 Stefanie Dukowic-Schulze
Stefanie Dukowic-Schulze Changbin Chen
Changbin Chen- Department of Horticultural Science, University of Minnesota, St. Paul, MN, USA
Although a number of genes that play key roles during the meiotic process have been characterized in great detail, the whole process of meiosis is still not completely unraveled. To gain insight into the bigger picture, large-scale approaches like RNA-seq and microarray can help to elucidate the transcriptome landscape during plant meiosis, discover co-regulated genes, enriched processes, and highly expressed known and unknown genes which might be important for meiosis. These high-throughput studies are gaining more and more popularity, but their beginnings in plant systems reach back as far as the 1960's. Frequently, whole anthers or post-meiotic pollen were investigated, while less data is available on isolated cells during meiosis, and only few studies addressed the transcriptome of female meiosis. For this review, we compiled meiotic transcriptome studies covering different plant species, and summarized and compared their key findings. Besides pointing to consistent as well as unique discoveries, we finally draw conclusions what can be learned from these studies so far and what should be addressed next.
Introduction
Knowledge regarding meiosis in plants and other species has been obtained over decades (by now even more than a century), mainly from cytological observations and mutant analysis. Many excellent reviews have been written about what is happening in plant meiosis at the chromosome level, and which key players orchestrate recombination, progression and division (for example see Jones and Franklin, 2008; Harrison et al., 2010). However, there is still a lot to explore, especially at the molecular level, facilitated by more recent high-throughput technologies like microarray or RNA sequencing. Massive data obtained by large-scale transcriptome studies allows in depth examinations of enriched processes and of genes that are up-regulated during meiosis, many of which remain uncharacterized.
To gain insight into the gene expression landscape during meiosis, large-scale meiotic transcriptome studies have been performed in various organisms like Saccharomyces cerevisiae (Chu et al., 1998; Primig et al., 2000), Schizosaccharomyces pombe (Mata et al., 2002), Drosophila melanogaster (Andrews et al., 2000), Caenorhabditis elegans (Reinke et al., 2000), Mus musculus (Pang et al., 2006), and Rattus norvegicus (Schlecht et al., 2004). Except in yeast systems, obtaining pure cells undergoing meiosis (meiocytes) can be tricky, and thus, often whole reproductive organs are used. As a consequence, although many transcriptome studies have been performed on reproductive tissues, only a handful used isolated meiocytes. To study gametophyte development, examining transcriptomes of whole reproductive organs is not ideal. An improvement are so-called ablation studies, which profile mutants defective in meiosis in comparison to wild type. This helped to detect key genes for meiosis due to their absent or down-regulated expression in mutants, (Johnston et al., 2007; Ma et al., 2007, 2012; Wijeratne et al., 2007; Nan et al., 2011; Feng et al., 2012; Wei et al., 2013). However, many of these plant studies paid their attention especially to post-meiotic male gametophyte development, starting after meiosis is completed (reviewed in Mascarenhas, 1990; McCormick, 2004; Twell et al., 2006; Becker and Feijó, 2007; Singh and Bhalla, 2007; Borg et al., 2009; Berger and Twell, 2011).
Very little is known of regulatory mechanisms of gene expression during meiosis although a few meiotic transcription factors such as Male Meiocyte Death 1 (MMD1) in Arabidopsis (Yang et al., 2003) and Meiosis-associated Zinc-finger Protein (MEZ1) in Petunia (Kapoor and Takatsuji, 2006) have already been characterized. Transcriptome profiles of these could provide insight into their target genes but have not been reported yet. In this review paper, we concentrate on the insight we gain from the existent studies that focused on the plant meiotic transcriptome, starting with a brief historical overview.
Reproductive Transcriptome Studies
Historical Progress in Transcriptome Studies
Although we will later focus on gene expression, the term transcriptome extends beyond this and includes different types of RNA that are transcribed; besides the protein coding messenger RNA (mRNA), total RNA contains structural ribosomal RNA (rRNA), transfer RNA (tRNA), and diverse kinds of non-coding regulatory small or microRNA (sRNA, miRNA) as well as long non-coding RNA. Early work on the plant reproductive transcriptome looked at the total RNA amount in developing pollen, using methods like micro-spectrophotometry, autoradiography, and differential extraction of DNA and RNA with perchloric acid at ambient temperatures (Ogur et al., 1951; Taylor, 1953; Woodard, 1958; reviewed in Mascarenhas, 1971). The total amount of RNA per pollen varied between different species, but did so in a reasonable range, with 196 pg in Tradescantia (5.1 pg with polyA, equalling ~6 million mRNA molecules; Mascarenhas and Mermelstein, 1981), 230 pg in Nicotiana tabaccum (6.2 pg with polyA; Tupy, 1982), and 352–705 pg in Zea mays (8.9–17.8 pg with polyA; Mascarenhas et al., 1984). An early study aimed to distinguish between RNA classes occurring in pollen was conducted by Steffensen (1966) in lily: most RNA was found to be synthesized between post-meiotic DNA replication and the first pollen mitosis, with a second smaller peak after pollen mitosis I, and ~75% of the RNA being ribosomal RNA. Thus, studies that measured the whole RNA amount in reproductive tissues reported substantially the changes in rRNA synthesis. To examine gene expression, other studies applied hybridization of 3H-labeled cDNA with RNA in excess (Galau et al., 1976). In contrast to routine large-scale transcriptome analyses done today, this technique was most often performed on polyA-RNA captured from ribosomes, thus giving an even better approximation of which genes are translated and their quantity. Estimated amounts of expressed pollen genes from these experiments (see Mascarenhas, 1975, 1990, 1993) are similar to current numbers obtained with microarray technology (Becker et al., 2003; Honys and Twell, 2003). For maize, Willing and Mascarenhas (1984) reported a total of ~24,000 genes expressed in pollen: 35% of those were very abundantly expressed, including ~240 genes with ~32,000 copies per cell, ~50% of the genes were in the moderately abundant class (~6000 genes with ~1700 copies), and 15% of the genes were in the weakly expressed class (~17,000 genes with ~200 copies); in shoots ~31,000 genes were detected in total, but genes were less abundantly expressed (Willing and Mascarenhas, 1984). For Tradescantia, gene numbers reported by Willing et al. (1988) were in a similar range, with ~20,000 genes in total, and again, compared with shoots (~30,000 genes total), the abundant and moderately abundant classes were far larger in pollen (75% of the genes instead of just 35% in shoots). In addition, the average copy number of the low abundant class was far higher (100 copies per cell instead of just 5–10 in shoots). Another lab (Kamalay and Goldberg, 1980), found ~24,000–27,000 transcripts in anther, ovary, leaf, stem, root and petal tissue, and the highest number of specific transcripts (~10,000) in the cases of anther and ovary. They reported that non-translated mRNA is also present in the nucleus, concluding that post-transcriptional regulation might play an important role for gene expression. Taken together, at that time 10–20% of transcripts detected in pollen were suggested to be pollen-specific (Willing and Mascarenhas, 1984; Stinson et al., 1987; Willing et al., 1988).
After these pioneering studies of quantitative assessment of pollen gene expression, studies followed up to identify abundant or specific transcripts by differential screening, sequencing and characterization of cDNA library clones (Koltunow et al., 1990; Scott et al., 1991; Tsuchiya et al., 1994). This method is of course not as high-throughput as RNA-seq or microarray, and usually yielded less than 50 specific cDNAs, but was still used some 10 years ago, e.g., for maize and tobacco generative cell cDNA libraries (Xu et al., 2002; Engel et al., 2003). The real large-scale, high-throughput era only arrived with the emergence of commercial microarrays and RNA-sequencing platforms.
General Remarks—Data Validity, Comparability and Usability
Advancement of new technologies propelled transcriptome research forward, with Affymetrix developing a microarray gene chip available for the model plant Arabidopsis thaliana, covering ~8000 probe sets, followed by the ATH1 gene chip with ~24,000 probe sets (compared in Hennig et al., 2003). Although microarrays allow for comparison between multiple samples, it has to be considered that not all genes might be represented on a chip, and that expression levels between genes are not directly comparable due to different hybridization strengths and background signals (Naef and Magnasco, 2003; Zhang et al., 2003; Gautier et al., 2004). Data analysis involving t-tests applied to anthers of different stages from wheat and rice microarray identified ~500 genes putatively involved in meiosis for wheat, in contrast to ~7500 for rice, including only 2 out of ~50 known meiotic genes (Crismani et al., 2011). The authors suggested that this might be due to (i) the wheat genome not being sequenced yet at that time, (ii) specific genes missing on the wheat chip, and/or (iii) to different research groups doing the sampling and processing. In a similar review as our current one, the drawbacks of most studies were mentioned to be as follows: (i) less genes than thought were detected because the ATH1 gene-chip contains mostly sporophytic genes (which can be avoided by using TILLING microarrays instead), (ii) most often whole anthers were used, which might drown gametophyte-specific genes, and (iii) in ablation studies (mutant-wild type comparisons), mutations could cause un-anticipated effects on the transcriptome (Schmidt et al., 2012). The last two drawbacks also apply to RNA sequencing. Hoewver, in contrast to microarrays, RNA sequencing does not rely on hybridization, but is a direct measurement of RNA levels, has excellent technical reproducibility (Marioni et al., 2008; Mortazavi et al., 2008; Bullard et al., 2010), and RNA-seq identifies ~30% more differentially expressed genes in human samples than a standard array (Marioni et al., 2008). Still, there are also minor issues with RNA sequencing, like read number bias due to GC content or fragments with preferential expression (Pickrell et al., 2010; Hansen et al., 2012), making downstream analysis including normalization an important factor. Furthermore, the percentage of alignment to the reference genome depends on RNA and library quality, as well as sequence completeness (Chen et al., 2010). Taken together, results from both microarrays and RNA-seq always depend not only on the quality of the reference genome and gene annotation, but also on the algorithms, software and parameters used, not to mention differences between sampling and processing in different research groups. As an additional note of precaution, we want to point out that neither do transcriptional induction and the amount of transcripts always correlate with protein amount and activity, nor does transcript abundance necessarily imply an essential role of the gene (Kaback and Feldberg, 1985). Nonetheless, transcriptome studies are a valuable tool to detect novel specific or abundant transcripts or prevalent pathways when comparing specific cell types and developmental stages, but have to be connectable with other datasets. Excellent ways to provide this connectivity are publically available datasets of raw data together with detailed description of how they were obtained, as well as studies that produce a whole gene expression atlas, e.g., in rice with 25 reproductive stages (Fujita et al., 2010), and later with 33 laser-assisted micro-dissected anther samples and 143 spatiotemporal samples (Aya et al., 2011), in barley with 15 tissues of different developmental stages (Druka et al., 2006), in Arabidopsis with 79 samples through development (Schmid et al., 2005), or in maize with 60 diverse tissues (Sekhon et al., 2011). Data from Arabidopsis, maize and more plant species can be easily accessed and mined using the eFP Browser (http://bar.utoronto.ca/welcome.htm; Winter et al., 2007). Figure 1 shows examples of how gene expression details can be examined using the eFP browser, i.e., genes identified in a comparative transcriptome study of Arabidopsis and maize data (Dukowic-Schulze et al., 2014a) as up-regulated in isolated meiocytes vs. seedlings (Figure 1A) or the other way around (Figure 1B). Figure 1C shows the well-known recombinase gene RAD51 which acts in both meiotic and somatic tissue.
