 Gabriel Guillén†
Gabriel Guillén† Claudia Díaz-Camino† Carlos A. Loyola-Torres Rosaura Aparicio-Fabre Alejandrina Hernández-López Mauricio Díaz-Sánchez
Claudia Díaz-Camino† Carlos A. Loyola-Torres Rosaura Aparicio-Fabre Alejandrina Hernández-López Mauricio Díaz-Sánchez Federico Sanchez*
Federico Sanchez*- Departamento de Biología Molecular de Plantas, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Morelos, México
Diverse plant genome sequencing projects coupled with powerful bioinformatics tools have facilitated massive data analysis to construct specialized databases classified according to cellular function. However, there are still a considerable number of genes encoding proteins whose function has not yet been characterized. Included in this category are small proteins (SPs, 30–150 amino acids) encoded by short open reading frames (sORFs). SPs play important roles in plant physiology, growth, and development. Unfortunately, protocols focused on the genome-wide identification and characterization of sORFs are scarce or remain poorly implemented. As a result, these genes are underrepresented in many genome annotations. In this work, we exploited publicly available genome sequences of Phaseolus vulgaris, Medicago truncatula, Glycine max, and Lotus japonicus to analyze the abundance of annotated SPs in plant legumes. Our strategy to uncover bona fide sORFs at the genome level was centered in bioinformatics analysis of characteristics such as evidence of expression (transcription), presence of known protein regions or domains, and identification of orthologous genes in the genomes explored. We collected 6170, 10,461, 30,521, and 23,599 putative sORFs from P. vulgaris, G. max, M. truncatula, and L. japonicus genomes, respectively. Expressed sequence tags (ESTs) available in the DFCI Gene Index database provided evidence that ~one-third of the predicted legume sORFs are expressed. Most potential SPs have a counterpart in a different plant species and counterpart regions or domains in larger proteins. Potential functional sORFs were also classified according to a reduced set of GO categories, and the expression of 13 of them during P. vulgaris nodule ontogeny was confirmed by qPCR. This analysis provides a collection of sORFs that potentially encode for meaningful SPs, and offers the possibility of their further functional evaluation.
Introduction
Legumes comprise one of the largest plant families in the world, mainly as a result of the ability of its members to establish mutually beneficial root symbioses with soil bacteria and fungi that provide the plants with nutrients that are scarce in many soils. Leguminosae are second only to the Gramineae with respect to agricultural production and human and animal consumption (Udvardi, 2002; Graham and Vance, 2003).
The societal relevance of legumes has motivated considerable investment in legume genomics research in recent years. Although most resources have primarily focused on the development of genomic tools and biological investigation of the model legumes barrel medic (Medicago truncatula) and birdsfoot trefoil (Lotus japonicus), over the past years additional efforts have allowed the advance of soybean (Glycine max) and common bean (Phaseolus vulgaris) genomics (Gepts et al., 2005). Virtually complete genome sequences of G. max, M. truncatula, and L. japonicus were published in 2009 (Cannon et al., 2009), and the P. vulgaris genome is forthcoming. Considering that the final goal of legume genomics is to understand the organization and function of a unified legume genome in all its diversity, in this study we designed a strategy to uncover the neglected sector of potential genes encoding small proteins (SPs) in G. max, L. japonicus, M. truncatula, and P. vulgaris genomes.
Short open reading frames (sORF) are translated into SPs of 30–150 amino acids (aa) that play essential roles in eukaryotes (Kastenmayer et al., 2006; Kondo et al., 2010; Hanada et al., 2013). In plants, SPs are involved in a variety of processes, e.g., modulation of cell division and differentiation (Fletcher et al., 1999; Mortier et al., 2012; Hanada et al., 2013), stabilization, assembly, and dimerization of the PSII complex (Shi and Schröder, 2004), priming plant defenses (Silverstein et al., 2007; Gleason et al., 2008; Jung et al., 2009; Van de Velde et al., 2010), and regulating flowering time (Notaguchi et al., 2008). These examples illustrate that SPs are ubiquitous and function in plant physiology, growth and development. However, the identification and characterization of many other SPs remain largely unexplored.
Whole-genome tiling array studies (Stolc et al., 2005) revealed that sORFs have been underestimated in plants at a genome-wide level. In practice, sORFs are generally eliminated during genome annotation due to the arbitrary minimum open reading frame (ORF) cutoff of 100–150 aa used to reduce the likelihood of falsely categorizing protein-coding (mRNA) and non-coding (ncRNA) RNA (Dinger et al., 2008). Hence, the development of effective methods to recognize potentially functional sORFs is critical.
In this work, we collected all predicted ORFs coding for proteins in the available genome sequences of P. vulgaris, M. truncatula, G. max, and L. japonicus. We analyzed annotated sORFs equal to or smaller than 120 aa in length in each legume genome. For evidence of functionality, we compared each potential sORF by sequence similarity against the EST Gene Index database. Legume sORFs potentially encoding non-coding RNAs (ncRNAs) were predicted by the Infernal program (Nawrocki et al., 2009) and eliminated from our sORFs collections. Sequence similarity of annotated SPs to larger proteins in the genome was evaluated as well as their evolutionary conservation within plants. Additionally, potential functional sORFs were classified according to Gene Ontology (GO) (McCarthy et al., 2006). Finally, a time-course study of a group of selected sORFs was carried out during P. vulgaris nodule ontogeny by qPCR. This study demonstrates that is possible to identify functional sORFs in legume plant genomes, even in cases where the genome annotation is not yet complete.
Materials and Methods
Strategy for Large-Scale Discovery of sORFs in Legume Genomes
ORFs of P. vulgaris, M. truncatula (Young et al., 2011), G. max (Schmutz et al., 2010) and L. japonicus (Sato et al., 2008) were collected. For comparison with non-legume plants, ORFs from Arabidopsis thaliana (Swarbreck et al., 2008) and maize (Zea mays) (Schnable et al., 2009) were also gathered. Plant genome databases utilized for this were Phytozome (http://www.phytozome.com, Goodstein et al., 2012) and PlantGDB (http://www.plantgdb.org/LjGDB, Duvick et al., 2008). To calculate protein length frequencies, annotated proteins were downloaded in FASTA format. Protein lengths were summed within 40-aa bins. Amino acid bins were plotted as a function of protein size. ORF prediction was further confirmed by comparing non-coding regions (1 kb) immediately downstream from stop codons against the A. thaliana proteome by BLASTX. The average protein length for the four legume species occurs around 120 aa, therefore sORF candidate sets encoding proteins with a length equal to or less than 120 aa were compiled. Considering that not all putative ORFs will be of functional significance, we focused on properties that could be assessed using bioinformatics tools. Using the rationale that a functional gene should be transcribed, we compared each potential sORF by sequence similarity against the DFCI Gene Index database (http://compbio.dfci.harvard.edu/tgi/plant.html, Quackenbush et al., 2001; Tsai et al., 2005). BLASTN searches were conducted using default parameters (Altschul et al., 1990). Only those with an expectation value cutoff of 10 (e-10) were considered as positive matches. Potentially, ncRNAs were identified in the sORF candidate set by using the Infernal program (Nawrocki et al., 2009). To find common protein domains within each legume, predicted SPs were compared to larger proteins (longer than 120 aa) in the genome. Based on the supposition that potential sORFs are more likely to represent “true” genes if an ortholog can be found in another plant genome, we also evaluated the presence of each sORF in the P. vulgaris, M. truncatula, G. max, L. japonicus, A. thaliana, and Z. mays genomes (BLASTP, e-10). Finally, legume sORFs were classified on the basis of GO annotation (http://www.agbase.msstate.edu/cgi-bin/tools/GOanna.cgi, McCarthy et al., 2006), and the expression of 13 selected sORFs was tested during P. vulgaris nodule ontogeny by qPCR.
