

- Corn Insects and Crop Genetics Research, Department of Plant Pathology and Microbiology, US Department of Agriculture - Agricultural Research Service, Center for Plant Responses to Environmental Stresses, Iowa State University, Ames, IA, USA
Rusts are one of the most severe threats to cereal crops because new pathogen races emerge regularly, resulting in infestations that lead to large yield losses. In 1999, a new race of stem rust, Puccinia graminis f. sp. tritici (Pgt TTKSK or Ug99), was discovered in Uganda. Most of the wheat and barley cultivars grown currently worldwide are susceptible to this new race. Pgt TTKSK has already spread northward into Iran and will likely spread eastward throughout the Indian subcontinent in the near future. This scenario is not unique to stem rust; new races of leaf rust (Puccinia triticina) and stripe rust (Puccinia striiformis) have also emerged recently. One strategy for countering the persistent adaptability of these pathogens is to stack complete- and partial-resistance genes, which requires significant breeding efforts in order to reduce deleterious effects of linkage drag. These varied resistance combinations are typically more difficult for the pathogen to defeat, since they would be predicted to apply lower selection pressure. Genetical genomics or expression Quantitative Trait Locus (eQTL) analysis enables the identification of regulatory loci that control the expression of many to hundreds of genes. Integrated deployment of these technologies coupled with efficient phenotyping offers significant potential to elucidate the regulatory nodes in genetic networks that orchestrate host defense responses. The focus of this review will be to present advances in genetical genomic experimental designs and analysis, particularly as they apply to the prospects for discovering partial disease resistance alleles in cereals.
Introduction
The heteroecious rust fungi are some of the most important pathogens of cereal crops. These comprise 3000 Puccinia species (Van Der Merwe et al., 2007), including stem rust [Puccinia graminis f. sp. tritici (Pgt)], wheat leaf rust (Puccinia triticina), stripe rust (Puccinia striiformis), barley leaf rust (Puccinia hordei), and oat crown rust (Puccinia coronata). All of these species can infect a large range of cereal hosts: 365 grass species for Puccinia graminis alone.
Rust fungi pose a serious threat to cereal production because new races continue to emerge, and because infestation almost invariably leads to dramatic yield losses across large geographic areas (Leonard and Szabo, 2005; Bolton et al., 2008b). Breeding for genetic resistance to rusts reduces negative environmental impacts to agrosystems. In practice however, this approach is not infallible; the adaptability of rusts allows them to routinely overcome resistance gene (R) alleles bred into elite varieties (Singh et al., 2004a). Moreover, breeding efforts in response to new rust races does not always prevent crop loss, since wind dispersal of urediniospores can cause additional infections across continents in a short period of time (Hovmoller et al., 2008). Indeed, new races of leaf rust (Singh et al., 2004a), stripe rust (Milus et al., 2009), and stem rust (Stokstad, 2007) became widespread well before genetic resistance could be delivered in elite cultivars. Thus, the central challenge to the small grains industry is to reduce the periodicity of such outbreaks through renewed breeding efforts and continued management of epidemiological parameters that affect the evolution of pathogen virulence.
Stem rust, caused by the obligate fungal biotroph Pgt, has been a periodic, but serious problem wherever wheat and barley are grown (Roelfs, 1985; Leonard and Szabo, 2005). In North America, severe epidemics of stem rust have occurred from the late 1800's through the 1950's (http://www.globalrust.org). A new threat to wheat and barley production is the discovery of a novel race (Pgt TTKSK) of wheat stem rust in East Africa (Wanyera et al., 2006; Stokstad, 2007). This race, commonly called Ug99, is virulent to the majority of wheat varieties grown as well as advanced lines in current breeding programs (Jin and Singh, 2006; Bonman et al., 2007; Jin et al., 2007). Pgt TTKSK, and races of this lineage, infect barley as well as wheat, and has the potential to spread throughout the Middle East and to the Indian subcontinent in the near future (Singh et al., 2008). To overcome these new threats, additional resistances are needed in the short term, but more importantly, substantial new research efforts will be required in order to identify durable resistance to rusts over the long term (Ayliffe et al., 2008).
Genetic and/or molecular identification of novel sources of rust resistance in small grains will be greatly facilitated by recent gains in our basic knowledge of plant defense mechanisms. Plants detect the presence of the pathogen by two interconnected mechanisms (Jones and Dangl, 2006). One mechanism takes advantage of a specific response of the plant host induced by pathogen effectors; historically, these have has been referred to as gene-for-gene interactions (Flor, 1971), or in current terms, effector triggered immunity (ETI) (Jones and Dangl, 2006). This mechanism depends on direct or indirect recognition between pathogen effector (avirulence) proteins and plant R proteins (Innes, 2004; Deyoung and Innes, 2006; Deyoung et al., 2012). Another mechanism, designated PAMP triggered immunity (PTI), is induced by general elicitors or PAMPs (pathogen-associated-molecular-patterns), and is characterized by basal defense responses (Chisholm et al., 2006; Bent and Mackey, 2007). Based on this general doctrine, different strategies have been used to implement disease resistance in crops.
Genetic Strategies for Deployment of Host Resistance in Crops
The two primary genetic strategies for identification of disease resistance alleles useful for breeding are to focus on complete resistance conferred by R genes, or to focus on partial resistance that can be identified using quantitative genetic approaches. Significant progress has been made in identifying R genes in wheat and barley. More than 40 R genes that activate defense in response to Puccinia triticina and 40 R genes against Pgt have been mapped in wheat, as well as 20 R genes against Puccinia striiformis that have been mapped in wheat or barley (McIntosh et al., 1995; Ayliffe et al., 2008). Despite the dramatic success of these longstanding efforts, only a few R genes conferring resistance to a cereal rust have been cloned and functionally characterized: Rpg1 (Brueggeman et al., 2002) and the rpg4/Rpg5 complex (Brueggeman et al., 2008, 2009; Kleinhofs et al., 2009; Wang et al., 2013) in barley, Rp1 (Collins et al., 1999) and Rp3 (Webb et al., 2002) in maize, as well as Lr21 (Huang et al., 2003) and Lr10 (Feuillet et al., 2003) in wheat. While R genes tend to confer quite strong resistance to rusts in these cereal hosts, their efficacy in agronomic systems has the potential to be overcome by dynamic and rapidly evolving pathogen populations.
As such, a good strategy for countering the persistent adaptability of rusts is to deploy a combination of quantitative and qualitative resistance alleles. This strategy is typically more complicated to implement, but has the advantage of being more difficult to defeat, given that the various combinations are effective against a broader spectrum of races and thus, are believed to apply lower selection pressures (Singh et al., 2004b). Consequently, an agronomic phenotype often sought by breeders is non-specific partial resistance, or “slow rusting.”
Partial resistance loci are difficult to identify for three reasons. First, quantitative measurement of symptoms such as the length of latent period, pustule size and spore production require significant expertise and effort. Second, Quantitative trait locus (QTL) analyses aimed at identifying partial resistance loci require large population sizes in order to detect these less obvious effects (Singh et al., 2004b). Finally, specific combinations of alleles are often required in order for a partial resistance locus to display sufficient penetrance, which makes the parentage of the experimental populations critically important (Simmonds, 1988). Despite these challenges, two such partial resistance loci have recently been cloned, Yr36 (Fu et al., 2009) and Lr34 (Krattinger et al., 2009). These genes both define new classes of resistance genes, encoding a kinase with a putative START lipid-binding domain and an adenosine triphosphate-binding cassette transporter, respectively. Under field conditions, Yr36 and Lr34 confer quantitative levels of adult plant resistance and are expected to provide durable resistance to rusts in wheat. It should be noted that cloning these partial resistance genes required careful planning and large-scale execution of breeding strategies designed specifically for this type of effort (Simmonds, 1988).
Molecular Approaches to Identify Host Defense Genes and Regulators
The identification of genes that have the capacity to confer quantitative levels of disease resistance to multiple pathogen races is an important step toward reliable crop protection over the long term (Poland et al., 2009). Although much of our understanding of PAMP- and ETI-mediated defense has been achieved through classic forward genetic approaches (Shirasu et al., 1999; Deyoung and Innes, 2006), there is great potential to combine these strategies with genome- or population-wide analysis of host transcriptomes during interactions with pathogens (Wise et al., 2007).
