

- 1RIKEN Plant Science Center, Yokohama, Kanagawa, Japan
- 2Department of Genome System Sciences, Graduate School of Nanobioscience, Kihara Institute for Biological Research, Yokohama, Kanagawa, Japan
Metabolomics has grown greatly as a functional genomics tool, and has become an invaluable diagnostic tool for biochemical phenotyping of biological systems. Over the past decades, a number of databases involving information related to mass spectra, compound names and structures, statistical/mathematical models and metabolic pathways, and metabolite profile data have been developed. Such databases complement each other and support efficient growth in this area, although the data resources remain scattered across the World Wide Web. Here, we review available metabolome databases and summarize the present status of development of related tools, particularly focusing on the plant metabolome. Data sharing discussed here will pave way for the robust interpretation of metabolomic data and advances in plant systems biology.
Introduction
Metabolomics, i.e., the measurement of the full suite of metabolites in a living tissue, has expanded greatly over the last decade, especially in the context of biochemical phenotyping. Specifically, in plant science, metabolomic approaches are increasingly used for understanding regulatory networks involved in genotype comparison (Roessner et al., 2001; Weckwerth et al., 2004), measurement of diurnal/circadian rhythms (Urbanczyk-Wochniak et al., 2005; Gibon et al., 2006; Fukushima et al., 2009a; Espinoza et al., 2010), evaluation of genetically modified plants (Catchpole et al., 2005; Baker et al., 2006; Kusano et al., 2011a; Ricroch et al., 2011), uncovering relationships between metabolites associated with carbon and nitrogen metabolism (Stitt and Fernie, 2003; Sato et al., 2008; Kusano et al., 2011b), stress responses (Kaplan et al., 2004; Urano et al., 2009; Caldana et al., 2011; Kusano et al., 2011c; Obata and Fernie, 2012), characterization of many bioresources (Meyer et al., 2007; Rowe et al., 2008; Sulpice et al., 2009), and identifying metabolite quantitative trait loci (mQTLs) (Morreel et al., 2006; Schauer et al., 2006; Lisec et al., 2008; Carreno-Quintero et al., 2012; Matsuda et al., 2012).
It is estimated that approximately 200,000 metabolites are produced in the plant kingdom (Fiehn, 2002). There is no single technique suitable for measurement of all metabolites because of the chemical diversity of cellular metabolites and their broad dynamic range, particularly as this pertains to plants (Hall, 2006; Fukushima et al., 2009b; Saito and Matsuda, 2010; Lei et al., 2011; Weckwerth, 2011). Because of this, an array of analytical methods and extraction procedures has been developed for the detection of a broad spectrum of metabolites. Most procedures are based on either mass spectrometry (MS) or nuclear magnetic resonance (NMR). Metabolite data are typically generated through the following processes (Figure 1): (1) Sample preparation, (2) Data acquisition, and (3) Data pre-processing (the first half of the cycle in Figure 1). The resultant data are then subjected to multi-step downstream processes, including (4) Statistical data analysis approaches such as principal component analysis (PCA) and (5) Data interpretation using methods such as pathway analysis, which facilitate (6) The generation of testable hypotheses and the construction of models that best represent the biological phenomenon (the second half of the cycle in Figure 1). Experimental validation (7) of hypotheses and models [(7) in Figure 1] is necessary for closing the systems biology research cycle (Kitano, 2002; Fernie, 2012).
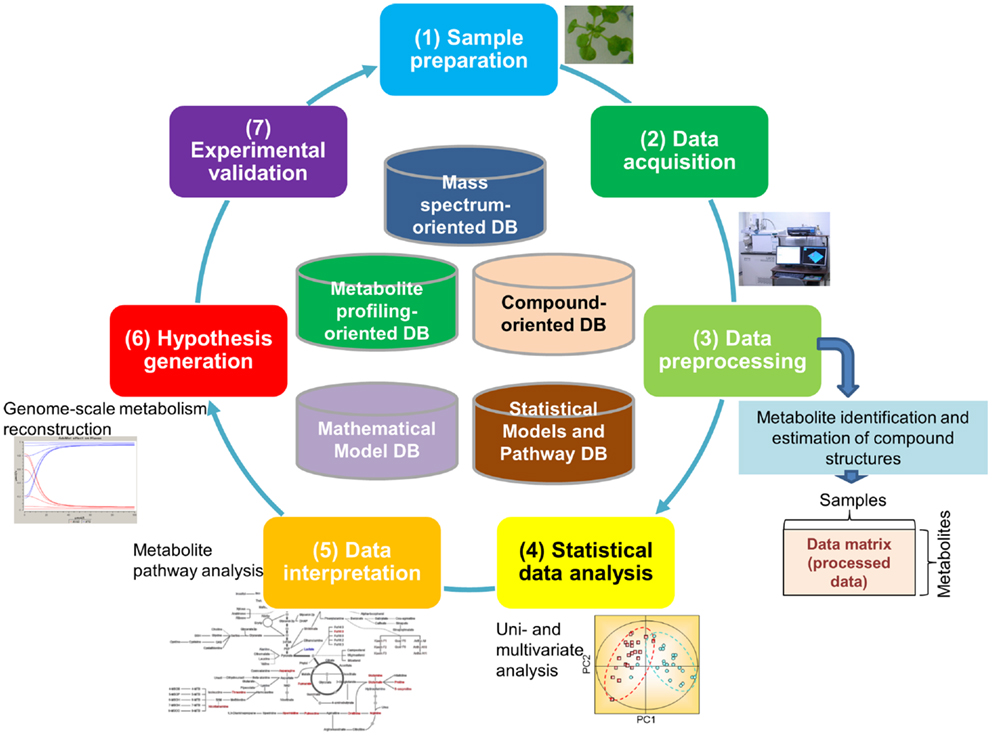
Figure 1. Major processes generating metabolomic data. The processes include (1) sample preparation, (2) data acquisition, and (3) data pre-processing. The resultant data go through multi-step downstream processes including (4) statistical data analysis such as principal component analysis, (5) data interpretation such as pathway analysis, (6) generation of testable hypothesis and construction of the model representing the biological phenomenon, and (7) experimental validation of hypothesis and building models.