Plant Material, RNA Extraction, and qRT-PCR Analysis of Expression
Phaseolus vulgaris L. cv. Negro Jamapa seeds were surface-sterilized in 10% (v/v) commercial sodium hypochlorite, rinsed with sterile water and germinated in the dark for three days on two layers of filter paper saturated sterile water at 28°C. Seedlings were inoculated with Rhizobium tropici CIAT899 (Martínez-Romero et al., 1991), transferred to vermiculite and grown in the greenhouse. Nodules and nodule-stripped roots were harvested at the indicated times, immediately frozen in liquid nitrogen, and stored at −70°C until use. Total RNA was isolated using the Fermentas GeneJET™ RNA purification kit according to the manufacturer's instructions (www.thermoscientificbio.com/fermentas/). RNA quantity was measured spectrophotometrically, and only the RNA samples with a 260/280 ratio between 1.9 and 2.1 and a 260/230 ratio greater than 2.0 were used for the analysis. The integrity of RNA samples was confirmed by agarose gel electrophoresis. For reverse transcription, 3 μg total RNA was treated with DNaseI (Fermentas), and 1 μg total RNA was reverse transcribed using the RevertAid™ H Minus First-strand cDNA synthesis kit (Fermentas) with anchored-oligo (dT) 18 primer according to manufacturer's instructions. For qPCR, 15 μl qPCR reactions using Maxima SYBR® Green qPCR Master Mix (Fermentas) were performed on an iCycle iQ5 apparatus (BioRad, www.bio-rad.com). The cycling conditions were: preheating for 5 min at 95°C followed by 30 cycles (denaturing for 15 s at 95°C, annealing and elongation for 15 s at 57°C and data acquisition at 81°C). A negative control reaction without template was also included for each primer combination. The melting curve protocol began immediately after amplification and consisted of 1 min at 55°C followed by 80 10 s steps with a 0.5°C increase in temperature at each step. The relative numbers for Ct of each gene (Table 1) were normalized to the house keeping gene Elongation factor 1-alpha (Ef1-α, Nawrocki et al., 2005). Data was analyzed using iQ™ 5 Optical System Software version 2.1 (BioRad). Three biological replicates were pooled and analyzed. At least six replicate PCR amplifications were performed for each sample.
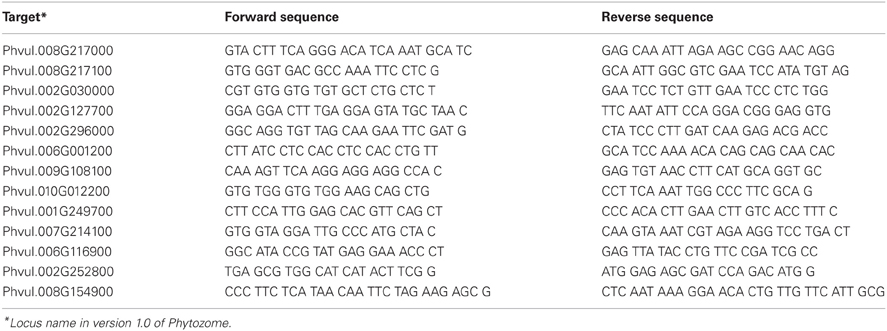
Table 1. List of oligonucleotides used in this work.
Results
Length Distribution of Protein Sequences in the sORF Sets of Analyzed Legumes
We collected 31,578, 55,715, 53,424, and 42,399 ORFs of P. vulgaris, G. max, M. truncatula, and L. japonicus genomes, respectively, and 27,414 (A. thaliana) and 63,540 (Z. mays) ORFs from non-legume plant genomes [http://www.phytozome.com (Goodstein et al., 2012) and PlantGDB (http://www.plantgdb.org/, Duvick et al., 2008)] (Tables 2, S1). Recent annotations of the A. thaliana genome include more sORFs relative to the earlier versions (Yang et al., 2011), indicating that the annotation of SPs is a key feature of improved annotations. Considering that a similar gene-calling procedure was followed to annotate all the legume genomes consulted in this work (Table 3), to evaluate the accuracy of this procedure, we retrieved 1 kb up- and downstream from each predicted ORF using the BioMart tool at the Phytozome website (http://www.phytozome.net/). We evaluated ORF prediction by comparing 1 kb of sequence downstream from the predicted stop codon of each putative P. vulgaris ORF against the A. thaliana proteome by BLASTX (Figure 1), an algorithm able to search a translated nucleotide sequence against a given protein sequence database. The lack of sequence similarity of these regions (represented by a low e value and low gene coverage) to known A. thaliana proteins indicated that most sORFs are not incorrectly annotated ORFs that are actually parts of longer ORFs.
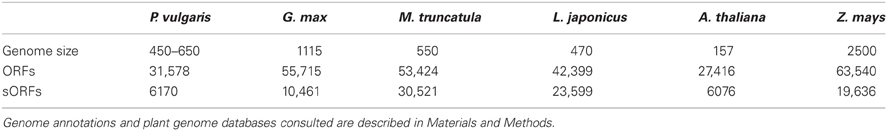
Table 2. Comparative values of genome size (in Mb), total open reading frames (ORFs) and short open reading frames (sORFs) encoding small proteins or peptides (SPs) in Phaseolus vulgaris (P. vulgaris), Glycine max (G. max), Medicago truncatula (M. truncatula), Lotus japonicus (L. japonicus), Arabidopsis thaliana (A. thaliana), and Zea mays (Z. mays).

Table 3. Analyzed plant genomes in version 9.0 of Phytozome.
