



- 1Institute of Population Genetics, Heinrich-Heine Universität, Düsseldorf, Germany
- 2Molecular and Computational Biology, University of Southern California, Los Angeles, CA, USA
- 3School of Biosciences, University of Exeter, Exeter, UK
- 4School of Life Sciences, University of Warwick, Wellesbourne, UK
Dimorphism at the Resistance to Pseudomonas syringae pv. maculicola 1 (RPM1) locus is well documented in natural populations of Arabidopsis thaliana and has been portrayed as a long-term balanced polymorphism. The haplotype from resistant plants contains the RPM1 gene, which enables these plants to recognize at least two structurally unrelated bacterial effector proteins (AvrB and AvrRpm1) from bacterial crop pathogens. A complete deletion of the RPM1 coding sequence has been interpreted as a single event resulting in susceptibility in these individuals. Consequently, the ability to revert to resistance or for alternative R-gene specificities to evolve at this locus has also been lost in these individuals. Our survey of variation at the RPM1 locus in a large species-wide sample of A. thaliana has revealed four new loss-of-function alleles that contain most of the intervening sequence of the RPM1 open reading frame. Multiple loss-of-function alleles may have originated due to the reported intrinsic cost to plants expressing the RPM1 protein. The frequency and geographic distribution of rpm1 alleles observed in our survey indicate the parallel origin and maintenance of these loss-of-function mutations and reveal a more complex history of natural selection at this locus than previously thought.
Introduction
Plants use two evolutionarily conserved mechanisms to induce defense responses against potential pathogens. The first mechanism is provided by plant receptor-like proteins in the plasma membrane that detect conserved microbe molecules outside the host cell, often referred to as pathogen associated molecular patterns (PAMPs). This PAMP-triggered immunity (PTI) is robust and encompasses recognition of a wide variety of molecules such as bacterial flagellin, heat shock proteins and peptidoglycans, and fungal chitin oligomers (reviewed in Schwessinger and Zipfel, 2008). The second mechanism is triggered by predominately cytoplasmic localized receptor-like proteins that enable detection of numerous microbial effector proteins, which have been released within the host cell and have specific functions which collectively suppress defense and/or re-configure host metabolism to nourish pathogen growth. The suite of pathogen effectors varies considerably among pathogens, with bacterial pathogens capable of delivering tens of effectors, whereas fungi and oomycetes are predicted to deliver hundreds of effectors. The plant membrane bound and cytosolic receptor-like proteins typically share a structurally conserved leucine-rich repeat (LRR) domain.
The bacterial resistance gene Resistance to Pseudomonas syringae pv. maculicola 1 (RPM1), encodes a classic cytoplasmic receptor-like protein that is characterized by an amino terminal coiled-coil domain, a central nucleotide binding (NB) site and a carboxyl terminal LRR domain. RPM1 was one of the first plant resistance genes to be molecularly described from studies of natural variation in Arabidopsis thaliana (Grant et al., 1995). Moreover, RPM1 was the first example of a dual specificity R-protein, recognizing the sequence-unrelated AvrB and AvrRpm1 effector proteins derived from bacterial pathogens of soybean and Brassica species, respectively (Bisgrove et al., 1994). Actual dual specificity recognition is through effector-mediated modifications of RPM1-interacting protein 4 (RIN4) in the presence of the bacterial type III effector proteins AvrRpm1 or AvrB (Mackey et al., 2002). Functional characterization of RPM1 and its interacting components have provided tremendous insight into underlying plant disease resistance mechanisms, and has also provided an important precedent for theoretical consideration of evolutionary and ecological genetics of plant innate defense (Stahl et al., 1999; Tian et al., 2003).
The RPM1 gene was shown to have originated prior to split between the ancestors of the Brassica and Arabidopsis lineages because a single homologous copy was found at two of six homologous loci in Brassica napus, a cultivated polyploid relative of A. thaliana (Grant et al., 1998). Susceptibility to bacteria expressing AvrRpm1 has previously only been observed as a complete deletion of the coding sequence, replaced by a much shorter fragment of unknown origin in susceptible A. thaliana plants, with similar but independent RPM1-deletions in two of the four rpm1-null loci of B. napus (Grant et al., 1998).
This evidence, combined with analysis of the divergence in the flanking sequence between the null and functional RPM1 haplotypes in A. thaliana, has led to the conclusion that this locus represents an example of a long-term balanced polymorphism (Stahl et al., 1999). A mechanistic explanation of the maintenance of this polymorphism was provided by evidence for a cost of RPM1 in the absence of the pathogen (Tian et al., 2003). Notably, no evidence has been found to indicate that alternative functional alleles segregate at this locus.
Assuming a cost of RPM1 resistance in the absence of the pathogen, we hypothesized that more intensive sampling of A. thaliana within a defined geographic range could reveal other loss-of-function alleles arising from independent mutation events, unless sufficient gene flow existed among populations for this single deletion null allele to spread throughout the species range.
To test this prediction, we first surveyed variation at the RPM1 locus in a UK-wide diversity collection of 89 A. thaliana accessions. Surprisingly, specific recognition of AvrRpm1 occurred at a much lower frequency of ca. 37%, suggesting lower disease pressure in the UK than reported in a global sample (Stahl et al., 1999; Aranzana et al., 2005). Molecular analysis of the RPM1 locus was undertaken in these accessions and in an expanded global sample of 92 accessions. This revealed four unique RPM1 protein variants that fail to confer disease resistance to bacteria expressing AvrRpm1 or AvrB effectors. Our data suggests a more complex evolutionary history at the RPM1 locus than previously indicated, in which multiple loss-of-resistance alleles are maintained in addition to RPM1-resistance specificity.
Results
The Majority of AvrRpm1 Susceptible Accessions Do Not have the rpm1-Null Deletion
Screening of a collection of 89 UK A. thaliana accessions for recognition of AvrRpm1 and AvrB by inoculation of the normally virulent P. syringae pv. tomato DC3000 (DC3000) modified to carry either AvrRpm1 or AvrB revealed a significantly higher occurrence of RPM1 compatible interactions (i.e., hosts were susceptible) than previously reported (Figure 1; Tables A1 and A2 in Appendix). In total, 63% of UK accessions were susceptible, based upon failure to restrict bacterial growth and absence of a visible macroscopic hypersensitive response. Examination of the susceptible accessions by PCR using primers specific for either the RPM1 gene or the deletion null allele revealed that not all susceptible accessions had a deletion of RPM1.
