediction just below the protein sequence. Positive score values indicate positive prediction, while increasing value indicates increasing probability of phosphorylation. Predicted Pfam domain structures are mapped onto the protein sequence and displayed in a yellow background, allowing the user to put the experimental and predicted phosphorylation sites in functional context (Durek et al., 2010). Below the sequence display, a list of experimentally identified phosphopeptides is available with icons signifying MS spectrum availability and quantitative information.
In the list of experimental data, phosphorylation sites are marked as defined if the precise location of the phosphorylated amino acid has been unambiguously determined by mass spectrometric analysis. Clear identification of the phosphorylated amino acid in the phosphopeptides often requires manual interpretation of mass spectra and use of additional fragment ion scoring algorithms (Olsen et al., 2006; MacLean et al., 2008). These defined sites in PhosPhAt are marked with brackets and a lowercase p, such as (pS), (pT), (pY). Phosphorylation sites marked as ambiguous were not clearly resolved by the mass spectrometric experiments. These sites are marked as lowercase letters in brackets, e.g., (s), (t), (y). The undefined sites are usually putatively phosphorylated amino acids in close proximity. In PhosPhAt, the remark “site undetermined” on the modified tryptic peptide is used to mark those situations where no statement could be made on the location of the phosphorylation site based on the mass spectrum (Heazlewood et al., 2008).
Upon double-clicking on a peptide-row in the list of experimentally validated phosphopeptides, the fragment spectrum of this ion, experimental origin, and available quantitative information are displayed. The annotated ions in the fragment spectrum are indicated by blue bars, and clicking one of them displays the fragment-specific information. The mass list of each particular peptide ion can be exported as peak list (.csv format). Also at the level of primary query result, custom information can be exported as tab delimited tables, Mascot compatible .mgf format, or in Motif-X format (Schwartz and Gygi, 2005). In all view pages, information displayed can be custom-adjusted by clicking on the column title and selecting desired information for display. A complete tab-delimited table of all database contents can be downloaded from the PhosPhAt main page (phosphat.mpimp-golm.mpg.de).
Using PhosPhAt Resource for Design of Targeted Experiments
The experimental and predicted phosphorylation sites available in PhosPhAt and particularly the spectra deposited in the database provide a valuable resource for targeted in-depth analysis of the role of protein phosphorylation in physiological contexts.
The use of fragment spectrum libraries for the design of targeted analyses has previously been described in detail (Gillet et al., 2012). Examples for targeted analysis of metabolic pathways are already available from yeast, providing a detailed dynamic proteome profile of the glycolytic pathway or microbial proteomes (Carroll et al., 2011; Schmidt et al., 2011). However, the combination of targeted protein analysis with monitoring of phosphorylation stoichiometry has not yet been widely applied in plant science. The commonly used methods for targeted protein quantification can also be well applied to phosphopeptides (Johnson et al., 2009) and synthetic standard (phospho)-peptides can be used to determine phosphorylation stoichiometry (Steen et al., 2005). Both approaches require reliable information of phosphopeptide identity and fragmentation properties.
The starting point for a targeted phosphorylation site analysis is a limited set of proteins of interest. The query will return experimentally identified phosphorylation sites from the desired proteins, and ideally an experimentally acquired fragment spectrum is hosted in the PhosPhAt database. By clicking on the peptide-rows, the individual spectra can be assessed, and a combined export of the peak list is available from the first query result page. The information from PhosPhAt may be complemented by additional literature information about the biological relevance of particular phosphorylation sites.