Traditionally, the elucidation of the structure of an unknown natural chemical compound has typically required the study of physicochemical properties, including the accurate mass and chemical shifts in 1H- and 13C-NMR spectra when the compound was first isolated. In contrast, metabolite identification in metabolomics using gas chromatography–mass spectrometry (GC-MS) and liquid chromatography–mass spectrometry (LC-MS) is not unambiguous. There are two venues to the identification of a chromatographic peak: (1) purification and NMR analysis or (2) interpretation of the spectra yielding a putative structure, followed by synthesizing or buying the compound and spiking. Detectable peaks using GC-MS and LC-MS are thought to be abundant and often authentic standards exist to identify them by spiking. Nonetheless, researchers commonly attempt to provisionally identify these peaks by comparing their mass spectra and/or the retention time or retention indices (RIs) with those present in a database build from the data of authentic standards. To validate the metabolite identification rigorously, the Metabolomics Standard Initiative (MSI) (Fiehn et al., 2007; Sansone et al., 2007) recommends different levels of identification (Sumner et al., 2007). Fernie et al. (2011) have also stated additional practical recommendations for reporting large-scale metabolite data.
It is quite evident that databases cataloging mass spectra and compounds give great support to metabolomic studies (for example, see Tohge and Fernie, 2009; Scalbert et al., 2011). Here, we highlight a wide range of metabolome databases, especially those that are widely used in MS-based metabolite profiling for rapid, but accurate, quantification, and identification of metabolites (Fiehn, 2002). We also discuss further steps to develop future databases facilitating metabolomic analyses and to improve bioinformatics tools in plant systems biology.
Mass Spectrum-Oriented Information
Since non-targeted metabolite profiling using GC-MS for plant extracts was established in the early 2000s (Fiehn, 2001; Lisec et al., 2006), many software packages and databases for electron impact (EI) mass spectra and RIs of compounds analyzed by GC-MS have been created (Table 1). The NIST/EPA/NIH mass spectral database represents the largest database commercially available for metabolite identification, containing mainly EI mass spectra RIs (Stein, 1999). Recently, the database also stores a set of MS/MS spectra of metabolites, drugs, peptides, and other compounds which are obtained by using ion trap-as well as tandem-MS instruments. The Golm Metabolome Database (GMD) provides GC-EI-MS mass spectral and RI (MSRI) libraries (Kopka et al., 2005; Schauer et al., 2005). It also contains mass spectral tags (MSTs) (Schauer et al., 2005), i.e., MS spectra of putative biological molecules which remain largely unidentified due to the lack of authentic standard compounds. GMD uses both alkanes and fatty acid methyl esters (FAMEs) for RI calculation whereas FiehnLib (Kind et al., 2009), a commercial MSRI library, uses FAMEs rather than alkanes. The Spectral Database for Organic Compounds1 (SDBS) includes a wide range of mass spectra for organic compounds, such as polysaccharides. MassBase2 is a mass spectral archive for LC-, GC-, and Capillary electrophoresis–MS (CE-MS). SetupX3 and BinBase4 are a Laboratory Information Management System (LIMS)/database system for automated metabolite annotation and mass spectra, respectively. The Adams library (Adams, 2007), Terpenoids Library5, and VocBinBase (Skogerson et al., 2011) are GC-specific MSRI libraries for volatile compounds. The former two are commercially available, while the VocBinBase database is freely available for their provisional identification (Skogerson et al., 2011). For MS data management and data sharing, MetabolomeExpress (Carroll et al., 2010) and MetaboLights (Haug et al., 2012; Steinbeck et al., 2012) were developed. The former is an ftp server that acts as a public data repository and web application for online data pre-processing and meta-analysis of publicly available metabolomic datasets analyzed by GC-MS. The latter is a general metabolomics repository; users can browse publicly available metabolomic datasets, search and see experimental meta-data, and re-use associated data files.
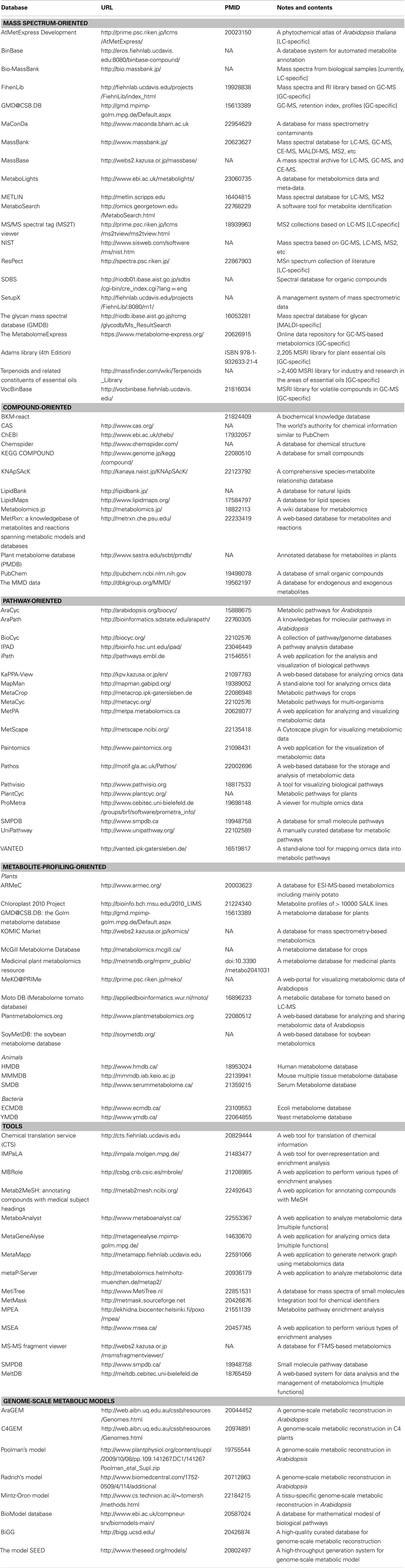
Table 1. Metabolome databases involving mass spectra, compounds, metabolic pathways, metabolite profiles, and statistical/mathematical tools.