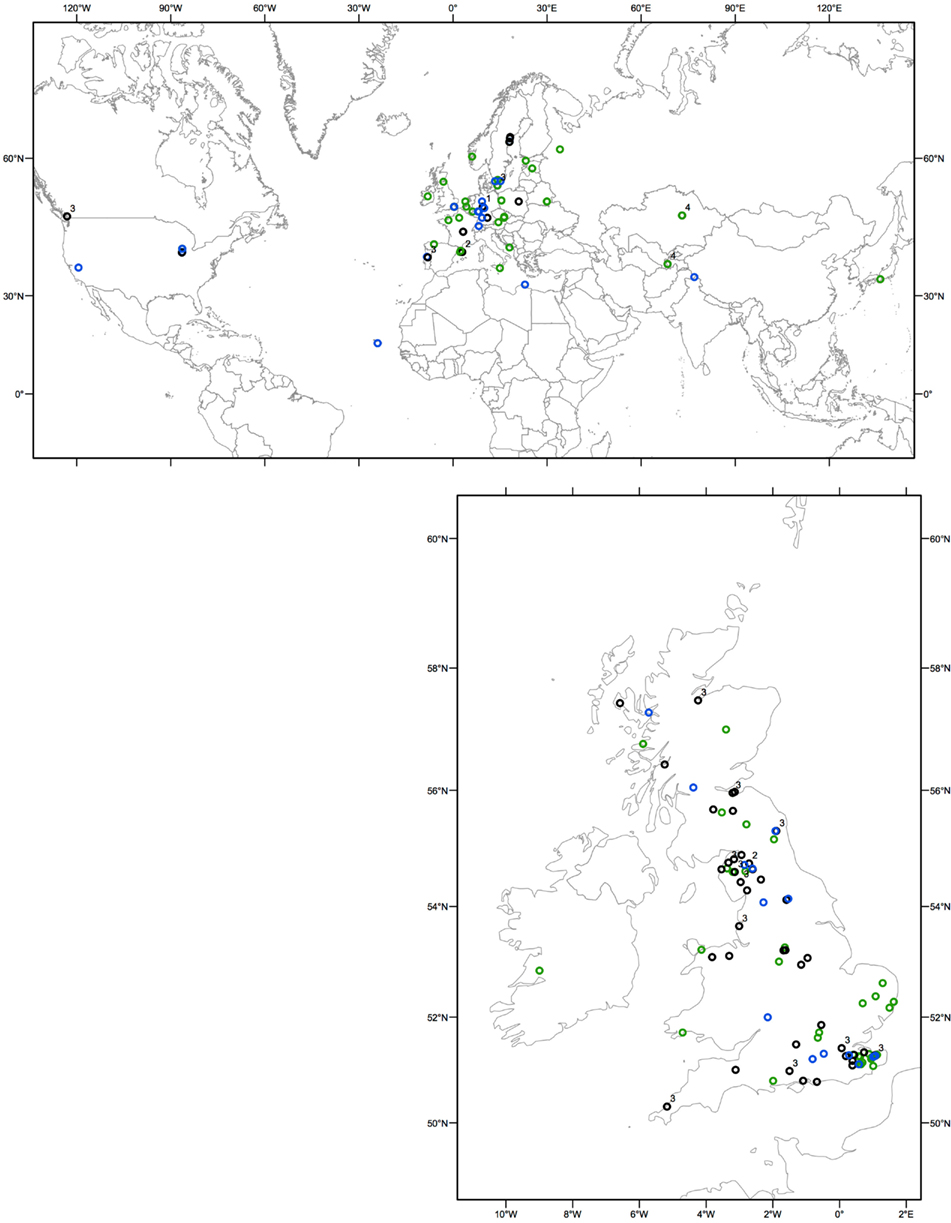
Figure 1. Geographical distribution of resistant and susceptible individuals determined from HR and PCR. Resistant accessions are represented by light green open circles, rpm1-deletion nulls by blue open circles. The susceptible accessions containing RPM1 genes with premature stop codons are represented by black open circles. Next to these circles, the type of the loss-of-function allele (allele types 1 through 4 as defined in Figure 2; Table 1) is indicated.
In the UK sample, of the accessions that lacked resistance, only 25% contained the previously described deletion null allele (Figure 1; Table A1 in Appendix). Based on these unexpected observations, we expanded our study to include 92 global A. thaliana accessions. PCR based genotyping revealed that a larger proportion (43%) of the susceptible individuals from the global sample had the deletion null allele. This still represented <20% of all accessions in the combined UK and global samples (Figure 1; Tables A1 and A2 in Appendix).
A total of 38% of all accessions phenotyped were found to be susceptible yet contain the RPM1 coding sequence (Figure 1; Tables A1 and A2 in Appendix). Our subsequent analysis of the entire RPM1 coding region and more than 500 nucleotides upstream and 178 nucleotides downstream of RPM1 in a sample of 31 A. thaliana accessions revealed loss-of-function (susceptibility), resulting from premature stop codons caused by single nucleotide substitutions or small indels (Table 1).
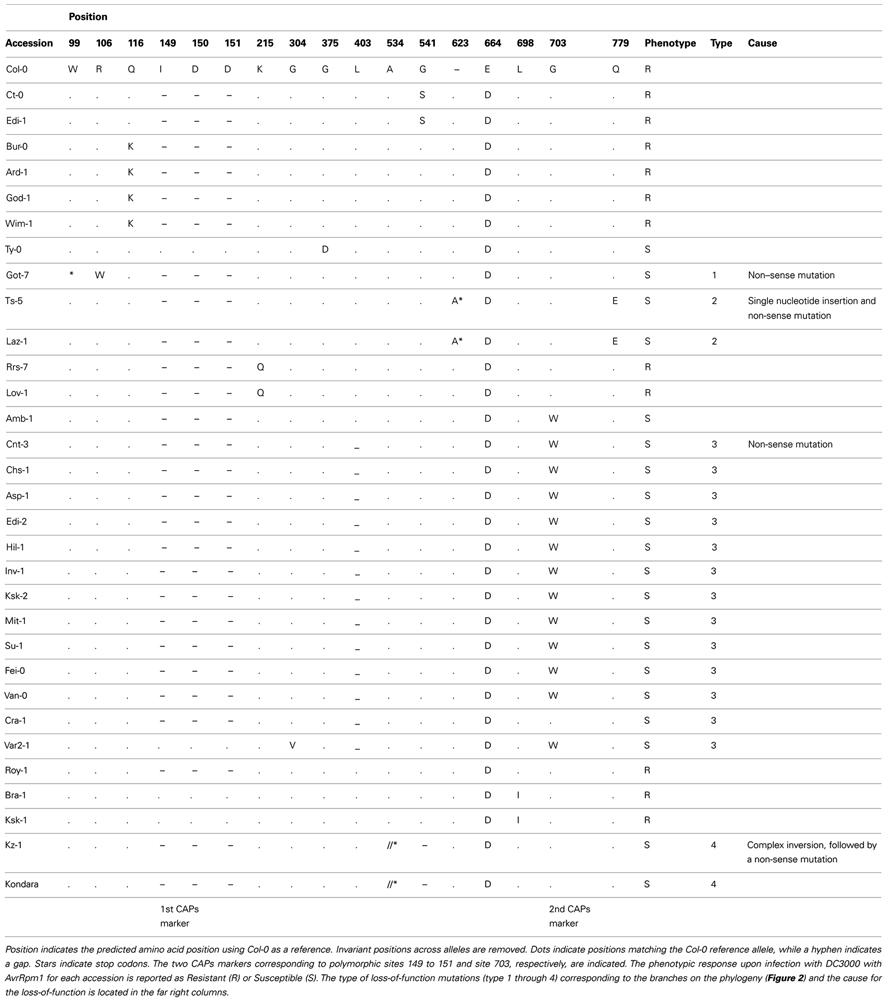
Table 1. Protein sequence variation among RPM1 alleles.