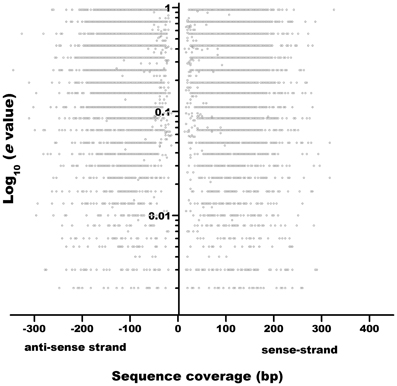
Figure 1. Sequence similarity of 3′-non-coding sequences of putative P. vulgaris sORFs to the Arabidopsis thaliana protein collection. 1 kb non-coding sequences (sense- and anti-sense strands) downstream stop codons of putative P. vulgaris sORFs are plotted as a function of similarity to A. thaliana proteins (e-10 value).
Protein length distribution analysis (Figure 2) indicated that the highest frequency of predicted SPs was in the genomes of M. truncatula (57.2%) and L. japonicus (55.6%). By contrast, P. vulgaris (19.5%), G. max (18.8%), A. thaliana (22.2%), and Z. mays (30.9%) showed an abundance of SPs that was relatively homogeneous compared to the whole protein collection (Tables 2, S1, and Figure 2). Interestingly, protein abundance declined abruptly in all cases below 40 aa. This observation may suggest that in the cellular context, a minimum length is required to achieve a properly functioning protein.
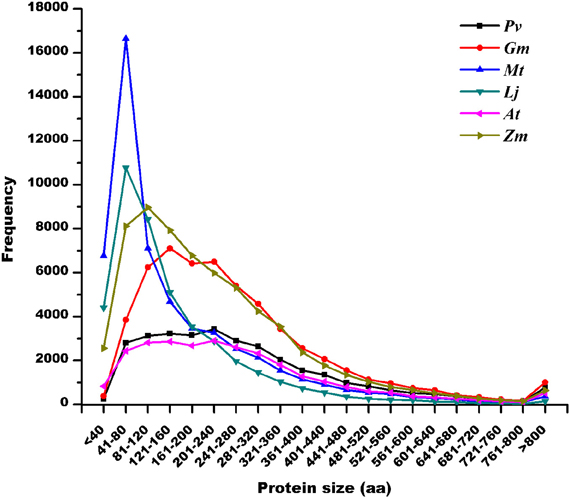
Figure 2. Length distribution of predicted protein sequences in legume and non-legume plant genomes. Pv, Phaseolus vulgaris protein sizes in P. vulgaris v0.9 annotation; Gm, Glycine max protein sizes in G. max v1.0 annotation; Mt, M. truncatula protein sizes in Mt3.0 annotation; Lj, L. japonicus protein sizes in Lj1.0 annotation; At, Arabidopsis thaliana protein sizes in genome release 9; and Zm, Zea mays protein sizes in Maize Golden Path B73 RefGen_v2.
Evaluating sORF Functionality by Evidence of Transcription
Compared to genomes, ESTs and other sources of transcript information are the most reliable evidence for gene expression and gene identification; introns and most pseudogenes are absent, the searched space is reduced compared to eukaryotic genes, and, typically, an mRNA encodes one protein (Frith et al., 2006). Unfortunately, the large volume of ESTs experimentally generated in each study (including a high number of short sequences) and the lack of functional annotation are frequent barriers for using ESTs for gene modeling and gene structure identification (Tsai et al., 2005).
To reduce the likelihood of falsely categorizing ncRNAs or transposable elements as mRNAs, many cDNA collections exclude transcripts under 500 nucleotides (nt) in length. Given that proteins of 120 aa or less may be encoded by transcripts of around 300 nt, this introduces a bias against evaluating the expression of short proteins. In this work, to assess whether sORFs were well represented in the EST libraries consulted (Tables 4, S1), we analyzed the lengths of ORFs that encode proteins of different sizes in legume species (Figure 3). For each protein size range (grouped in 40 aa bins) the RNA length varied between 180 and 800 base pairs, indicating that RNAs encoding potential SPs in those legumes would not be completely excluded from the publicly available EST libraries analyzed and that the databases could be used to evaluate sORF expression.
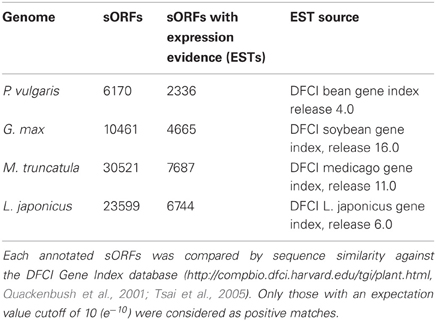
Table 4. Evidence of transcription of legume sORFs based on ESTs.
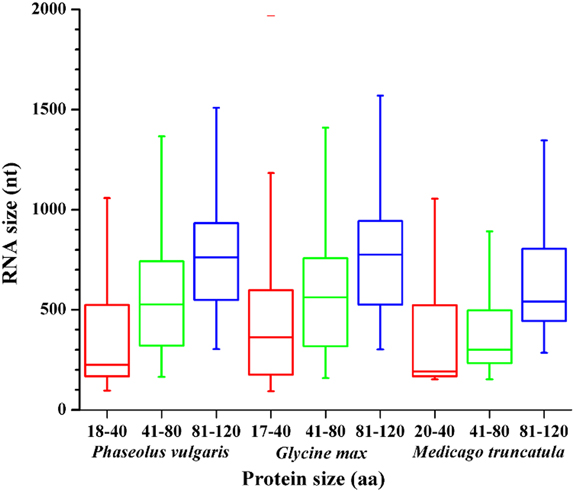
Figure 3. RNA sizes for different ranges of protein size represented in a box and whisker plot. The center lines indicate the medians, the top and bottom of each box indicate the first and third quartiles, and the whiskers extend to the most extreme data points.
Although ESTs are not sufficient to predict whether a gene is translated into a functional protein, their detection constitutes strong evidence of gene expression. BLASTN searches were conducted using default parameters (Altschul et al., 1990) and only those ESTs with an e value cutoff of 10 (e-10) were considered to be positive matches for any legume sORFs. We found 2334, 4665, 7687, and 6744 EST counterparts to sORFs predicted in P. vulgaris, G. max, M. truncatula, and L. japonicus genomes, which represent 37.82, 44.59, 25.18, and 28.57% of the total sORFs, respectively (Tables 4, S1).
A disadvantage of determining sORFs functionality exclusively based on transcript evidence is that these may be ncRNAs (Frith et al., 2006), which are difficult to distinguish from mRNAs encoding short proteins. ncRNA genes do not encode proteins but produce functional RNA molecules that play important biological functions in the cell. To determine whether candidate sORFs below 120 aa in length could be ncRNAs, an Rfam-based search with all legume sORFs using the Infernal program (Nawrocki et al., 2009) was performed. Just a proportion (10.5, 14.9, 0.06, and 5%, Table S2) of annotated as sORFs in the P. vulgaris, G. max, L. japonicus, and M. truncatula genomes were predicted as potential ncRNAs and eliminated.
