 Àngel Fernández i Martí1,2*
Àngel Fernández i Martí1,2* Michelle Wirthensohn3
Michelle Wirthensohn3 José M. Alonso1 Rafel Socias i Company1 Maria Hrmova4
José M. Alonso1 Rafel Socias i Company1 Maria Hrmova4- 1 Unidad de Fruticultura, Centro de Investigación y Tecnología Agroalimentaria, Zaragoza, Spain
- 2 Laboratory of Plant Breeding and Molecular Biology, Parque Científico Tecnológico Campus de Aula Dei, Zaragoza, Spain
- 3 School of Agriculture, Food and Wine, Waite Research Institute, University of Adelaide, Glen Osmond, SA, Australia
- 4 Australian Centre for Plant Functional Genomics, University of Adelaide, Glen Osmond, SA, Australia
Gametophytic self-incompatibility (GSI) is a mechanism in flowering plants, to prevent inbreeding and promote outcrossing. GSI is under the control of a specific locus, known as the S-locus, which contains at least two genes, the RNase and the SFB. Active S-RNases in the style are essential for rejection of haploid pollen, when the pollen S-allele matches one of two S-alleles of the diploid pistil. However, the nature of their mutual interactions at genetic and biochemical levels remain unclear. Thus, detailed understanding of the protein structure involved in GSI may help in discovering how the proteins involved in GSI may function and how they fulfill their biological roles. To this end, 3D models of the SC (Sf) and two SI (S8 and S23) S-RNases of almond were constructed, using comparative modeling tools. The modeled structures consisted of mixed α and β folds, with six helices and six β-strands. However, the self-compatible (Sf) RNase contained an additional extended loop between the conserved domains RC4 and C5, which may be involved in the manifestation of self-compatibility in almond.
Introduction
Most almond (Prunus amygdalus Batsch) cultivars are self-incompatible (SI; Socias i Company, 1990). SI in the Prunus species shows the gametophytic self-incompatibility (GSI) system, controlled by a single polymorphic locus containing at least two linked genes, one specifically expressed in the pistil and the other in the pollen (Kao and Tsukamoto, 2004). Pollen tube growth is arrested in the style whenever the single S allele expressed in the haploid pollen matches one of the two S haplotypes expressed in the diploid pistil tissue. The pistil component of SI in Rosaceae, Solanaceae, and Plantaginaceae has been determined to be an S-RNase (McClure et al., 1989). The Prunus S-RNase is of the T2-type (Igic and Kohn, 2001), with five conserved domains (C1, C2, C3, RC4, and C5) and one hyper-variable region (Sassa et al., 1997). The candidate gene for the pollen component in almond has been identified to be an SFB by Ushijima et al. (2003), showing a tight association with the S-RNase gene (Ikeda et al., 2005).
In spite of the knowledge on the genetic structure of the female and male determinants in SI, the nature of their interactions remains unclear. The S-RNases are proteins and, as such, are built from sequences of amino acid residues, encoded by the corresponding gene. The linked amino acid residues bond in space to form a 3D structure. The knowledge of the 3D structure has been useful in order to understand how some proteins work and which molecular mechanisms underpin their function. Thus, the 3D structure of the S-RNase proteins involved in SI may shed light on elucidating the recognition mechanism of GSI in the Rosaceous species at the molecular levels to understand how these proteins mediate the GSI function to fulfill their biological roles.
Protein structure can be determined experimentally using X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryoelectron microscopy, but these approaches are time-consuming (Ida et al., 2001). Consequently, predictive computer molecular modeling has been considered as a useful alternative. Molecular modeling may be defined as the science and/or art that defines molecular structure and function and that yields a 3D model through computation. Protein structures are guided by two sets of principles operating on vastly different time scales. The first set of principles is defined by the laws of physics, while the second set is directed by the theory of evolution. Each of these two sets of principles has led to the development of predictive methods to build 3D protein models (Hrmova and Fincher, 2009).