One of the exciting outcomes of these fundamental advances on host-pathogen interaction is the degree to which this basic knowledge is transferrable from one system to another. Regulators implicated in a specific interaction have been shown to play important roles in pathogen interactions across several species (Bent and Mackey, 2007; Shirasu, 2009). For example, Rar1, first identified in barley (Shirasu et al., 1999), has functional orthologs in Arabidopsis (Muskett et al., 2002), tobacco (Liu et al., 2002), rice (Thao et al., 2007), and wheat (Tai, 2008). Another example is the Arabidopsis PBS1 kinase, which is targeted by AvrPphB, a cysteine protease effector from Pseudomonas syringae pv. phaseolicola (Zhu et al., 2004). Cleavage of PBS1 by AvrPphB activates RPS5-specified resistance (Deyoung et al., 2012). PBS1 is widely conserved in monocots and dicots. Hence, a clear challenge is to accelerate discovery of such regulators, such that natural variants or transgenic alleles of these genes can be exploited (Innes, 2004; Deyoung and Innes, 2006; Shirasu, 2009; Deyoung et al., 2012).
Several cereal rust interactions have been investigated using parallel expression approaches (Table 1); not only to identify genes in particular defense pathways, but also to compare transcriptome reprogramming between mutants and their progenitors in order to clone defense regulators (Zhang et al., 2006) or genes involved in broad-spectrum resistance to stem rust (Zhang et al., 2008b). Upon comparison of the genes identified in these different experiments (Table 1), it was observed that the major classes of rust-responsive genes are similar with genes that are responsive during other biotic interactions. These include, but are not limited to genes that encode peroxidases, chitinases, pathogenesis related (PR) proteins, MAPK kinases, kinases, WRKY transcription factors, transport proteins, and proteins transported to chloroplasts. However, the regulation and the kinetics of expression for these genes may be vastly different, depending on the specific host-pathogen interaction that has been interrogated (Wise et al., 2007). A deeper comparative approach should facilitate discovery of common defense pathways between barley and wheat during interactions with rusts.
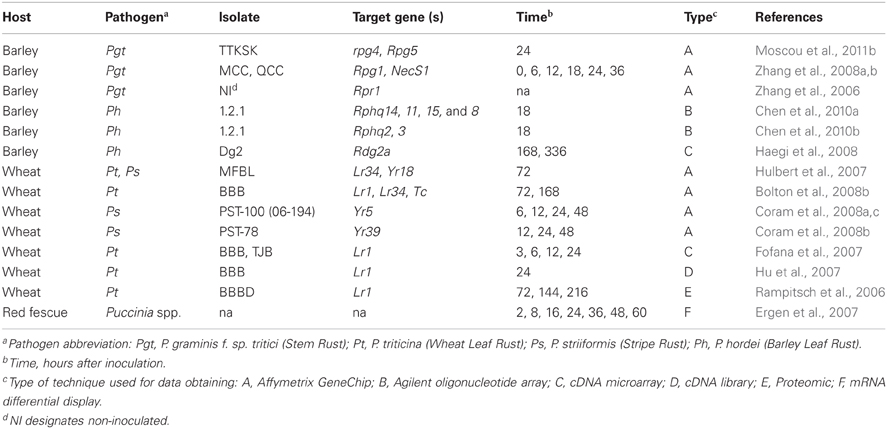
Table 1. Investigations of differentially expressed genes or proteins during interaction between rust and cereal crops.
Approaches such as expression correlation and protein-protein interaction studies have facilitated the construction of defense pathways, but a clear understanding of the larger network of defense pathways and how they overlap remains elusive. The best documented defense network is based on work performed on Arabidopsis thaliana (Consortium, 2011; Mukhtar et al., 2011). Several reviews approach the defense response as a whole (Hammond-Kosack and Parker, 2003; Hofius et al., 2007), while others take a more focused approach (Mittler et al., 2004). Unfortunately, in cereals, graphical representations that efficiently foster a collective understanding are for the most part lacking, although several authors have reported pathway inferences from microarray experiments in rice (Cooper et al., 2003; Qiu et al., 2008). However, in these types of experiments, it is not possible to distinguish between cause and effect among correlated nodes. Several questions typically arise when co-expression networks are viewed: How can the key regulators of the pathway be identified? Are there connections between these regulators? Can the results of this network help a breeder make decisions? If so, what type of follow-up efforts should be pursued? Fortunately, a genetical genomic approach can provide the solution to these shortcomings. Surprisingly though, there has been little utilization of this approach in plant-pathogen interaction studies, despite overwhelming success when applied in human and animal disease research (de Koning and Haley, 2005; Mozhui et al., 2008).
Genetical Genomics Offers New Horizons to Investigate Plant Defense Mechanisms
QTL mapping finds statistical associations between genotypes and phenotypes, allowing regions of the genome harboring allelic differences that cause variation in the phenotype to be identified; these regions are called QTLs (reviewed by Mackay, 2001). Transcript abundance of a single gene is a quantitative trait and its regulation can be genetically interrogated. This is often called genetical genomics, or expression Quantitative Trait Locus (eQTL) mapping because the phenotypes in question are the expression of individual genes (Kendziorski and Wang, 2006; Rockman and Kruglyak, 2006). With the availability of high quality gene-expression platforms for barley (Close et al., 2004; Chen et al., 2010b), wheat (Schreiber et al., 2009), Puccinia spp. genome sequences (Cantu et al., 2011; Duplessis et al., 2011), as well as emerging next generation sequencing technologies (Li et al., 2010, 2013; Mayer et al., 2012), new strategies can be envisaged that interrogate both host and pathogen on a genome-wide, as well as a population-based scale.
The use of an eQTL strategy to identify and/or clone phenotypic QTL is well documented (Hansen et al., 2008). By profiling gene expression in each member of a segregating population, it is possible to use linkage analyses to identify key regulators of gene expression for a particular condition (Jansen and Nap, 2001; Rockman and Kruglyak, 2006; Williams et al., 2007; Kliebenstein, 2009; Li et al., 2013). For example, eQTL analysis of transcript abundance in embryo-derived tissues in barley has been combined with a QTL experiment on stem rust resistance in the same population (Druka et al., 2008). Three major QTL were detected in this population: two of them correspond to the known resistance genes Rpg1 and Rpg5/rpg4, on chromosome 7H and 5H, respectively, and a third QTL was found on chromosome 2H. The cloned resistance gene Rpg1 was detected as one of the best candidate genes to underlie the QTL detected on chromosome 7H, thereby substantiating the eQTL strategy for the candidate gene approach. Other strong candidates were detected for the two other loci. In another study, Moscou et al. (2011b) used an eQTL approach to identify a master regulator on chromosome 2H that controls hundreds of genes in response to Ug99 stem rust. In collaboration with U.S. and Kenyan partners, they showed that rpg4/Rpg5-mediated adult plant resistance is enhanced by allelic variants of the regulator. Thus, eQTL analysis is a useful strategy to identify and clone genes whose allelic variation results in phenotypic variation.
The eQTL strategy can also be applied to characterize gene networks or to confirm biological pathways (Keurentjes et al., 2007; Sonderby et al., 2007). For example, the Arabidopsis gene ERECTA is known to act pleiotropically on several pathways, including flowering time and resistance to bacterial wilt (Godiard et al., 2003). The role played by ERECTA in flowering time was confirmed using an eQTL mapping approach; in addition, new connections and regulatory nodes in ERECTA-specified pathways were identified (Keurentjes et al., 2007). Another interesting example is the MYB transcription factor MYB28, which was confirmed as a regulator of aliphatic glucosinolate, a defense metabolite in Brassicales (Sonderby et al., 2007). Several regulators have now been identified in plant/pathogen interactions. Network analysis should confirm the role played by these regulators, thus, developing a more complete picture of plant defense pathways.