In an example, we are interested in studying phosphorylation stoichiometry of proteins involved in nitrogen uptake and assimilation. A query of ammonium and nitrate transporters as well as nitrate reductase reveals spectral information for nitrate reductase (AT1G37130.1), a nitrate transporter NRT2.1 (AT1G08090), and an ammonium transporter AMT 1.1 (AT4G13510.1) among others. We have selected peptides found in phosphorylated and non-phosphorylated form for each one of these proteins. They are: SV(pS)TPFMNTTAK/SVSTPFMNTTAK for nitrate reductase; EQSFAFSVQ(pS)PIVHTDK/EQSFAFSVQSPIVHTDK for nitrate transporter NRT2.1, and ISSEDEMAGMDM(pT)R/ISSEDEMAGMDMTR for ammonium transporter AMT 1.1. Independent studies show that phosphorylation of S534 in nitrate reductase (Kaiser and Huber, 2001) is involved in the activity regulation of nitrate reductase NIA2 and especially in its interaction with 14-3-3 proteins (Kaiser and Huber, 2001; Lillo et al., 2004). Conveniently this region is covered by the experimentally identified phosphopeptide. To our knowledge, there is no precise information about the function of the phosphorylation site of the NRT2.1 transporter, although the level of phosphorylation of this peptide has been found to change upon nitrate re-supply to starved seedlings (Engelsberger and Schulze, 2011). For the ammonium transporter peptide it has been shown that it is subject to inactivation by C-terminal phosphorylation at the threonine residue in the experimentally confirmed phosphopeptide (Yuan et al., 2007). Thus, on one hand among these three proteins we have clear examples of experimentally verified phosphopeptides, where phosphorylation has been shown to influence protein activity and can be used for diagnostics purposes in various mutants. On the other hand there are novel phosphopeptides, like the NRT2.1 peptide, where we know that the level of phosphorylation changes but we are not sure yet how this influences the protein itself. Following the choice of target peptides, a selected reaction monitoring method is designed as described (Lange et al., 2008) and applied to mutant or wildtype plants subjected to various treatments. When selecting the transitions to be monitored for each peptide, we could use the annotated fragment spectra available from PhosPhAt, and select a number of reliable ions that can be reproducibly monitored, as in the example of NIA2 shown in Figure 2. PhosPhAt therefore also serves as a phosphopeptide library resource.
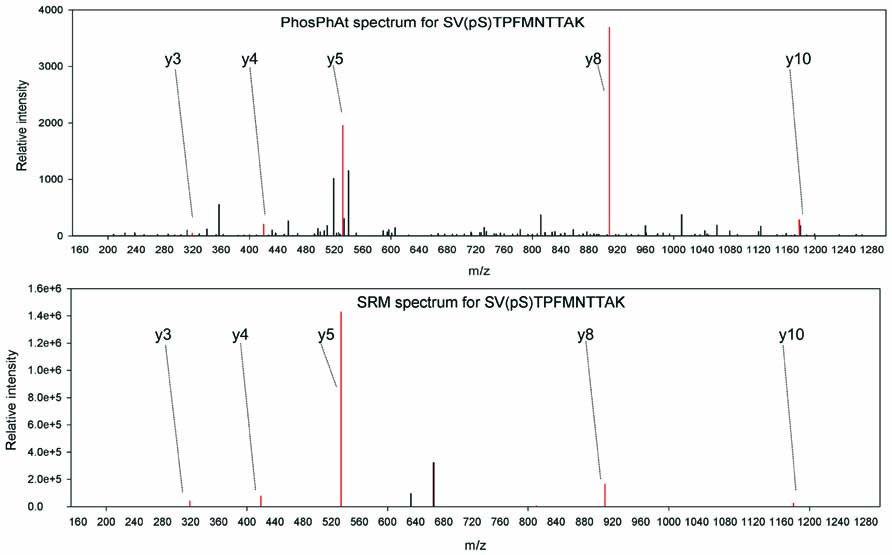
FIGURE 2.Example spectra from PhosPhAt and selected transitions for quantification in targeted SRM analyses for NIA2.
Summary
The PhosPhAt database was initiated to provide a resource that consolidates our current knowledge of mass spectrometry-based identified phosphorylation sites in the model plant Arabidopsis. It is combined with a phosphorylation site prediction tool specifically trained on plant type phosphorylation motifs. Thus, PhosPhAt not only serves as a searchable knowledge base for experimentally identified phosphorylation sites, but also provides a powerful resource for the characterization and annotation of yet unidentified phosphorylation sites in plant proteins. Furthermore, the stored spectra for large numbers of phosphorylation sites provide a direct resource for the design of additional targeted experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Camoni, L., Iori, V., Marra, M., and Aducci, P. (2000). Phosphorylation-dependent interaction between plant plasma membrane H(+) ATPase and 14-3-3 proteins. J. Biol. Chem. 275, 99919–99923.
Carroll, K. M., Simpson, D. M., Eyers, C. E., Knight, C. G., Brownridge, P., Dunn, W. B., Winder, C. L., Lanthaler, K., Pir, P., Malys, N., Kell, D. B., Oliver, S. G., Gaskell, S. J., and Beynon, R. J. (2011). Absolute quantification of the glycolytic pathway in yeast: deployment of a complete QconCAT approach. Mol. Cell. Proteomics 10, M111.007633.
Chen, Y., Hoehenwarter, W., and Weckwerth, W. (2010). Comparative analysis of phytohormone-responsive phosphoproteins in Arabidopsis thaliana using TiO2-phosphopeptide enrichment and mass accuracy precursor alignment. Plant J. 63, 1–17.
Chung, H. J., Sehnke, P. C., and Ferl, R. J. (1999). The 14-3-3 proteins: cellular regulators of plant metabolism. Trends Plant Sci. 4, 367–371.