Currently, one of the most popular comparative modeling programs is MODELLER (Sali and Blundell, 1993). It is a computer program that models 3D structures of proteins and their assemblies by satisfaction of spatial restraints. The user provides an alignment of a sequence to be modeled with related 3D structures already known and MODELLER will calculate a new 3D model of a target protein. The array of 10–50 models typically produced by MODELLER can be evaluated to assess the stereo-chemical quality and the energy profiles of protein models. Thus, after selecting the best model, the structure needs to be put in perspective with a biological function and tested to see if the model is helpful in proposing a useful hypothesis in biology.
Consequently, our objective was to identify the 3D structures of the almond S-RNases and SFBs through molecular modeling tools and to investigate a link between their 3D structures and the SI mechanism.
Materials and Methods
Three different S-RNases from two almond cultivars were modeled because their sequences and physiological activity were available (Fernández i Martí et al., 2009). The S-RNase sequences have been deposited in the EMBL/DDBJ/GenBank under AB467371 (Sf-RNase from “Blanquerna”), AB481108 (S8-RNase from “Blanquerna”), and AB488496 (S23-RNase from “Vivot”).
The modeling procedure started with the alignment of the sequence to be modeled (target) with related known 3D structures (template) derived from the Protein Data Bank (PDB) using FASTA and BLAST (EMBL nucleotide database). In this procedure, the template to be selected among all possibilities must show the highest identity with the target, at least higher than 35%. The coordinates of this template protein were used as a template for further modeling.
Once the best candidate template was selected, the sequence adjustment between the S-RNase sequences and the template was performed manually to minimize the number of gaps and insertions/deletions. The frame of the 3D model was constructed by MODELLER 9v5. A total of 40 models were constructed for each S-RNase. The four models with the lowest value of the Modeller objective function were chosen for further refinement. Energy function was evaluated through PROSAIIv3 (Sippl, 1993). This program detects errors in protein structures and thus serves to indicate their quality.
On the other hand, stereo-chemical quality and the overall G-factors of the protein models were calculated using PROCHECK (Laskowski et al., 1993). This software compares the residue-by-residue geometry of a set of closely related structures. The models with lower number of amino acid residues in disallowed regions were selected as the most suitable models. A Ramachandran plot (also known as a Ramachandran map or a Ramachandran diagram) outputted by PROCHECK visualizes dihedral angles Ψ against φ of amino acid residues in the protein structure, thus showing the possible conformation of Ψ and φ angles for a polypeptide (Ramachandran et al., 1963).
During a further modeling, the loop refinement protocol was used to generate 40 new models from the previously best model. The same steps as described above were followed, selecting the best four models according to their lowest values of the Modeller objective function, and then selecting the “best of the best” from the results obtained by PROSAIIv3 and PROCHECK. Finally, the molecular graphics were generated with PYMOL, which visualizes protein structures (http://www.pymol.org).
Results and Discussion
The 3D models of the Sf-, S23-, and S8-RNases were compared with the related known 3D structures derived from PDB. The best candidate template selected was the RNase MC1 mutant with accession 1J1G (Numata et al., 2003), because the identity between this template and the target sequences was 42%. On the other hand, the SFBf, SFB8, and SFB23 models could not be generated because sequence identity higher than 30% was not found in PDB.
Protein structures represent combinations of secondary structural elements, α-helices and β-strands that are inter-connected by loops. These structural elements form the core regions (the inside of the molecule) and are connected by loop regions on the protein surface with surface-exposed α-helices and β-strands The structure of the S-RNases belonged to the α and β class, with six α-helices and six β-strands connected by loops. The folding topologies of its main chains were very similar to the topologies of the RNase T2 family enzymes. Their overall dimensions were approximately 40Å × 50 Å × 30 Å.