Genetical genomics can also be used to investigate the heritability of gene expression, as well as the basis for transgressive segregation, where progeny phenotypes are statistically outside the range that would be predicted by parental phenotypes (Keurentjes et al., 2007; West et al., 2007; Li et al., 2013). Transgressive segregation has been measured in two plant studies (Keurentjes et al., 2007; West et al., 2007). In both cases ~50% of the genes show a significant difference in gene expression between parents and progeny in the population. This difference can be ascribed to the reassortment of additive genetic effects contributed by both parents, or to epistatic interactions among eQTL. Transgressive segregation is one of the reasons why some loci are difficult to use by breeders. Indeed, there are several cases of resistance genes that do not show the same phenotype in parental varieties as compared to their progenies. Understanding this phenomenon was required in order identify the suppressor of the Lr34 leaf rust resistance gene in wheat (Vanegas et al., 2008). In another case, the Lr13 leaf rust resistance gene is known to enhance resistance only in the presence of Lr17 (Kolmer, 1992). The strength of eQTL analysis is that it provides the capacity to explain such complexities in a single experiment, rather than merely identifying the challenge.
Challenges to Designing, Executing, and Analyzing an eQTL Experiment
Several recent reviews focus on the advances in genetical genomics (Doerge, 2002; Rockman and Kruglyak, 2006; Rosa et al., 2006; Williams et al., 2007; Wise et al., 2007; Gilad et al., 2008; Kliebenstein, 2009, 2010). To rapidly and efficiently discover partial or quantitative resistance alleles, one goal for future studies should be to understand how key regulators can modify gene expression, both temporally and spatially, during pathogen challenge and subsequent infection. In this section, we will outline parameters of experimental design to investigate this question, including factors that can influence the mapping process such as: population, experimental procedures and statistical analysis.
Population Types
Several factors determine the utility of a population for any given QTL mapping application, all of which are related to pedigree and/or population size (Lauter et al., 2008). Pedigree determines which alleles are contrasted, how many alleles per locus are tested, which modes of action can be investigated, and the degree of genetic resolution that is achievable. Population size partially determines genetic resolution and largely controls the level of statistical power that is available to accurately determine modes of allele action, including the detection of epistasis.
Most QTL studies in plants investigate allelic contrasts between only two alleles, which at some point in the pedigree were present in a single F1 plant. Common population types include F2, recombinant inbred line (RIL), intermated recombinant inbred lines (iRIL), double haploids (DHs), and back cross (BC). Based on simulation studies, a well-developed RIL population appears to be the most efficient for accurate QTL mapping (Ferreira et al., 2006). This makes sense, because during development of the population, each generation produces an additional round of meiotic recombination: i.e., R = 2r × (1 + 2r)−1, where r is the recombination frequency in the corresponding F2 (Burr and Burr, 1991). RIL populations, as well as DH and iRIL populations, also have the advantage of isogenicity, permitting experimental replication and testing of treatment effects without further genotyping.
A limitation of working with isogenic and true breeding lines is that the mode of allele action can not be determined, such that effects of recessive, dosage dependent, and dominant QTL alleles are indistinguishable without further analysis. This is a bigger limitation for breeding hybrid crops such as maize, where discovery of dominant QTL is preferred, than it is for inbred crops such as wheat or barley. Although limiting genotypic complexity is a drawback for allele characterization, it is experimentally beneficial in other regards. More statistical power to detect recessive and epistatic effects exists in a RIL population than in an F2 population of equivalent size. Imagine an extreme phenotype conditioned only by recessive allele action at three independent loci: in an F2 population, only one in 64 plants will have this genotype, compared to one in every eight plants in a RIL population. Detection of the main effects as well as the epistatic interdependence of these three hypothetical loci could only be revealed in an F2 study if a very large number of plants were used.
Population Size
Increasing the population size for a QTL experiment increases statistical power for both detection and localization of effects. Improved statistical power comes from larger numbers of lines or plants representing a particular genotype, while increased resolving power comes from additional recombination events that more closely flank the loci of interest. However, increased population size comes at a price. Thus, the challenge is to optimize population size as a function of price per significant gain in understanding the trait. Unfortunately, a priori determination of a minimum population size required for a particular level of success is largely an intuitive exercise. In combination with the population type, the mode of inheritance for the trait in question plays a prominent role in determining the genetic tractability of a trait. For example, a polygenic trait (in the classical sense) is much more difficult to dissect than one whose inheritance architecture is oligogenic (Lauter et al., 2004). For this reason, it is helpful to know the phenotypic distribution of the trait in order to have some indication of the underlying genotypic cause of something other than a normal phenotypic distribution.
Are the Genes Determining Resistance Qualitative or Quantitative?
A common case in plant pathological investigations, the presence of an effective R gene, illustrates this point quite clearly. It is common for partial resistance alleles to be more easily detected when they enhance the function of a resistance gene that has a major effect. There are many possible explanations for this, but a simple and intuitive way to look at it is that it is easier to distinguish between completely disease free and slightly diseased than between “mostly dead” and “all dead.” Suppose then that detection (for whatever reason) of a partial resistance allele epistatically depends on the presence of the R gene allele that confers resistance (Wise et al., 1996; Yu et al., 2001): in a RIL population, only half of the lines are useful for isolating the effect of this locus. The phenotypic distribution for such a trait should be strongly bimodal, which could be used to improve the experimental design prior to spending the money for genotyping. Further breeding and selection of a subset of lines for use are two of the simple solutions for this case.
Optimizing Populations
There have been some efforts to empirically determine the point of diminishing returns for manipulation of population size. Several simulation studies have predicted that a population of 200 RILs is required for a statistically accurate analysis (Kim et al., 2005; Silva et al., 2007). However, most of the time, such populations only allow the detection of phenotypic QTL with major effects, a severe limitation when partial resistance alleles are the target for discovery (de Koning and Haley, 2005). There are several useful tricks for overcoming the population size limitation without breaking the bank. One use of a genetical genomics approach is to identify key transcriptional regulators that likely harbor the genetic variation underlying phenotypic QTL. Accomplishing this feat requires good resolution of both the QTL and eQTL effects. The best way to globally improve genetic resolution without increasing population size is to intermate progenies during population construction. This breaks up linkage blocks without introducing additional alleles by capitalizing on successive rounds of recombination. The addition of four generations of intermating to a maize population breeding effort has been shown to provide up to 50-fold gains in genetic resolution (Balint-Kurti et al., 2007; Lauter et al., 2008). Another way to enhance resolution is to capitalize on evolutionary recombination events, as has been done with the nested association mapping (NAM) population for maize (Yu et al., 2008). However, the per-line gain in resolution of this approach is not yet clear. Moreover, the simultaneous use of many alleles in a partial resistance search is not advisable unless the nesting parent (B73 in the case of maize NAM) harbors an allele for which suppressors and enhancers are sought.
Increasing global resolution and power is often not the focus of an investigator's effort. Many pathologists already have ideal allelic contrasts ready for eQTL dissection with appropriate genetic materials, but simply wish to limit the effective population size in favor of improving replication. There are tricks for this as well. Potokina and associates (2008b) show that when the focus is on a particular phenotypic QTL, subsets of lines can be selected based on known recombination events and allelic composition to improve efficiency. To some degree, this strategy is generalizable any time an excess of previously genotyped lines exists (Rosa et al., 2006). Such selective phenotyping approaches sample the population in a way that minimizes segregation distortion while maximizing global recombination rate, thereby increasing both power and precision on a per line basis (de Koning et al., 2007). Indeed, the power of eQTL detection can be increased if a subset of the population is chosen for its genetic dissimilarity without a commensurate decrease in mapping power (Yan et al., 2006). Another appealing approach is to select two different subsets of the population for two different treatments (Li et al., 2008). In this way, it's possible to divide the population into two subsets with similar genetic background. During subsequent statistical analysis, the subset of the population used in one treatment (mock inoculated) can be considered as a reference for the other subset of the population used in the other treatment (inoculated), effectively doubling the number of lines compared to a classical experimental design; significantly increasing mapping power, as has been shown recently by Moscou and colleagues (2011b).
Heritability
Since statistical power depends in part on heritability, the delicate balance of population size vs. replication needs to be optimized based on experimental goals. In general, when de novo detection of minor QTL effects is a primary goal, replication is much more important than when the aim is to more finely map a known minor effect locus. In our view, increasing population size in an eQTL experiment is its own form of replication, similar to how replicated evaluations of F3 families had previously been a norm for QTL mapping in plants (Cowen, 1988). The additional lines provide the benefit of new recombination events, while population measures such as mean and variance can be used to assess what proportion of total variance should be heritable. If a QTL allele cannot be reliably detected in a good experimental design, it is difficult to imagine how it will be of agronomic value.