Durek, P., Schmidt, R., Heazlewood, J. L., Jones, A., MacLean, D., Nagel, A., Kersten, B., and Schulze, W. X. (2010). PhosPhAt: the Arabidopsis thaliana phosphorylation site database. An update. Nucleic Acids Res. 38, D828–D834.
Engelsberger, W. R., and Schulze, W. X. (2011). Nitrate and ammonium lead to distinct global dynamic phosphorylation patterns when resupplied to nitrogen-starved Arabidopsis seedlings. Plant J. 69, 978–995.
Gillet, L. C., Navarro, P., Tate, S., Roest, H., Selevsek, N., Reiter, L., Bonner, R., and Aebersold, R. (2012). Targeted data extraction of the MS/MS spectra generated by data independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics. doi: 10.1074/mcp.O111.016717 [Epub ahead of print].
Hardin, S. C., Larue, C. T., Oh, M. H., Jain, V., and Huber, S. C. (2009). Coupling oxidative signals to protein phosphorylation via methionine oxidation in Arabidopsis. Biochem. J. 422, 305–312.
Heazlewood, J. L., Durek, P., Hummel, J., Selbig, J., Weckwerth, W., Walther, D., and Schulze, W. X. (2008). PhosPhAt: a database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 36, D1015–D1021.
Hrabak, E. M., Chan, C. W., Gribskov, M., Harper, J. F., Choi, J. H., Halford, N., Kudla, J., Luan, S., Nimmo, H. G., Sussman, M. R., Thomas, M., Walker-Simmons, K., Zhu, J. K., and Harmon, A. C. (2003). The Arabidopsis CDPK-SnRK superfamily of protein kinases. Plant Physiol. 132, 666–680.
Hunter, T. (2007). The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738.
Johnson, H., Eyers, C. E., Eyers, P. A., Beynon, R. J., and Gaskell, S. J. (2009). Rigorous determination of the stoichiometry of protein phosphorylation using mass spectrometry. J. Am. Soc. Mass Spectrom. 20, 2211–2220.
Joshi, H. J., Hirsch-Hoffmann, M., Baerenfaller, K., Gruissem, W., Baginsky, S., Schmidt, R., Schulze, W. X., Sun, Q., van Wijk, K. J., Egelhofer, V., Wienkoop, S., Weckwerth, W., Bruley, C., Rolland, N., Toyoda, T., Nakagami, H., Jones, A. M., Briggs, S. P., Castleden, I., Tanz, S. K., Millar, A. H., and Heazlewood, J. L. (2011). MASCP Gator: an aggregation portal for the visualization of Arabidopsis proteomics data. Plant Physiol. 155, 259–270.
Kaiser, W. M., and Huber, S. C. (2001). Post-translational regulation of nitrate reductase: mechanism, physiological relevance and environmental triggers. J. Exp. Bot. 52, 1981–1989.
Kline, K. G., Barrett-Wilt, G. A., and Sussman, M. R. (2010). In planta changes in protein phosphorylation induced by the plant hormone abscisic acid. Proc. Natl. Acad. Sci. U.S.A. 107, 15986–15991.
Lamesch, P., Berardini, T. Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R., Muller, R., Dreher, K., Alexander, D. L., Garcia-Hernandez, M., Karthikeyan, A. S., Lee, C. H., Nelson, W. D., Ploetz, L., Singh, S., Wensel, A., and Huala, E. (2012). The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210.
Lange, V., Picotti, P., Domon, B., and Aebersold, R. (2008). Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222.
Li, H., Wong, W. S., Zhu, L., Guo, H. W., Ecker, J., and Li, N. (2009). Phosphoproteomic analysis of ethylene-regulated protein phosphorylation in etiolated seedlings of Arabidopsis mutant ein2 using two-dimensional separations coupled with a hybrid quadrupole time-of-flight mass spectrometer. Proteomics 9, 1646–1661.
Lillo, C., Meyer, C., Lea, U. S., Provan, F., and Oltedal, S. (2004). Mechanism and importance of post-translational regulation of nitrate reductase. J. Exp. Bot. 55, 1275–1282.
MacLean, D., Burrell, M. A., Studholme, D. J., and Jones, A. M. (2008). PhosCalc: a tool for evaluating the sites of peptide phosphorylation from mass spectrometer data. BMC Res. Notes 1, 30. doi: 10.1186/1756-0500-1-30
Nakagami, H., Sugiyama, N., Mochida, K., Daudi, A., Yoshida, Y., Toyoda, T., Tomita, M., Ishihama, Y., and Shirasu, K. (2010). Large-scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants. Plant Physiol. 153, 1161–1174.