Ramachandran plot statistics for the S-RNases showed than 97% amino acid residues were positioned in the “allowed” regions. In fact, when structures place 95–97% or more of the amino acid residues in the “allowed” positions, they are considered to be reliable in modeling experiments, and this indicates how well the structures fits with the expected main chain length and torsion angle distributions (Laskowski et al., 1993; Kleywegt and Jones, 1996). The best four models of the Sf-, S23-, and S8-RNases were selected for further modeling. As shown in Figure 1, in the model sfi1_BL00040001 (Figure 1A), all residues were positioned in the allowed region (red arrow), whereas in the model sfi1_BL00010001 (Figure 1B), 1.6% of the residues were in a disallowed region (green arrow). Thus, the model BL00040001 was selected as the best model to be analyzed. Higher numbers of residues in the disallowed region reflect a distorted geometry in the models, because there are higher proportions of residues falling outside the limits of main chain bond length and torsion angles derived from the small molecule library (Engh and Huber, 1991). These results indicate that our models were optimal.
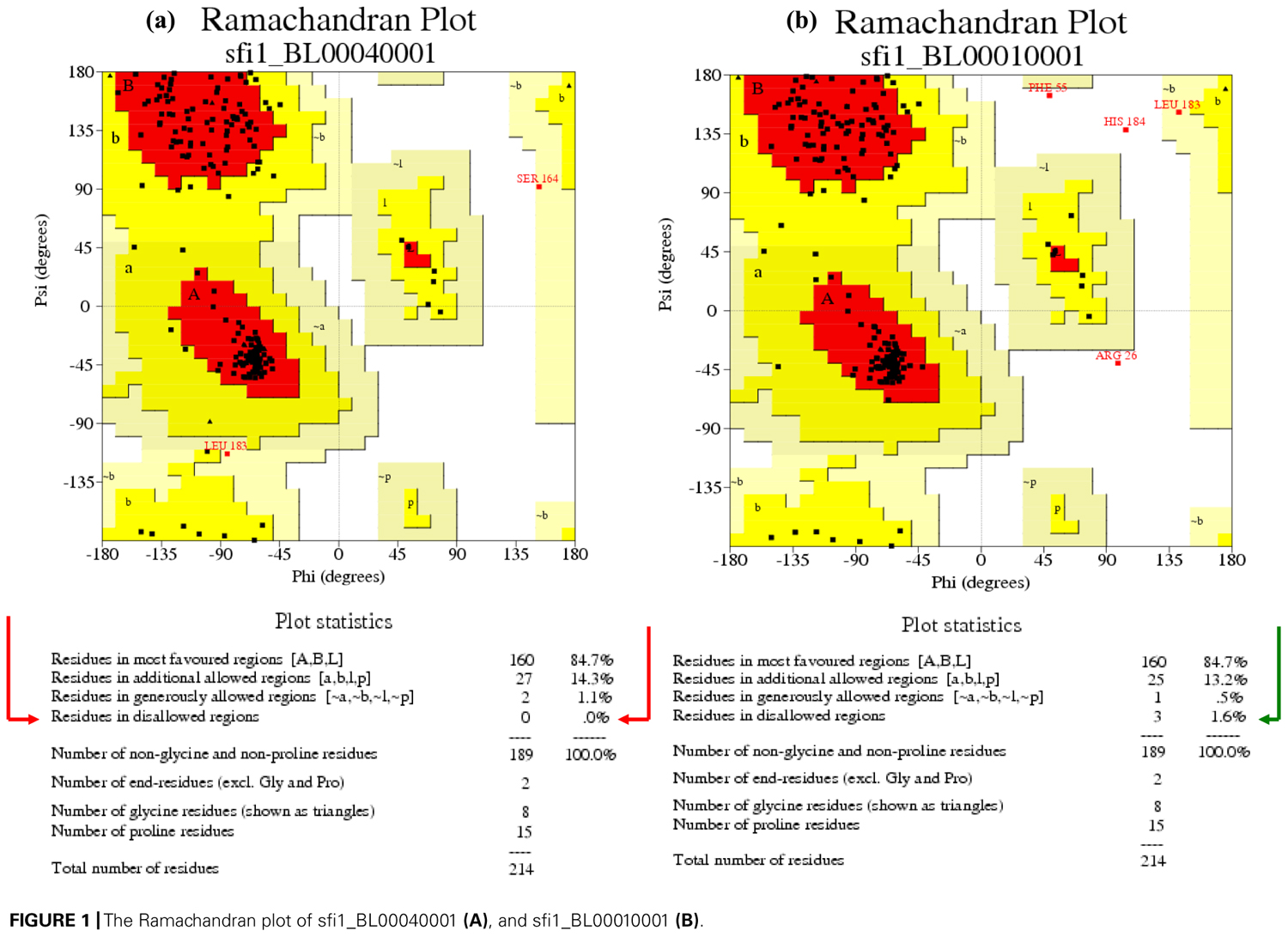
FIGURE 1. The Ramachandran plot of sfi1_BL00040001 (A), and sfi1_BL00010001 (B).