Several wheat and barley populations have been used to map QTL for stem rust, leaf rust and stripe rust (Singh et al., 2004b; Druka et al., 2008; Chen et al., 2010a; Moscou et al., 2011b). However, small population sizes restrict the utility of these populations at present. A wider sampling of wheat and barley alleles would also provide stronger foundations for future research. Therefore, additional populations, preferably iRILs, should be created to allow discovery and fine mapping of new partial resistance loci. Particular attention must be paid to the R gene and QTL alleles carried when parents for these populations are selected.
Treatment and Statistical Design for an eQTL Experiment
Considering the statistical design details of an eQTL experiment, two principle questions should be resolved early: what alleles will be contrasted and under what conditions, where conditions need to include treatment, the tissue to be dissected, as well as the selection of the desired timepoint.
The scientific objectives generally pursued in molecular plant pathology are to find genes that capacitate critical steps in the specific interaction between host and pathogen. There are many possible experimental approaches available, including contrasts of host vs. non-host interactions, virulent vs. avirulent isolates, inoculated vs. mock-inoculated treatments, mutant vs. wild-type genotypes. Treatment design strongly affects the selection of alleles to be contrasted for pathological studies, as virulence vs. avirulence is so often controlled by gene-for-gene interactions between the pathogen and the host. Control treatments can also be beneficial, but are not necessarily required if the goal is to identify allelic differences that affect the regulation of transcription, rather than to characterize under what conditions the observed regulation occurs. In pathological experiments, non-inoculated control treatment often allow the researchers to distinguish between consequences of inoculation vs. infection, which can be essential for limiting the number of candidate genes that appear to regulate whether or not infection occurs (Moscou et al., 2011a,b).
In pathological experiments, timepoint needs to be considered as a function of plant development as well as a function of infection kinetics following controlled inoculation. It is important to know the kinetics of the interaction prior to designing an eQTL experiment, particularly if reducing costs is essential. The timing of events after inoculation have been measured for stem rust (Zhang et al., 2008a,b), leaf rust (Bolton et al., 2008a; Chen et al., 2010a), and stripe rust (Coram et al., 2008c). Penetration begins 12 hours after inoculation (HAI), haustorium formation at 18 HAI, and intercellular hyphal growth at 24 HAI (Sellam and Wilcoxson, 1976; Lin et al., 1998). The choice of timepoint within these parameters affects the outcome of the experiment. If resources were not limiting, an attractive option for a complete linkage and network analysis of infection would be to measure the expression of each individual of the population at each time point. In this way, one could track the eQTL actions as a temporal function of the interaction.
The tissue taken for RNA extraction should also be chosen carefully. Indeed, because only a small portion of the cells interact directly with the pathogen, the transcriptomic approach generally considers a population of cells, and thus, of an RNA mixture between interacting and non-interacting cells. For practical reasons, most microarray studies have used a seedling leaf 1- or 2-weeks old, with the assumption that the use of replicates would increase statistical power of small differences in expression. Yet, separation of even different leaf parts can be enlightening; in one study involving the interactions among wheat and wheat leaf rust and/or wheat stripe rust, significant differential gene expression was observed between basal and apical sections of each leaf (Hulbert et al., 2007).
Similarly, but not surprisingly, Potokina et al. (2008a) demonstrated that different eQTLs could be identified in embryo vs. seedling leaf tissue in barley. Wherever possible, one should collect tissue that is as narrowly defined as possible while minimizing perturbations to homeostasis (Li et al., 2010). In barley and wheat, epidermal peels are quite easy to isolate from the rest of the leaf; one can do so quickly and without wounding them such that the gain in specificity potentially outweighs the potential error introduced by manipulation. While this approach has proven useful for powdery mildew investigations (Zierold et al., 2005), no simple approach exists for rust research. Laser capture micro-dissection would be an option, but it would seem that this would be more appropriate if the experiment was focused on how infection and defense signaling are propagated across cell types.
Challenges in Statistical Analysis
Several reviews have described methods for statistical analysis of microarray data and subsequent eQTL mapping procedures (Manly et al., 2004; Kendziorski and Wang, 2006; Williams et al., 2006; Jiang et al., 2008). First, microarray data are normalized in order to provide the phenotypic data for the expression level of each gene. If a linkage map already exists, eQTL mapping can commence immediately. Alternatively, microarray data can be used to create transcript-derived markers (TDMs) that can in turn be used to construct a genetic linkage map (Luo et al., 2007; Potokina et al., 2008b; Druka et al., 2010; Moscou et al., 2011b). This feature of eQTL mapping alone can often make the effort worthwhile; even with inexpensive genotyping, it is often cost effective in the long term to generate a TDM map of several thousand markers, which usually ensures that any two recombination events have a marker between them and that locations of genetic cross-overs are well-defined. TDMs can be integrated with Genotyping by Sequencing (GBS) approaches to anchor genetic maps to physical maps (Poland et al., 2012; Sonah et al., 2013).
Surprisingly, eQTL mapping is actually the simple portion of eQTL analysis work. The more difficult part is to figure out how to identify trends and meaningful patterns and in such a large volume of data, typically >10,000 regulatory relationships between an eQTL locus and the transcript it regulates. One of the most important things to establish is whether an eQTL acts in cis or trans, i.e., does the regulatory difference that leads to differential expression exist at the locus where the gene resides (cis) or elsewhere (trans). This level of characterization helps define the putative function of the eQTL, promoter difference vs. transcription factor difference, for example. Also, when searching for genes that act as capacitors of a significant process, it is helpful to know if many genes involved in a process are regulated by a locus where few or none of them reside. Such loci are termed eQTL hotspots and can regulate more than 1000 genes in some cases (West et al., 2007; Potokina et al., 2008a; Moscou et al., 2011b). Identification of eQTL hotspots is an effective way toward building gene networks, especially if one can identify a locus that regulates a cluster of genes associated with the biological phenomenon of interest, such as disease defense (Chen et al., 2010a; Moscou et al., 2011b). In barley and wheat, determining cis vs. trans is becoming clearer, with the recent emergence of genome sequence resources with many genes tied to a genetic and/or physical position (Brenchley et al., 2012; Mayer et al., 2012). Synteny coordinates in rice or Brachypodium can assist in such analyses. In addition, such an approach to hotspot detection can be enhanced by seeding the analysis only with genes that have at least two eQTL, which ensures that at least one of them must be acting in trans.
Beyond the many possible methods to identify eQTL hotspots (Williams et al., 2007), the nature of their action remains unclear: could they be gene-dense regions where recombination is limited, or do they actually exist as the signature of a high-level regulatory gene (Breitling et al., 2008)? Notably, the statistical models described to date do not consider the influence of gene function, co-location patterns or co-expression. Plant resistance and defense genes are often clustered in the genome; the influence of such genomic organization on eQTL detection needs to be integrated into the statistical analysis. Indeed, simulations have shown that expression correlation can explain a large part of eQTL co-localization (Wang et al., 2007). Fortunately, permutation tests should be able to assess the reliability of putative eQTL hotspots (Breitling et al., 2008).
Some of the statistical challenges could also be viewed as opportunities. When it is not possible to determine which force is acting, it should be true that tracking of either phenomena can lead to the source. For example, co-location of several hotspots that regulate genes with correlated expression patterns could lead to more robust eQTL detection. Indeed, within an eQTL hotspot, one of the best candidates for the “master regulator” may be a gene whose expression correlates with the other genes whose eQTL have mapped to the same locus. This parsimonious inference has been applied to further test the predicted flowering time network in Arabidopsis (Keurentjes et al., 2007) and also between particular eQTL and transcription factors in yeast (Sun et al., 2007). For plant pathological applications, many transcription factors regulating defense are known, facilitating application of this approach.