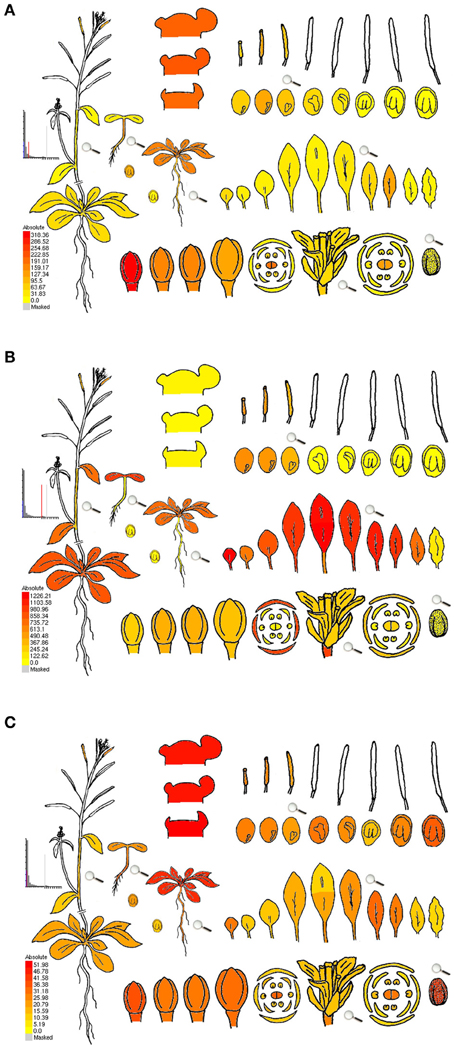
Figure 1. eFP browser view of gene expression during Arabidopsis development. (A) DMC1 (At3g22880), encoding a well-characterized meiotic recombinase, confirmed as up-regulated in isolated meiocytes vs. seedlings in Arabidopsis and in maize (>5-fold and >1000-fold respectively). (B) NDF1 (At1g15980), encoding a chloroplast NAD(P)H dehydrogenase subunit, identified as more than 35-fold up-regulated in seedlings vs. isolated meiocytes in Arabidopsis (and by more than 700-fold in maize homologs). (C) RAD51 (At2g20850), encoding a well-characterized recombinase, operating in both meiotic and somatic tissue; slightly up-regulated in isolated meiocytes vs. seedlings in Arabidopsis and in maize (>2-fold and <2-fold respectively). Expression strength coded by color: yellow = low, red = high. The Arabidopsis eFP Browser is located at bar.utoronto.ca, published in Winter et al. (2007), developed by B. Vinegar, drawn by J. Alls and N. Provart. Data from Gene Expression Map of Arabidopsis Development by Schmid et al. (2005) and the Nambara lab for seed stages.
What We Have Learned from Reproductive Transcriptome Studies So Far
Overview
Around a 100 large-scale studies on plant reproductive transcriptomes have been published so far (Figures 2A–E), at least half of them with pre- or post-meiotic samples, many on mature pollen which are easy to obtain (reviewed in Mascarenhas, 1990; McCormick, 2004; Twell et al., 2006; Becker and Feijó, 2007; Singh and Bhalla, 2007; Borg et al., 2009; Berger and Twell, 2011). The remaining around 50 publications focused on or at least included samples during meiosis; 25 studies used whole anthers, 13 studies whole female organs, and 13 studies used isolated male meiocytes (Table 1 and Figure 2). Female studies conducted are almost all on post-meiotic stages, only one study was performed with isolated megaspore mother cells (Schmidt et al., 2011). Whole anthers contain different specialized cell layers surrounding the meiocytes, and the obtained expression profile is thus a mix of these; indeed, in an expression atlas in barley, the anther was found to be the most complex tissue regarding its transcriptome (Druka et al., 2006), and the same was found for maize anthers (Ma et al., 2008). In maize, the examination of different anther developmental stages showed that the transcriptome is most diverse in pre-meiosis (Ma et al., 2007), with reduced expression and few changes during meiosis (Ma et al., 2008). In accordance with this, male Arabidopsis meiocytes exhibited less DNA and RNA metabolic activity and more signal transduction when compared to either whole anthers of the same developmental stage or to young seedlings (Chen et al., 2010). Deveshwar et al. (2011) suggested the existence of a two-way molecular switch which shuts down sporophytic genes and activates gametophytic ones toward the end of meiosis. Although most studies were performed on male reproductive transcriptomes, comparison with the few studies from female showed shared and special transcriptome features, as reviewed in Schmidt et al. (2012). Given the excellent reviews on transcriptome studies on post-meiotic gametophytic development (reviewed in Mascarenhas, 1990; McCormick, 2004; Twell et al., 2006; Becker and Feijó, 2007; Singh and Bhalla, 2007; Borg et al., 2009; Berger and Twell, 2011), we concentrate on studies that included or solely used meiotic samples.
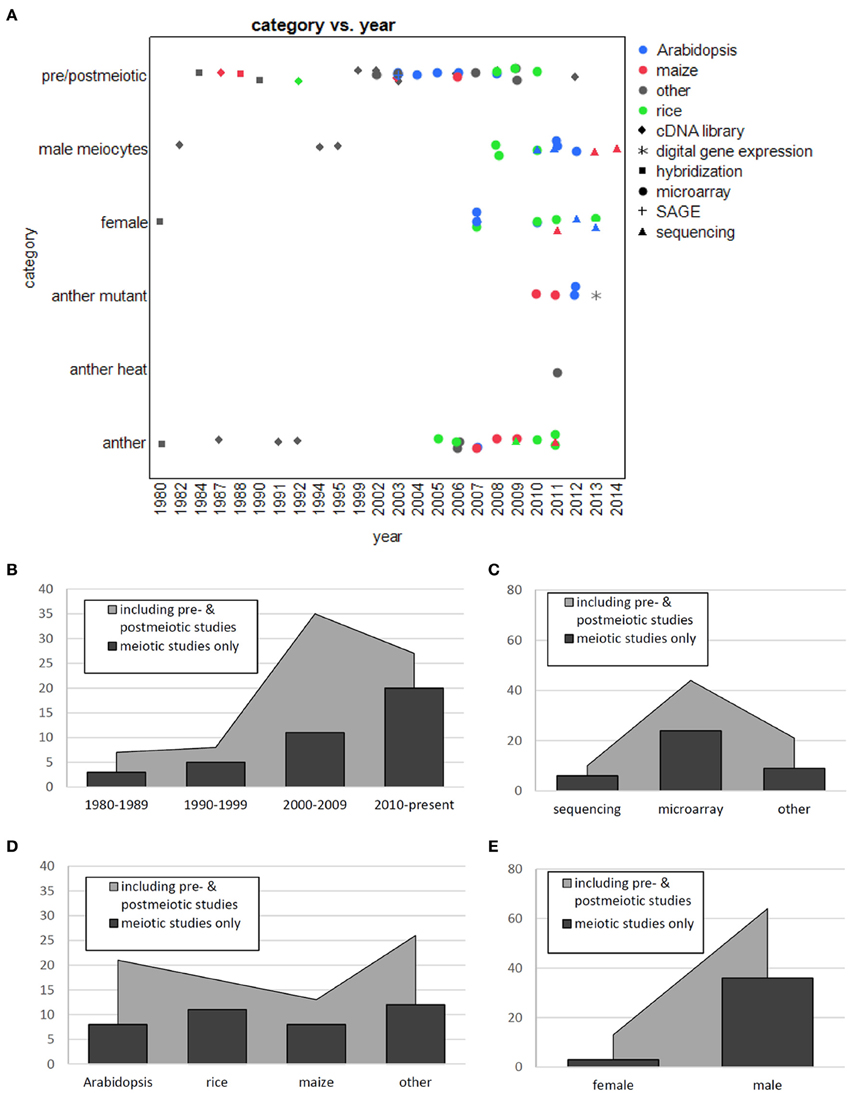
Figure 2. Distribution of primary literature on transcriptomes of reproductive structures. (A) Scatter plot with species coded by color, techniques by shape. (B) Transcriptome studies per decade. (C) Transcriptome studies per technology. (D) Transcriptome studies per species. (E) Transcriptome studies per gender.
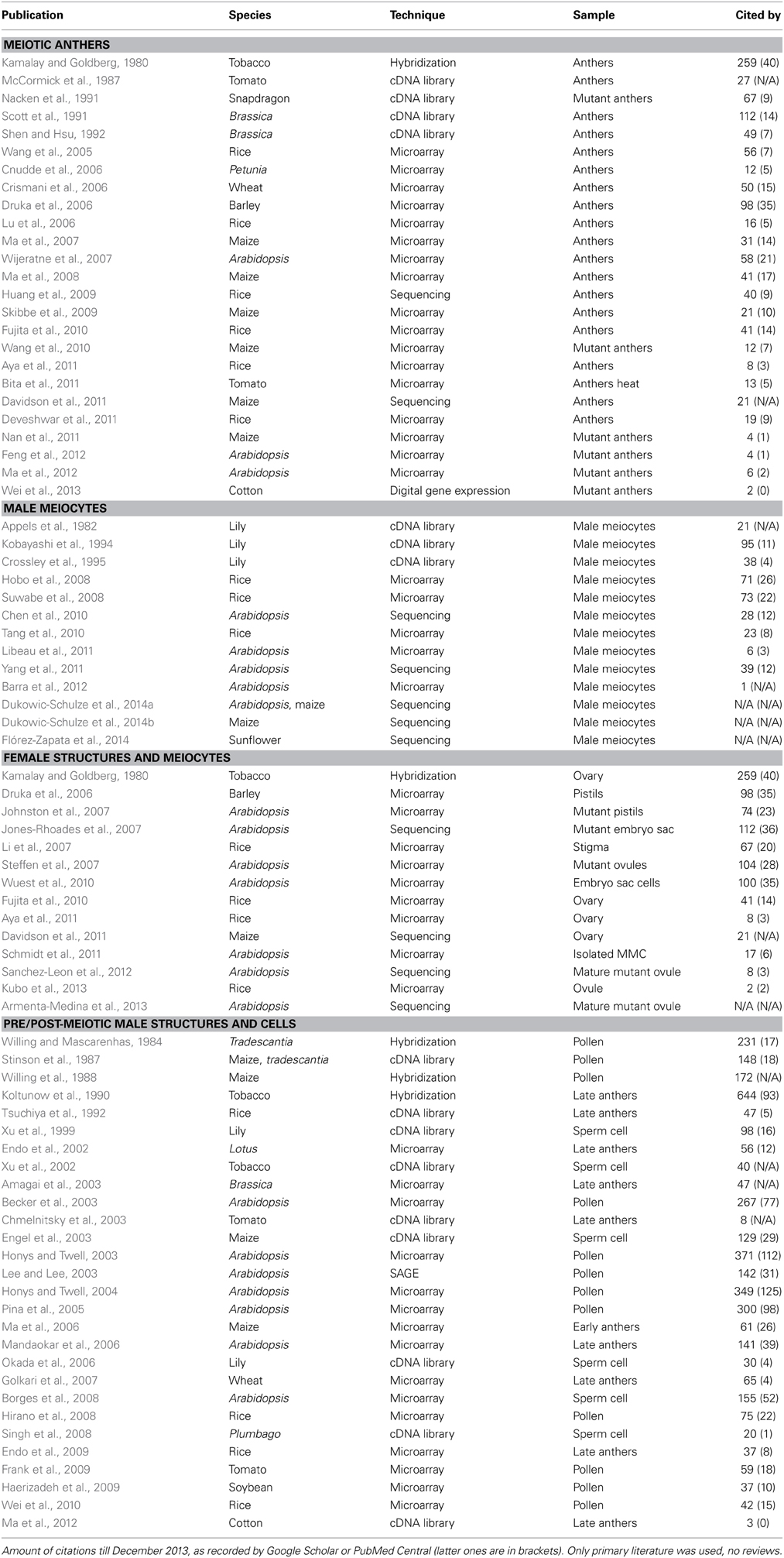
Table 1. List of published literature regarding the transcriptome of reproductive stages.
Results and Insight from Whole Anther Studies
We will first recapitulate on the insight obtained from whole anther studies. There are additional ablation studies on which we do not elaborate here. These usually focused especially on the genes down-regulated in mutants while we concentrate here on the gene expression landscape in wild-type anthers. Studies using microarray contributed most of the knowledge (Figures 2A,C), but the use of other pioneering techniques earlier on had generated comparable results: Kamalay and Goldberg (1980) had used ribosome-bound mRNA for hybridization with DNA to detect ~26,000 genes in tobacco anthers, of which ~10,000 seemed to be anther-specific. In another non-model organism without available sequence data at that time, Cnudde et al. (2006) used a cDNA-AFLP (Amplified Fragment Length Polymorphism) strategy on meiotic anthers from Petunia, and found ~8000 sequence tags of which ~6% were modulated during meiosis, including known meiotic genes.