Four novel independent loss-of-function alleles were identified (Figure 2B; Table 1) and their occurrence varied in frequency in our sample (Table 1). At one extreme, a single accession from Germany had a premature stop codon at amino acid position 99. At the other extreme, 13 susceptible individuals shared a premature stop codon at amino acid position 403. These 13 individuals included accessions from both Europe and North America. The other two loss-of-function mutations were each found in a pair of accessions. One allelic type contains a simple frameshift mutation that leads to a premature stop codon at amino acid position 625. This mutation is found in one accession from the UK and one accession from Spain.
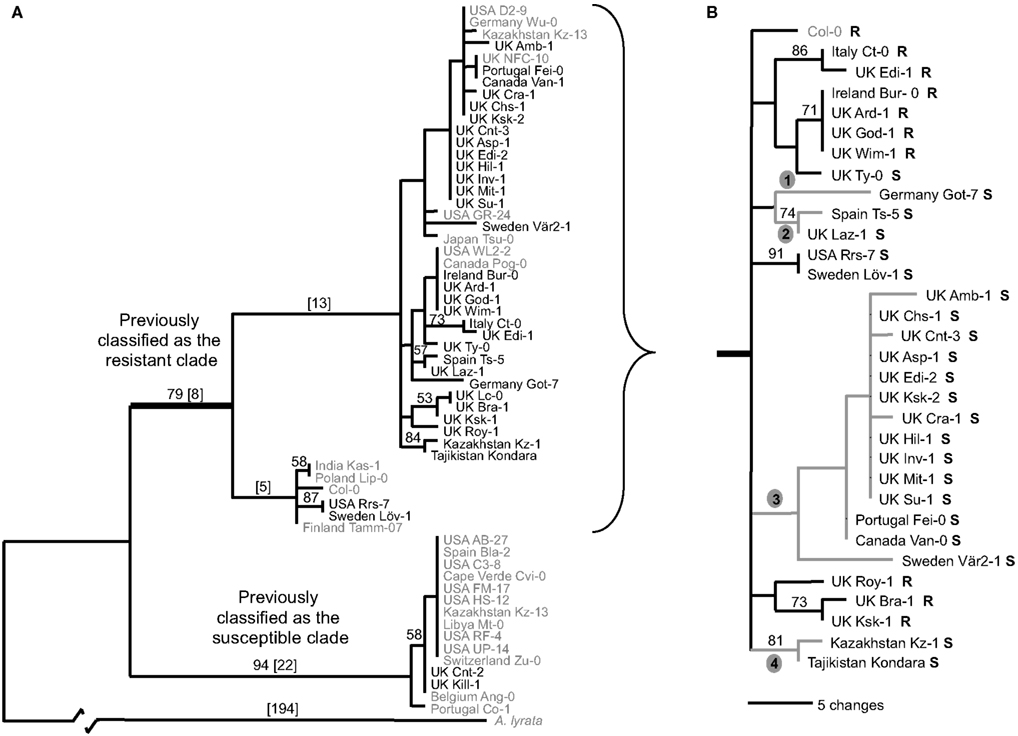
Figure 2. Maximum parsimony analysis of RPM1 region from a worldwide collection of A. thaliana indicating recurrent loss-of-function at the RPM1 locus. (A) Analysis based on the RPM1 coding region (2781 nucleotides), 2190 nucleotides upstream, and 176 nucleotides downstream of coding region. (B) Analysis based on coding region and 500 nucleotides upstream and 176 nucleotides downstream of coding region. Allele sequences new in this study are black; sequences from Stahl et al. (1999) are gray. No sequence data was available for the accessions from Stahl et al. from 1743 bp onward. These positions were treated as missing data in the analysis. Bootstrap values are indicated above branches; branch lengths are in square brackets. Resistance (R) or susceptibility (S) based on HR to DC3000 carrying AvrRpm1 indicated next to accession name. The four new branches on which loss-of-function substitutions occurred are indicated. The specific substitutions are found in Table 1.
A more complicated mutational scenario underlies the loss-of-function mutation found in the accessions from Kazakhstan and Tajikistan. The RPM1 alleles from these two individuals encode a premature codon at amino acid position 509. This stop codon is embedded in a large insertion of 225 nucleotides at this part of the RPM1 gene. At the site of the insertion, these two alleles are missing 130 nucleotides of the RPM1 gene relative to the other alleles. However, the 225 bp insert contains these missing 130 nucleotides in reverse complement form. In fact the entire 225 insert is a 100% match to the reverse complement of this region of the other RPM1 alleles sampled, and includes the missing 130 nucleotides plus an additional 12 bp from the left border and 83 nucleotides matching the right border of the indel position. In contrast to the other loss-of-function mutations that involve single nucleotide substitutions, this loss-of-function allele appears to be derived from a complex mutational event, possibly via a double strand break and repair mechanism.
The Loss-of-Function Alleles Arose Independently and are Younger than the Deletion Null Allele
Orthologs of RPM1 are present in close relatives of A. thaliana, indicating that the ancestral allele at this locus was most likely a functional version of RPM1. The significant haplotype divergence between the deletion null and RPM1 containing haplotypes indicates that the deletion null predates the other loss-of-function alleles. Phylogenetic analysis of the flanking region and the RPM1 coding sequence revealed that the novel loss-of-function alleles cluster within the clade previously described to be the “resistance haplotype” (Figure 2). This implies that these loss-of-function mutations are derived from previously functional alleles. Furthermore, each loss-of-function represents a unique mutation and these losses are predicted to have occurred independently.
To survey the sequence diversity in our larger collection of plants, we developed two diagnostic Cleaved Amplified Polymorphic Sequences (CAPs) markers (Table A3 in Appendix). The presence of either marker alone was not associated with susceptibility, however all individuals carrying both CAPs markers (44 out of 172 screened individuals) were susceptible. This combination of markers was found in nearly all individuals of clade 3 of the RPM1 gene tree (Figure 2B) and this loss-of-function allele appears to be widespread in its distribution (Figure A1 in Appendix).
Haplotype Diversity Around the RPM1-Deletion Junction
The haplotype differentiation, which is visible on the phylogenetic tree and in the sliding window analysis (Figures 2 and 3), results in statistically significant positive values of Tajima’s D within the flanking regions (Table 2). This marked haplotype differentiation at the deletion junction has been reported in previous analyses and was interpreted as a hallmark of balancing selection (Stahl et al., 1999). Upon combined examination of the previously reported sequences and of our new sequences, we observed that a substantial portion (92%) of the previously analyzed region upstream of RPM1 contains a region predicted to encode the NSN1 gene (Figure 3). The coding region of this gene shows evidence of evolutionary constraint with πnon/πsyn = 0.181 (within A. thaliana) and Ka/Ks = 0.15 (divergence to A. lyrata). However, immediately downstream of the NSN1 gene, silent polymorphism within A. thaliana shows a sharp increase (πsil in coding region = 0.02015 < πsil downstream of NSN1 = 0.07622). Furthermore, of the 21 fixed differences between the two A. thaliana haplotypes, 11 reside in the short 137 bp interval downstream of the stop codon, while the other 10 are distributed across the 1600 nucleotide coding region of the NSN1 gene. Models of long-term balancing selection would predict a gradual increase in allelic divergence over the entire interval, yet these data show divergence is 12 times higher immediately downstream of the stop codon of the NSN1, compared to within the NSN1 gene.