Epistasis
Epistasis has been shown to have an impact on numerous major phenotypic QTL and will likely explain significant variance components of plant gene expression (Rowe and Kliebenstein, 2008). However, the epistatic interdependencies of gene expression have generally been neglected in plant eQTL analysis studies (Sun et al., 2008). While such tests require more statistical power than is usually available, future populations and experimental designs are sure to be more powerful, so a challenge to build the analysis infrastructure and to improve definition of the statistical underpinnings for large-scale tests for epistasis lie clearly before us. It will be necessary to define common criteria and performance measures for such analyses in order to permit the establishment of a collective intuition that is meaningful for evaluation of inferences. To this end, the routine sharing of data and analysis methods in a database such as PLEXdb (http://plexdb.org/) (Dash et al., 2012), WebQTL, or GeneNetwork (http://www.genenetwork.org/) facilitates these goals.
Concluding Remarks
Genetical genomics offers a new approach to the study of plant pathogen interactions. This systems biology approach leverages the complementary strengths of classical genetics and transcriptomics to connect loci that confer resistance with gene expression networks that are responsive to infection. Given these complementary strengths, it is incumbent upon the community to have the vision to perform definitive experiments to associate cause and effect. In this way, these experiments will facilitate the identification and cloning of new loci as well as known phenotypic QTL.
For the near term, the community has only some of these resources in hand, so the challenge is to carry out beneficial experiments with existing resources, while continuing to develop the next generation of tools to answer critical questions. These include: To what extent do polymorphisms in transcription factors and the promoter regions with which they interact govern the outcome of plant defense? What has the highest influence on gene regulation: polymorphism in regulators, polymorphism in downstream pathway, or variability in the environment? What is the evolution of defense regulators and how are they maintained in populations?
Suggested experiments to begin to answer these questions should connect kinetics of pathogen infection with responsive host genes and regulatory networks. In order to best track individual eQTL through the interaction, an optimal experimental plan would require: (1) high resolution population(s) that harbor genetic variation for resistance to the pathogen—intermated RILs should provide the resolution, while simultaneously providing a reasonable number of individuals for downstream molecular work; (2) all-genes expression-profiling platforms for the hosts and pathogens in question—with NextGen sequencing technologies, these are becoming possible at a reasonable cost; (3) high-throughput genotyping—several platforms offer the possibility of genotyping individuals with multiplex capability (Poland et al., 2012; Sonah et al., 2013), and (4) detailed infection phenotyping—with a reasonable number of intermated RILs, response to multiple pathogens or isolates could realistically be accomplished. In addition to these host parameters, one could dramatically increase the power of the investigation if equivalent resources (population, expression profiling, genotyping) were in place for the pathogen. In that case, exploration of “all by all” (segregating host by segregating pathogen) could be pursued.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Matthew Moscou for helpful advice and ideas on genetical genomics. This research was supported in part by National Science Foundation Plant Genome grants 05-00461 and 09-22746, and USDA-ARS CRIS project 3625-21000-057-00D. Elsa Ballini was supported by a USDA-ARS Postdoctoral Research Associateship. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Abbreviations
eQTL, (gene) expression quantitative trait locus; PAMP, pathogen-associated-molecular-pattern; ETI, effector triggered immunity; PTI, PAMP triggered immunity; DH, doubled haploid; RIL, recombinant inbred line, TDM, transcript-derived marker.
References
Ayliffe, M., Singh, R., and Lagudah, E. (2008). Durable resistance to wheat stem rust needed. Curr. Opin. Plant Biol. 11, 187–192.
Balint-Kurti, P. J., Zwonitzer, J. C., Wisser, R. J., Carson, M. L., Oropeza-Rosas, M. A., Holland, J. B., et al. (2007). Precise mapping of quantitative trait loci for resistance to southern leaf blight, caused by Cochliobolus heterostrophus race O, and flowering time using advanced intercross maize lines. Genetics 176, 645–657.
Bent, A. F., and Mackey, D. (2007). Elicitors, effectors, and R genes: the new paradigm and a lifetime supply of questions. Annu. Rev. Phytopathol. 45, 399–436.
Bolton, M. D., Kolmer, J. A., and Garvin, D. F. (2008a). Wheat leaf rust caused by Puccinia triticina. Mol. Plant Pathol. 9, 563–575.
Bolton, M. D., Kolmer, J. A., Xu, W. W., and Garvin, D. F. (2008b). Lr34-mediated leaf rust resistance in wheat: transcript profiling reveals a high energetic demand supported by transient recruitment of multiple metabolic pathways. Mol. Plant Microbe Interact. 21, 1515–1527.
Bonman, J. M., Bockelman, H. E., Jin, Y., Hijmans, R. J., and Gironella, A. I. N. (2007). Geographic distribution of stem rust resistance in wheat landraces. Crop Sci. 47, 1955–1963.
Breitling, R., Li, Y., Tesson, B. M., Fu, J., Wu, C., Wiltshire, T., et al. (2008). Genetical genomics: spotlight on QTL hotspots. PLoS Genet. 4:e1000232. doi: 10.1371/journal.pgen.1000232
Brenchley, R., Spannagl, M., Pfeifer, M., Barker, G. L. A., D'Amore, R., Allen, A. M., et al. (2012). Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705–710.
Brueggeman, R., Druka, A., Nirmala, J., Cavileer, T., Drader, T., Rostoks, N., et al. (2008). The stem rust resistance gene Rpg5 encodes a protein with nucleotide-binding-site, leucine-rich, and protein kinase domains. Proc. Natl. Acad. Sci. U.S.A. 105, 14970–14975.
Brueggeman, R., Rostoks, N., Kudrna, D., Kilian, A., Han, F., Chen, J., et al. (2002). The barley stem rust-resistance gene Rpg1 is a novel disease-resistance gene with homology to receptor kinases. Proc. Natl. Acad. Sci. U.S.A. 99, 9328–9333.
Brueggeman, R., Steffenson, B. J., and Kleinhofs, A. (2009). The rpg4/Rpg5 stem rust resistance locus in barley: resistance genes and cytoskeleton dynamics. Cell Cycle 8, 977–981.
Burr, B., and Burr, F. A. (1991). Recombinant inbreds for molecular mapping in maize: theoretical and practical considerations. Trends Genet. 7, 55–60.
Cantu, D., Govindarajulu, M., Kozik, A., Wang, M., Chen, X., Kojima, K. K., et al. (2011). Next generation sequencing provides rapid access to the genome of Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust. PLoS ONE 6:e24230. doi: 10.1371/journal.pone.0024230
Chen, X., Hackett, C., Niks, R., Hedley, P., Booth, C., Druka, A., et al. (2010a). An eQTL analysis of partial resistance to Puccinia hordei in barley. PLoS ONE 5:e8598. doi: 10.1371/journal.pone.0008598
Chen, X., Niks, R., Hedley, P., Morris, J., Druka, A., Marcel, T., et al. (2010b). Differential gene expression in nearly isogenic lines with QTL for partial resistance to Puccinia hordei in barley. BMC Genomics 11:629. doi: 10.1186/1471-2164-11-629
Chisholm, S. T., Coaker, G., Day, B., and Staskawicz, B. J. (2006). Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124, 803–814.
Close, T. J., Wanamaker, S. I., Caldo, R. A., Turner, S. M., Ashlock, D. A., Dickerson, J. A., et al. (2004). A new resource for cereal genomics: 22K barley GeneChip comes of age. Plant Physiol. 134, 960–968.
Collins, N., Drake, J., Ayliffe, M., Sun, Q., Ellis, J., Hulbert, S., et al. (1999). Molecular characterization of the maize Rp1-D rust resistance haplotype and its mutants. Plant Cell 11, 1365–1376.
Consortium, A. I. M. (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607.
Cooper, B., Clarke, J. D., Budworth, P., Kreps, J., Hutchison, D., Park, S., et al. (2003). A network of rice genes associated with stress response and seed development. Proc. Natl. Acad. Sci. U.S.A. 100, 4945–4950.
Coram, T., Settles, M., Wang, M., and Chen, X. (2008a). Surveying expression level polymorphism and single-feature polymorphism in near-isogenic wheat lines differing for the Yr5 stripe rust resistance locus. Theor. Appl. Genet. 117, 401–411.
Coram, T. E., Settles, M. L., and Chen, X. (2008b). Transcriptome analysis of high-temperature adult-plant resistance conditioned by Yr39 during the wheat Puccinia striiformis f. sp. tritici interaction. Mol. Plant Pathol. 9, 479–493.