Comparison of Common Gene Regions Encoding Putative SPs with Genes Encoding Larger Proteins, and Evidence for Orthologs
Finding common regions or domains among proteins is a valid approach to distinguish protein-coding from non-coding genes (Frith et al., 2006; Kastenmayer et al., 2006). To test for sequence similarity between annotated SPs and larger proteins in the genomes of the legumes analyzed, we first compared P. vulgaris, G. max, M. truncatula, and L. japonicus SP to all other polypeptides (longer than 120 aa) included in the respective genome (Figure 4 and Table 5). With the exception of M. truncatula (2.4%), most annotated SPs in legumes share counterpart regions or domains with larger proteins Figure 4C, suggesting they may encode for functional SPs. Other potential SPs, particularly abundant in P. vulgaris, although identical in sequence, differ slightly in length among them Figure 4A or are partially related to other proteins Figure 4B. These groups could represent different members of protein families that have a common or related biological function (Marszalek et al., 1999; Schwaiger et al., 2003). Finally, there is a considerable number of SPs that lack similarity to other proteins in the same organism (Table 5, subsets <e−5 and “no hit”). This is particularly evident in M. truncatula and L. japonicus, where most sORFs (66 and 79%, respectively) are unique, and have no similarity to any longer proteins encoded in the genome. Although at least part of these sORFs may still encode genuine proteins, i.e., proteins that evolve at faster rates, in general these sORFs are listed as random ORFs arising in non-coding transcripts (Frith et al., 2006; Kastenmayer et al., 2006; Clamp et al., 2007). The P. vulgaris sORFs GC-content is similar to the average for the P. vulgaris genome (30–40%), which suggests that these sORFs may be actually protein-coding genes (Table S1 and Figure S1).
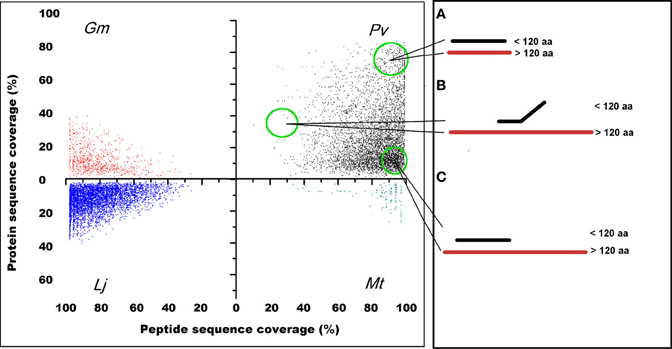
Figure 4. Legume sORFs display common aa regions or domains with larger polypeptides of the same genome. The identity level of P. vulgaris, G. max, M. truncatula, and L. japonicus predicted sORFs (peptide sequence coverage) is spread across several homologous proteins of variable size (protein sequence coverage) of the respective genome. As an example, (A) illustrates the distribution pattern of sORFs in P. vulgaris that are identical in sequence to other small proteins (slightly larger than 120 aa); in (B) sORFs that share a domain with larger polypeptides are included; and in (C) sORFs that are completely equivalent to regions or domains found in larger proteins are indicated.
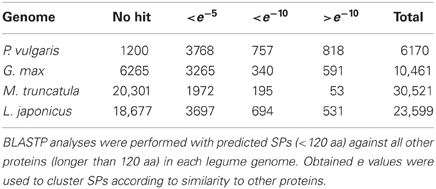
Table 5. Frequency of potential sORFs sharing domains with larger polypeptides of the same genome.
Since sORFs are more likely to represent functional proteins if an ortholog can be found, sORFs detected in each legume genome were compared to each other based on sequence similarity (BLASTP, e−10). We also included in this analysis sORFs from A. thaliana and Z. mays to assess which sORFs might have evolved from ancestral genes present in a common plant ancestor, but lost in other legumes. A large percentage of P. vulgaris and G. max SPs were found to have orthologs in model legumes, and also in other plants (Table 6). In contrast to P. vulgaris and G. max, only a small fraction of SPs from M. truncatula and L. japonicus (less than 10% in all comparisons) shared sORFs or had orthologs in non-legume plants (Table 6).
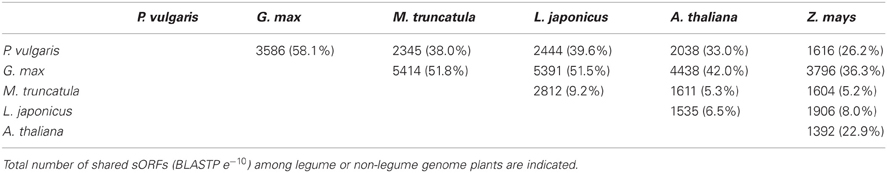
Table 6. Number of sORFs from legume species predicted to be orthologous to each other.
SP Classification Based on Gene Ontology
One of the most important tools to establish ontologies is GO analysis (Ashburner et al., 2001), which depicts the potential function of a gene product in a cellular context; thus, annotation of putative SPs encoded by sORFs in legume genomes could provide valuable information to interpret their biological role. Out of 6170 potential sORFs in P. vulgaris, 4590 are homologous to A. thaliana proteins, and 2670 of them were associated with a GO “biological process.” The Fisher's exact test (Routledge, 1998) was applied to determine which GO categories were statistically over-represented compared to all proteins of the genome (p < 0.05, corrected by Benjamini adjustment). Interestingly, 14% of the total sORFs were preliminary classified into “response to stress” (Figure 5 and Table S3). A similar ratio was obtained after analyzing the SPs contained in the genomes of G. max, M. truncatula, and L. japonicus (Figure 5 and Table S3). Remarkably, only in P. vulgaris were a considerable percentage of sORFs (6.4%) grouped into “developmental process.”
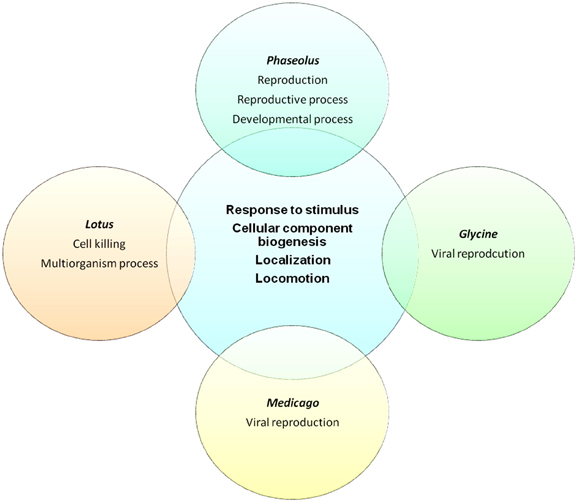
Figure 5. Venn diagram representing the distribution of GO categories found in each legume genome. Around 15% of all sORFs in legumes were included in “response to stimulus” and close to 20% were related to “localization” GO categories. The Fisher's exact test (Routledge, 1998) was applied to determine which GO categories were statistically over-represented compared to all proteins of the genome (p < 0.05, corrected by Benjamini adjustment).