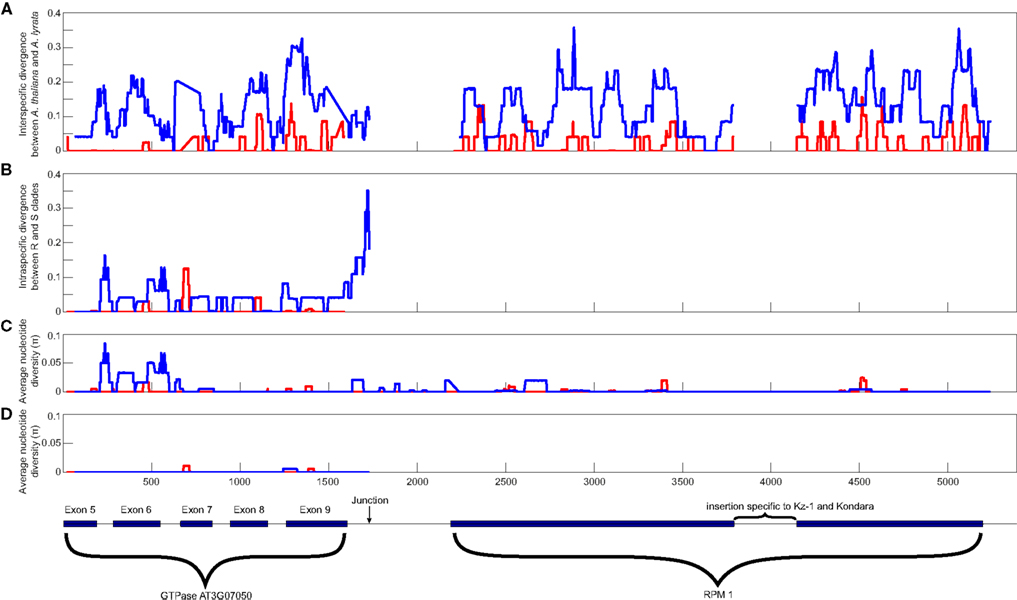
Figure 3. Sliding window analyses across the RPM1 and NSN1 region. (A) Interspecific divergence between A. thaliana and A. lyrata. (B) Intraspecific divergence between A. thaliana alleles belonging to the R and S clades. (C) Intraspecific polymorphism among alleles carrying the RPM1 gene. (D) Intraspecific polymorphism among alleles carrying the RPM1-deletion null. Silent polymorphisms include both synonymous polymorphisms and polymorphisms in non-coding regions, such as introns. The structure of the genes is shown below the graph. Boxes indicate exons, solid lines indicate introns. Sliding window analyses were conducted using DnaSP v. 5 (Librado and Rozas, 2009). Values are midpoints of 25 bp windows.
While this peak in divergence between haplotypes in A. thaliana in the vicinity of the deletion junction has previously been interpreted as evidence of a long-term balanced polymorphism, new information about the gene content in this segment of the A. thaliana genome reveals that this marked peak may be partially related to differences in the form and strength of natural selection operating in parallel on the NSN1 gene. Haplotype differentiation between the resistant haplotype and deletion null haplotype extends across the NSN1 coding region and the jump of divergence between haplotypes occurs specifically in the non-coding region between these genes. Combined with the observation of multiple, independent loss-of-function mutations at the RPM1 locus in A. thaliana, our data suggest a much more dynamic situation exists at the RPM1 disease resistance locus than previously proposed and we can reject strict bi-allelic balanced polymorphism at RPM1.
Protein Sequence Variation within RPM1 Coding Region
Previous studies of this locus focused primarily on the sequence evolution flanking the RPM1-deletion junction and did not address sequence variation within RPM1. Based on analyses of an interspecific pair of RPM1 alleles, Stahl et al. state that the “selectively driven turnover of resistance alleles does not appear to be important in RPM1 protein evolution.” However, in our analyses of 31 RPM1 alleles (plus the reference Col-0 sequence) we observe a significant excess of amino acid polymorphism relative to the neutral expectation in the RPM1 alleles (McDonald–Kreitman test, p-value = 0.028, Table 2). Elevated amino acid polymorphism within the RPM1 coding region cannot be attributed exclusively to the presence of loss-of-function alleles, because functional and non-functional alleles contribute nearly equal numbers of non-synonymous segregating sites (five versus six respectively), although there are nearly 1.5 times as many non-functional alleles in our sample (Table 2). The maintenance of multiple amino acid variants of both functional and non-functional alleles is consistent with a history of diversifying selection, similar to that reported at Pto in wild tomatoes (Rose et al., 2005, 2007).
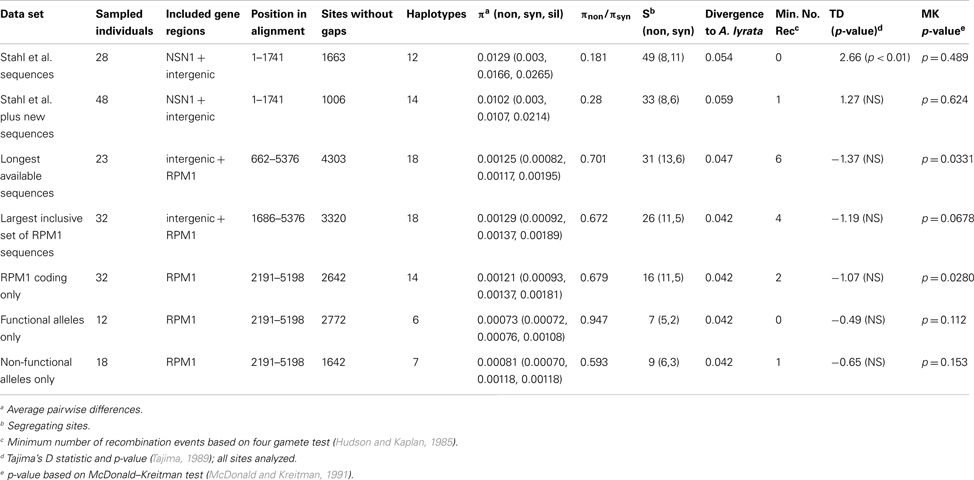
Table 2. Summary statistics and tests of neutrality.
Discussion
Evidence for parallel loss-of-resistance and maintenance of multiple mutant alleles from this study provides a new dimension to our understanding of how natural selection shapes genetic variation of resistance genes in plants. Although loss-of-function mutations are not uncommon for traits that are no longer required in an organism (for example eyesight in cave dwelling animals), it is surprising to observe the maintenance of multiple loss-of-function alleles at a locus that is expected to help protect the organism against disease.
An intriguing question highlighted in the original RPM1 study was the discrepancy between the levels of within haplotype diversity and the actual observed frequency of the two haplotypes within A. thaliana. The deletion null haplotype was sampled at a frequency of 0.48, although it harbored 10-fold less polymorphism than the resistant haplotype at a frequency of 0.52. Furthermore, since the branches separating these two haplotypes carried nearly equivalent numbers of derived polymorphisms relative to an outgroup, the polymorphism appeared to be stable over a long period of time and the alleles were assumed to be nearly the same age. Theoretical modeling revealed that cyclical fluctuations of the two haplotypes over a very long period of time could account for these observations, although the mean effective population size of the resistance allele in these models was required to exceed the sampled frequency to fit the data. With the acquisition of additional data, we now know that multiple loss-of-function alleles segregate within A. thaliana and therefore, the long-term effective population size of a single loss-of-function class may be indeed accurately reflected in the level of within haplotype diversity. If multiple, alternative loss-of-function alleles segregate in the species, then theoretical models do not need to account for this extreme discrepancy in within class polymorphism.