It has been demonstrated that metabolite profiling using LC-MS has the potential to reveal secondary metabolites produced by plants, but most of the detected peaks in LC-MS profile data are largely unknown (Moco et al., 2006; De Vos et al., 2007; Iijima et al., 2008; Matsuda et al., 2010). Compared to EI, LC-MS ionization methods such as electrospray ionization (ESI) does hardly fragment the molecular ions. Even if authentic standards do not exist, putative metabolite identification can be done via MS/MS fragmentation and recording of accurate masses using ultra-high resolution MS such as Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) (Lenz et al., 2004; Nakabayashi et al., 2013). Different collision energy/analytical conditions cause different fragment patterns in mass spectra and should also be noted. Several databases have been developed for the sharing of ESI mass spectral information. METLIN stores high-resolution MS/MS spectra at four different collision energies (Tautenhahn et al., 2012). MassBank is a publicly available database of ESI-MS/MS spectra of authentic metabolite standards obtained under five collision energies as well as EI spectra (Horai et al., 2010). Bio-MassBank catalogs those obtained from biological samples6. In analogy with the MST spectra obtained from GC-MS data, the MS/MS spectral tag (MS2T) of detectable metabolites using LC-ESI-quadrupole-time-of-flight/MS is also available (Matsuda et al., 2009, 2010; Sakurai et al., 2013). This library contains MS2T obtained from species such as Arabidopsis thaliana, rice, soybean, and wheat. Compatible LC-MS settings with those used for the MS2T recording can be used for annotating detected peaks. Based on literature surveys, a plant-specific MS/MS spectra database was constructed by the same group (Sawada et al., 2012). MetaboSearch can be used to simultaneously retrieve mass-based metabolite data from multiple metabolite databases (Zhou et al., 2012). This contains tools to query and/or comprehensively analyze LC-MS-based metabolomic data. Together, these mass spectral databases make an important contribution to metabolite identification and also facilitate the development of bioinformatic tools (e.g., mining unknown metabolites) in metabolomics.
Compound-Oriented Information and Structure Characterization
Compound Databases
There are also compound databases (Table 1), such as Chemical Abstract Service (CAS)7. CAS is the oldest database of chemical information (e.g., journal abstracts); substances in the CAS registry database are each assigned a unique ID number. The PubChem database in NCBI (Wang et al., 2009), ChEBI (Degtyarenko et al., 2008), and ChemSpider (Pence and Williams, 2010) are freely available and can be used to retrieve chemical structures of small molecules. Well-curated chemical information including compounds and pathways are available in KEGG database (Kanehisa et al., 2012), KNApSAcK database (Afendi et al., 2012), and Metabolomics.jp8 (Arita and Suwa, 2008). The Plant Metabolome Database (PMDB) is a freely available database of secondary metabolites in plants (Udayakumar et al., 2012). For bioactive lipids, LipidBank (Watanabe et al., 2000) and LipidMap (Fahy et al., 2007) are available. The Manchester Metabolomics Database (MMD) has been developed to simultaneously utilize genome-scale data from the Human Metabolome Database (HMDB) (Wishart et al., 2009), KEGG, and LipidMaps. Other well-organized databases of biochemical knowledge are also available, such as BKM-react (Lang et al., 2011) and MetRxn (Kumar et al., 2012).
Structural Characterization
One of the known bottlenecks in metabolomics is in the identification process of unknown metabolites, which can be classified as either “known unknowns” or “unknown unknowns” (Wishart, 2009). The former corresponds to a metabolite that has been previously detected but has not yet been identified, while the latter corresponds to a truly novel metabolite that has never been formally identified. Schymanski et al. (2012) have shown that consensus structure elucidation using a combination of GC-EI-MS, structure generation, and physicochemical properties calculated from unknown compounds may be applicable to the characterization of unknown metabolites. Kumari et al. (2011) evaluated a novel de novo workflow for the annotation of unknown metabolites using accurate mass data, PubChem queries, RI matching, and structure constraints. To predict elemental compositions from accurate mass data collected from high-resolution mass spectrometers, “Seven Golden Rules” (Kind and Fiehn, 2007) and MFSearcher9 are available. Krumsiek et al. (2012) demonstrated that the integration of metabolite profiling with genome-wide association studies (GWAS) on metabolic quantitative traits is very useful for deriving biochemical pathways for unknown metabolites. In addition, several groups have attempted to classify unidentified MSTs using supervised machine learning approaches, including decision tree (Hummel et al., 2010) and soft independent modeling of class analogies (SIMCA) (Tsugawa et al., 2011). For structural characterization, there are recent powerful approaches by comparing mass spectral fragmentation trees (Rasche et al., 2011; Hufsky et al., 2012; Rojas-Cherto et al., 2012) (see also the review by Xiao et al., 2012). To evaluate whether detected peaks are biochemically produced by organisms, an in vivo13C-labeling system has been used with 13CO2 in metabolite profiling using both GC-MS (Huege et al., 2007) and LC-MS (Giavalisco et al., 2009). The method allows for the rejection of non-biological peaks and improved annotation of elemental composition. Because artificial biological gradients developed by Redestig et al. (2011) can evaluate actual concentration differences of metabolite peaks detected in two different types of samples (e.g., leaves and fruits), this allows to filter out all unavoidable artifacts in MST/MS2T data. Such a method will make it possible to reject analytical artifacts and prioritize unknown candidate metabolites for further characterization.
Statistical Models, Pathway Information, and Data Interpretation
Uni- and Multivariate Analysis
To perform extensive data analysis such as PCA, MetaGeneAlyse (Daub et al., 2003) and MetaboAnalyst (Xia et al., 2012) are available. Conceptually, these are very similar web-based applications for the analysis of high-throughput omics data. MetaGeneAlyse implements standard normalization/clustering methods, e.g., k-means, and independent component analysis (ICA). MetaboAnalyst provides many statistical methods, including t-tests, partial least square discriminant analysis (PLSDA), pathway enrichment analysis, and additional machine learning methods. Please note that several tools and databases presented in this review have multiple functions. Furthermore, several web-based applications for metabolomic data are available (Table 1), such as MetaMapp (Barupal et al., 2012), metaP-Server (Kastenmuller et al., 2011), MeltDB (Neuweger et al., 2008), and MetiTree (Rojas-Cherto et al., 2012). They cover multiple steps from data pre-processing to biological interpretation.
Metabolite Pathway Analysis
Metabolite data, which contain information about metabolite name and changes of metabolite levels/relationships, can be described in pathways or networks. For example, the nodes represent metabolites and the edges represent biochemical reactions. Well-curated database for metabolic pathways in plants are available, such as KEGG (Kanehisa et al., 2012) and AraCyc (Zhang et al., 2005). MetaCrop stores well-curated information for 60 major metabolic pathways in eight crop plants, as well as Arabidopsis (Schreiber et al., 2012). UniPathway (Morgat et al., 2012) and SMPDB (Frolkis et al., 2010) also provide well-curated information about metabolic pathways. Tools involving pathway analysis and enrichment analysis are also available, such as AraPath (Lai et al., 2012), Kappa-view (Tokimatsu et al., 2005; Sakurai et al., 2011), and MapMan (Usadel et al., 2009) (Table 1). For detailed information about these tools, see the excellent review by Chagoyen and Pazos (2012).