Coram, T. E., Wang, M. N., and Chen, X. M. (2008c). Transcriptome analysis of the wheat - Puccinia striiformis f. sp tritici interaction. Mol. Plant Pathol. 9, 157–169.
Cowen, N. M. (1988). The use of replicated progenies in marker-based mapping of QTLs. Theor. Appl. Genet. 75, 857–862.
Dash, S., Van Hemert, J., Hong, L., Wise, R. P., and Dickerson, J. A. (2012). PLEXdb: gene expression resources for plants and plant pathogens. Nucleic Acids Res. 40, D1194–D1201.
de Koning, D. J., Cabrera, C. P., and Haley, C. S. (2007). Genetical genomics: combining gene expression with marker genotypes in poultry. Poult. Sci. 86, 1501–1509.
de Koning, D. J., and Haley, C. S. (2005). Genetical genomics in humans and model organisms. Trends Genet. 21, 377–381.
Deyoung, B. J., and Innes, R. W. (2006). Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 7, 1243–1249.
Deyoung, B. J., Qi, D., Kim, S. H., Burke, T. P., and Innes, R. W. (2012). Activation of a plant nucleotide binding-leucine rich repeat disease resistance protein by a modified self protein. Cell Microbiol. 14, 1071–1084.
Doerge, R. W. (2002). Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 3, 43–52.
Druka, A., Potokina, E., Luo, Z., Bonar, N., Druka, I., Zhang, L., et al. (2008). Exploiting regulatory variation to identify genes underlying quantitative resistance to the wheat stem rust pathogen Puccinia graminis f. sp. tritici in barley. Theor. Appl. Genet. 117, 261–272.
Druka, A., Potokina, E., Luo, Z., Jiang, N., Chen, X., Kearsey, M., et al. (2010). Expression quantitative trait loci analysis in plants. Plant Biotechnol. J. 8, 10–27.
Duplessis, S., Cuomo, C. A., Lin, Y.-C., Aerts, A., Tisserant, E., Veneault-Fourrey, C., et al. (2011). Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. 108, 9166–9171.
Ergen, N. Z., Dinler, G., Shearman, R. C., and Budak, H. (2007). Identifying, cloning and structural analysis of differentially expressed genes upon Puccinia infection of Festuca rubra var. rubra. Gene 393, 145–152.
Ferreira, A., Silva, M. F. D., Silva, L. D. C. E., and Cruz, C. D. (2006). Estimating the effects of population size and type on the accuracy of genetic maps. Genet. Mol. Biol. 29, 187–192.
Feuillet, C., Travella, S., Stein, N., Albar, L., Nublat, A. L., and Keller, B. (2003). Map-based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc. Natl. Acad. Sci. U.S.A. 100, 15253–15258.
Fofana, B., Banks, T. W., McCallum, B. D., Strelkov, S. E., and Cloutier, S. (2007). Temporal gene expression profiling of the wheat leaf rust pathosystem using cDNA microarray reveals differences in compatible and incompatible defence pathways. Int. J. Plant Genomics, 2007:17542. doi: 10.1155/2007/17542
Fu, D. L., Uauy, C., Distelfeld, A., Blechl, A., Epstein, L., Chen, X. M., et al. (2009). A Kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science 323, 1357–1360.
Gilad, Y., Rifkin, S. A., and Pritchard, J. K. (2008). Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 24, 408–415.
Godiard, L., Sauviac, L., Torii, K. U., Grenon, O., Mangin, B., Grimsley, N. H., et al. (2003). ERECTA, an LRR receptor-like kinase protein controlling development pleiotropically affects resistance to bacterial wilt. Plant J. 36, 353–365.
Haegi, A., Bonardi, V., Dall'aglio, E., Glissant, D., Tumino, G., Collins, N. C., et al. (2008). Histological and molecular analysis of Rdg2a barley resistance to leaf stripe. Mol. Plant Pathol. 9, 463–478.
Hammond-Kosack, K. E., and Parker, J. E. (2003). Deciphering plant-pathogen communication: fresh perspectives for molecular resistance breeding. Curr. Opin. Biotechnol. 14, 177–193.
Hansen, B. G., Halkier, B. A., and Kliebenstein, D. J. (2008). Identifying the molecular basis of QTLs: eQTLs add a new dimension. Trends Plant Sci. 13, 72–77.
Hofius, D., Tsitsigiannis, D. I., Jones, J. D. G., and Mundy, J. (2007). Inducible cell death in plant immunity. Semin. Cancer Biol. 17, 166–187.
Hovmoller, M. S., Yahyaoui, A. H., Milus, E. A., and Justesen, A. F. (2008). Rapid global spread of two aggressive strains of a wheat rust fungus. Mol. Ecol. 17, 3818–3826.
Hu, G., Linning, R., McCallum, B. D., Banks, T. W., Cloutier, S., Butterfield, Y., et al. (2007). Generation of a wheat leaf rust, Puccinia triticina, EST database from stage-specific cDNA libraries. Mol. Plant Pathol. 8, 451–467.
Huang, L., Brooks, S. A., Li, W., Fellers, J. P., Trick, H. N., and Gill, B. S. (2003). Map-based cloning of leaf rust resistance gene Lr21 from the large and polyploid genome of bread wheat. Genetics 164, 655–664.
Hulbert, S. H., Bai, J., Fellers, J. P., Pacheco, M. G., and Bowden, R. L. (2007). Gene expression patterns in near isogenic lines for wheat rust resistance gene Lr34/Yr18. Phytopathology 97, 1083–1093.
Innes, R. W. (2004). Guarding the goods. New insights into the central alarm system of plants. Plant Physiol. 135, 695–701.
Jansen, R. C., and Nap, J.-P. (2001). Genetical genomics: the added value from segregation. Trends Genet. 17, 388–391.
Jiang, N., Leach, L., Hu, X., Potokina, E., Jia, T., Druka, A., et al. (2008). Methods for evaluating gene expression from Affymetrix microarray datasets. BMC Bioinformatics 9:284. doi: 10.1186/1471-2105-9-284
Jin, Y., Pretorius, Z. A., and Singh, R. P. (2007). New virulence within race TTKS (Ug99) of the stem rust pathogen and effective resistance genes. Phytopathology 97, S137.
Jin, Y., and Singh, R. P. (2006). Resistance in U.S. wheat to recent eastern African isolates of Puccinia graminis f. sp. tritici with virulence to resistance gene Sr31. Plant Dis. 90, 476–480.
Kendziorski, C., and Wang, P. (2006). A review of statistical methods for expression quantitative trait loci mapping. Mamm. Genome 17, 509–517.
Keurentjes, J. J. B., Fu, J., Terpstra, I. R., Garcia, J. M., Van Den Ackerveken, G., Snoek, L. B., et al. (2007). Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. U.S.A. 104, 1708–1713.
Kim, K., West, M. A. L., Michelmore, R. W., Clair, D. A. S., and Doerge, R. W. (2005). “Old methods for new ideas: Genetic dissection of the determinants of gene expression levels,” in Genome exploitation: data mining the genome. The 23rd volume in the Stadler Symposia, eds J. P. Gustafson, R. Shoemaker, and J. W. Snape (New York, NY: Springer), 89–105.
Kleinhofs, A., Brueggeman, R., Nirmala, J., Zhang, L., Mirlohi, A., Druka, A., et al. (2009). Barley stem rust resistance genes: structure and function. Plant Genome 2, 109–120.
Kliebenstein, D. (2009). Quantitative genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTLs. Annu. Rev. Plant Biol. 60, 93–114.
Kliebenstein, D. J. (2010). Systems biology uncovers the foundation of natural genetic diversity. Plant Physiol. 152, 480–486.
Kolmer, J. A. (1992). Enhanced leaf rust resistance in wheat conditioned by resistance gene pairs with Lr13. Euphytica 61, 123–130.
Krattinger, S. G., Lagudah, E. S., Spielmeyer, W., Singh, R. P., Huerta-Espino, J., McFadden, H., et al. (2009). A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323, 1360–1363.
Lauter, N., Gustus, C., Westerbergh, A., and Doebley, J. (2004). The inheritance and evolution of leaf pigmentation and pubescence in teosinte. Genetics 167, 1949–1959.