The amount of genes expressed in anthers was reported as more than 30,000 in maize and wheat (Crismani et al., 2006; Ma et al., 2008; Wang et al., 2010), and ~22,000 in rice (Deveshwar et al., 2011). These numbers vary depending on the stages used for comparison and the criteria used to define expression. More important and informative are lists of genes that are specific to anthers or temporally up-regulated in anthers during meiosis. Of these, 1350 were reported in wheat, 30 of which displayed a more than 8-fold expression change during meiosis, others showing more subtle changes in their expression modulation as seen by hierarchical clustering (Crismani et al., 2006). These weak differences in expression were suggested to be due to the anther dilution effect, and possible additional somatic functions (Crismani et al., 2006). In rice, a similar number, 1000 genes, were found to be anther-specific, most of them due to specificity in mature pollen stages, with only 78 specific during meiosis (Deveshwar et al., 2011). In maize, ~10,000 genes in total might be specific to one or few anther stages, for example ~200 stage-specific genes in 2 mm long anthers (containing cells in meiosis), ~700–900 transcripts shared with the following stage (Ma et al., 2008).
Of the 1350 temporally regulated genes in wheat, many were annotated for chromatin association, synaptonemal complex, recombination, mismatch repair and fertility; and concurrent with anther developmental progression, most functional categories decreased in their expression level, especially meiosis/cell division, ribosomes, transcription factors, organelle activity, signal transduction and lipid and protein metabolism (Crismani et al., 2006). GO (Gene Ontology) terms enriched in genes abundantly expressed in rice anthers during meiosis were transcription factors, protein folding, sorting and degradation, as well as cell structure components (Deveshwar et al., 2011). In maize, transcription factors were prevalent in anthers, and 131 cell wall associated transcripts were found to be enriched (Ma et al., 2008).
Since one intention of these studies was to find out more about meiotic genes, it is interesting which known meiotic genes could be identified by their expression profile. In wheat, the 1350 anther-regulated transcripts included ASY1, MSH2, 6 and 7, RAD51B and C, DMC1, and RPA (Crismani et al., 2006). In rice, MSH2, 6 and 7, RAD51B and DMC1, as well as SPO11, MND1, RAD50, ZYP1A/B, and MUS81 showed elevated expression levels in meiotic anthers (Deveshwar et al., 2011). In maize, anther genes included genes similar to EXS1, KU70, and MMD1 (Ma et al., 2008), and clustering of genes revealed genes required for meiosis initiation with DMC1, RAD51 and an ARGONAUTE/PIWI homolog (Ma et al., 2007). Interestingly, Deveshwar et al. (2011) pointed out that many meiotic genes such as HOP2 and RAD51 were not specifically expressed in meiosis in rice, and concluded that they might play other roles in addition. Another rice study by Tang et al. (2010) on isolated pollen mother cells (PMCs) had found 1158 PMC preferential genes, including many known meiotic genes, but the study on whole rice anthers claimed only 372 core meiotic anther genes since most of the 1158 were also expressed at earlier or later anther stages (Deveshwar et al., 2011).
A data analysis study of the rice and wheat anther data described above found only two out of around 50 known meiotic genes as enriched in both sets concomitantly, namely ASY1 and MLH3 (Crismani et al., 2011). Whether other known meiotic genes were missed due to more static expression levels or because they were not included on the wheat chip, a take-home message here is the conserved up-regulation of ASY1 and MLH3, meiotic factors shown to be involved in meiotic recombination and progression (Caryl et al., 2000; Jackson et al., 2006; Sanchez-Moran et al., 2008). Inter-specific conservation of anther-expressed genes could be shown for homologs of maize transcripts in rice and Arabidopsis flowers (Ma et al., 2007, 2008), and for some meiotic anther-specific wheat-rice-homolog pairs in reproductive organs of Arabidopsis and poplar, though there were exceptions (Crismani et al., 2011). A summary of the efforts to identify early meiotic genes in cereals and an overview table can be found in Bovill et al. (2009). Known genes in early anther development in Arabidopsis and maize are also summarized in a supplemental table in Wang et al. (2010). Gene expression during anther development is orchestrated by diverse transcription factors, extensively recounted in a recent review by Khurana et al. (2012). We already mentioned the broad-scale gene expression atlas studies which can be mined for co-expression networks or for detailed information for a gene of interest; in addition, diverse reproductive transcriptomes were analyzed with RNA sequencing in maize (Davidson et al., 2011).
Results and Insight from Isolated Male Meiocytes
In contrast to a vast literature record for mature pollen, there are only few studies that examined the transcriptome of isolated meiocytes. This is probably due to the difficulty and effort involved in obtaining isolated meiotic cells; only recently did feasible techniques become established, such as FACS-based purification techniques applied to pollen (Borges et al., 2012), laser-assisted microdissection (LAM) and capillary collection of meiocytes (CCM) (Schmidt et al., 2012; Chen and Retzel, 2013; Dukowic-Schulze et al., 2014c). An impressive study conducted in lily around 30 years ago is worth mentioning here since it appears to be the first one that used isolated meiocytes for examination of the transcriptome with diverse methods (Appels et al., 1982): autoradiography to detect RNA synthesis of cultured meiocytes implied that most RNA remained on chromosomes in lily pachytene cells, and melting curves of polyA RNA from mitotic and zygotene-pachytene cells were indistinguishable, but little or no ribosomal RNA was detected in meiocytes. Furthermore, they extracted polyA RNA from isolated and cultured meiocytes from interphase till pachytene to generate a cDNA library; of ~5000 cDNA clones, 49 were specifically found in the meiotic samples, and 13 of them were very abundant with 11 sharing a repeated sequence, found to be homologous to sequences of wheat, rye and maize. In addition, they produced polypeptides from lily meiocyte polyA RNA of various stages in an in vitro wheat-translation system, which revealed a larger portion of low molecular weight proteins than from somatic polyA RNA samples (Appels et al., 1982). Three more studies on lily meiocytes followed, with Bouchard (1990) continuing analysis of the detected sequence repeats, concluding that these are common with heat-shock proteins. Another lily study used zygotene meiocytes with new cDNA library screening and found 18 genes specific or abundant, including RAD51 related, as well as heat-shock and serine protease genes, many with hydrophobic N-terminus (Kobayashi et al., 1994). Still another lily meiocyte cDNA library was produced from cells ranging from metaphase I to telophase II, and unexpectedly, the 3 clones that were found all expressed far more in tapetal cells than in meiocytes (Crossley et al., 1995). This is a phenomenon also detected in other studies: gene expression from tapetum appears to sometimes (i) contribute overly to the whole anther transcriptome, and also (ii) “leak” to meiocytes, meaning a gene is highly expressed in meiocytes although it has been shown to have a specific function in tapetal cells. Both scenarios can be inspected by in situ hybridization for up-regulated genes from either whole anthers or isolated meiocytes (Nacken et al., 1991; Scott et al., 1991; Shen and Hsu, 1992; Rubinelli et al., 1998; Wang et al., 2005; Cnudde et al., 2006; Dukowic-Schulze et al., 2014b). That none of the other lily studies seemed to capture these tapetum-prevalent genes might be dependent on the stages examined: the transcriptome landscape of meiocytes is generally more unique and richer in early prophase stages than in later stages, when the transcriptome activity and complexity in meiocytes decreases (Mackenzie et al., 1967; Crismani et al., 2006; Ma et al., 2008). After these initial studies, the next set of studies used rice: 140 anther-specific genes, identified before in anthers containing pollen (Endo et al., 2004), were further classified by microarray of laser-assisted micro-dissected tapetum and microspores from 5 different developmental stages as 71 being gametophyte-specific, 7 tapetum-specific and 62 common to both (Suwabe et al., 2008). A following publication isolated pre-meiotic PMCs with laser-assisted micro-dissection and analyzed their transcriptome with microarray (Tang et al., 2010). Among the 1158 PMC preferential genes were known meiotic genes such as SPO11 and DMC1, and enriched processes included DNA replication and repair, ubiquitin-dependent proteolysis and MADS-box transcription factors.
Very recent Arabidopsis studies concentrated on isolated meiocytes during meiosis, and on what the core meiotic transcriptome can tell us. In all three cases, pools of meiocytes from various stages were collected by micro-capillary collection, and the extracted RNA processed by Illumina sequencing (Chen et al., 2010), SOLID sequencing (Yang et al., 2011), or CATMA microarray (Libeau et al., 2011). In spite of using different technologies and downstream analysis tools, the numbers of genes found up-regulated in meiocytes were quite close, with over 1000 genes in Chen et al. (2010), over 800 in Yang et al. (2011), and 1586 or 2155 (with an overlap of 752 genes in both sets) in Libeau et al. (2011), depending on whether leaves or root tips were used for comparison. Two of the groups reported up-regulated transposable elements (TEs) in meiocytes, ~1000 located preferentially in the pericentromeric region in one case (Chen et al., 2010), vs. a generally low TE activity and only ~50 up-regulated TEs, mostly concurrent with high neighboring gene activity, in the other case (Yang et al., 2011). Such discrepancies between results in different publications can occur, or only one group investigated a specific aspect which was neglected by other groups. Attentive readers might thus have some open questions, and can in case of further interest consult the original data which is usually publically available. Examples for meiocyte transcriptome specialties only reported by one group are the high up-regulation of 55 genes that mapped to a mitochondrion-derived insert on chromosome 2 (Chen et al., 2010), the transcription of introns of more than 400 annotated genes which might be due to annotation errors or special functions (Yang et al., 2011), and the detection of small RNAs (Yang et al., 2011).
One area covered by all three Arabidopsis meiocyte studies was the analysis for known meiosis genes. Libeau et al. (2011) inferred that the expression of known meiotic genes is not strongly up-regulated, but that it might be due to a dilution effect from using a population of various stages. An attempt to cluster meiosis genes together was only partly successful, with one cluster (out of 220) with 110 genes that contained 8 meiosis genes. Using a different approach, Chen et al. (2010) found most of the 68 known meiotic genes analyzed to be expressed at 2-fold or higher in meiocytes and/or anthers when compared with seedlings, including prominent players like SPO11, DMC1, RAD51C, XRCC3, MSH4 and 5, MER3/RCK, PTD, MUS81, and SDS. Looking at known meiotic genes from different perspectives, Yang et al. (2011) found SDS, RCK, and MMD1 included in their meiocyte up-regulated gene list, and expression of many mismatch repair gene family members during meiosis. In addition, they used a broader analysis to compare the genes expressed in meiocytes with their homologs' expression in mouse and yeast and concluded that Arabidopsis shows more similarity to mouse than to yeast (Yang et al., 2011). Functional analysis for GO terms enriched in genes up-regulated in meiocytes vs. non-meiotic tissues or whole anthers was carried out in both sequencing-based studies, but with different tools, implying less DNA and RNA metabolism, but more signaling activity regarding biological processes, as well as less kinase but more nucleotide binding activity regarding molecular function (Chen et al., 2010), and pointing to prevalence of transcription factors, cell-cycle progression genes, proteolysis, DNA replication, repair and pairing (Yang et al., 2011).
After these initial studies on isolated Arabidopsis meiocytes, an additional study used LCM and microarray to compare the transcriptome of isolated meiocytes between wild type and the histone acetylation mutant Atmcc1 (Barra et al., 2012). Besides detecting candidate AtMCC1 target genes, they found up-regulation of ASK1 and RAD51C in the mutant, and effects on expression levels of genes for the meiotic or mitotic cell cycle, proteolysis and chromatin (Barra et al., 2012).