Mathematical Model Information and Other Tool
Genome-Scale Metabolism Reconstruction
Over the past few decades, a significant number of metabolic reconstructions have been performed in many organisms, for example, SEED servers (Aziz et al., 2012). Currently, several genome-scale metabolic models in plants are available for evaluating metabolic behavior based on the alteration of metabolic pathways (Table 1) (Collakova et al., 2012; De Oliveira Dal’molin and Nielsen, 2012; Seaver et al., 2012). Poolman et al. (2009) constructed such a metabolism model in Arabidopsis to characterize possible flux behaviors using flux balance analysis (FBA) (Orth et al., 2010;Sweetlove and Ratcliffe, 2011). Instead of using metabolic flux analysis (MFA) (for example, see the reviews by Libourel and Shachar-Hill, 2008; Allen et al., 2009), this analysis can predict steady-state flux distribution by using a linear programing. AraGEM is also another metabolic reconstruction of Arabidopsis metabolism (De Oliveira Dal’molin et al., 2010). Radrich et al. (2010) semi-automatically integrated multiple databases involving metabolic pathways to reconstruct Arabidopsis metabolism. A compartmentalized, reconstructed metabolic model of Arabidopsis is also currently available (Mintz-Oron et al., 2012). Combinations of theoretical and experimental approaches will pave the way for robust interpretation of metabolomic data and practical metabolic engineering in plants.
Tools for Metabolite Identifiers
Managing compound identifiers in metabolomic data analysis is important. MSI also proposed the use of database identifiers for peer-reviewed papers, for example, the most common compound identifiers, including CAS, KEGG COMPOUND, CHEBI, and HMDB. The Chemical Translation Service (CTS) (Wohlgemuth et al., 2010) and MetMask (Redestig et al., 2010) are a conversion tool for chemical identifiers (Table 1). The former is a web-based tool for performing batch conversions of compound identifiers, while the latter is a stand-alone command line program for integrating the most common compound identifiers. Metab2MeSH (Sartor et al., 2012) is a web application for annotating compounds with Medical Subject Headings (MeSH), which is a controlled vocabulary. Controlled vocabulary means well defined index term is used for indexing journal articles. Metab2MeSH links from metabolites to the biomedical research literature, PubChem, and HMDB. These tools in this subsection are helpful for reporting metabolomic data.
Metabolite-Profiling-Oriented Information
In addition to mass spectrum and compound databases, several metabolite-profiling databases have also been developed in the past few years (Table 1). Among these, PlantMetabolomics.org (Bais et al., 2010, 2012; Quanbeck et al., 2012) and Medicinal Plant Metabolomics Resource (MPMR) (Wurtele et al., 2012) are one of the most important databases. These contain metabolomic information for >140 Arabidopsis mutants and 14 medicinal plants based on MS data from multiple laboratories (Bais et al., 2012). Their profiling broadly covers a wide range of metabolites relating to amino and fatty acids, organic acids, phytosterols, isoprenoids, lipids, and secondary metabolites. PlantMetabolomics.org and MPMR also provide multiple data analysis tools including data normalization and visualization. Using these tools investigators can generate testable hypotheses with respect to gene functions in Arabidopsis (Quanbeck et al., 2012). Another example is Chloroplast 2010, which contains data related to large-scale phenotypic screening of Arabidopsis chloroplast mutants (Lu et al., 2011), based on assays of amino acids and fatty acids in leaves and seeds using GC-MS and LC-MS (Gu et al., 2007; Bell et al., 2012). Recently, we constructed the MeKO database10 (Fukushima et al., submitted), which is similar in concept to PlantMetabolomics.org. MeKO contains metabolomic information on 50 Arabidopsis mutants, including plants with uncharacterized gene functions. The website also provides MSI-compliant data, experimental meta-data, and the results of statistical data analyses such as differential accumulation compared with wild-type plants (Columbia ecotype). These databases are very useful for functional genomics and make it possible to develop additional bioinformatic tools for pre-processing of metabolomics raw data, extraction of biologically meaningful mass spectra, and reduction/correction of unwanted variation in large-scale metabolomic data.
Conclusion
In this review, we have highlighted an extensive list of databases that incorporate both MS-based metabolomics, as well as data analysis tools. Clearly, a small, but significant, number of integrated databases, including the full annotation of metabolites, metabolite profiling, and data analysis tools are emerging, such as PlantMetabolomics.org (Quanbeck et al., 2012). In addition to those for plants, metabolome databases for bacteria and animals also exist (see Table 1). Increases in metabolomic data sharing and the improvement of technological capabilities, such as database integration, are likely to play important roles in the future development of plant metabolomics, and facilitate advances plant systems biology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research activity of Atsushi Fukushima is partly supported by a Grant-in-Aid for Young Scientists (B; grant no. 23700355 to Atsushi Fukushima) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Footnotes
- ^http://riodb01.ibase.aist.go.jp/sdbs/
- ^http://webs2.kazusa.or.jp/massbase/
- ^http://code.google.com/p/setupx/
- ^http://sourceforge.net/projects/binbase/
- ^http://massfinder.com/wiki/Terpenoids_Library
- ^http://bio.massbank.jp/
- ^http://www.cas.org/
- ^http://metabolomics.jp/
- ^http://webs2.kazusa.or.jp/mfsearcher/index.html
- ^http://prime.psc.riken.jp/meko
References
Adams, R. P. (2007). Identification of Essential Oil Components by Gas Chromatography-Quadrupole Mass Spectroscopy, 4th Edn. Carol Stream, IL: Allured Pub Corp.
Afendi, F. M., Okada, T., Yamazaki, M., Hirai-Morita, A., Nakamura, Y., Nakamura, K., et al. (2012). KNApSAcK family databases: integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 53, e1.
Allen, D. K., Libourel, I. G., and Shachar-Hill, Y. (2009). Metabolic flux analysis in plants: coping with complexity. Plant Cell Environ. 32, 1241–1257.
Arita, M., and Suwa, K. (2008). Search extension transforms wiki into a relational system: a case for flavonoid metabolite database. BioData Min. 1, 7.
Aziz, R. K., Devoid, S., Disz, T., Edwards, R. A., Henry, C. S., Olsen, G. J., et al. (2012). SEED servers: high-performance access to the seed genomes, annotations, and metabolic models. PLoS ONE 7:e48053. doi:10.1371/journal.pone.0048053
Bais, P., Moon, S. M., He, K., Leitao, R., Dreher, K., Walk, T., et al. (2010). PlantMetabolomics.org: a web portal for plant metabolomics experiments. Plant Physiol. 152, 1807–1816.