Confirming sORF Functionality by Analysis of Transcript Expression in P. vulgaris
The P. vulgaris genome has 6170 annotated SPs equal or smaller than 120 aa (Tables 2, S1). A high proportion of these genes are exclusively present in the P. vulgaris genome (Figure 6, Pv), whereas others have a counterpart in other legume [G. max (Gm), M. truncatula (Mt) and L. japonicus (Lj)] or non-legume [A. thaliana (At), and Z. mays (Zm)] genomes (Figure 6). As expected, a higher proportion of the predicted SPs in P. vulgaris were also identified in legumes that form determinate nodules (Figure 6, LegDN), such as G. max, L. japonicus, and Vignia unguiculata. However, an important number of these sORFs are also found in legumes that form indeterminate nodules, such as M. truncatula, Pisum sativum, and Trifolium repens (Figure 6, Leg).
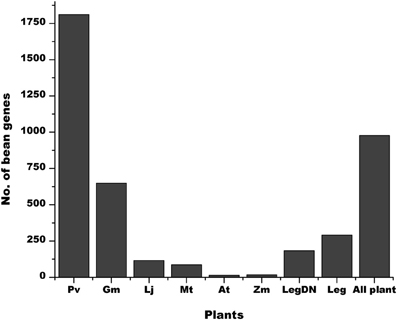
Figure 6. Some sORFs in P. vulgaris are shared with other plants. The graph shows the number of P. vulgaris sORFs exclusively found in this plant (Pv) compared to those that are also present in G. max (Gm), M. truncatula (Mt), L. japonicus (Lj), A. thaliana (At) and Z. mays (Zm). It also shows the number of sORFs of legumes that form determinate (LegDN) or undeterminate (Leg) nodules. The All plants bar represents the number of sORFs that are common to all plant species evaluated.
Based on ESTs, 2336 sORFs had evidence of gene expression (Tables 4, S1). By comparing the sORFs of P. vulgaris with larger proteins of the P. vulgaris genome and with sORFs of other legume (G. max, M. truncatula, and L. japonicus) and non-legume plants (A. thaliana and Z. mays) we determined that 5521 had counterpart regions or domains found in larger proteins of the P. vulgaris genome (Figure 4) and that 3914 contained a high level of identity (BLASTP e−10) to sORFs found in other plant species (Table 6). Many of the sORFs in P. vulgaris were detected by more than one approach (Figure 8). For example, a large number of sORFs were transcribed and contained common regions found in larger P. vulgaris proteins, or were transcribed and had potential orthologs in other legume or non-legume plants (Figures 6, 8). sORFs detected by all techniques were deemed likely to be bona fide genes.
To test the efficiency of our method in validating legume sORFs, we explored by qPCR the expression of 13 sORFs selected from a group consisting of 186 that are exclusively present in determinate nodules (Gene Index database, http://compbio.dfci.harvard.edu/tgi/plant.html, Quackenbush et al., 2001; Tsai et al., 2005; Figure 8, LegDN). P. vulgaris roots were inoculated with Rhizobium tropici CIAT899 (Martínez-Romero et al., 1991). Results from each nodule developmental stage were compared to age equivalent un-inoculated roots. With the sole exception of Phvul.008G217000, all other selected sORFs have more than one evidences of functionality (Table 7). Interestingly, all selected sORFs (even Phvul.008G217000) are expressed in P. vulgaris root nodules of 10 and 14 days after inoculation (d.p.i.) compared to age equivalent un-inoculated roots (Table 7 and Figure 7).
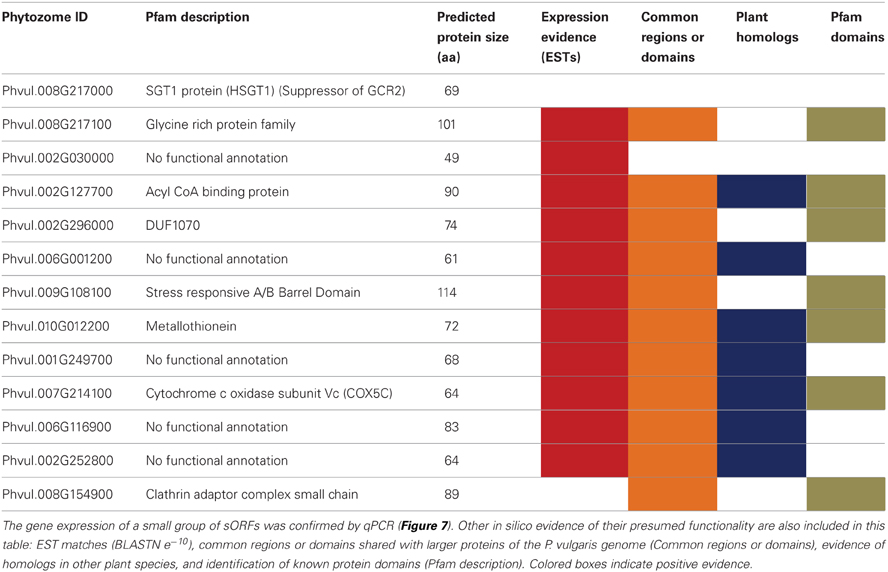
Table 7. sORF expression during nodule ontogeny.
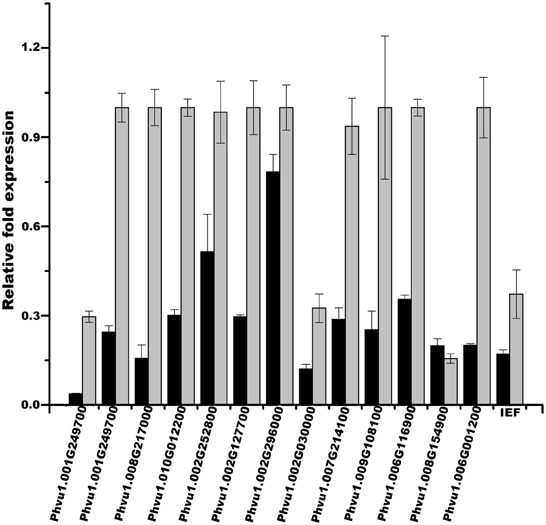
Figure 7. sORFs expression during nodule ontogeny. The gene expression of a small group of sORFs was confirmed by qPCR. Relative expression levels of a selected group of sORFs (Table 7) were determined in nodules and nodule-stripped roots at the indicated times by qPCR. Total RNA was isolated from each biological sample. First strand cDNA was synthesized and subjected to qPCR as described in Materials and Methods. Expression levels were normalized against Elongation factor 1-alpha (Ef1-α) values. Ratios of expression in nodule-stripped roots to nodules are graphed. These values represent the mean and SD of triplicate experiments.