Very recently, comprehensive transcriptome profiling of isolated meiocytes of two other plant species was carried out: in maize meiocytes from early prophase, ~2000 genes were preferentially expressed, most of them uncharacterized (Dukowic-Schulze et al., 2014b). These up-regulated genes included many genes whose products function in mitochondria, and GO analysis indicated enrichment of carbohydrate metabolism, proteolysis, protein targeting, chromatin modification and redox homeostasis. In addition, candidate homologs of most known meiotic genes were up-regulated in meiocytes in comparison to seedlings, for example ~5-fold for MSH2, RAD51 and RPA1, ~10-fold for ASY1, ZYP1, MND1 and SPO11, and even ~1000-fold for DMC1 (Dukowic-Schulze et al., 2014b). A comparison of this maize data and new Arabidopsis data with only early prophase meiocytes from the same lab and methods showed that an increased amount of reads mapped to unannotated features in meiocytes in both species (Figure 3; Dukowic-Schulze et al., 2014a). Furthermore, it could be demonstrated, that mitochondrial transcripts were elevated in both species and derived from the mitochondria themselves. Known meiotic gene homolog-pairs could be clustered together, and common enriched GO terms included organelle organization, protein import, targeting and localization, as well as DNA and double-strand break repair. There were also unique features and GO terms, which might be biologically relevant, but could also be due to differences in annotation quality and quantity. Meiocytes of both Arabidopsis and maize were enriched for transcription factors, especially of the MADS-box family, including homeotic genes like APETALA3, PISTILLATA, and AGAMOUS; and in accordance with common transcription factors, promoters of meiocyte up-regulated genes also possessed common cis-regulatory motifs (Dukowic-Schulze et al., 2014a), which had also been reported in Arabidopsis before (Li et al., 2012).
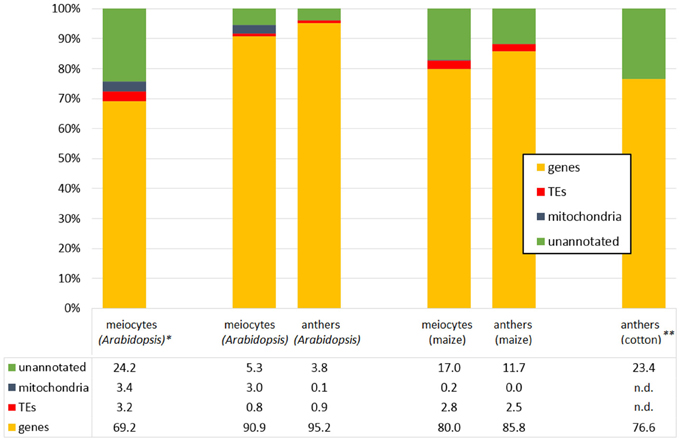
Figure 3. Example distributions of aligned RNA-seq reads. Meiocyte or anther RNA-seq reads, mapping to genes, mitochondria sequence, transposable elements (TEs) and unannotated features. Data from Dukowic-Schulze et al. (2014a,b), *Chen et al. (2010), **Wei et al. (2013).
In contrast to Arabidopsis and maize which are excellent model organisms with many resources and a huge research community, another study with isolated meiocytes dedicated its efforts toward a less used and more challenging species, sunflower (Flórez-Zapata et al., 2014). They found 7755 transcripts with higher expression in meiocytes than in somatic tissue. These were enriched for GO terms related to reproduction, and included homologs of genes with known meiotic function. Furthermore, transcription factors specific to meiocytes were detected, as well as many genes related to gene silencing (Flórez-Zapata et al., 2014).
Results and Insight from Female Pistils and Meiocytes
In contrast to the relatively well-studied male meiotic transcriptome, female meiosis was less examined so far, probably due to the difficulties to obtain the corresponding material of whole early pistils or even isolated megaspore mother cells. One study examined the stigma transcriptome (Li et al., 2007), and genetic ablation was used to study genes preferentially expressed in the mature ovule (Sanchez-Leon et al., 2012; Armenta-Medina et al., 2013) and in the mature embryo sac (Johnston et al., 2007; Jones-Rhoades et al., 2007; Steffen et al., 2007). Furthermore, individual cell types in the mature embryo sac were examined (Wuest et al., 2010; Ohnishi et al., 2011), but all these studies looked at post-meiotic transcriptomes.
A study focused on the meiotic transcriptome was performed by applying microarray on laser-dissected megaspore mother cells (MMCs) of Arabidopsis, which indicated the expression of ~9000 genes, including known key meiosis genes like MLH1, MSH4, REC8/SYN1, PTD1, and TAM (Schmidt et al., 2011). Around 800 genes were significantly up-regulated in MMCs, enriched for processes related to structural components and ribosome biogenesis, translation control, ion transport and chromatin constituents. Another, very recent study was performed with whole rice ovules of different stages containing MMCs up to the mature embryo sac, including integument tissue, which still required laser-dissection (Kubo et al., 2013). They found ~37,000 probes expressed in ovules, ~450 only in ovules, including 8 cytochrome P450 genes and 24 transcription factors. In total, 85 transcription factors were up-regulated in ovules, especially of the MADS-box, AP2-EREBP and ABI3VP1 families. Known meiosis-related genes were found, e.g. DMC1B, MEL1, MSP1, SDS, PAIR1, and 3 were highly expressed during both male and female meiosis, and DMC1A, MEL2, MER3/RCK, TOP6A, PAIR2, ZEP1, and RAD21-4 showed lower and more constant expression in ovules than in anthers. Since they examined different stages, they could show that a major down-regulation of genes takes place between zygotene-pachytene and later stages, with many genes more than 10-fold decreased, which might be due to genome-wide silencing. During pachytene, a temporal and dramatic up-regulation of a subgroup of TE elements was detected, especially of the mudrA-like, snapdragon TNP2-like and retrotransposon-like classes. After pachytene, genes involved in epigenetic modifications that might be involved in TE repression were up-regulated namely MET1-2, MET2a, and RDR3 (Kubo et al., 2013).
Special Types of RNA
We mentioned in a previous paragraph that the earliest studies on reproductive plant tissue measured whole RNA content, and that most of it was due to rRNA. Interestingly, there appears to be an oscillating production curve for rRNA. An observation that was made by molecular and cytological means during meiosis was that the total RNA content decreases in prophase I till diplotene/diakinesis, and increases again afterwards; in accordance with this, pachytene cytoplasm showed fewer ribosomes than for example pre-leptotene (Mackenzie et al., 1967). The ribosome depletion during meiosis has also been observed by other groups (Williams et al., 1973; Stern and Hotta, 1977; Dickinson, 1987).
Most rRNA synthesis in microspores was detected between post-meiotic DNA replication and pollen mitosis I in lily, with a second but smaller peak after pollen mitosis I (Steffensen, 1966). Another study had also shown an increase in total RNA late in pollen maturation, and that total RNA content was higher during late meiosis than during the first free microspore stages (Ogur et al., 1951). A similar ribosome synthesis pattern was seen in Tradescantia with most RNA synthesis before anther dehiscence (Mascarenhas and Bell, 1970). Mascarenhas (1990) confirmed that rRNA transcription happened prior to pollen mitosis I and that no synthesis occurred in mature pollen or the developing tube in both Tradescantia and lily. The pattern of tRNA synthesis was similar (Peddada and Mascarenhas, 1975). In agreement with low or no rRNA synthesis in mature pollen, nucleoli—the structure where rRNA transcription takes place—are very small or absent late in pollen maturation in Tradescantia (Woodard, 1958). Taken together, the described oscillation of rRNA synthesis and related structures is very intriguing, and might imply changing requirements and resource allocation during pollen development.
While rRNA production was investigated more thoroughly early on, recent interest shifted to another transcriptome component, namely small RNAs. These small regulatory molecules, connected with silencing or gene regulation in general, were shown to affect both male and female reproduction (Millar and Gubler, 2005; Wu et al., 2006). Some of the transcriptome studies on female meiosis examined small RNAs during megasporogenesis (Schmidt et al., 2011), and found enrichment for PAZ and PIWI domain proteins in egg cells (Wuest et al., 2010). Studies on post-meiotic male gametophyte development detected the presence of small RNAs and proteins involved in small RNA pathways (Chambers and Shuai, 2009; Grant-Downton et al., 2009a,b; Slotkin et al., 2009; Wei et al., 2010). These and other studies already revealed exciting possibilities for regulation of post-meiotic gene expression and transposable element silencing, and the interested reader is referred to comprehensive reviews on this topic (Borges et al., 2011; Trionnaire et al., 2011; Van Ex et al., 2011; Gutierrez-Marcos and Dickinson, 2012; Feng et al., 2013). Here, we concentrate our attention on findings from studies during meiosis, though only few clues are existent at this time, most of which do not stem from large-scale transcriptomic analyses: AGO1 is one of the main regulators of microRNA (miRNA, 21–24 nucleotides long) directed target cleavage in Arabidopsis, and mutants show multiple developmental defects, including sterile flowers in some instances (Vaucheret et al., 2004). An AGO5 homolog in rice, MEL1 was shown to be required for correct progression through meiosis (Nonomura et al., 2007). AGO9 is required to specify cell fate in Arabidopsis ovules, controlling the amount of gametes (Olmedo-Monfil et al., 2010), and a mutant of an AGO9 homolog in maize could produce gametes without meiosis, while showing chromatin condensation defects and failure in chromosome segregation in meiosis (Singh et al., 2011). Taken together, the progress in research of small RNA pathways resembles the one for meiotic genes: at first, mutant analysis supported the characterization of single genes, now moving to large-scale approaches. A few recent studies on meiotic transcriptomes mentioned small RNAs or ARGONAUTE genes: in whole anther transcriptome studies, Ma et al. (2006; pre-meiotic), as well as Huang et al. (2009; including meiotic anthers) detected antisense transcripts at around 10% of sense transcripts level, suggesting their role to be regulatory or RNA splice intermediates. One of the studies conducted on isolated male meiocytes in Arabidopsis (Yang et al., 2011) reported miRNAs, for example mi163, which was shown to act against SAM-dependent methyltransferases (Allen et al., 2004); it was pointed out that the approach can only detect small RNAs with polyA, so called pri-mRNA (Kurihara and Watanabe, 2004). In the transcriptome of isolated male meiocytes, another clue for the importance of small RNA pathways was found, since AGO3 and AGO8 were highly expressed in meiocytes (Chen et al., 2010).
Beyond the Transcriptome—What's Next?
When writing this review, we observed a trend that studies done around the same time focused on a certain area. In the very beginning, most attention was on rRNA, which is now not thoroughly examined in the context of meiosis anymore. Then, different techniques enabled mRNA-focused studies. While the first of these were done on meiocytes or reproductive organs during meiosis, afterwards for around a decade around the turn of the century, there was a substantial gap before attention shifted back to meiosis (Figures 2A,B). In the meantime, lots of transcriptome studies had focused on pre- or even more frequently on post-meiotic reproductive organs and processes.
With this in mind, we want to share our thoughts about where the future of meiotic studies will probably lead us. One big emerging topic surely are regulatory RNAs, encompassing both long non-coding RNA and small RNA. Many different kinds of small RNAs exist, defined in part by their nucleotide length, and they act in multiple roles in gene regulation. Sequencing of small RNA from meiotic stages will certainly give us even more data that can be integrated into whole regulatory networks. As a proof of concept, pioneer studies on small RNA profiling in plant reproductive tissue have been already performed on mature pollen and mature ovules, revealing novel regulation mechanisms in these late stages (Slotkin et al., 2009; Olmedo-Monfil et al., 2010). Further approaches which can help elucidate the underlying mechanisms of the meiosis transcriptome architecture include profiling of epigenetic marks such as DNA methylation and histone modification, and should be performed with isolated meiocytes in the future.