Bais, P., Moon-Quanbeck, S. M., Nikolau, B. J., and Dickerson, J. A. (2012). Plantmetabolomics.org: mass spectrometry-based Arabidopsis metabolomics – database and tools update. Nucleic Acids Res. 40, D1216–D1220.
Baker, J. M., Hawkins, N. D., Ward, J. L., Lovegrove, A., Napier, J. A., Shewry, P. R., et al. (2006). A metabolomic study of substantial equivalence of field-grown genetically modified wheat. Plant Biotechnol. J. 4, 381–392.
Barupal, D. K., Haldiya, P. K., Wohlgemuth, G., Kind, T., Kothari, S. L., Pinkerton, K. E., et al. (2012). MetaMapp: mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinformatics 13:99. doi:10.1186/1471-2105-13-99
Bell, S. M., Burgoon, L. D., and Last, R. L. (2012). MIPHENO: data normalization for high throughput metabolite analysis. BMC Bioinformatics 13:10. doi:10.1186/1471-2105-13-10
Caldana, C., Degenkolbe, T., Cuadros-Inostroza, A., Klie, S., Sulpice, R., Leisse, A., et al. (2011). High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 67, 869–884.
Carreno-Quintero, N., Acharjee, A., Maliepaard, C., Bachem, C. W., Mumm, R., Bouwmeester, H., et al. (2012). Untargeted metabolic quantitative trait loci analyses reveal a relationship between primary metabolism and potato tuber quality. Plant Physiol. 158, 1306–1318.
Carroll, A. J., Badger, M. R., and Harvey Millar, A. (2010). The MetabolomeExpress project: enabling web-based processing, analysis and transparent dissemination of GC/MS metabolomics datasets. BMC Bioinformatics 11:376. doi:10.1186/1471-2105-11-376
Catchpole, G. S., Beckmann, M., Enot, D. P., Mondhe, M., Zywicki, B., Taylor, J., et al. (2005). Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proc. Natl. Acad. Sci. U.S.A. 102, 14458–14462.
Chagoyen, M., and Pazos, F. (2012). Tools for the functional interpretation of metabolomic experiments. Brief. Bioinformatics (in press).
Collakova, E., Yen, J. Y., and Senger, R. S. (2012). Are we ready for genome-scale modeling in plants? Plant Sci. 19, 53–70.
Daub, C. O., Kloska, S., and Selbig, J. (2003). MetaGeneAlyse: analysis of integrated transcriptional and metabolite data. Bioinformatics 19, 2332–2333.
De Oliveira Dal’molin, C. G., and Nielsen, L. K. (2012). Plant genome-scale metabolic reconstruction and modelling. Curr. Opin. Biotechnol. 24, 271–277.
De Oliveira Dal’molin, C. G., Quek, L. E., Palfreyman, R. W., Brumbley, S. M., and Nielsen, L. K. (2010). AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 152, 579–589.
De Vos, R. C., Moco, S., Lommen, A., Keurentjes, J. J., Bino, R. J., and Hall, R. D. (2007). Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2, 778–791.
Degtyarenko, K., De Matos, P., Ennis, M., Hastings, J., Zbinden, M., Mcnaught, A., et al. (2008). ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 36, D344–D350.
Espinoza, C., Degenkolbe, T., Caldana, C., Zuther, E., Leisse, A., Willmitzer, L., et al. (2010). Interaction with diurnal and circadian regulation results in dynamic metabolic and transcriptional changes during cold acclimation in Arabidopsis. PLoS ONE 5:e14101. doi:10.1371/journal.pone.0014101
Fahy, E., Sud, M., Cotter, D., and Subramaniam, S. (2007). LIPID MAPS online tools for lipid research. Nucleic Acids Res. 35, W606–W612.
Fernie, A. R. (2012). Grand challenges in plant systems biology: closing the circle(s). Front. Plant Sci. 3:35. doi:10.3389/fpls.2012.00035
Fernie, A. R., Aharoni, A., Willmitzer, L., Stitt, M., Tohge, T., Kopka, J., et al. (2011). Recommendations for reporting metabolite data. Plant Cell 23, 2477–2482.
Fiehn, O. (2001). Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genomics 2, 155–168.
Fiehn, O. (2002). Metabolomics – the link between genotypes and phenotypes. Plant Mol. Biol. 48, 155–171.
Fiehn, O., Robertson, D., Griffin, J. L., Van Der Werf, M., Nikolau, B., Morrison, N., et al. (2007). The metabolomics standards initiative (MSI). Metabolomics 3, 175–178.
Frolkis, A., Knox, C., Lim, E., Jewison, T., Law, V., Hau, D. D., et al. (2010). SMPDB: the small molecule pathway database. Nucleic Acids Res. 38, D480–D487.
Fukushima, A., Kusano, M., Nakamichi, N., Kobayashi, M., Hayashi, N., Sakakibara, H., et al. (2009a). Impact of clock-associated Arabidopsis pseudo-response regulators in metabolic coordination. Proc. Natl. Acad. Sci. U.S.A. 106, 7251–7256.
Fukushima, A., Kusano, M., Redestig, H., Arita, M., and Saito, K. (2009b). Integrated omics approaches in plant systems biology. Curr. Opin. Chem. Biol. 13, 532–538.
Giavalisco, P., Kohl, K., Hummel, J., Seiwert, B., and Willmitzer, L. (2009). 13C isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Anal. Chem. 81, 6546–6551.
Gibon, Y., Usadel, B., Blaesing, O. E., Kamlage, B., Hoehne, M., Trethewey, R., et al. (2006). Integration of metabolite with transcript and enzyme activity profiling during diurnal cycles in Arabidopsis rosettes. Genome Biol. 7, R76.
Gu, L., Jones, A. D., and Last, R. L. (2007). LC-MS/MS assay for protein amino acids and metabolically related compounds for large-scale screening of metabolic phenotypes. Anal. Chem. 79, 8067–8075.
Hall, R. D. (2006). Plant metabolomics: from holistic hope, to hype, to hot topic. New Phytol. 169, 453–468.
Haug, K., Salek, R. M., Conesa, P., Hastings, J., De Matos, P., Rijnbeek, M., et al. (2012). MetaboLights – an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 41, D781–D786.
Horai, H., Arita, M., Kanaya, S., Nihei, Y., Ikeda, T., Suwa, K., et al. (2010). MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass. Spectrom. 45, 703–714.