Lauter, N., Moscou, M. J., Habiger, J., and Moose, S. P. (2008). Quantitative genetic dissection of shoot architecture traits in maize: towards a functional genomics approach. Plant Genome 1, 99–110.
Leonard, K. J., and Szabo, L. J. (2005). Stem rust of small grains and grasses caused by Puccinia graminis. Mol. Plant Pathol. 6, 99–111.
Li, L., Petsch, K., Shimizu, R., Liu, S., Xu, W. W., Ying, K., et al. (2013). Mendelian and non-mendelian regulation of gene expression in maize. PLoS Genet. 9:e1003202. doi: 10.1371/journal.pgen.1003202
Li, P., Ponnala, L., Gandotra, N., Wang, L., Si, Y., Tausta, S. L., et al. (2010). The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 42, 1060–1067.
Li, Y., Breitling, R., and Jansen, R. C. (2008). Generalizing genetical genomics: getting added value from environmental perturbation. Trends Genet. 24, 518–524.
Lin, K. C., Bushnell, W. R., Smith, A. G., and Szabo, L. J. (1998). Temporal accumulation patterns of defence response gene transcripts in relation to resistant reactions in oat inoculated with Puccinia graminis. Physiol. Mol. Plant Pathol. 52, 95–114.
Liu, Y., Schiff, M., Marathe, R., and Dinesh-Kumar, S. P. (2002). Tobacco Rar1, EDS1 and NPR1/NIM1 like genes are required for N-mediated resistance to tobacco mosaic virus. Plant J. 30, 415–429.
Luo, Z. W., Potokina, E., Druka, A., Wise, R., Waugh, R., and Kearsey, M. J. (2007). SFP genotyping from Affymetrix arrays is robust but largely detects cis-acting expression regulators. Genetics 176, 789–800.
Mackay, T. F. C. (2001). The genetic architecture of quantitative traits. Annu. Rev. Genet. 35, 303–339.
Manly, K. F., Nettleton, D., and Hwang, J. T. G. (2004). Genomics, prior probability, and statistical tests of multiple hypotheses. Genome Res. 14, 997–1001.
Mayer, K., Waugh, R., Langridge, P., Close, T. J., Wise, R. P., Graner, A., et al. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716.
McIntosh, R. A., Wellings, C. R., and Park, R. F. (1995). Wheat rusts: an Atlas of Resistance Genes. London: Kluwer Academic Publishers.
Milus, E. A., Kristensen, K., and Hovmoller, M. S. (2009). Evidence for increased aggressiveness in a recent widespread strain of Puccinia striiformis f. sp tritici causing stripe rust of wheat. Phytopathology 99, 89–94.
Mittler, R., Vanderauwera, S., Gollery, M., and Van Breusegem, F. (2004). Reactive oxygen gene network of plants. Trends Plant Sci. 9, 490–498.
Moscou, M. J., Lauter, N., Caldo, R. A., Nettleton, D., and Wise, R. P. (2011a). Quantitative and temporal definition of the Mla transcriptional regulon during barley-powdery mildew interactions. Mol. Plant Microbe Interact. 24, 694–705.
Moscou, M. J., Lauter, N., Steffenson, B., and Wise, R. P. (2011b). Quantitative and qualitative stem rust resistance factors in barley are associated with transcriptional suppression of defense regulons. PLoS Genet. 7:e1002208. doi: 10.1371/journal.pgen.1002208
Mozhui, K., Ciobanu, D. C., Schikorski, T., Wang, X., Lu, L., and Williams, R. W. (2008). Dissection of a QTL hotspot on mouse distal chromosome 1 that modulates neurobehavioral phenotypes and gene expression. PLoS Genet. 4:e1000260. doi: 10.1371/journal.pgen.1000260
Mukhtar, M. S., Carvunis, A.-R., Dreze, M., Epple, P., Steinbrenner, J., Moore, J., et al. (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333, 596–601.
Muskett, P. R., Kahn, K., Austin, M. J., Moisan, L. J., Sadanandom, A., Shirasu, K., et al. (2002). Arabidopsis RAR1 exerts rate-limiting control of R gene-mediated defenses against multiple pathogens. Plant Cell 14, 979–992.
Parlevliet, J. E. (1978). Further evidence of polygenic inheritance of partial resistance in barley to leaf rust, Puccinia hordei. Euphytica 27, 369–379.
Poland, J. A., Balint-Kurti, P. J., Wisser, R. J., Pratt, R. C., and Nelson, R. J. (2009). Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 14, 21–29.
Poland, J. A., Brown, P. J., Sorrells, M. E., and Jannink, J.-L. (2012). Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253. doi: 10.1371/journal.pone.0032253
Potokina, E., Druka, A., Luo, Z., Moscou, M., Wise, R., Waugh, R., et al. (2008a). Tissue-dependent limited pleiotropy affects gene expression in barley. Plant J. 56, 287–296.
Potokina, E., Druka, A., Luo, Z., Wise, R., Waugh, R., and Kearsey, M. (2008b). Gene expression quantitative trait locus analysis of 16 000 barley genes reveals a complex pattern of genome-wide transcriptional regulation. Plant J. 53, 90–101.
Qiu, D., Xiao, J., Xie, W., Liu, H., Li, X., Xiong, L., et al. (2008). Rice gene network inferred from expression profiling of plants overexpressing OsWRKY13, a positive regulator of disease resistance. Mol. Plant 1, 538–551.
Rampitsch, C., Bykova, N. V., McCallum, B., Beimcik, E., and Ens, W. (2006). Analysis of the wheat and Puccinia triticina (leaf rust) proteomes during a susceptible host-pathogen interaction. Proteomics 6, 1897–1907.
Rockman, M. V., and Kruglyak, L. (2006). Genetics of global gene expression. Nat. Rev. Genet. 7, 862–872.
Roelfs, A. P. (1985). “Wheat and rye stem rust,” in The Cereal Rusts, eds A. P. Roelfs and W. R. Bushnell (Orlando, FL: Academic Press), 3–37.
Rosa, G. J., De Leon, N., and Rosa, A. J. (2006). Review of microarray experimental design strategies for genetical genomics studies. Physiol. Genomics 28, 15–23.
Rowe, H. C., and Kliebenstein, D. J. (2008). Complex genetics control natural variation in Arabidopsis thaliana resistance to Botrytis cinerea. Genetics 180, 2237–2250.
Schreiber, A. W., Sutton, T., Caldo, R. A., Kalashyan, E., Lovell, B., Mayo, G., et al. (2009). Comparative transcriptomics in the Triticeae. BMC Genomics 10:285. doi: 10.1186/1471-2164-10-285
Sellam, M. A., and Wilcoxson, R. D. (1976). Development of Puccinia graminis f. sp. tritici on resistant and susceptible barley cultivars. Phytopathology 66, 667–668.
Shirasu, K. (2009). The HSP90-SGT1 chaperone complex for NLR immune sensors. Ann. Rev. Plant Biol. 60, 139–164.
Shirasu, K., Lahaye, T., Tan, M. W., Zhou, F., Azevedo, C., and Schulze-Lefert, P. (1999). A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell 99, 355–366.
Silva, L. D. C. E., Cruz, C. D., Moreira, M. A., and Barros, E. G. D. (2007). Simulation of population size and genome saturation level for genetic mapping of recombinant inbred lines (RILs). Genet. Mol. Biol. 30, 1101–1108.
Simmonds, N. W. (1988). “Synthesis: the strategy of rust resistance breeding,” in Breeding Strategies for Resistance to the Rusts of Wheat, eds N. W. Simmonds and S. Rajaram (Mexico: Cimmyt), 119–136.
Singh, R. P., Hodson, D. P., Huerta-Espino, J., Jin, Y., Njau, P., Wanyera, R., et al. (2008). Will stem rust destroy the world's wheat crop? Adv. Agron. 98, 271–309.
Singh, R. P., Huerta-Espino, J., Pfeiffer, W., and Figueroa-Lopez, P. (2004a). Occurrence and impact of a new leaf rust race on durum wheat in northwestern Mexico from 2001 to 2003. Plant Dis. 88, 703–708.