Although a lot of reproductive transcriptome data have been produced till date, the data from large-scale transcriptome studies are neither fully exploited nor should they be seen as the gold standard for high-throughput examination of gene expression: first, conclusions that can be drawn reproducibly from different species should be pursuit in more detail, like the enrichment for certain transcription factors and cis-regulatory elements, proteolysis as an important regulatory component, and the abundance of mitochondrial transcripts. Discoveries made in one or multiple species should be verified by re-analyzing more data sets simultaneously, and then examining more details by exploiting the data for this particular aspect and adding additional experiments such as those involved in mutant characterization. Second, we should keep in mind that we cannot absolutely rely on conclusions we draw from mere polyA RNA-sequencing data because (i) the splicing landscape might be globally reprogrammed as in mouse meiosis (Schmid et al., 2013), having consequences for expression or function, and (ii) translation regulation can still change the resulting amount of gene products, as an translational initiation factor in Drosophila spermatogenesis was shown to be important for segregation, cytokinesis and shaping protein levels (Hernández et al., 2012). Two approaches to get closer to the relevant gene expression are ribosome profiling and proteomics—both challenging due to the difficulty to obtain enough pure meiotic material. An extensive ribosome profiling study in yeast investigated RNA bound to ribosomes at 25 time points during meiosis (Brar et al., 2012), and for ~200 genes they demonstrated an ~10-fold change in translation efficiency (calculated by dividing the reads per kilobase per million mapped reads—RPKM—from ribosome-bound RNA by the RPKM of total polyA RNA). Besides the very early studies done in lily and tobacco, to our knowledge no newer large-scale plant meiotic study has used ribosome-profiling. The second approach to get more insight into processes occurring during meiosis is to look even further, to the protein level. Indeed, first proteomics studies have been conducted in meiotic anthers and pollen, and though their resolution is not that high yet, they yield valuable information about major factors present during meiosis. As in the case of transcriptome studies, most proteomics studies used mature pollen (Holmes-Davis et al., 2005; Noir et al., 2005; Dai et al., 2006, 2007; Sheoran et al., 2006, 2007; Lopez-Casado et al., 2012). However, two associated studies in rice included early anthers containing meiotic cells (Imin et al., 2001; Kerim et al., 2003), and found global differences in the proteome during microspore development, with 150 proteins identified. An interesting aspect was revealed by Wang et al. (2010), who performed both transcriptomics and proteomics on anthers and found additional changes in timing or differential expression when looking at the proteome instead of the transcriptome.
Concluding Remarks
Why are all these transcriptome studies of reproductive plant tissue important? Besides the academic questions about how organs as sophisticated as anthers and pistils arise, develop, function, and are regulated, there are also direct uses feasible for horticulture and agriculture, namely breeding and ensuring crop production. Both are intertwined—basics have to be understood and unraveled by fundamental research, and real-life observations, issues and goals can lead to more application-based research, aiming toward better selection and manipulation of plants.
Examining the effects of extreme temperatures on meiosis is one example and appealing for two reasons, (i) climate change (especially since elevated temperature can cause sterility and thus result in crop yield reduction), and (ii) recombination-pattern changes in elevated temperatures (Lu, 1974; Higgins et al., 2012), which could lead to broader variety in plant phenotypes for breeding. A recent review (Giorno et al., 2013) nicely sums up the findings from transcriptome studies of plant reproductive tissues where heat was applied. The amount of genes that was found to be differentially regulated by temperature influence in the anther ranged from 31 to 3353, and might contain candidates for targeted breeding to cope with changing climate.
Facilitating hybrid creation with a male-sterile parent can be accomplished by genetic engineering for male sterility. For this, knowledge is needed about candidate genes essential for male fertility as well as meiosis-specific promoters. Some studies already followed up on large-scale transcriptome profiling to identify and test meiosis-specific promoters (Steffen et al., 2007; Li et al., 2012).
Although there are a lot of published studies on the plant meiotic transcriptome by now, most of the obtained data are not yet exploited to a satisfactory level. Besides the promoter studies mentioned above, another study followed up on high expression of AGO9 in Arabidopsis ovules with a detailed cytological characterization and sequencing of AGO9-associated small RNAs (Olmedo-Monfil et al., 2010). We hope to see more research of this kind in the future, that examines existent data sets and focuses on specific findings to reveal more detail. In addition, there is a need for more studies that compare conclusions from different studies, concentrating on conserved findings as well as discrepancies that might be interesting to pursue by first re-examining raw data.
What is now needed are more specific (meaning single gene or process) examinations, further aspects (like ribosome profiling, small RNA sequencing and proteomics), as well as improved connectivity (between different species and platforms). In addition, far more studies address male meiosis than female meiosis. For the future, research will hopefully catch up on neglected aspects like female meiosis, the differences in gene expression and processes between male and female meiosis, and the importance or consequences of up-regulated transposons or mitochondria genes. We are looking forward to a time of combined efforts that will hopefully lead to better fundamental understanding of meiosis, and to innovative applications and adaptations to ensure meiotic success in a changing environment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by funding from the National Science Foundation (IOS: 1025881) to C.C. and others, and from BRDC to C.C.
References
Allen, E., Xie, Z., Gustafson, A. M., Sung, G.-H., Spatafora, J. W., and Carrington, J. C. (2004). Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat. Genet. 36, 1282–1290. doi: 10.1038/ng1478
Amagai, M., Ariizumi, T., Endo, M., Hatakeyama, K., Kuwata, C., Shibata, D., et al. (2003). Identification of anther-specific genes in a cruciferous model plant, Arabidopsis thaliana, by using a combination of Arabidopsis macroarray and mRNA derived from Brassica oleracea. Sex. Plant Reprod. 15, 213–220. doi: 10.1007/s00497-002-0159-z
Andrews, J., Bouffard, G. G., Cheadle, C., Lü, J., Becker, K. G., and Oliver, B. (2000). Gene discovery using computational and microarray analysis of transcription in the Drosophila melanogaster testis. Genome Res. 10, 2030–2043. doi: 10.1101/gr.10.12.2030
Appels, R., Bouchard, R. A., and Stern, H. (1982). cDNA clones from meiotic-specific poly(A)+ RNA in Lilium: homology with sequences in wheat, rye, and maize. Chromosoma 85, 591–602. doi: 10.1007/BF00327353
Armenta-Medina, A., Huanca-Mamani, W., Sanchez-Leon, N., Rodriguez-Arevalo, I., and Vielle-Calzada, J.-P. (2013). Functional analysis of sporophytic transcripts repressed by the female gametophyte in the ovule of Arabidopsis thaliana. PLoS ONE 8:e76977. doi: 10.1371/journal.pone.0076977
Aya, K., Suzuki, G., Suwabe, K., Hobo, T., Takahashi, H., Shiono, K., et al. (2011). Comprehensive network analysis of anther-expressed genes in rice by the combination of 33 laser microdissection and 143 spatiotemporal microarrays. PLoS ONE 6:e26162. doi: 10.1371/journal.pone.0026162
Barra, L., Aiese-Cigliano, R., Cremona, G., De Luca, P., Zoppoli, P., Bressan, R. A., et al. (2012). Transcription profiling of laser microdissected microsporocytes in an Arabidopsis mutant (Atmcc1) with enhanced histone acetylation. J. Plant Biol. 55, 281–289. doi: 10.1007/s12374-011-0268-z
Becker, J. D., Boavida, L. C., Carneiro, J., Haury, M., and Feijó, J. A. (2003). Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiol. 133, 713–725. doi: 10.1104/pp.103.028241
Becker, J. D., and Feijó, J. A. (2007). How many genes are needed to make a pollen tube? Lessons from transcriptomics. Ann. Bot. 100, 1117–1123. doi: 10.1093/aob/mcm208
Berger, F., and Twell, D. (2011). Germline specification and function in plants. Annu. Rev. Plant Biol. 62, 461–484. doi: 10.1146/annurev-arplant-042110-103824
Bita, C. E., Zenoni, S., Vriezen, W. H., Mariani, C., Pezzotti, M., and Gerats, T. (2011). Temperature stress differentially modulates transcription in meiotic anthers of heat-tolerant and heat-sensitive tomato plants. BMC Genomics 12:384. doi: 10.1186/1471-2164-12-384
Borg, M., Brownfield, L., and Twell, D. (2009). Male gametophyte development: a molecular perspective. J. Exp. Bot. 60, 1465–1478. doi: 10.1093/jxb/ern355
Borges, F., Gardner, R., Lopes, T., Calarco, J. P., Boavida, L. C., Slotkin, R. K., et al. (2012). FACS-based purification of Arabidopsis microspores, sperm cells and vegetative nuclei. Plant Methods 8:44. doi: 10.1186/1746-4811-8-44
Borges, F., Gomes, G., Gardner, R., Moreno, N., McCormick, S., Feijó, J. A., et al. (2008). Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol. 148, 1168–1181. doi: 10.1104/pp.108.125229
Borges, F., Pereira, P. A., Slotkin, R. K., Martienssen, R. A., and Becker, J. D. (2011). MicroRNA activity in the Arabidopsis male germline. J. Exp. Bot. 62, 1611–1620. doi: 10.1093/jxb/erq452
Bouchard, R. A. (1990). Characterization of expressed meiotic prophase repeat transcript clones of Lilium: meiosis-specific expression, relatedness, and affinities to small heat shock protein genes. Genome Natl. Res. Counc. Can. Génome Cons. Natl. Rech. Can. 33, 68–79.
Bovill, W. D., Deveshwar, P., Kapoor, S., and Able, J. A. (2009). Whole genome approaches to identify early meiotic gene candidates in cereals. Funct. Integr. Genomics 9, 219–229. doi: 10.1007/s10142-008-0097-4
Brar, G. A., Yassour, M., Friedman, N., Regev, A., Ingolia, N. T., and Weissman, J. S. (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557. doi: 10.1126/science.1215110
Bullard, J. H., Purdom, E., Hansen, K. D., and Dudoit, S. (2010). Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11:94. doi: 10.1186/1471-2105-11-94
Caryl, A. P., Armstrong, S. J., Jones, G. H., and Franklin, F. C. (2000). A homologue of the yeast HOP1 gene is inactivated in the Arabidopsis meiotic mutant asy1. Chromosoma 109, 62–71. doi: 10.1007/s004120050413
Chambers, C., and Shuai, B. (2009). Profiling microRNA expression in Arabidopsis pollen using microRNA array and real-time PCR. BMC Plant Biol. 9:87. doi: 10.1186/1471-2229-9-87
Chen, C., Farmer, A. D., Langley, R. J., Mudge, J., Crow, J. A., May, G. D., et al. (2010). Meiosis-specific gene discovery in plants: RNA-Seq applied to isolated Arabidopsis male meiocytes. BMC Plant Biol. 10:280. doi: 10.1186/1471-2229-10-280
Chen, C., and Retzel, E. F. (2013). “Analyzing the meiotic transcriptome using isolated meiocytes of Arabidopsis thaliana,” in Plant Meiosis: Methods and Protocols, eds W. Pawlowski, M. Grelon, and S. Armstrong (New York, NY: Humana Press), 203–213.