Huege, J., Sulpice, R., Gibon, Y., Lisec, J., Koehl, K., and Kopka, J. (2007). GC-EI-TOF-MS analysis of in vivo carbon-partitioning into soluble metabolite pools of higher plants by monitoring isotope dilution after 13CO2 labelling. Phytochemistry 68, 2258–2272.
Hufsky, F., Rempt, M., Rasche, F., Pohnert, G., and Bocker, S. (2012). De novo analysis of electron impact mass spectra using fragmentation trees. Anal. Chim. Acta 739, 67–76.
Hummel, J., Strehmel, N., Selbig, J., Walther, D., and Kopka, J. (2010). Decision tree supported substructure prediction of metabolites from GC-MS profiles. Metabolomics 6, 322–333.
Iijima, Y., Nakamura, Y., Ogata, Y., Tanaka, K., Sakurai, N., Suda, K., et al. (2008). Metabolite annotations based on the integration of mass spectral information. Plant J. 54, 949–962.
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114.
Kaplan, F., Kopka, J., Haskell, D. W., Zhao, W., Schiller, K. C., Gatzke, N., et al. (2004). Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol. 136, 4159–4168.
Kastenmuller, G., Romisch-Margl, W., Wagele, B., Altmaier, E., and Suhre, K. (2011). metaP-server: a web-based metabolomics data analysis tool. J. Biomed. Biotechnol. 2011:839862. doi:10.1155/2011/839862
Kind, T., and Fiehn, O. (2007). Seven golden rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics 8:105. doi:10.1186/1471-2105-8-105
Kind, T., Wohlgemuth, G., Do, Y., Lu, Y., Palazoglu, M., Shahbaz, S., et al. (2009). FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 81, 10038–10048.
Kopka, J., Schauer, N., Krueger, S., Birkemeyer, C., Usadel, B., Bergmuller, E., et al. (2005).R01EQENTQi5EQg==: the Golm metabolome database. Bioinformatics 21, 1635–1638.
Krumsiek, J., Suhre, K., Evans, A. M., Mitchell, M. W., Mohney, R. P., Milburn, M. V., et al. (2012). Mining the unknown: a systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 8:e1003005. doi:10.1371/journal.pgen.1003005
Kumar, A., Suthers, P. F., and Maranas, C. D. (2012). MetRxn: a knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinformatics 13:6. doi:10.1186/1471-2105-13-6
Kumari, S., Stevens, D., Kind, T., Denkert, C., and Fiehn, O. (2011). Applying in-silico retention index and mass spectra matching for identification of unknown metabolites in accurate mass GC-TOF mass spectrometry. Anal. Chem. 83, 5895–5902.
Kusano, M., Redestig, H., Hirai, T., Oikawa, A., Matsuda, F., Fukushima, A., et al. (2011a). Covering chemical diversity of genetically-modified tomatoes using metabolomics for objective substantial equivalence assessment. PLoS ONE 6:e16989. doi:10.1371/journal.pone.0016989
Kusano, M., Tabuchi, M., Fukushima, A., Funayama, K., Diaz, C., Kobayashi, M., et al. (2011b). Metabolomics data reveal a crucial role of cytosolic glutamine synthetase 1;1 in coordinating metabolic balance in rice. Plant J. 66, 456–466.
Kusano, M., Tohge, T., Fukushima, A., Kobayashi, M., Hayashi, N., Otsuki, H., et al. (2011c). Metabolomics reveals comprehensive reprogramming involving two independent metabolic responses of Arabidopsis to UV-B light. Plant J. 67, 354–369.
Lai, L., Liberzon, A., Hennessey, J., Jiang, G., Qi, J., Mesirov, J. P., et al. (2012). AraPath: a knowledgebase for pathway analysis in Arabidopsis. Bioinformatics 28, 2291–2292.
Lang, M., Stelzer, M., and Schomburg, D. (2011). BKM-react, an integrated biochemical reaction database. BMC Biochem. 12:42. doi:10.1186/1471-2091-12-42
Lei, Z., Huhman, D. V., and Sumner, L. W. (2011). Mass spectrometry strategies in metabolomics. J. Biol. Chem. 286, 25435–25442.
Lenz, E. M., Bright, J., Knight, R., Wilson, I. D., and Major, H. (2004). Cyclosporin A-induced changes in endogenous metabolites in rat urine: a metabonomic investigation using high field 1H NMR spectroscopy. J. Pharm. Biomed. Anal. 35, 599–608.
Libourel, I. G., and Shachar-Hill, Y. (2008). Metabolic flux analysis in plants: from intelligent design to rational engineering. Annu. Rev. Plant Biol. 59, 625–650.
Lisec, J., Meyer, R. C., Steinfath, M., Redestig, H., Becher, M., Witucka-Wall, H., et al. (2008). Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J. 53, 960–972.
Lisec, J., Schauer, N., Kopka, J., Willmitzer, L., and Fernie, A. R. (2006). Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 1, 387–396.
Lu, Y., Savage, L. J., Larson, M. D., Wilkerson, C. G., and Last, R. L. (2011). Chloroplast 2010: a database for large-scale phenotypic screening of Arabidopsis mutants. Plant Physiol. 155, 1589–1600.
Matsuda, F., Hirai, M. Y., Sasaki, E., Akiyama, K., Yonekura-Sakakibara, K., Provart, N. J., et al. (2010). AtMetExpress development: a phytochemical atlas of Arabidopsis development. Plant Physiol. 152, 566–578.
Matsuda, F., Okazaki, Y., Oikawa, A., Kusano, M., Nakabayashi, R., Kikuchi, J., et al. (2012). Dissection of genotype-phenotype associations in rice grains using metabolome quantitative trait loci analysis. Plant J. 70, 624–636.
Matsuda, F., Yonekura-Sakakibara, K., Niida, R., Kuromori, T., Shinozaki, K., and Saito, K. (2009). MS/MS spectral tag-based annotation of non-targeted profile of plant secondary metabolites. Plant J. 57, 555–577.
Meyer, R. C., Steinfath, M., Lisec, J., Becher, M., Witucka-Wall, H., Torjek, O., et al. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 104, 4759–4764.
Mintz-Oron, S., Meir, S., Malitsky, S., Ruppin, E., Aharoni, A., and Shlomi, T. (2012). Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 339–344.
Moco, S., Bino, R. J., Vorst, O., Verhoeven, H. A., De Groot, J., Van Beek, T. A., et al. (2006). A liquid chromatography-mass spectrometry-based metabolome database for tomato. Plant Physiol. 141, 1205–1218.