When the three S-RNases were superpositioned, the Sf-RNase structure contained an additional extended loop, which was not present in the S8 or S23 models. This loop, shown in Figures 2 and 3 contained the amino acid residues CKG NPQ RQA KSQ PKN RGK SQP KSQ ATT QFL, which were placed between the conserved domains RC4 and C5. Through the software PYMOL, it has been possible to visualize which amino acid residues comprised α-helices, β-strands, and loops. It has been suggested that loops in 3D structures serve to interconnect α-helices and β-strands, and also that longer surface-exposed loops could be susceptible to proteolytic degradation (Branden and Tooze, 1998). As the main structural difference found between the Sf-, S8-, and S23-RNases resides in the presence of this “extended looping region,” this long loop could be prone to degradation and, as a consequence, this S-RNase could be less stable. As a result of this possible degradation, the pollen tube could grow through its own pistil giving rise to SC.
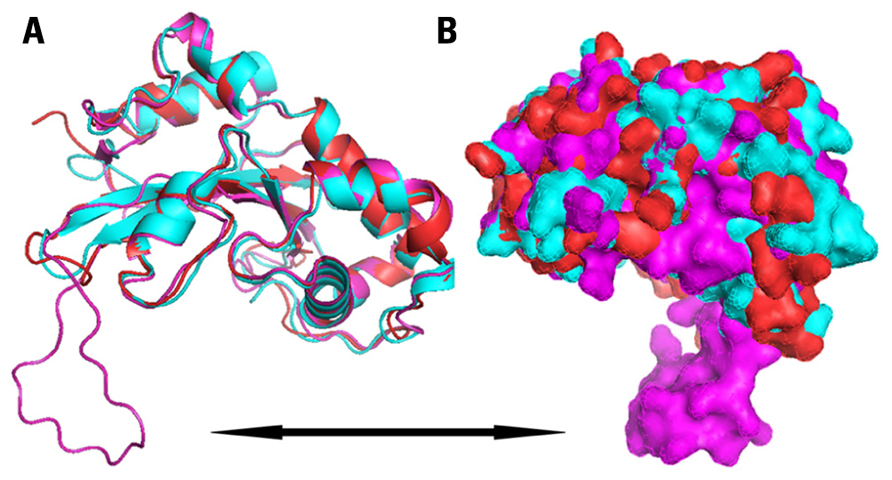
FIGURE 2. Ribbon diagram (A) and surface representation (B) of modeled structure of almond Sf-RNase (magenta), S8-RNase (cyan), and S23-RNase (red), showing secondary structural elements and surfaces. Black arrow indicates the “long loop” found in the Sf model.