Niittyla, T., Fuglsang, A. T., Palmgren, M. G., Frommer, W. B., and Schulze, W. X. (2007). Temporal analysis of sucrose-induced phosphorylation changes in plasma membrane proteins of Arabidopsis. Mol. Cell. Proteomics 6, 1711–1726.
Obayashi, T., Hayashi, S., Saeki, M., Ohta, H., and Kinoshita, K. (2009). ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res. 37, D987–D991.
Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., and Mann, M. (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648.
Pawson, T. (2004). Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203.
Pawson, T., and Gish, G. D. (1992). SH2 and SH3 domains: from structure to function. Cell 71, 359–362.
Reiland, S., Messerli, G., Baerenfaller, K., Gerrits, B., Endler, A., Grossmann, J., Gruissem, W., and Baginsky, S. (2009). Large-scale Arabidopsis phosphoproteome profiling reveals novel chloroplast kinase substrates and phosphorylation networks. Plant Physiol. 150, 889–903.
Schmidt, A., Beck, M., Malmstrom, J., Lam, H., Claassen, M., Campbell, D., and Aebersold, R. (2011). Absolute quantification of microbial proteomes at different states by directed mass spectrometry. Mol. Syst. Biol. 7, 510.
Schwacke, R., Schneider, A., van der Graaff, E., Fischer, K., Catoni, E., Desimone, M., Frommer, W. B., Flügge, U.-I., and Kunze, R. (2003). ARAMEMNON, a novel database for Arabidopsis integral membrane proteins. Plant Physiol. 131, 16–26.
Schwartz, D., and Gygi, S. P. (2005). An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398.
Smith, B. E., Hill, J. A., Gjukich, M. A., and Andrews, P. C. (2011). Tranche distributed repository and ProteomeCommons.org. Methods Mol. Biol. 696, 123–145.
Steen, H., Jebanathirajah, J. A., Springer, M., and Kirschner, M. W. (2005). Stable isotope-free relative and absolute quantitation of protein phosphorylation stoichiometry by MS. Proc. Natl. Acad. Sci. U.S.A. 102, 3948–3953.
Thimm, O., Blasing, O., Gibon, Y., Nagel, A., Meyer, S., Kruger, P., Selbig, J., Muller, L. A., Rhee, S. Y., and Stitt, M. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939.
Thomas, S. N., Cripps, D., and Yang, A. J. (2009). Proteomic analysis of protein phosphorylation and ubiquitination in Alzheimer's disease. Methods Mol. Biol. 566, 109–121.
van Noort, V., Seebacher, J., Bader, S., Mohammed, S., Vonkova, I., Betts, M. J., Kuhner, S., Kumar, R., Maier, T., O'Flaherty, M., Rybin, V., Schmeisky, A., Yus, E., Stulke, J., Serrano, L., Russell, R. B., Heck, A. J., Bork, P., and Gavin, A. C. (2012). Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol. Syst. Biol. 8, 571.
Wang, X., Goshe, M. B., Sonderblom, E. J., Phinney, B. S., Kuchar, J. A., Li, J., Asami, T., Yoshida, S., Huber, S. C., and Clouse, S. D. (2005). Identification and functional analysis of in vivo phosphorylation sites of the Arabidopsis BRASSINOSTEROID-INSENSITIVE1 receptor kinase. Plant Cell 17, 1685–1703.
Winter, D., Vinegar, B., Nahal, H., Ammar, R., Wilson, G. V., and Provart, N. J. (2007). An “electronic fluorescent pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2, e718. doi: 10.1371/journal.pone.0000718
Yaffe, M. B. (2002). Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 3, 177–186.
Keywords: PhosPhAt, protein phosphorylation, database, proteomics, Arabidopsis
Citation: Arsova B and Schulze WX (2012) Current status of the plant phosphorylation site database PhosPhAt and its use as a resource for molecular plant physiology. Front. Plant Sci. 3:132. doi:10.3389/fpls.2012.00132
Received: 28 April 2012; Accepted: 04 June 2012;
Published online: 19 June 2012.
Edited by:
Joshua L. Heazlewood, Lawrence Berkeley National Laboratory, USAReviewed by:
Andrew Carroll, Stanford University, USAJacqueline Monaghan, The Sainsbury Laboratory, UK
Copyright: © 2012 Arsova and Schulze.This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Waltraud X. Schulze, Max Planck Institute for Molecular Plant Physiology, Am Mühlenberg 1, 14476 Potsdam, Germany. e-mail:d3NjaHVsemVAbXBpbXAtZ29sbS5tcGcuZGU=