Discussion
The release of the Arabidopsis thaliana genome sequence in Arabidopsis Genome Initiative (2000) marked the beginning of the plant genomics era. In the last decade, diverse plant genome sequencing projects, including the Legume Crops Genome Initiative (Gepts et al., 2005), coupled with the development of powerful bioinformatics tools has facilitated massive data analysis. However, there are still a considerable number of proteins without assigned functions.
It has been reported that some SPs (30–150 aa in length) are involved in plant cell signaling and other processes in plants, but the overall scope of their abundance and biological relevance is still unknown. Here, sORFs encoding proteins of 120 aa or less in length in P. vulgaris, G. max, M. truncatula, and L. japonicus were analyzed and compared to those from two non-legume genomes (A. thaliana and Z. mays).
Our data indicate that the frequency distribution of potential SPs in the genomes of P. vulgaris and G. max are similar to that in A. thaliana (Figure 2), a vastly explored non-legume plant genome. Interestingly, the highest frequency of sORFs was found in the genomes of M. truncatula and L. japonicus, which are the two best-studied genomes in leguminous plants, and just a small proportion of these sORFs (0.06 and 5% in L. japonicus and M. truncatula, respectively) were identified as potential ncRNAs (Infernal program; Nawrocki et al., 2009). However, the existence of an ORF in genomic sequence does not necessarily demonstrate the existence of a functional gene.
We evaluated ORF prediction by comparing 1 kb of sequence downstream from the predicted stop codon of each putative P. vulgaris ORF against the A. thaliana proteome by BLASTX (Figure 1). In P. vulgaris genome, the average intron length is 400–500 bp and 75% of all introns are below 875 bp (data not shown). Therefore, comparing 1 kb downstream of the predicted stop codon against the protein database of Arabidopsis thaliana, (in the six possible reading frames), should be sufficient to reduce false positives created by truncated gene models. In other species, such as maize, that contains larger introns, a larger window would need to be analyzed to resolve this potential annotation issue.
As the first evidence for sORF functionality, we searched for evidence of expression in EST collections (Tables 4, S1). By this method, we estimated that between 25 and 45% of all potential SPs encoded in the genomes analyzed are represented by at least one EST (Tables 4, S1). These results imply that the majority of sORFs predicted in legume genomes are under-represented in the EST collections consulted, particularly those that encode for tiny proteins (less than 40 aa). Although unlikely, these data could indicate that some of the sORFs are simply random ORFs, rather than valid protein-coding genes.
Domains are the structural and functional building blocks of proteins. Given that most protein-encoding genes share conserved domains, we compared P. vulgaris, G. max, M. truncatula, or L. japonicus annotated SPs against longer polypeptides in their respective genomes (Figure 4 and Table 5). Most potential SPs in legumes were found to be equivalent to regions or domains found in larger proteins Figure 4C. Interestingly, the distribution pattern of a large number of SPs in P. vulgaris indicated a remarkable abundance of proteins that are identical in sequence but vary slightly in length Figure 4A. Domain length variations in proteins can result in functional differences such as in some actin-binding protein families, where domain length variations are related to their mechanical stability in binding F-actin (Marszalek et al., 1999; Schwaiger et al., 2003). Therefore, it is possible that these groups of sORFs share similar biological functions.
A considerable number of sORFs in all legumes analyzed showed “low-identity” (Table 5, subsets <e−5 and “no hit”). This was particularly evident among sORFs of M. truncatula and L. japonicus, where most (66 and 79%, respectively) were unique. Although it is unclear to what extent these sORFs encode real proteins, proteins that are both short and dissimilar to any known protein in the genome could be acquired by horizontal gene transfer or could represent novel genes that arose after divergence. Both possibilities should be evaluated in greater detail.
Most sORFs of P. vulgaris and G. max had orthologs in other legumes and plants (Table 6). In general, orthologs retain the same function through evolution (Tatusov et al., 1997); thus, sORFs found in non-legume plants are likely to be related to common biological and chemical processes in plants (Hanada et al., 2013), whereas sORF orthologs present uniquely in Medicago or Lotus may reflect a distinctive function of legumes, such as nodulation. Again, just a small fraction of sORFs from M. truncatula and L. japonicus (less than 10% in all comparisons) were shared or had orthologs in non-legume plants (Table 6). This fact, together with the absence of domains shared with larger polypeptides (Figure 4 and Table 5), could reflect speciation events leading to a variety of large gene families. Indeed, synteny comparisons between Medicago and Lotus indicate that a genome duplication event occurred after speciation (Cannon et al., 2006). Alternatively, these proteins could arise from ancestral genes present in a common ancestor, but lost in other legumes. An example of a remarkable family of proteins that share this feature is the nodule-specific cysteine-rich (NCR) legume peptides involved in regulating the differentiation of soil nitrogen-fixing bacteria during symbiosis (Van de Velde et al., 2010).
Leguminous plants are able to establish symbiotic relationships with soil nitrogen-fixing bacteria (commonly called rhizobia), an association that leads to the formation of a new organ in the plant, the symbiotic nodule. Nodulation in legumes provides a major conduit of available nitrogen into the biosphere; therefore, its study is of great importance in sustainable agriculture. We are particularly interested in studying diverse signaling mechanisms during the organogenesis of nitrogen-fixing nodules in P. vulgaris. For this reason, we explored the expression of sORFs that are exclusively present in determinate nodules (Figure 6). We selected 13 sORFs whose expression was related to nodulation, i.e., for which we found evidence of expression only in EST libraries generated from modulated plants (Gene Index database, http://compbio.dfci.harvard.edu/tgi/plant.html, Quackenbush et al., 2001; Tsai et al., 2005). All of these sORFs were homologous to other larger P. vulgaris proteins and some of them had potential orthologs in other plant species. As expected, all tested sORFs were expressed during nodule ontogeny (Table 7 and Figure 7).