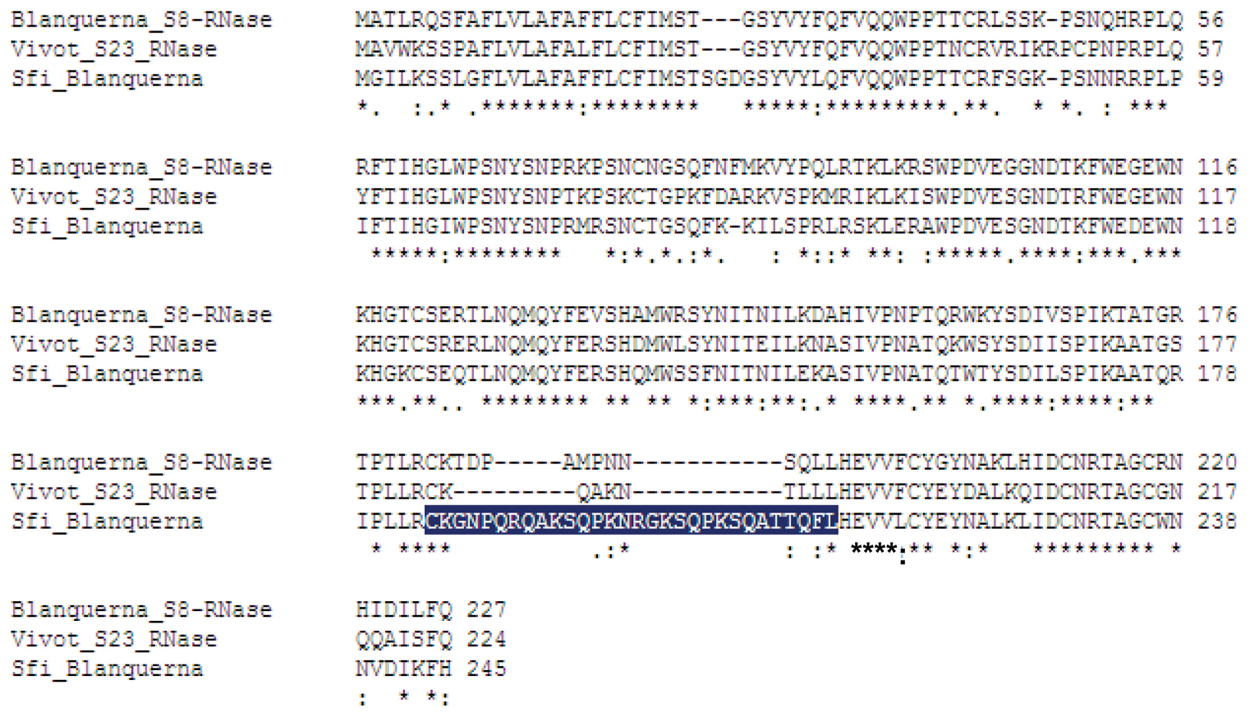
FIGURE 3. Multiple sequence alignment of the Sf,S8, and S23 sequences of RNases, indicating the amino acid residues that belong to the “extended loop” in the Sf 3D structure (marked in blue).
Additionally, the 3D models of the Sf-, S8-, and S23-RNases were compared with that of another Rosaceous species, the Pyrus pyrifolia S3-RNase (Matsuura et al., 2001). The structure of the pear S3-RNase was consistent with the models of the almond S8 and S23 RNase. The fact that both the pear and almond S-RNases confer SI, and that their models did not contain this extended loop, the main structural differences between the SI and the SC RNases could reside in the presence of the loop. Therefore, the amino acid residues that form the extended loop positioned at the surface of the Sf-RNase (Figure 2), between the conserved domains RC4 and C5, could be responsible for the differences in function of RNases. Further studies are required to ascertain how this loop mediates SI and to reveal if it indeed is involved in the SI mechanism in plants.
Conclusion
The molecular nature of the S-locus has been widely studied in many species. In spite of the knowledge on the genetic structure of the female (RNase) and male (SFB) determinants of SI, the nature of their interaction remains unclear. The presenceof a loop in the predicted structure of Sf is a remarkable difference between this S-RNase allele and the other two analyzed in this work, thus we may suggest that this extended loop may be involved in the manifestation of self-(in)compatibility in almond. However, further details and approaches are strongly needed in order to better understand the role of this loop in the SI complex.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the grant AGL2010-22197-C02-01 (Spain) and the Research Group A12 of Aragón (Spain). Àngel Fernández i Martí acknowledges a scholarship co-funded by the Spanish “Ministerio de Educación y Ciencia” and the European Social Fund (FSE), under projects AGL 2004-06674-C02-01 and BES-2006-12621.
References
Engh, R. A., and Huber, R. (1991). Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A 47, 392–400.
Fernández i Martí, A., Hanada, T., Alonso, J. M., Yamane, H., Tao, R., and Socias i Company, R. (2009). A modifier locus affecting the expression of the S-RNase gene could be the cause of breakdown of self-incompatibility in almond. Sex. Plant Reprod. 22, 179–186.