Based on analysis of the flanking sequence of these alleles, we have established that the additional loss-of-function alleles are younger than the RPM1-deletion null first reported, and are independently derived from individuals carrying the full-length RPM1 allele. Segregation of multiple alleles that appear to be selectively equivalent is one hallmark of soft selective sweeps, in which independent (putatively selectively advantageous) mutations increase in frequency simultaneously. Soft sweeps are likely when the effective population size is large or the allelic mutation rate is high. In particular, when the adaptive allele involves a loss-of-function, a high proportion of these adaptations may result from recurrent mutation (Pennings and Hermisson, 2006). This may specifically apply in the case of RPM1 loss-of-function mutations.
A further question is whether to view these loss-of-function alleles as advantageous (since these individuals are consequently no longer able to recognize certain pathogen strains) or whether this is simply an example of relaxed selective constraint. Since the RPM1 gene is reported to carry a fitness cost in the absence of pathogen infection (Tian et al., 2003), it may be possible to consider the loss of RPM1 resistance as a specific adaptation to local environmental conditions. Since pathogen populations are notoriously variable in time and space, natural selection may have played a role in driving the loss of the RPM1 function in the absence of pathogen pressure. The limited outcrossing in A. thaliana may also restrict the spread of alleles throughout the species’ range. This would explain why the original RPM1-deletion allele is not the only “solution” to the fitness cost in the absence of pathogen pressure.
Another consideration is that alternative loss-of-function alleles may not be functionally equivalent, especially in the face of recurrent changes in the adaptive landscape typical of host-parasite interactions. Clearly, a complete gene deletion does not have the same probability of reverting to a functional allele as an allele with a single or few nucleotide changes. After a gene’s function has been altered (perhaps through a missense, non-sense, or frameshift mutation), it may serve as a repository of genetic variation for future bouts of selection. Gene disruption by relatively simple nucleotide changes may be only transient, while a complete deletion is typically not reversible.
This interpretation is in line with the “less is more” hypothesis of Olson (1999). He hypothesized that simple loss-of-function alleles may represent a reservoir of “near-functional” alleles and if the adaptive landscape is subject to frequent changes, reversion of previously disrupted alleles will be a common means of adaptation. He envisioned this hypothesis originally to explain examples of adaptive loss-of-function that apparently spread rapidly through early human populations. However, it now appears to be widespread, as cases for multiple, independent loss-of-function alleles have been shown to occur extensively throughout the microbial, animal, and plant kingdoms (Olson, 1999; Johanson et al., 2000; Protas et al., 2006; Kivimaki et al., 2007; Jeong et al., 2008; Ordonez et al., 2008; Dworkin and Jones, 2009; Kingsley et al., 2009; Kovach et al., 2009; Stergiopoulos and de Wit, 2009).
Indeed, the “less is more” proposal provides a more accurate description of evolution at the RPM1 locus than previously reported. Unlike the original null allele described at the RPM1 locus that resulted from a complete deletion of the coding sequence, the novel alleles we discovered are more recent in origin and have independently arisen from functional RPM1 alleles. An intrinsic cost of expressing the RPM1-resistance protein in the absence of selection pressure by the corresponding pathogen may have been an important factor allowing the persistence of these independent mutant alleles in wild A. thaliana populations (Tian et al., 2003).
An exemplification of the adaptive potential of a single copy R-gene in A. thaliana is the downy mildew resistance gene Recognition of Peronospora Parasitica 13 (RPP13; Rose et al., 2004; Hall et al., 2009). Multiple, functionally differentiated alleles are maintained at RPP13 and this diversity is matched by allelic variation in the corresponding proteins of an oomycete thought to be an endemic parasite in European populations of A. thaliana (Allen et al., 2004; Holub, 2008; Hall et al., 2009). Likewise, multiple protein variants (both functional and non-functional) segregate at RPM1. However, protein polymorphism is not reported in the two bacterial proteins that are known to interact with RPM1 (i.e., neither AvrB or AvrRpm1 show striking patterns of diversification across pathovars of P. syringae; Bisgrove et al., 1994). Instead, these bacterial proteins appear pathologically dispensable under laboratory conditions and can be readily excised from the pathogen. Therefore, the diversification in RPM1 protein sequence may not be directly driven by diversifying selection imposed by coevolution with these two pathogen proteins. Instead the selection may be on RPM1’s ability to recognize and respond to changes elicited by effectors such as AvrRpm1 and AvrB to RIN4 (Chung et al., 2011). Thus, RPM1 may have additional recognition specificities that have not been revealed through the narrow effector repertoire on which it has been tested to date. Alternatively, RPM1 may contribute other fitness benefits: R proteins are emerging as important components of the integrated stress response, and what we consider to be rpm1 loss-of-function mutations may in fact confer a selective advantage in other environments (Chini et al., 2004).
Our data supports previous reports that RPM1 resistance is an ancestral trait in the Brassicaceae. However, the frequency and geographic distribution of the multiple rpm1 alleles indicate the parallel origin and persistence of loss-of-function mutations at this locus in A. thaliana. Further empirical evidence is required to determine whether the novel alleles can in fact be maintained without positing a microbial selection pressure. In this respect, the expanding assortment of rpm1 alleles highlights the importance of this locus as an example for ecological genetics and the need for field experiments to compare the relative intrinsic costs of the new rpm1 alleles.
Materials and Methods
Plant Materials and Disease Resistance Phenotyping
The seed collection of A. thaliana accessions from the UK was assembled by E. Holub. Seeds from the global sample were obtained from the Nordberg collection (Aranzana et al., 2005; available from the Arabidopsis stock centers). Five-week old rosettes were phenotyped for RPM1 disease resistance using abaxial leaf inoculation of DC3000 modified to carry AvrRpm1 or AvrB (2 × 108 cfu ml−1) via needle-less syringe. Resistance (hypersensitive response, HR), evident as rapid collapse of the infiltrated leaf, was recorded after 6 h and susceptibility (no evident leaf collapse) at 20 h post inoculation. Response to carrier isolate (DC3000) was also determined after 6 h and 20 h to ensure that the phenotype was due to AvrRpm1 interaction and not recognition of effectors delivered by the carrier (none were observed). Response to AvrB was identical to AvrRpm1 and as such is not mentioned further.
In planta growth analysis was carried out using the same inoculation technique but at a reduced bacterial concentration (2 × 106 cfu ml−1). Three days post inoculation bacterial multiplication was determined as follows: Three leaf disks per plant were excised, homogenized in 10 mM MgCl2, and the suspension serially diluted. Diluted leaf disk homogenates were plated on King’s Broth (King et al., 1954) containing rifampicin (50 μg/ml; DC3000) or rifampicin and kanamycin (50 μg/ml; DC3000AvrRpm1 or DC3000AvrB), incubated for 36 h at 28°C, before colonies were counted. Resistant accessions had at least a 10-fold difference in bacterial growth between DC3000 carrying AvrRpm1 and the virulent DC3000 strain.