Chmelnitsky, I., Sobolev, I., Barg, R., Shabtai, S., and Salts, Y. (2003). Isolation and preliminary characterization of tomato petal- and stamen-specific cDNAs from a subtracted and equilibrated cDNA library. Euphytica 129, 229–236. doi: 10.1023/A:1021983806607
Chu, S., DeRisi, J., Eisen, M., Mulholland, J., Botstein, D., Brown, P. O., et al. (1998). The transcriptional program of sporulation in budding yeast. Science 282, 699–705. doi: 10.1126/science.282.5389.699
Cnudde, F., Hedatale, V., de Jong, H., Pierson, E. S., Rainey, D. Y., Zabeau, M., et al. (2006). Changes in gene expression during male meiosis in Petunia hybrida. Chromosome Res. 14, 919–932. doi: 10.1007/s10577-006-1099-5
Crismani, W., Baumann, U., Sutton, T., Shirley, N., Webster, T., Spangenberg, G., et al. (2006). Microarray expression analysis of meiosis and microsporogenesis in hexaploid bread wheat. BMC Genomics 7:267. doi: 10.1186/1471-2164-7-267
Crismani, W., Kapoor, S., and Able, J. A. (2011). Comparative transcriptomics reveals 129 transcripts that are temporally regulated during anther development and meiotic progression in both bread wheat (Triticum aestivum) and rice (Oryza sativa). Int. J. Plant Genomics 2011, 1–9. doi: 10.1155/2011/931898
Crossley, S. J., Greenland, A. J., and Dickinson, H. G. (1995). The characterisation of tapetum-specific cDNAs isolated from a Lilium henryi L. meiocyte subtractive cDNA library. Planta 196, 523–529. doi: 10.1007/BF00203652
Dai, S., Chen, T., Chong, K., Xue, Y., Liu, S., and Wang, T. (2007). Proteomics identification of differentially expressed proteins associated with pollen germination and tube growth reveals characteristics of germinated Oryza sativa pollen. Mol. Cell. Proteomics MCP 6, 207–230. doi: 10.1074/mcp.M600146-MCP200
Dai, S., Li, L., Chen, T., Chong, K., Xue, Y., and Wang, T. (2006). Proteomic analyses of Oryza sativa mature pollen reveal novel proteins associated with pollen germination and tube growth. Proteomics 6, 2504–2529. doi: 10.1002/pmic.200401351
Davidson, R. M., Hansey, C. N., Gowda, M., Childs, K. L., Lin, H., Vaillancourt, B., et al. (2011). Utility of RNA sequencing for analysis of maize reproductive transcriptomes. Plant Genome 4, 191–203. doi: 10.3835/plantgenome2011.05.0015
Deveshwar, P., Bovill, W. D., Sharma, R., Able, J. A., and Kapoor, S. (2011). Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice. BMC Plant Biol. 11:78. doi: 10.1186/1471-2229-11-78
Druka, A., Muehlbauer, G., Druka, I., Caldo, R., Baumann, U., Rostoks, N., et al. (2006). An atlas of gene expression from seed to seed through barley development. Funct. Integr. Genomics 6, 202–211. doi: 10.1007/s10142-006-0025-4
Dukowic-Schulze, S., Harris, A., Li, J., Sundarajan, A., Mudge, J., Retzel, E., et al. (2014a). Comparative transcriptomics of early meiosis in Arabidopsis and maize. J. Genet. Genomics 41, 139–152. doi: 10.1016/j.jgg.2013.11.007
Dukowic-Schulze, S., Sundararajan, A., Mudge, J., Ramaray, T., Farmer, A., Wang, M., et al. (2014b). The transcriptome landscape of early maize meiosis. BMC Plant Biol. 14:118. doi: 10.1186/1471-2229-14-118
Dukowic-Schulze, S., Sundararajan, A., Ramaraj, T., Mudge, J., and Chen, C. (2014c). Sequencing-based large-scale genomics approaches with small numbers of isolated maize meiocytes. Plant Genet. Genomics 5:57. doi: 10.3389/fpls.2014.00057
Endo, M., Matsubara, H., Kokubun, T., Masuko, H., Takahata, Y., Tsuchiya, T., et al. (2002). The advantages of cDNA microarray as an effective tool for identification of reproductive organ-specific genes in a model legume, Lotus japonicus. FEBS Lett. 514, 229–237. doi: 10.1016/S0014-5793(02)02371-2
Endo, M., Tsuchiya, T., Hamada, K., Kawamura, S., Yano, K., Ohshima, M., et al. (2009). High temperatures cause male sterility in rice plants with transcriptional alterations during pollen development. Plant Cell Physiol. 50, 1911–1922. doi: 10.1093/pcp/pcp135
Endo, M., Tsuchiya, T., Saito, H., Matsubara, H., Hakozaki, H., Masuko, H., et al. (2004). Identification and molecular characterization of novel anther-specific genes in Oryza sativa L. by using cDNA microarray. Genes Genet. Syst. 79, 213–226. doi: 10.1266/ggs.79.213
Engel, M. L., Chaboud, A., Dumas, C., and McCormick, S. (2003). Sperm cells of Zea mays have a complex complement of mRNAs. Plant J. 34, 697–707. doi: 10.1046/j.1365-313X.2003.01761.x
Feng, B., Lu, D., Ma, X., Peng, Y., Sun, Y., Ning, G., et al. (2012). Regulation of the Arabidopsis anther transcriptome by DYT1 for pollen development. Plant J. Cell Mol. Biol. 72, 612–624. doi: 10.1111/j.1365-313X.2012.05104.x
Feng, X., Zilberman, D., and Dickinson, H. (2013). A conversation across generations: soma-germ cell crosstalk in plants. Dev. Cell 24, 215–225. doi: 10.1016/j.devcel.2013.01.014
Flórez-Zapata, N. M. V., Reyes-Valdés, M. H., Hernandez-Godínez, F., and Martinez, O. (2014). Transcriptomic landscape of prophase I sunflower male meiocytes. Front. Plant Sci. 5:277. doi: 10.3389/fpls.2014.00277
Frank, G., Pressman, E., Ophir, R., Althan, L., Shaked, R., Freedman, M., et al. (2009). Transcriptional profiling of maturing tomato (Solanum lycopersicum L.) microspores reveals the involvement of heat shock proteins, ROS scavengers, hormones, and sugars in the heat stress response. J. Exp. Bot. 60, 3891–3908. doi: 10.1093/jxb/erp234
Fujita, M., Horiuchi, Y., Ueda, Y., Mizuta, Y., Kubo, T., Yano, K., et al. (2010). Rice expression atlas in reproductive development. Plant Cell Physiol. 51, 2060–2081. doi: 10.1093/pcp/pcq165
Galau, G. A., Klein, W. H., Davis, M. M., Wold, B. J., Britten, R. J., and Davidson, E. H. (1976). Structural gene sets active in embryos and adult tissues of the sea urchin. Cell 7, 487–505. doi: 10.1016/0092-8674(76)90200-2
Gautier, L., Cope, L., Bolstad, B. M., and Irizarry, R. A. (2004). affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315. doi: 10.1093/bioinformatics/btg405
Giorno, F., Wolters-Arts, M., Mariani, C., and Rieu, I. (2013). Ensuring reproduction at high temperatures: the heat stress response during anther and pollen development. Plants 2, 489–506. doi: 10.3390/plants2030489
Golkari, S., Gilbert, J., Prashar, S., and Procunier, J. D. (2007). Microarray analysis of Fusarium graminearum-induced wheat genes: identification of organ-specific and differentially expressed genes. Plant Biotechnol. J. 5, 38–49. doi: 10.1111/j.1467-7652.2006.00213.x
Grant-Downton, R., Hafidh, S., Twell, D., and Dickinson, H. G. (2009a). Small RNA pathways are present and functional in the angiosperm male gametophyte. Mol. Plant 2, 500–512. doi: 10.1093/mp/ssp003
Grant-Downton, R., Le Trionnaire, G., Schmid, R., Rodriguez-Enriquez, J., Hafidh, S., Mehdi, S., et al. (2009b). MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. BMC Genomics 10:643. doi: 10.1186/1471-2164-10-643
Gutierrez-Marcos, J. F., and Dickinson, H. G. (2012). Epigenetic reprogramming in plant reproductive lineages. Plant Cell Physiol. 53, 817–823. doi: 10.1093/pcp/pcs052
Haerizadeh, F., Wong, C. E., Bhalla, P. L., Gresshoff, P. M., and Singh, M. B. (2009). Genomic expression profiling of mature soybean (Glycine max) pollen. BMC Plant Biol. 9:25. doi: 10.1186/1471-2229-9-25
Hansen, K. D., Irizarry, R. A., and Wu, Z. (2012). Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216. doi: 10.1093/biostatistics/kxr054
Harrison, C. J., Alvey, E., and Henderson, I. R. (2010). Meiosis in flowering plants and other green organisms. J. Exp. Bot. 61, 2863–2875. doi: 10.1093/jxb/erq191
Hennig, L., Menges, M., Murray, J. A. H., and Gruissem, W. (2003). Arabidopsis transcript profiling on Affymetrix GeneChip arrays. Plant Mol. Biol. 53, 457–465. doi: 10.1023/B:PLAN.0000019069.23317.97
Hernández, G., Han, H., Gandin, V., Fabian, L., Ferreira, T., Zuberek, J., et al. (2012). Eukaryotic initiation factor 4E-3 is essential for meiotic chromosome segregation, cytokinesis and male fertility in Drosophila. Development 139, 3211–3220. doi: 10.1242/dev.073122
Higgins, J. D., Perry, R. M., Barakate, A., Ramsay, L., Waugh, R., Halpin, C., et al. (2012). Spatiotemporal asymmetry of the meiotic program underlies the predominantly distal distribution of meiotic crossovers in barley. Plant Cell 24, 4096–4109. doi: 10.1105/tpc.112.102483
Hirano, K., Aya, K., Hobo, T., Sakakibara, H., Kojima, M., Shim, R. A., et al. (2008). Comprehensive transcriptome analysis of phytohormone biosynthesis and signaling genes in microspore/pollen and tapetum of rice. Plant Cell Physiol. 49, 1429–1450. doi: 10.1093/pcp/pcn123
Hobo, T., Suwabe, K., Aya, K., Suzuki, G., Yano, K., Ishimizu, T., et al. (2008). Various spatiotemporal expression profiles of anther-expressed genes in rice. Plant. Cell. Physiol. 49, 1417–1428. doi: 10.1093/pcp/pcn128
Holmes-Davis, R., Tanaka, C. K., Vensel, W. H., Hurkman, W. J., and McCormick, S. (2005). Proteome mapping of mature pollen of Arabidopsis thaliana. Proteomics 5, 4864–4884. doi: 10.1002/pmic.200402011
Honys, D., and Twell, D. (2003). Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol. 132, 640–652. doi: 10.1104/pp.103.020925
Honys, D., and Twell, D. (2004). Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 5:R85. doi: 10.1186/gb-2004-5-11-r85
Huang, M.-D., Wei, F.-J., Wu, C.-C., Hsing, Y.-I. C., and Huang, A. H. C. (2009). Analyses of advanced rice anther transcriptomes reveal global tapetum secretory functions and potential proteins for lipid exine formation. Plant Physiol. 149, 694–707. doi: 10.1104/pp.108.131128
Imin, N., Kerim, T., Weinman, J. J., and Rolfe, B. G. (2001). Characterisation of rice anther proteins expressed at the young microspore stage. Proteomics 1, 1149–1161. doi: 10.1002/1615-9861(200109)1:9%3C1149::AID-PROT1149%3E3.3.CO;2-I
Jackson, N., Sanchez-Moran, E., Buckling, E., Armstrong, S. J., Jones, G. H., and Franklin, F. C. H. (2006). Reduced meiotic crossovers and delayed prophase I progression in AtMLH3-deficient Arabidopsis. EMBO J. 25, 1315–1323. doi: 10.1038/sj.emboj.7600992
Johnston, A. J., Meier, P., Gheyselinck, J., Wuest, S. E., Federer, M., Schlagenhauf, E., et al. (2007). Genetic subtraction profiling identifies genes essential for Arabidopsis reproduction and reveals interaction between the female gametophyte and the maternal sporophyte. Genome Biol. 8:R204. doi: 10.1186/gb-2007-8-10-r204
Jones, G. H., and Franklin, F. C. H. (2008). “Meiosis in Arabidopsis thaliana: recombination, chromosome organization and meiotic progression,” in Recombination and Meiosis. Genome Dynamics and Stability, eds R. Egel and D.-H. Lankenau (Berlin; Heidelberg: Springer-Verlag), 279–306.
Jones-Rhoades, M. W., Borevitz, J. O., and Preuss, D. (2007). Genome-wide expression profiling of the Arabidopsis female gametophyte identifies families of small, secreted proteins. PLoS Genet. 3:e171, 1848–1861. doi: 10.1371/journal.pgen.0030171
Kaback, D. B., and Feldberg, L. R. (1985). Saccharomyces cerevisiae exhibits a sporulation-specific temporal pattern of transcript accumulation. Mol. Cell. Biol. 5, 751–761.