Hrmova, M., and Fincher, G. B. (2009). “Functional genomics and structural biology in the definition of gene function,” in Plant Genomics, eds D. Somers, P. Langridge, and P. Gustafson (Totowa, NJ: Humana Press), 199–227.
Ida, K., Norioka, S., Yamamoto, M., Kumasaka, T., Yamashita, E., Newbigin, E., Clarke, A. E., Sakiyam, A. F., and Sato, M. (2001). The 1.55 Å resolution structure of Nicotiana alata SF11-RNase associated with gametophytic self-incompatibility. J. Mol. Biol. 314, 103–112.
Igic, B., and Kohn, J. R. (2001). Evolutionary relationships among self-incompatibility RNases. Proc. Natl. Acad. Sci. U.S.A. 98, 13167–13171.
Ikeda, K., Ushijima, K., Yamane, H., Tao, R., Hauck, N. R., Sebolt, A. M., and Iezzoni, A. F. (2005). Linkage and physical distances between the S-haplotype S-RNase and SFB genes in sweet cherry. Sex. Plant Reprod. 17, 289–296.
Kao, T., and Tsukamoto, T. (2004). The molecular and genetic bases of S-RNase based self-incompatibility. Plant Cell 16, S72–S83.
Kleywegt, G. J., and Jones, T. A. (1996). Phi/psi-chology: Ramachandran revisited. Structure 4, 1395–1400.
Laskowski, R. A., McArthur, M. W., Moss, D. S., and Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291.
Matsuura, T., Unno, M., Sakai, H., Tsukihara, T., and Norioka, S. (2001). Purification and crystallization of Japanese pear S-RNase associated with gametophytic self-incompatibility. Acta Crystallogr. D Biol. Crystallogr. 57, 172–173.
McClure, B. A., Haring, V., Ebert, P. R., Anderson, M. A., Simpson, R. J., Sakiyama, F., and Clarke, A. E. (1989). Style self incompatibility gene products of Nicotiana alata are ribonucleases. Nature 342, 955–957.
Numata, T., Suzuki, A., Kakuta, Y., Kimura, K., Yao, M., Tanaka, I., Yoshida, Y., Ueda, T., and Kimura, M. (2003). Crystal structures of the ribonuclease MC1 mutants N71T and N71S in complex with 5′-GMP: structural basis for alterations in substrate specificity. Biochemistry 42, 5270–5278.
Ramachandran, G. N., Ramakrishnan, C., and Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99.
Sali, A., and Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815.
Sassa, H., Hirano, H., Nishio, T., and Koba, T. (1997). Style-specific self-incompatibility mutation caused by deletion of the S-RNase gene in Japanese pear (Pyrus serotina). Plant Cell Physiol. 12, 223–227.
Sippl, M. J. (1993). Recognition of errors in 3-dimensional structures of proteins. Proteins Struct. Funct. Genet. 17, 355–362.
Keywords: almond, self-(in)compatibility, 3D modeling, RNase T2
Citation: Fernández i Martí À, Wirthensohn M, Alonso JM, Socias i Company R and Hrmova M (2012) Molecular modeling of S-RNases involved in almond self-incompatibility. Front. Plant Sci. 3:139. doi: 10.3389/fpls.2012.00139
Received: 23 May 2012; Accepted: 09 June 2012;
Published online: 27 June 2012.
Edited by:
Sun Hee Woo, Chungbuk National University, South KoreaReviewed by:
Nnadozie Oraguzie, Washington State University, USAAbu Hena Mostafa Kamal, Korea Research Institute of Bioscience and Biotechnology, South Korea
Copyright: © 2012 Fernández i Martí, Wirthensohn, Alonso, Socias i Company and Hrmova. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Angel Fernández i Marti, Laboratory of Plant Breeding and Molecular Biology, Parque Científico Tecnológico Aula Dei, Zaragoza, Spain. e-mail:YWZlcm5hbmRlekBwY3RhZC5jb20=