PCR Amplification and Sequencing
Alleles of the RPM1 region were amplified by PCR using Taq (Promega, Madison WI). The RPM1 locus was amplified in all accessions from 194 bp upstream of the start codon to 697 bp downstream of the termination codon. RPM1 specific primers were designed to amplify this region in seven overlapping products (Table 3). To identify accessions containing the rpm1-deletion null, primers located 1700 bp upstream and 990 bp downstream from the RPM1-deletion junction were used. Expected amplicon length was 2733 bp in accessions lacking the RPM1 gene and 5513 bp in accessions with the full-length RPM1 gene. The upstream region of the RPM1 gene was also amplified and sequenced. This region contains the 3′ half of the NSN1 gene and a non-coding region directly upstream of RPM1. This region was amplified using two sets of primer pairs.

Table 3. Sequences and annealing positions of primers used in this study.
Pooled PCR samples (four reactions per primer combination) from the seven RPM1 specific products and the NSN1 gene and 5′ RPM1 region from each of the 31 accessions were gel purified on 1.5% agarose gel and subsequently purified using QIA quick PCR Purification Kit (Qiagen). PCR products were directly sequenced on an ABI3700 capillary sequencer (Applied Biosystems). Sequence traces were aligned to the original RPM1 sequence obtained from Col-0 and base-calling was completed manually (Grant et al., 1995; NCBI Gene ID: 819889). All sequences are available from GenBank (Accession numbers: KC211311–KC211321 and KC249727–KC249748).
Sequence Analysis
Phylogenetic analysis, implemented in PAUP 4.0 beta 10, was used to infer the likely ancestry of the novel rpm1 loss-of-function alleles (Swofford, 1999). The phylogenetic relationships between these sequences were inferred using maximum parsimony and neighbor joining (using the HKY85 model of substitution). These methods yielded similar topologies. Two datasets were analyzed. The first consisted of the RPM1 coding region (2781 nucleotides), 2190 nucleotides upstream, and 176 nucleotides downstream of coding region. The second dataset was based on the RPM1 coding region and 500 nucleotides upstream and 176 nucleotides downstream of coding region. No sequence data was available for the accessions from Stahl et al. (1999) from 1743 bp onward. These positions were treated as missing data in the analysis. The standard summary statistics, tests of neutrality, and sliding window analyses were performed using DnaSP v. 5 (Librado and Rozas, 2009). For the sliding window analysis, window size was set to 25 sites and step size was set to 1.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported in part by BBSRC Grant D16978 to Susanna Atwell, Murray Grant, and Eric B. Holub and NSF DEB-0519961 to Susanna Atwell. The authors would like to acknowledge M. Cheetham for the geographical figures, T. Klösges for technical assistance, and J. Parsch for editing advice.
References
Allen, R. L., Bittner-Eddy, P. D., Grenvitte-Briggs, L. J., Meitz, J. C., Rehmany, A. P., Rose, L. E., et al. (2004). Host-parasite coevolutionary conflict between Arabidopsis and downy mildew. Science 306, 1957–1960.
Aranzana, M. J., Kim, S., Zhao, K. Y., Bakker, E., Horton, M., Jakob, K., et al. (2005). Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 1:e60.
Bisgrove, S. R., Simonich, M. T., Smith, N. M., Sattler, A., and Innes, R. W. (1994). A disease resistance gene in Arabidopsis with specificity for 2 different pathogen avirulence genes. Plant Cell 6, 927–933.
Chini, A., Grant, J. J., Seki, M., Shinozaki, K., and Loake, G. J. (2004). Drought tolerance established by enhanced expression of the CC-NBS-LRR gene, ADR1, requires salicylic acid, EDS1 and ABI1. Plant J. 38, 810–822.
Chung, E. H., Da Cunha, L., Wu, A. J., Gao, Z., Cherkis, K., Afzal, A. J., et al. (2011). Specific threonine phosphorylation of a host target by two unrelated type III effectors activates a host innate immune receptor in plants. Cell Host Microbe 9, 125–136.
Dworkin, I., and Jones, C. D. (2009). Genetic changes accompanying the evolution of host specialization in Drosophila sechellia. Genetics 181, 721–736.
Grant, M. R., Godiard, L., Straube, E., Ashfield, T., Lewald, J., Sattler, A., et al. (1995). Structure of the Arabidopsis Rpm1 gene enabling dual-specificity disease resistance. Science 269, 843–846.
Grant, M. R., McDowell, J. M., Sharpe, A. G., Zabala, M. D. T., Lydiate, D. J., Dangl, J. L., et al. (1998). Independent deletions of a pathogen-resistance gene in Brassica and Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 95, 15843–15848.
Hall, S. A., Allen, R. L., Baumber, R. E., Baxter, L. A., Fisher, K., Bittner-Eddy, P. D., et al. (2009). Maintenance of genetic variation in plants and pathogens involves complex networks of gene-for-gene interactions. Mol. Plant Pathol. 10, 449–457.
Holub, E. B. (2008). Natural history of Arabidopsis thaliana and oomycete symbioses. Eur. J. Plant Pathol. 122, 91–109.
Hudson, R. R., and Kaplan, N. L. (1985). Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111, 147–164.
Jeong, S., Rebeiz, M., Andolfatto, P., Werner, T., True, J., Carroll, S. B., et al. (2008). The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell 132, 783–793.
Johanson, U., West, J., Lister, C., Michaels, S., Amasino, R., Dean, C., et al. (2000). Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 290, 344–347.
King, E. O., Ward, K. M., and Raney, D. E. (1954). Two simple media for the demonstration of pyocyanin and fluroesin. J. Lab. Clin. Med. 44, 301–307.
Kingsley, E. P., Manceau, M., Wiley, C. D., and Hoekstra, H. E. (2009). Melanism in Peromyscus is caused by independent mutations in Agouti. PLoS ONE 4:e6435.
Kivimaki, M., Karkkainen, K., Gaudeul, M., Loe, G., and Agren, J. (2007). Gene, phenotype and function, GLABROUS1 and resistance to herbivory in natural populations of Arabidopsis lyrata. Mol. Ecol. 16, 453–462.
Kovach, M. J., Calingacion, M. N., Fitzgerald, M. A., and McCouch, S. R. (2009). The origin and evolution of fragrance in rice (Oryza sativa l). Proc. Natl. Acad. Sci. U.S.A. 106, 14444–14449.
Librado, P., and Rozas, J. (2009). DnaSP v5, a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452.
Mackey, D., Holt, B. F., Wiig, A., and Dangl, J. L. (2002). Rin4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell 108, 743–754.
McDonald, J. H., and Kreitman, M. (1991). Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654.
Olson, M. V. (1999). When less is more: gene loss as an engine of evolutionary change. Am. J. Hum. Genet. 64, 18–23.
Ordonez, G. R., Hillier, L. W., Warren, W. C., Gruetzner, F., Lopez-Otin, C., Puente, X. S., et al. (2008). Loss of genes implicated in gastric function during platypus evolution. Genome Biol. 9, R81.
Pennings, P. S., and Hermisson, J. (2006). Soft sweeps II – molecular population genetics of adaptation from recurrent mutation or migration. Mol. Biol. Evol. 23, 1076–1084.