Morgat, A., Coissac, E., Coudert, E., Axelsen, K. B., Keller, G., Bairoch, A., et al. (2012). UniPathway: a resource for the exploration and annotation of metabolic pathways. Nucleic Acids Res. 40, D761–D769.
Morreel, K., Goeminne, G., Storme, V., Sterck, L., Ralph, J., Coppieters, W., et al. (2006). Genetical metabolomics of flavonoid biosynthesis in Populus: a case study. Plant J. 47, 224–237.
Nakabayashi, R., Sawada, Y., Yamada, Y., Suzuki, M., Hirai, M. Y., Sakurai, T., et al. (2013). Combination of liquid chromatography-fourier transform ion cyclotron resonance-mass spectrometry with (13)c-labeling for chemical assignment of sulfur-containing metabolites in onion bulbs. Anal. Chem. 85, 1310–1315.
Neuweger, H., Albaum, S. P., Dondrup, M., Persicke, M., Watt, T., Niehaus, K., et al. (2008). MeltDB: a software platform for the analysis and integration of metabolomics experiment data. Bioinformatics 24, 2726–2732.
Obata, T., and Fernie, A. R. (2012). The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 69, 3225–3243.
Orth, J. D., Thiele, I., and Palsson, B. O. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248.
Pence, H. E., and Williams, A. (2010). ChemSpider: an online chemical information resource. J. Chem. Educ. 87, 1123–1124.
Poolman, M. G., Miguet, L., Sweetlove, L. J., and Fell, D. A. (2009). A genome-scale metabolic model of Arabidopsis and some of its properties. Plant Physiol. 151, 1570–1581.
Quanbeck, S. M., Brachova, L., Campbell, A. A., Guan, X., Perera, A., He, K., et al. (2012). Metabolomics as a hypothesis-generating functional genomics tool for the annotation of Arabidopsis thaliana genes of “unknown function.” Front. Plant Sci. 3:15. doi:10.3389/fpls.2012.00015
Radrich, K., Tsuruoka, Y., Dobson, P., Gevorgyan, A., Swainston, N., Baart, G., et al. (2010). Integration of metabolic databases for the reconstruction of genome-scale metabolic networks. BMC Syst. Biol. 4:114. doi:10.1186/1752-0509-4-114
Rasche, F., Svatos, A., Maddula, R. K., Bottcher, C., and Bocker, S. (2011). Computing fragmentation trees from tandem mass spectrometry data. Anal. Chem. 83, 1243–1251.
Redestig, H., Kobayashi, M., Saito, K., and Kusano, M. (2011). Exploring matrix effects and quantification performance in metabolomics experiments using artificial biological gradients. Anal. Chem. 83, 5645–5651.
Redestig, H., Kusano, M., Fukushima, A., Matsuda, F., Saito, K., and Arita, M. (2010). Consolidating metabolite identifiers to enable contextual and multi-platform metabolomics data analysis. BMC Bioinformatics 11:214. doi:10.1186/1471-2105-11-214
Ricroch, A. E., Berge, J. B., and Kuntz, M. (2011). Evaluation of genetically engineered crops using transcriptomic, proteomic, and metabolomic profiling techniques. Plant Physiol. 155, 1752–1761.
Roessner, U., Luedemann, A., Brust, D., Fiehn, O., Linke, T., Willmitzer, L., et al. (2001). Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 13, 11–29.
Rojas-Cherto, M., Peironcely, J. E., Kasper, P. T., Van Der Hooft, J. J., De Vos, R. C., Vreeken, R., et al. (2012). Metabolite identification using automated comparison of high-resolution multistage mass spectral trees. Anal. Chem. 84, 5524–5534.
Rowe, H. C., Hansen, B. G., Halkier, B. A., and Kliebenstein, D. J. (2008). Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell 20, 1199–1216.
Saito, K., and Matsuda, F. (2010). Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 61, 463–489.
Sakurai, N., Ara, T., Ogata, Y., Sano, R., Ohno, T., Sugiyama, K., et al. (2011). KaPPA-View4: a metabolic pathway database for representation and analysis of correlation networks of gene co-expression and metabolite co-accumulation and omics data. Nucleic Acids Res. 39, D677–D684.
Sakurai, T., Yamada, Y., Sawada, Y., Matsuda, F., Akiyama, K., Shinozaki, K., et al. (2013). PRIMe update: innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol. 54, e5.
Sansone, S. A., Fan, T., Goodacre, R., Griffin, J. L., Hardy, N. W., Kaddurah-Daouk, R., et al. (2007). The metabolomics standards initiative. Nat. Biotechnol. 25, 846–848.
Sartor, M. A., Ade, A., Wright, Z., States, D., Omenn, G. S., Athey, B., et al. (2012). Metab2MeSH: annotating compounds with medical subject headings. Bioinformatics 28, 1408–1410.
Sato, S., Arita, M., Soga, T., Nishioka, T., and Tomita, M. (2008). Time-resolved metabolomics reveals metabolic modulation in rice foliage. BMC Syst. Biol. 2:51. doi:10.1186/1752-0509-2-51
Sawada, Y., Nakabayashi, R., Yamada, Y., Suzuki, M., Sato, M., Sakata, A., et al. (2012). RIKEN tandem mass spectral database (ReSpect) for phytochemicals: a plant-specific MS/MS-based data resource and database. Phytochemistry 82, 38–45.
Scalbert, A., Andres-Lacueva, C., Arita, M., Kroon, P., Manach, C., Urpi-Sarda, M., et al. (2011). Databases on food phytochemicals and their health-promoting effects. J. Agric. Food Chem. 59, 4331–4348.
Schauer, N., Semel, Y., Roessner, U., Gur, A., Balbo, I., Carrari, F., et al. (2006). Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 24, 447–454.
Schauer, N., Steinhauser, D., Strelkov, S., Schomburg, D., Allison, G., Moritz, T., et al. (2005). GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 579, 1332–1337.
Schreiber, F., Colmsee, C., Czauderna, T., Grafahrend-Belau, E., Hartmann, A., Junker, A., et al. (2012). MetaCrop 2.0: managing and exploring information about crop plant metabolism. Nucleic Acids Res. 40, D1173–D1177.
Schymanski, E. L., Gallampois, C. M., Krauss, M., Meringer, M., Neumann, S., Schulze, T., et al. (2012). Consensus structure elucidation combining GC/EI-MS, structure generation, and calculated properties. Anal. Chem. 84, 3287–3295.