The identification of novel genes is an urgent requirement for gene investigation in the age of genomics. The strategy for discovery of potential sORFs at a large-scale in legume genomes described here will contribute to their annotation and identifies new potential regulators of diverse biological processes in plants that should improve our understanding of plant biology. Our analysis revealed that in P. vulgaris, 2336 potential sORFs are transcribed, 2929 potential SPs sharing common regions or domains with other proteins of P. vulgaris and 3274 were homologous to other SPs found in different plant species (Figure 8). Remarkably, 2553 putative SPs in P. vulgaris have at least one evidence of functionality, 2321 have two of them and a total of 776 sORFs have all of them. sORFs detected by all techniques are likely to be bona fide protein coding genes.
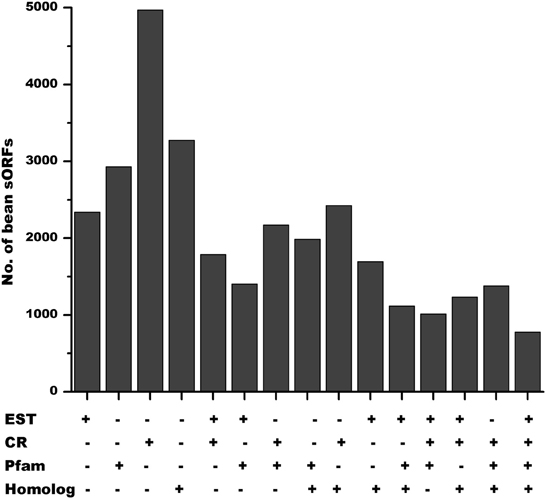
Figure 8. Evidence of functional SPs in P. vulgaris. Out of 6170 annotated sORFs in the genome of P. vulgaris, 2336 had expression evidence (DFCI Gene Index database), 2929 shared common regions or domains with other proteins (larger than 120 aa) of P. vulgaris and 3274 were homologous to SPs found in different plant species. According to the Phytozome annotation, 4970 belong to one or more protein families. 2553 sORFs in P. vulgaris have at least one of these types of evidence of functionality, whereas 2321 have two of them and a total of 776 sORFs have all of them.
Caveat to this approach: SPs below 40 aa in length, those that are encoded by genes with low expression, SPs that fall into protein families that are entirely species-specific or that contain unknown protein domains are not here represented. However, the functionality of sORFs could also be validated by highly sensitive methods to detect gene expression, such as qPCR, LC-MS, or HPLC-MS in a particular tissue or cell compartment (Wienkoop and Saalbach, 2003), in a wide range of growth stages or stress conditions (Zhang et al., 2006; Yang et al., 2011), and by gain-of- and loss-of-function (Hanada et al., 2013) approaches.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Phaseolus vulgaris sequence data was produced by the US Department of Energy Joint Genome Institute http://www.jgi.doe.gov/ in collaboration with the user community. This work was supported in part by CONACYT 177207 and DGAPA IN201412 grants to Claudia Díaz-Camino and by CONACYT 177744 and DGAPA IN106012 grants to Federico Sanchez. We thank Olivia Santana and Juan Elias Olivares for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Plant_Genetics_and_Genomics/10.3389/fpls.2013.00208/abstract
Table S1 | List of sORFs in P. vulgaris and evidences of functionality.
Table S2 | Infernal data.
Table S3 | Venn diagram data.
Figure S1 | GC-content of promoters, ORFs and sORFs in P. vulgaris.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Arabidopsis Genome Initiative. (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Ashburner, M., Ball, C. A., Blake, J. A., Butler, H., Cherry, J. M., Corradi, J., et al. (2001). Creating the gene ontology resource: design and implementation. Genome Res. 11, 1425–1433. doi: 10.1101/gr.180801
Cannon, S. B., May, G. D., and Jackson, S. A. (2009). Three sequenced legume genomes and many crop species: rich opportunities for translational genomics. Plant Physiol. 151, 970–977. doi: 10.1104/pp.109.144659
Cannon, S. B., Sterck, L., Rombauts, S., Sato, S., Cheung, F., Gouzy, J., et al. (2006). Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. U.S.A. 103, 14959–14964. doi: 10.1073/pnas.0603228103
Clamp, M., Fry, B., Kamal, M., Xie, X., Cuff, J., Lin, M. F., et al. (2007). Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. U.S.A. 104, 19428–19433. doi: 10.1073/pnas.0709013104
Dinger, M. E., Pang, K. C., Mercer, T. R., and Mattick, J. S. (2008). Differentiating protein coding and non coding RNA: challenges and ambiguities. PLoS Comput. Biol. 4:e1000176. doi: 10.1371/journal.pcbi.1000176
Duvick, J., Fu, A., Muppirala, U., Sabharwal, M., Wilkerson, M. D., Lawrence, C. J., et al. (2008). PlantGDB: a resource for comparative plant genomics. Nucleic Acids Res.36, D959–D996. doi: 10.1093/nar/gkm1041
Fletcher, J. C., Brand, U., Running, M. P., Simon, R., and Meyerowitz, E. M. (1999). Signaling of cell fate decisions by CLAVATA3 in Arabidopsis shoot meristems. Science 283, 1911–1914. doi: 10.1126/science.283.5409.1911
Frith, M. C., Forrest, A. R., Nourbakhsh, E., Pang, K. C., Kai, C., Kawai, et al. (2006). The abundance of short proteins in the mammalian proteome. PLoS Genet. 2:e52. doi: 10.1371/journal.pgen.0020052
Gepts, P., Beavis, W. D., Brummer, E. C., Shoemaker, R. C., Stalker, H. T., Weeden, N. F., et al. (2005). Legumes as a model plant family. Genomics for food and feed report of the cross-legume advances through genomics conference. Plant Physiol. 137, 1228–1235. doi: 10.1104/pp.105.060871
Gleason, C. A., Liu, Q. L., and Williamson, V. M. (2008). Silencing a candidate nematode effector gene corresponding to the tomato resistance gene Mi-1 leads to acquisition of virulence. Mol. Plant Microbe Interact. 21, 576–585. doi: 10.1094/MPMI-21-5-0576
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Graham, P. H., and Vance, C. P. (2003). Legumes: importance and constraints to greater use. Plant Physiol. 131, 872–877. doi: 10.1104/pp.017004
Hanada, K., Higuchi-Takeuchi, M., Okamoto, M., Yoshizumi, T., Shimizu, M., Nakaminami, K., et al. (2013). Small open reading frames associated with morphogenesis are hidden in plant genomes. Proc. Natl. Acad. Sci. USA 110, 2395–2400. doi: 10.1073/pnas.1213958110
Jung, H. W., Tschaplinski, T. J., Wang, L., Glazebrook, J., and Greenberg, J. T. (2009). Priming in systemic plant immunity. Science 324, 89–91. doi: 10.1126/science.1170025