Kamalay, J. C., and Goldberg, R. B. (1980). Regulation of structural gene expression in tobacco. Cell 19, 935–946. doi: 10.1016/0092-8674(80)90085-9
Kapoor, S., and Takatsuji, H. (2006). Silencing of an anther-specific Zinc-finger gene, MEZ1, causes aberrant meiosis and pollen abortion in Petunia. Plant Mol. Biol. 61, 415–430. doi: 10.1007/s11103-006-0020-0
Kerim, T., Imin, N., Weinman, J. J., and Rolfe, B. G. (2003). Proteome analysis of male gametophyte development in rice anthers. Proteomics 3, 738–751. doi: 10.1002/pmic.200300424
Khurana, R., Kapoor, S., and Tyagi, A. K. (2012). Anthology of anther/pollen-specific promoters and transcription factors. Crit. Rev. Plant Sci. 31, 359–390. doi: 10.1080/07352689.2012.664986
Kobayashi, T., Kobayashi, E., Sato, S., Hotta, Y., Miyajima, N., Tanaka, A., et al. (1994). Characterization of cDNAs induced in meiotic prophase in lily microsporocytes. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 1, 15–26. doi: 10.1093/dnares/1.1.15
Koltunow, A. M., Truettner, J., Cox, K. H., Wallroth, M., and Goldberg, R. B. (1990). Different temporal and spatial gene expression patterns occur during anther development. Plant Cell Online 2, 1201–1224. doi: 10.1105/tpc.2.12.1201
Kubo, T., Fujita, M., Takahashi, H., Nakazono, M., Tsutsumi, N., and Kurata, N. (2013). Transcriptome analysis of developing ovules in rice isolated by laser microdissection. Plant Cell Physiol. 54, 750–765. doi: 10.1093/pcp/pct029
Kurihara, Y., and Watanabe, Y. (2004). Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. U.S.A. 101, 12753–12758. doi: 10.1073/pnas.0403115101
Lee, J.-Y., and Lee, D.-H. (2003). Use of serial analysis of gene expression technology to reveal changes in gene expression in Arabidopsis pollen undergoing cold stress. Plant Physiol. 132, 517–529. doi: 10.1104/pp.103.020511
Li, J., Farmer, A. D., Lindquist, I. E., Dukowic-Schulze, S., Mudge, J., Li, T., et al. (2012). Characterization of a set of novel meiotically-active promoters in Arabidopsis. BMC Plant Biol. 12:104. doi: 10.1186/1471-2229-12-104
Li, M., Xu, W., Yang, W., Kong, Z., and Xue, Y. (2007). Genome-wide gene expression profiling reveals conserved and novel molecular functions of the stigma in rice. Plant Physiol. 144, 1797–1812. doi: 10.1104/pp.107.101600
Libeau, P., Durandet, M., Granier, F., Marquis, C., Berthomé, R., Renou, J. P., et al. (2011). Gene expression profiling of Arabidopsis meiocytes. Plant Biol. Stuttg. 13, 784–793. doi: 10.1111/j.1438-8677.2010.00435.x
Lopez-Casado, G., Covey, P. A., Bedinger, P. A., Mueller, L. A., Thannhauser, T. W., Zhang, S., et al. (2012). Enabling proteomic studies with RNA-Seq: the proteome of tomato pollen as a test case. Proteomics 12, 761–774. doi: 10.1002/pmic.201100164
Lu, B. C. (1974). Genetic recombination in Coprinus. IV. a kinetic study of the temperature effect on recombination frequency. Genetics 78, 661–677.
Lu, X.-C., Gong, H.-Q., Huang, M.-L., Bai, S.-L., He, Y.-B., Mao, X., et al. (2006). Molecular analysis of early rice stamen development using organ-specific gene expression profiling. Plant Mol. Biol. 61, 845–861. doi: 10.1007/s11103-006-0054-3
Ma, J., Duncan, D., Morrow, D. J., Fernandes, J., and Walbot, V. (2007). Transcriptome profiling of maize anthers using genetic ablation to analyze pre-meiotic and tapetal cell types. Plant J. 50, 637–648. doi: 10.1111/j.1365-313X.2007.03074.x
Ma, J., Morrow, D. J., Fernandes, J., and Walbot, V. (2006). Comparative profiling of the sense and antisense transcriptome of maize lines. Genome Biol. 7:R22. doi: 10.1186/gb-2006-7-3-r22
Ma, J., Skibbe, D. S., Fernandes, J., and Walbot, V. (2008). Male reproductive development: gene expression profiling of maize anther and pollen ontogeny. Genome Biol. 9:R181. doi: 10.1186/gb-2008-9-12-r181
Ma, X., Feng, B., and Ma, H. (2012). AMS-dependent and independent regulation of anther transcriptome and comparison with those affected by other Arabidopsis anther genes. BMC Plant Biol. 12:23. doi: 10.1186/1471-2229-12-23
Mackenzie, A., Heslop-Harrison, J., and Dickinson, H. G. (1967). Elimination of ribosomes during meiotic prophase. Nature 215, 997–999. doi: 10.1038/215997a0
Mandaokar, A., Thines, B., Shin, B., Lange, B. M., Choi, G., Koo, Y. J., et al. (2006). Transcriptional regulators of stamen development in Arabidopsis identified by transcriptional profiling. Plant J. Cell Mol. Biol. 46, 984–1008. doi: 10.1111/j.1365-313X.2006.02756.x
Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., and Gilad, Y. (2008). RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517. doi: 10.1101/gr.079558.108
Mascarenhas, J. P. (1971). “RNA and protein synthesis during pollen development and tube growth,” in Pollen: Development and Physiology, ed J. Heslop-Harrison (London: Butterworths), 201–222.
Mascarenhas, J. P. (1975). The biochemistry of angiosperm pollen development. Bot. Rev. 41, 259–314. doi: 10.1007/BF02860839
Mascarenhas, J. P. (1990). Gene activity during pollen development. Annu. Rev. Plant Physiol. Plant Mol. Biol. 41, 317–338. doi: 10.1146/annurev.pp.41.060190.001533
Mascarenhas, J. P. (1993). Molecular mechanisms of pollen tube growth and differentiation. Plant Cell 5, 1303–1314. doi: 10.1105/tpc.5.10.1303
Mascarenhas, J. P., and Bell, E. (1970). RNA synthesis during development of the male gametophyte of Tradescantia. Dev. Biol. 21, 475–490. doi: 10.1016/0012-1606(70)90073-4
Mascarenhas, J. P., and Mermelstein, J. (1981). Messenger RNAs: their utilization and degradation during pollen germination and tube growth. Acta Soc. Bot. Pol. 50, 13–20. doi: 10.5586/asbp.1981.002
Mascarenhas, N. T., Bashe, D., Eisenberg, A., Willing, R. P., Xiao, C. M., and Mascarenhas, J. P. (1984). Messenger RNAs in corn pollen and protein synthesis during germination and pollen tube growth. Theor. Appl. Genet. Theor. Angew. Genet. 68, 323–326. doi: 10.1007/BF00267885
Mata, J., Lyne, R., Burns, G., and Bähler, J. (2002). The transcriptional program of meiosis and sporulation in fission yeast. Nat. Genet. 32, 143–147. doi: 10.1038/ng951
McCormick, S. (2004). Control of male gametophyte development. Plant Cell 16(Suppl.) S142–S153. doi: 10.1105/tpc.016659
McCormick, S., Smith, A., Gasser, C., Sachs, K., Hinchee, M., Horsch, R., et al. (1987). “Identification of genes specifically expressed in reproductive organs of tomato,” in Tomato Biotechnology, eds D. Nevins and R. Jones, (New York, NY: Wiley & Sons Inc.) 255–265.
Millar, A. A., and Gubler, F. (2005). The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 17, 705–721. doi: 10.1105/tpc.104.027920
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nacken, W. K., Huijser, P., Beltran, J. P., Saedler, H., and Sommer, H. (1991). Molecular characterization of two stamen-specific genes, tap1 and fil1, that are expressed in the wild type, but not in the deficiens mutant of Antirrhinum majus. Mol. Gen. Genet. MGG 229, 129–136. doi: 10.1007/BF00264221
Naef, F., and Magnasco, M. O. (2003). Solving the riddle of the bright mismatches: labeling and effective binding in oligonucleotide arrays. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 68:011906. doi: 10.1103/PhysRevE.68.011906
Nan, G.-L., Ronceret, A., Wang, R. C., Fernandes, J. F., Cande, W. Z., and Walbot, V. (2011). Global transcriptome analysis of two ameiotic1 alleles in maize anthers: defining steps in meiotic entry and progression through prophase I. BMC Plant Biol. 11:120. doi: 10.1186/1471-2229-11-120
Noir, S., Bräutigam, A., Colby, T., Schmidt, J., and Panstruga, R. (2005). A reference map of the Arabidopsis thaliana mature pollen proteome. Biochem. Biophys. Res. Commun. 337, 1257–1266. doi: 10.1016/j.bbrc.2005.09.185
Nonomura, K.-I., Morohoshi, A., Nakano, M., Eiguchi, M., Miyao, A., Hirochika, H., et al. (2007). A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell 19, 2583–2594. doi: 10.1105/tpc.107.053199
Ogur, M., Erickson, R. O., Rosen, U., Sax, K. B., and Holden, C. (1951). Nucleic acids in relation to cell division in Lilium longiflorum. Exptl. Cell Res. 2, 73–89. doi: 10.1016/0014-4827(51)90007-9
Ohnishi, T., Takanashi, H., Mogi, M., Takahashi, H., Kikuchi, S., Yano, K., et al. (2011). Distinct gene expression profiles in egg and synergid cells of rice as revealed by cell type-specific microarrays. Plant Physiol. 155, 881–891. doi: 10.1104/pp.110.167502
Okada, T., Singh, M. B., and Bhalla, P. L. (2006). Histone H3 variants in male gametic cells of lily and H3 methylation in mature pollen. Plant Mol. Biol. 62, 503–512. doi: 10.1007/s11103-006-9036-8
Olmedo-Monfil, V., Durán-Figueroa, N., Arteaga-Vázquez, M., Demesa-Arévalo, E., Autran, D., Grimanelli, D., et al. (2010). Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature 464, 628–632. doi: 10.1038/nature08828
Pang, A. L. Y., Johnson, W., Ravindranath, N., Dym, M., Rennert, O. M., and Chan, W.-Y. (2006). Expression profiling of purified male germ cells: stage-specific expression patterns related to meiosis and postmeiotic development. Physiol. Genomics 24, 75–85. doi: 10.1152/physiolgenomics.00215.2004
Peddada, L. B., and Mascarenhas, J. P. (1975). The synthesis of 5S ribosomal RNA during pollen development. Dev. Growth Differ. 17, 1–8. doi: 10.1111/j.1440-169X.1975.00001.x
Pickrell, J. K., Marioni, J. C., Pai, A. A., Degner, J. F., Engelhardt, B. E., Nkadori, E., et al. (2010). Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464, 768–772. doi: 10.1038/nature08872
Pina, C., Pinto, F., Feijó, J. A., and Becker, J. D. (2005). Gene family analysis of the Arabidopsis pollen transcriptome reveals biological implications for cell growth, division control, and gene expression regulation. Plant Physiol. 138, 744–756. doi: 10.1104/pp.104.057935
Primig, M., Williams, R. M., Winzeler, E. A., Tevzadze, G. G., Conway, A. R., Hwang, S. Y., et al. (2000). The core meiotic transcriptome in budding yeasts. Nat. Genet. 26, 415–423. doi: 10.1038/82539
Reinke, V., Smith, H. E., Nance, J., Wang, J., Van Doren, C., Begley, R., et al. (2000). A global profile of germline gene expression in C. elegans. Mol. Cell 6, 605–616. doi: 10.1016/S1097-2765(00)00059-9
Rubinelli, P., Hu, Y., and Ma, H. (1998). Identification, sequence analysis and expression studies of novel anther-specific genes of Arabidopsis thaliana. Plant Mol. Biol. 37, 607–619. doi: 10.1023/A:1005964431302
Sanchez-Leon, N., Arteaga-Vazquez, M., Alvarez-Mejia, C., Mendiola-Soto, J., Duran-Figueroa, N., Rodriguez-Leal, D., et al. (2012). Transcriptional analysis of the Arabidopsis ovule by massively parallel signature sequencing. J. Exp. Bot. 63, 3829–3842. doi: 10.1093/jxb/ers075