Singh, R. P., William, H. M., Huerta-Espino, J., and Rosewarne, G. (2004b). “Wheat rust in Asia: meeting the challenges with old and new technologies,” in New Directions for a Diverse Planet: Proceedings for the 4th International Crop Science Congress, eds T. Fisher, N. Turner, J. Angus, L. McIntyre, M. Robertson, A. Borrell, and D. Lloyd (Gosford, NSW: The Regional Institute Ltd.), 13.
Sonah, H., Bastien, M., Iquira, E., Tardivel, A. L., Légaré, G., Boyle, B., et al. (2013). An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 8:e54603. doi: 10.1371/journal.pone.0054603
Sonderby, I. E., Hansen, B. G., Bjarnholt, N., Ticconi, C., Halkier, B. A., and Kliebenstein, D. J. (2007). A systems biology approach identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PLoS ONE 2:e1322. doi: 10.1371/journal.pone.0001322
Stokstad, E. (2007). Plant pathology: deadly wheat fungus threatens world's breadbaskets. Science 315, 1786–1787.
Sun, W., Yu, T., and Li, K.-C. (2007). Detection of eQTL modules mediated by activity levels of transcription factors. Bioinformatics 23, 2290–2297.
Sun, W., Yuan, S., and Li, K.-C. (2008). Trait-trait dynamic interaction: 2D-trait eQTL mapping for genetic variation study. BMC Genomics 9:242. doi: 10.1186/1471-2164-9-242
Tai, Y. S. (2008). Interactome of signaling networks in wheat: the protein-protein interaction between TaRAR1 and TaSGT1. Mol. Biol. Rep. 35, 337–343.
Thao, N. P., Chen, L., Nakashima, A., Hara, S. I., Umemura, K., Takahashi, A., et al. (2007). RAR1 and HSP90 form a complex with Rac/Rop GTPase and function in innate-immune responses in rice. Plant Cell 19, 4035–4045.
Van Der Merwe, M., Ericson, L., Walker, J., Thrall, P. H., and Burdon, J. J. (2007). Evolutionary relationships among species of Puccinia and Uromyces (Pucciniaceae, Uredinales) inferred from partial protein coding gene phylogenies. Mycol. Res. 111, 163–175.
Vanegas, C. D. G., Garvin, D. F., and Kolmer, J. A. (2008). Genetics of stem rust resistance in the spring wheat cultivar Thatcher and the enhancement of stem rust resistance by Lr34. Euphytica 159, 391–401.
Wang, S., Zheng, T., and Wang, Y. (2007). Transcription activity hot spot, is it real or an artifact? BMC Proc. 1:S94. Available online at: http://www.biomedcentral.com/1753-6561/1/S1/S94
Wang, X., Richards, J., Gross, T., Druka, A., Kleinhofs, A., Steffenson, B., et al. (2013). The rpg4-mediated resistance to wheat stem rust (Puccinia graminis) in barley (Hordeum vulgare) requires Rpg5, a second NBS-LRR gene, and an actin depolymerization factor. Mol. Plant Microbe Interact. 26, 407–418.
Wanyera, R., Kinyua, M. G., Jin, Y., and Singh, R. P. (2006). The spread of stem rust caused by Puccinia graminis f. sp. tritici, with virulence on Sr31 in wheat in eastern Africa. Plant Dis. 90, 113.
Webb, C. A., Richter, T. E., Collins, N. C., Nicolas, M., Trick, H. N., Pryor, T., et al. (2002). Genetic and molecular characterization of the maize rp3 rust resistance locus. Genetics 162, 381–394.
West, M. A., Kim, K., Kliebenstein, D. J., Van Leeuwen, H., Michelmore, R. W., Doerge, R. W., et al. (2007). Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 175, 1441–1450.
Williams, R. B. H., Chan, E. K. F., Cowley, M. J., and Little, P. F. R. (2007). The influence of genetic variation on gene expression. Genome Res. 17, 1707–1716.
Williams, R. B. H., Cotsapas, C. J., Cowley, M. J., Chan, E., Nott, D. J., and Little, P. F. R. (2006). Normalization procedures and detection of linkage signal in genetical-genomics experiments. Nat. Genet. 38, 855–856.
Wise, R. P., Lee, M., and Rayapati, P. J. (1996). Recombination within a 5-centimorgan region in diploid Avena reveals multiple specificities conferring resistance to Puccinia coronata. Phytopathology 86, 340–346.
Wise, R. P., Moscou, M. J., Bogdanove, A. J., and Whitham, S. A. (2007). Transcript profiling in host-pathogen interactions. Annu. Rev. Phytopathol. 45, 329–369.
Yan, J.-B., Tang, J.-H., Meng, Y.-J., Ma, X.-Q., Teng, W.-T., Chander, S., et al. (2006). Improving QTL mapping resolution based on genotypic sampling–a case using a RIL population. Acta Genetica Sinica 33, 617–624.
Yu, G.-X., Braun, E., and Wise, R. P. (2001). Rds and Rih Mediate hypersensitive cell death independent of gene-for-gene resistance to the oat crown rust pathogen Puccinia coronata f. sp. avenae. Mol. Plant Microbe Interact. 14, 1376–1383.
Yu, J., Holland, J. B., McMullen, M. D., and Buckler, E. S. (2008). Genetic design and statistical power of Nested Association Mapping in maize. Genetics 178, 539–551.
Zhang, L., Castell-Miller, C., Dahl, S., Steffenson, B., and Kleinhofs, A. (2008a). Parallel expression profiling of barley-stem rust interactions. Funct. Integr. Genomics 8, 187–198.
Zhang, L., Lavery, L., Gill, U., Gill, K., Steffenson, B., Yan, G., et al. (2008b). A cation/proton-exchanging protein is a candidate for the barley NecS1 gene controlling necrosis and enhanced defense response to stem rust. Theor. Appl. Genet. 118, 385–397.
Zhang, L., Fetch, T., Nirmala, J., Schmierer, D., Brueggeman, R., Steffenson, B., et al. (2006). Rpr1, a gene required for Rpg1-dependent resistance to stem rust in barley. Theor. Appl. Genet. 113, 847–855.
Zhu, M., Shao, F., Innes, R. W., Dixon, J. E., and Xu, Z. (2004). The crystal structure of Pseudomonas avirulence protein AvrPphB: a papain-like fold with a distinct substrate-binding site. Proc. Natl. Acad. Sci. U.S.A. 101, 302–307.
Zierold, U. W. E., Scholz, U. W. E., and Schweizer, P. (2005). Transcriptome analysis of mlo-mediated resistance in the epidermis of barley. Mol. Plant Pathol. 6, 139–151.
Glossary of Definitions
Partial resistance is a form of resistance, phenotypically characterized by a temporally and/or spatially reduced rate of pathogen development on the host (Parlevliet, 1978); this also may be referred to as Quantitative resistance (Poland et al., 2009). Genetical Genomics refers to the use of quantitative genetic mapping to dissect the regulatory underpinnings of molecular phenotypes collected en masse using high-throughput genomic technologies (Jansen and Nap, 2001). expression Quantitative Trait Locus (eQTL) mapping is the treatment of transcript abundance as a quantitative trait.
Keywords: eQTL, parallel expression, wheat, barley, Puccinia, Triticeae, cereal rusts
Citation: Ballini E, Lauter N and Wise R (2013) Prospects for advancing defense to cereal rusts through genetical genomics. Front. Plant Sci. 4:117. doi: 10.3389/fpls.2013.00117
Received: 19 March 2013; Accepted: 15 April 2013;
Published online: 01 May 2013.
Edited by:
Corné M. J. Pieterse, Utrecht University, NetherlandsReviewed by:
Nils Rostoks, University of Latvia, LatviaBrande B. Wulff, The Sainsbury Laboratory, UK
Copyright © 2013 Ballini, Lauter and Wise. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Roger Wise, Corn Insects and Crop Genetics Research, Department of Plant Pathology and Microbiology, US Department of Agriculture - Agricultural Research Service, Center for Plant Responses to Environmental Stresses, Iowa State University, 351 Bessey Hall, Ames, IA 50011-1020, USA. e-mail:cnB3aXNlQGlhc3RhdGUuZWR1
†Present address: Elsa Ballini, UMR BGPI - TA A-54/K- Equipe riz, SupAgro Montpellier, Département Biologie et Ecologie, Campus International de Baillarguet, 34 398 Montpellier Cedex 5, France.