Kastenmayer, J. P., Ni, L., Chu, A., Kitchen, L. E., Au, W. C., Yang, H., et al. (2006). Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Res. 16, 365–373. doi: 10.1101/gr.4355406
Kondo, T., Plaza, S., Zanet, J., Benrabah, E., Valenti, P., Hashimoto, H., et al. (2010). Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science 329, 336–339. doi: 10.1126/science.1188158
Marszalek, P. E., Lu, H., Li, H., Carrion-Vazquez, M., Oberhauser, A. F., Schulten, K., et al. (1999). Mechanical unfolding intermediates in titin modules. Nature 402, 100–103. doi: 10.1038/47083
Martínez-Romero, E., Segovia, L., Mercante, F. M., Franco, A. A., Graham, P., and Pardo, M. A. (1991). Rhizobium tropici, a novel species nodulating Phaseolus vulgaris L. beans and Leucaena sp. trees. Int. J. Syst. Bacteriol. 41, 417–426. doi: 10.1099/00207713-41-3-417
McCarthy, F. M., Wang, N., Magee, G. B., Nanduri, B., Lawrence, M. L., Camon, E. B., et al. (2006). AgBase: a functional genomics resource for agriculture. BMC Genomics 7:229. doi: 10.1186/1471-2164-7-229
Mortier, V., De Wever, E., Vuylsteke, M., Holsters, M., and Goormachtig, S. (2012). Nodule numbers are governed by interaction between CLE peptides and cytokinin signaling. Plant J. 70, 367–376. doi: 10.1111/j.1365-313X.2011.04881.x
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Nicot, N., Hausman, J. F., Hoffmann, L., and Evers, D. (2005). Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 56, 2907–2914. doi: 10.1093/jxb/eri285
Notaguchi, M., Abe, M., Kimura, T., Daimon, Y., Kobayashi, T., Yamaguchi, A., et al. (2008). Long-distance, graft transmissible action of Arabidopsis FLOWERING LOCUS T protein to promote flowering. Plant Cell Physiol. 49, 1645–1658. doi: 10.1093/pcp/pcn154
Quackenbush, J., Cho, J., Lee, D., Liang, F., Holt, I., Karamycheva, S., et al. (2001). The TIGR gene Indices: analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res. 29, 159–164. doi: 10.1093/nar/29.1.159
Routledge, R. (1998). “Fisher's exact test,” in Encyclopedia of Biostatistics, Vol. 2, eds P. Armitage and T. Colton (New York: John Wiley), 1519–1523.
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., Kato, T., Nakao, M., et al. (2008). Genome structure of the legume, Lotus japonicus. DNA Res. 15, 227–239. doi: 10.1093/dnares/dsn008
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115. doi: 10.1126/science.1178534
Schwaiger, I., Kardinal, A., Schleicher, M., Noegel, A. A., and Rief, M. (2003). A mechanical unfolding intermediate in an actin-crosslinking protein. Nat. Struct. Mol. Biol. 11, 81–85. doi: 10.1038/nsmb705
Shi, L. X., and Schröder, W. P. (2004). The low molecular mass subunits of the photosynthetic supracomplex, photosystem II. Biochim. Biophys. Acta 1608, 75–96. doi: 10.1016/j.bbabio.2003.12.004
Silverstein, K. A., Moskal, W. A., Wu, H. C., Underwood, B. A., Graham, M. A., Town, C. D., et al. (2007). Small cysteine-rich peptides resembling antimicrobial peptides have been under-predicted in plants. Plant J. 51, 262–280. doi: 10.1111/j.1365-313X.2007.03136.x
Stolc, V., Samanta, M. P., Tongprasit, W., Sethi, H., Liang, S., Nelson, D. C., et al. (2005). Identification of transcribed sequences in Arabidopsis thaliana by using high-resolution genome tiling arrays. Proc. Natl. Acad. Sci. U.S.A. 102, 4453–4458. doi: 10.1073/pnas.0408203102
Swarbreck, D., Wilks, C., Lamesch, P., Berardini, T. Z., Garcia-Hernandez, M., Foerster, H., et al. (2008). The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 36, D1009–D1014. doi: 10.1093/nar/gkm965
Tatusov, R. L., Koonin, E. V., and Lipman, D. J. (1997). A genomic perspective on protein families. Science 278, 631–637. doi: 10.1126/science.278.5338.631
Tsai, J., Sunkara, S., Karamycheva, S., Pertea, G., Sultana, R., Antonescu, V., et al. (2005). The TIGR gene indices: clustering and assembling EST and known genes and integration with eukaryotic genomes. Nucleic Acids Res. 33, D71–D74. doi: 10.1093/nar/gki064
Udvardi, M. K. (2002). Meeting report: legume genomes and discoveries in symbiosis research. Genome Biol. 3: Reports 4028.1–4028.3. doi: 10.1186/gb-2002-3-9-reports4028
Van de Velde, W., Zehirov, G., Szatmari, A., Debreczeny, M., Ishihara, H., Kevei, Z., et al. (2010). Plant peptides govern terminal differentiation of bacteria in symbiosis. Science 327, 1122–1126. doi: 10.1126/science.1184057
Wienkoop, S., and Saalbach, G. (2003). Proteome analysis. Novel proteins identified at the peribacteroid membrane from Lotus japonicus root nodules. Plant Physiol. 131, 1080–1109. doi: 10.1104/pp.102.015362
Yang, X., Tschaplinski, T. J., Hurst, G. B., Jawdy, S., Abraham, P. E., Lankford, P. K., et al. (2011). Discovery and annotation of small proteins using genomics, proteomics, and computational approaches. Genome Res. 21, 634–641. doi: 10.1101/gr.109280.110
Young, N. D., Debellé, F., Oldroyd, G. E., Geurts, R., Cannon, S. B., Udvardi, M. K., et al. (2011). The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524. doi: 10.1038/nature10625
Keywords: gene annotation, legume genomes, short open reading frames
Citation: Guillén G, Díaz-Camino C, Loyola-Torres CA, Aparicio-Fabre R, Hernández-López A, Díaz-Sánchez M and Sanchez F (2013) Detailed analysis of putative genes encoding small proteins in legume genomes. Front. Plant Sci. 4:208. doi: 10.3389/fpls.2013.00208
Received: 14 March 2013; Accepted: 01 June 2013;
Published online: 20 June 2013.
Edited by:
Rajeev K. Varshney, International Crops Research Institute for the Semi-Arid Tropics, IndiaCopyright © 2013 Guillén, Díaz-Camino, Loyola-Torres, Aparicio-Fabre, Hernández-López, Díaz-Sánchez and Sanchez. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Federico Sanchez, Departamento de Biología Molecular de Plantas, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Av. Universidad 2001, Colonia Chamilpa, CP 62210, Cuernavaca, Morelos, México e-mail:ZmVkZXJpY29AaWJ0LnVuYW0ubXg=
†These authors have contributed equally to this work.