Protas, M. E., Hersey, C., Kochanek, D., Zhou, Y., Wilkens, H., Jeffery, W. R., et al. (2006). Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nat. Genet. 38, 107–111.
Rose, L. E., Bittner-Eddy, P. D., Langley, C. H., Holub, E. B., Michelmore, R. W., and Beynon, J. L. (2004). The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana. Genetics 166, 1517–1527.
Rose, L. E., Langley, C. H., and Michelmore, R. W. (2007). Natural variation in the Pto pathogen resistance gene within species of wild tomato (Lycopersicon): II. Population genetics of Pto. Genetics 175, 1307–1319.
Rose, L. E., Langley, C. H., Bernal, A. J., and Michelmore, R. W. (2005). Natural variation in the Pto pathogen resistance gene within species of wild tomato (Lycopersicon): I. Functional analysis of Pto alleles. Genetics 171, 345–357.
Schwessinger, B., and Zipfel, C. (2008). News from the frontline: recent insights into PAMP-triggered immunity in plants. Curr. Opin. Plant Biol. 11, 389–395.
Stahl, E. A., Dwyer, G., Mauricio, R., Kreitman, M., and Bergelson, J. (1999). Dynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature 400, 667–671.
Stergiopoulos, I., and de Wit, P. J. G. M. (2009). Fungal effector proteins. Annu. Rev. Phytopathol. 47, 233–263.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–596.
Tian, D., Traw, M. B., Chen, J. Q., Kreitman, M., and Bergelson, J. (2003). Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423, 74–77.
Appendix
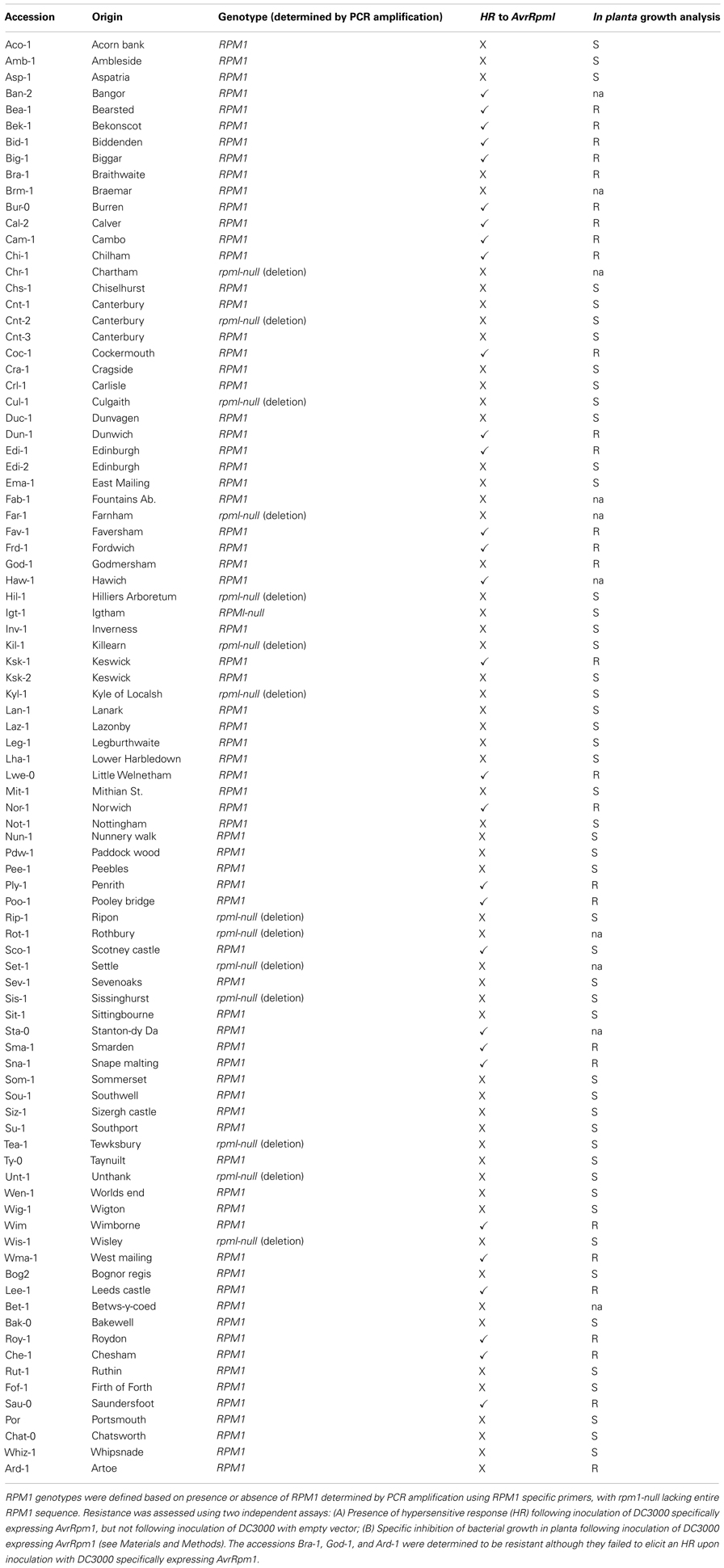
Table A1. RPM1 genotypes and resistance phenotypes across UK accessions.
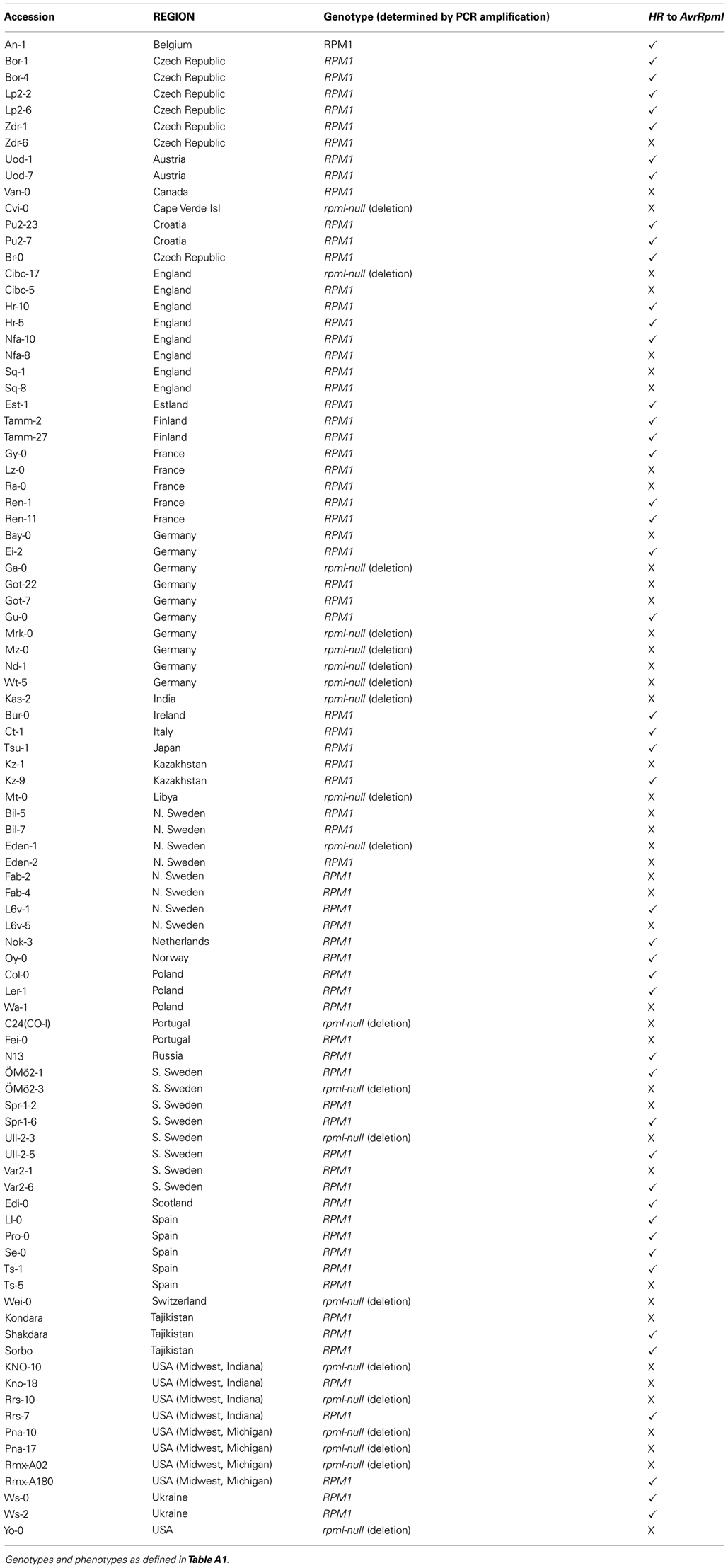
Table A2. RPM1 genotypes and resistance phenotypes across world accessions.