Seaver, S. M., Henry, C. S., and Hanson, A. D. (2012). Frontiers in metabolic reconstruction and modeling of plant genomes. J. Exp. Bot. 63, 2247–2258.
Skogerson, K., Wohlgemuth, G., Barupal, D. K., and Fiehn, O. (2011). The volatile compound BinBase mass spectral database. BMC Bioinformatics 12:321. doi:10.1186/1471-2105-12-321
Stein, S. E. (1999). An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry data. J. Am. Soc. Mass Spectrom. 10, 770–781.
Steinbeck, C., Conesa, P., Haug, K., Mahendraker, T., Williams, M., Maguire, E., et al. (2012). MetaboLights: towards a new COSMOS of metabolomics data management. Metabolomics 8, 757–760.
Stitt, M., and Fernie, A. R. (2003). From measurements of metabolites to metabolomics: an ‘on the fly’ perspective illustrated by recent studies of carbon-nitrogen interactions. Curr. Opin. Biotechnol. 14, 136–144.
Sulpice, R., Pyl, E. T., Ishihara, H., Trenkamp, S., Steinfath, M., Witucka-Wall, H., et al. (2009). Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. U.S.A. 106, 10348–10353.
Sumner, L. W., Amberg, A., Barrett, D., Beale, M. H., Beger, R., Daykin, C. A., et al. (2007). Proposed minimum reporting standards for chemical analysis. Metabolomics 3, 211–221.
Sweetlove, L. J., and Ratcliffe, R. G. (2011). Flux-balance modeling of plant metabolism. Front. Plant Sci. 2:38. doi:10.3389/fpls.2011.00038
Tautenhahn, R., Cho, K., Uritboonthai, W., Zhu, Z., Patti, G. J., and Siuzdak, G. (2012). An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 30, 826–828.
Tohge, T., and Fernie, A. R. (2009). Web-based resources for mass-spectrometry-based metabolomics: a user’s guide. Phytochemistry 70, 450–456.
Tokimatsu, T., Sakurai, N., Suzuki, H., Ohta, H., Nishitani, K., Koyama, T., et al. (2005). KaPPA-view: a web-based analysis tool for integration of transcript and metabolite data on plant metabolic pathway maps. Plant Physiol. 138, 1289–1300.
Tsugawa, H., Tsujimoto, Y., Arita, M., Bamba, T., and Fukusaki, E. (2011). GC/MS based metabolomics: development of a data mining system for metabolite identification by using soft independent modeling of class analogy (SIMCA). BMC Bioinformatics 12:131. doi:10.1186/1471-2105-12-131
Udayakumar, M., Chandar, D. P., Arun, N., Mathangi, J., Hemavathi, K., and Seenivasagam, R. (2012). PMDB: plant f database – a metabolomic approach. Med. Chem. Res. 21, 47–52.
Urano, K., Maruyama, K., Ogata, Y., Morishita, Y., Takeda, M., Sakurai, N., et al. (2009). Characterization of the ABA-regulated global responses to dehydration in Arabidopsis by metabolomics. Plant J. 57, 1065–1078.
Urbanczyk-Wochniak, E., Baxter, C., Kolbe, A., Kopka, J., Sweetlove, L. J., and Fernie, A. R. (2005). Profiling of diurnal patterns of metabolite and transcript abundance in potato (Solanum tuberosum) leaves. Planta 221, 891–903.
Usadel, B., Poree, F., Nagel, A., Lohse, M., Czedik-Eysenberg, A., and Stitt, M. (2009). A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species. Maize. Plant Cell Environ. 32, 1211–1229.
Wang, Y., Xiao, J., Suzek, T. O., Zhang, J., Wang, J., and Bryant, S. H. (2009). PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 37, W623–W633.
Watanabe, K., Yasugi, E., and Oshima, M. (2000). How to search the glycolipid data in LIPIDBANK for Web: the newly developed lipid database. Jpn. Trend Glycosci. Glycotechnol. 12, 175–184.
Weckwerth, W. (2011). Unpredictability of metabolism – the key role of metabolomics science in combination with next-generation genome sequencing. Anal. Bioanal. Chem. 400, 1967–1978.
Weckwerth, W., Loureiro, M. E., Wenzel, K., and Fiehn, O. (2004). Differential metabolic networks unravel the effects of silent plant phenotypes. Proc. Natl. Acad. Sci. U.S.A. 101, 7809–7814.
Wishart, D. S. (2009). Computational strategies for metabolite identification in metabolomics. Bioanalysis 1, 1579–1596.
Wishart, D. S., Knox, C., Guo, A. C., Eisner, R., Young, N., Gautam, B., et al. (2009). HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 37, D603–D610.
Wohlgemuth, G., Haldiya, P. K., Willighagen, E., Kind, T., and Fiehn, O. (2010). The chemical translation service – a web-based tool to improve standardization of metabolomic reports. Bioinformatics 26, 2647–2648.
Wurtele, E. S., Chappell, J., Jones, A. D., Celiz, M. D., Ransom, N., Hur, M., et al. (2012). Medicinal plants: a public resource for metabolomics and hypothesis development. Metabolites 2, 1031–1059.
Xia, J., Mandal, R., Sinelnikov, I. V., Broadhurst, D., and Wishart, D. S. (2012). MetaboAnalyst 2.0 – a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 40, W127–W133.
Xiao, J. F., Zhou, B., and Ressom, H. W. (2012). Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trends. Analyt. Chem. 32, 1–14.
Zhang, P., Foerster, H., Tissier, C. P., Mueller, L., Paley, S., Karp, P. D., et al. (2005). MetaCyc and AraCyc. Plant Physiol. 138, 27–37.
Keywords: metabolomics, metabolite profiling, plant metabolism, hypothesis generation, database
Citation: Fukushima A and Kusano M (2013) Recent progress in the development of metabolome databases for plant systems biology. Front. Plant Sci. 4:73. doi: 10.3389/fpls.2013.00073
Received: 19 December 2012; Accepted: 15 March 2013;
Published online: 04 April 2013.
Edited by:
Wolfram Weckwerth, University of Vienna, AustriaReviewed by:
Kris Morreel, University Ghent, BelgiumCamila Caldana, Brazilian Bioethanol Science and Technology Laboratory, Brazil
Copyright: © 2013 Fukushima and Kusano. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Atsushi Fukushima, RIKEN Plant Science Center, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan. e-mail:YS1mdWt1c2hAcHNjLnJpa2VuLmpw