Sanchez-Moran, E., Osman, K., Higgins, J. D., Pradillo, M., Cuñado, N., Jones, G. H., et al. (2008). ASY1 coordinates early events in the plant meiotic recombination pathway. Cytogenet. Genome Res. 120, 302–312. doi: 10.1159/000121079
Schlecht, U., Demougin, P., Koch, R., Hermida, L., Wiederkehr, C., Descombes, P., et al. (2004). Expression profiling of mammalian male meiosis and gametogenesis identifies novel candidate genes for roles in the regulation of fertility. Mol. Biol. Cell 15, 1031–1043. doi: 10.1091/mbc.E03-10-0762
Schmid, M., Davison, T. S., Henz, S. R., Pape, U. J., Demar, M., Vingron, M., et al. (2005). A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37, 501–506. doi: 10.1038/ng1543
Schmid, R., Grellscheid, S. N., Ehrmann, I., Dalgliesh, C., Danilenko, M., Paronetto, M. P., et al. (2013). The splicing landscape is globally reprogrammed during male meiosis. Nucleic Acids Res. 41, 10170–10184. doi: 10.1093/nar/gkt811
Schmidt, A., Schmid, M. W., and Grossniklaus, U. (2012). Analysis of plant germline development by high-throughput RNA profiling: technical advances and new insights. Plant J. 70, 18–29. doi: 10.1111/j.1365-313X.2012.04897.x
Schmidt, A., Wuest, S. E., Vijverberg, K., Baroux, C., Kleen, D., and Grossniklaus, U. (2011). Transcriptome analysis of the Arabidopsis megaspore mother cell uncovers the importance of RNA helicases for plant germline development. PLoS Biol. 9:e1001155. doi: 10.1371/journal.pbio.1001155
Scott, R., Dagless, E., Hodge, R., Paul, W., Soufleri, I., and Draper, J. (1991). Patterns of gene expression in developing anthers of Brassica napus. Plant Mol. Biol. 17, 195–207. doi: 10.1007/BF00039494
Sekhon, R. S., Lin, H., Childs, K. L., Hansey, C. N., Buell, C. R., de Leon, N., et al. (2011). Genome-wide atlas of transcription during maize development. Plant J. 66, 553–563. doi: 10.1111/j.1365-313X.2011.04527.x
Shen, J. B., and Hsu, F. C. (1992). Brassica anther-specific genes: characterization and in situ localization of expression. Mol. Gen. Genet. MGG 234, 379–389. doi: 10.1007/BF00538697
Sheoran, I. S., Ross, A. R. S., Olson, D. J. H., and Sawhney, V. K. (2007). Proteomic analysis of tomato (Lycopersicon esculentum) pollen. J. Exp. Bot. 58, 3525–3535. doi: 10.1093/jxb/erm199
Sheoran, I. S., Sproule, K. A., Olson, D. J. H., Ross, A. R. S., and Sawhney, V. K. (2006). Proteome profile and functional classification of proteins in Arabidopsis thaliana (Landsberg erecta) mature pollen. Sex. Plant Reprod. 19, 185–196. doi: 10.1007/s00497-006-0035-3
Singh, M., Goel, S., Meeley, R. B., Dantec, C., Parrinello, H., Michaud, C., et al. (2011). Production of viable gametes without meiosis in maize deficient for an ARGONAUTE protein. Plant Cell Online 23, 443–458. doi: 10.1105/tpc.110.079020
Singh, M. B., and Bhalla, P. L. (2007). Control of male germ-cell development in flowering plants. Bioessays 29, 1124–1132. doi: 10.1002/bies.20660
Singh, M. B., Bhalla, P. L., and Russell, S. D. (2008). Molecular repertoire of flowering plant male germ cells. Sex. Plant Reprod. 21, 27–36. doi: 10.1007/s00497-008-0067-y
Skibbe, D. S., Fernandes, J. F., Medzihradszky, K. F., Burlingame, A. L., and Walbot, V. (2009). Mutator transposon activity reprograms the transcriptomes and proteomes of developing maize anthers. Plant J. 59, 622–633. doi: 10.1111/j.1365-313X.2009.03901.x
Slotkin, R. K., Vaughn, M., Borges, F., Tanurdziæ, M., Becker, J. D., Feijó, J. A., et al. (2009). Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136, 461–472. doi: 10.1016/j.cell.2008.12.038
Steffen, J. G., Kang, I.-H., Macfarlane, J., and Drews, G. N. (2007). Identification of genes expressed in the Arabidopsis female gametophyte. Plant J. Cell Mol. Biol. 51, 281–292. doi: 10.1111/j.1365-313X.2007.03137.x
Steffensen, D. M. (1966). Synthesis of ribosomal RNA during growth and division in Lilium. Exp. Cell Res. 44, 1–12. doi: 10.1016/0014-4827(66)90407-1
Stern, H., and Hotta, Y. (1977). Biochemistry of meiosis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 277, 277–294. doi: 10.1098/rstb.1977.0018
Stinson, J. R., Eisenberg, A. J., Willing, R. P., Pe, M. E., Hanson, D. D., and Mascarenhas, J. P. (1987). Genes expressed in the male gametophyte of flowering plants and their isolation. Plant Physiol. 83, 442–447. doi: 10.1104/pp.83.2.442
Suwabe, K., Suzuki, G., Takahashi, H., Shiono, K., Endo, M., Yano, K., et al. (2008). Separated transcriptomes of male gametophyte and tapetum in rice: validity of a Laser Microdissection (LM) microarray. Plant Cell Physiol. 49, 1407–1416. doi: 10.1093/pcp/pcn124
Tang, X., Zhang, Z.-Y., Zhang, W.-J., Zhao, X.-M., Li, X., Zhang, D., et al. (2010). Global gene profiling of laser-captured pollen mother cells indicates molecular pathways and gene subfamilies involved in rice meiosis. Plant Physiol. 154, 1855–1870. doi: 10.1104/pp.110.161661
Taylor, J. H. (1953). Autoradiographic detection of incorporation of P32 into chromosomes during meiosis and mitosis. Exp. Cell Res. 4, 164–173. doi: 10.1016/0014-4827(53)90199-2
Trionnaire, G. L., Grant-Downton, R. T., Kourmpetli, S., Dickinson, H. G., and Twell, D. (2011). Small RNA activity and function in angiosperm gametophytes. J. Exp. Bot. 62, 1601–1610. doi: 10.1093/jxb/erq399
Tsuchiya, T., Toriyama, K., Ejiri, S., and Hinata, K. (1994). Molecular characterization of rice genes specifically expressed in the anther tapetum. Plant Mol. Biol. 26, 1737–1746. doi: 10.1007/BF00019488
Tsuchiya, T., Toriyama, K., Nasrallah, M. E., and Ejiri, S. (1992). Isolation of genes abundantly expressed in rice anthers at the microspore stage. Plant Mol. Biol. 20, 1189–1193. doi: 10.1007/BF00028907
Tupy, J. (1982). Alterations in polyadenylated RNA during pollen maturation and germination. Biol. Plant 24, 231–237. doi: 10.1007/BF02909098
Twell, D., Oh, S.-A., and Honys, D. (2006). “Pollen development, a genetic and transcriptomic view,” in The Pollen Tube Plant Cell Monographs, ed R. Malhó (Berlin; Heidelberg: Springer), 15–45. Available online at: http://link.springer.com/chapter/10.1007/7089_042 [Accessed December 6, 2013].
Van Ex, F., Jacob, Y., and Martienssen, R. A. (2011). Multiple roles for small RNAs during plant reproduction. Curr. Opin. Plant Biol. 14, 588–593. doi: 10.1016/j.pbi.2011.07.003
Vaucheret, H., Vazquez, F., Crété, P., and Bartel, D. P. (2004). The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 18, 1187–1197. doi: 10.1101/gad.1201404
Wang, D., Oses-Prieto, J. A., Li, K. H., Fernandes, J. F., Burlingame, A. L., and Walbot, V. (2010). The male sterile 8 mutation of maize disrupts the temporal progression of the transcriptome and results in the mis-regulation of metabolic functions. Plant J. 63, 939–951. doi: 10.1111/j.1365-313X.2010.04294.x
Wang, Z., Liang, Y., Li, C., Xu, Y., Lan, L., Zhao, D., et al. (2005). Microarray analysis of gene expression involved in anther development in rice (Oryza sativa L.). Plant Mol. Biol. 58, 721–737. doi: 10.1007/s11103-005-8267-4
Wei, L. Q., Xu, W. Y., Deng, Z. Y., Su, Z., Xue, Y., and Wang, T. (2010). Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. BMC Genomics 11:338. doi: 10.1186/1471-2164-11-338
Wei, M., Song, M., Fan, S., and Yu, S. (2013). Transcriptomic analysis of differentially expressed genes during anther development in genetic male sterile and wild type cotton by digital gene-expression profiling. BMC Genomics 14:97. doi: 10.1186/1471-2164-14-97
Wijeratne, A. J., Zhang, W., Sun, Y., Liu, W., Albert, R., Zheng, Z., et al. (2007). Differential gene expression in Arabidopsis wild-type and mutant anthers: insights into anther cell differentiation and regulatory networks. Plant J. 52, 14–29. doi: 10.1111/j.1365-313X.2007.03217.x
Williams, E., Heslop-Harrison, J., and Dickinson, H. G. (1973). The activity of the nucleolus organizing region and the origin of cytoplasmic nucleoids in meiocytes of Lilium. Protoplasma 77, 79–93. doi: 10.1007/BF01287293
Willing, R. P., Bashe, D., and Mascarenhas, J. P. (1988). An analysis of the quantity and diversity of messenger RNAs from pollen and shoots of Zea mays. Theor. Appl. Genet. 75, 751–753. doi: 10.1007/BF00265600
Willing, R. P., and Mascarenhas, J. P. (1984). Analysis of the complexity and diversity of mRNAs from pollen and shoots of Tradescantia. Plant Physiol. 75, 865–868. doi: 10.1104/pp.75.3.865
Winter, D., Vinegar, B., Nahal, H., Ammar, R., Wilson, G. V., and Provart, N. J. (2007). An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2:e718. doi: 10.1371/journal.pone.0000718
Woodard, J. W. (1958). Intracellular amounts of nucleic acids and protein during pollen grain growth in Tradescantia. J. Biophys. Biochem. Cytol. 4, 383–390. doi: 10.1083/jcb.4.4.383
Wu, M.-F., Tian, Q., and Reed, J. W. (2006). Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 133, 4211–4218. doi: 10.1242/dev.02602
Wuest, S. E., Vijverberg, K., Schmidt, A., Weiss, M., Gheyselinck, J., Lohr, M., et al. (2010). Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr. Biol. 20, 506–512. doi: 10.1016/j.cub.2010.01.051
Xu, H., Swoboda, I., Bhalla, P. L., and Singh, M. B. (1999). Male gametic cell-specific gene expression in flowering plants. Proc. Natl. Acad. Sci. U.S.A. 96, 2554–2558. doi: 10.1073/pnas.96.5.2554
Xu, H., Weterings, K., Vriezen, W., Feron, R., Xue, Y., Derksen, J., et al. (2002). Isolation and characterization of male-germ-cell transcripts in Nicotiana tabacum. Sex. Plant Reprod. 14, 339–346. doi: 10.1007/s00497-002-0128-6
Yang, H., Lu, P., Wang, Y., and Ma, H. (2011). The transcriptome landscape of Arabidopsis male meiocytes from high-throughput sequencing: the complexity and evolution of the meiotic process. Plant J. 65, 503–516. doi: 10.1111/j.1365-313X.2010.04439.x
Yang, X., Makaroff, C. A., and Ma, H. (2003). The Arabidopsis MALE MEIOCYTE DEATH1 gene encodes a PHD-finger protein that is required for male meiosis. Plant Cell 15, 1281–1295. doi: 10.1105/tpc.010447
Keywords: meiosis, transcriptome, meiocytes, anthers, RNA-seq, microarray
Citation: Dukowic-Schulze S and Chen C (2014) The meiotic transcriptome architecture of plants. Front. Plant Sci. 5:220. doi: 10.3389/fpls.2014.00220
Received: 20 February 2014; Paper pending published: 21 March 2014;
Accepted: 02 May 2014; Published online: 05 June 2014.
Edited by:
Ian Henderson, University of Cambridge, UKReviewed by:
Peter Langridge, Australian Centre for Plant Functional Genomics, AustraliaDaphné Autran, Institut de Recherche pour le Développement, France
Copyright © 2014 Dukowic-Schulze and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changbin Chen, Department of Horticultural Science, University of Minnesota, 260 Alderman Hall, 1970 Folwell Avenue, St. Paul, MN 55108, USA e-mail:Y2hlbng0ODFAdW1uLmVkdQ==