Table A3. Survey of sequence variation at RPM1 in worldwide sample using diagnostic markers.
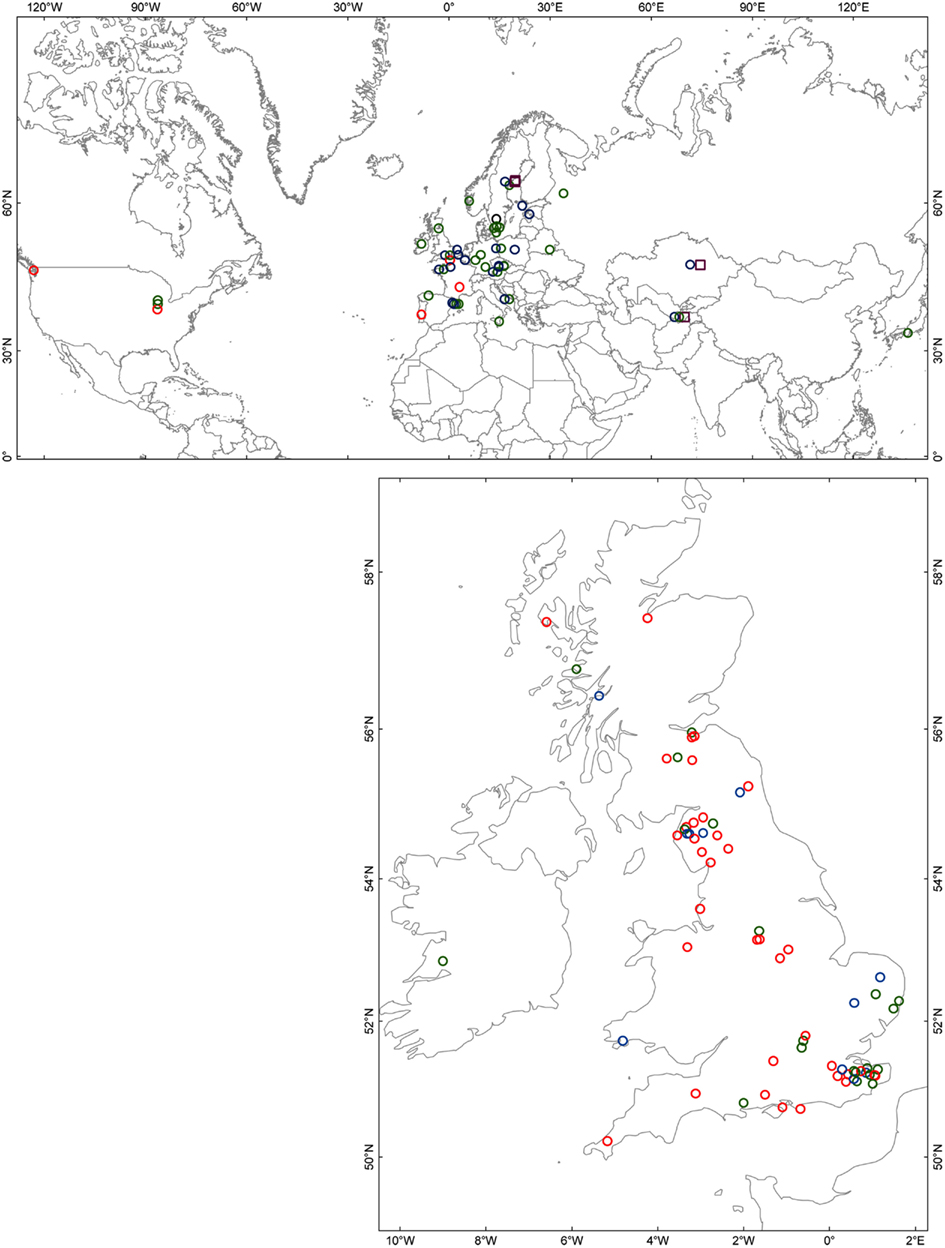
Figure A1. Geographical distribution of phenotyped accessions in which genotypes have been defined based on the diagnostic CAPs markers. Accessions with the I149D150D151 deletion (CAPs marker 1) and the G703 > W703 (CAPs marker 2) are represented by open red circles; presence of only CAPs marker 1 by open green circles; presence of only CAPs marker 2 by a black open circle; matching Col-0 reference (i.e., lacking both diagnostic markers) by blue open circles; presence of the larger indel/inversion as found in Kz-1 and Kondara by open purple squares. (This indel/inversion was detected by the digestion pattern of CAPs marker 2 in combination with a larger PCR fragment size).
Keywords: parallel evolution, host-parasite coevolution, adaptation, loss-of-function, balancing selection
Citation: Rose L, Atwell S, Grant M and Holub EB (2012) Parallel loss-of-function at the RPM1 bacterial resistance locus in Arabidopsis thaliana. Front. Plant Sci. 3:287. doi: 10.3389/fpls.2012.00287
Received: 27 August 2012; Accepted: 03 December 2012;
Published online: 26 December 2012.
Edited by:
Jean-Philippe Vielle-Calzada, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Ruairidh Sawers, Laboratorio Nacional de Genomica para la Biodiversidad, MexicoJianxin Ma, Purdue University, USA
Copyright: © 2012 Rose, Atwell, Grant and Holub. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Laura Rose, Institute of Population Genetics, Heinrich-Heine Universität, Universitätstr. 1, 40225 Düsseldorf, Germany. e-mail:bGF1cmEucm9zZUBoaHUuZGU=
†Laura Rose and Susanna Atwell have contributed equally to this work.