 Ya-Fen Lin
Ya-Fen Lin Edouard I. Severing
Edouard I. Severing Bas te Lintel Hekkert
Bas te Lintel Hekkert Elio Schijlen
Elio Schijlen Mark G. M. Aarts
Mark G. M. Aarts- 1Laboratory of Genetics, Wageningen University, Wageningen, Netherlands
- 2Laboratory of Bioinformatics, Wageningen University, Wageningen, Netherlands
- 3Business Unit Bioscience, Plant Research International, Wageningen University and Research Centres, Wageningen, Netherlands
Noccaea caerulescens is an extremophile plant species belonging to the Brassicaceae family. It has adapted to grow on soils containing high, normally toxic, concentrations of metals such as nickel, zinc, and cadmium. Next to being extremely tolerant to these metals, it is one of the few species known to hyperaccumulate these metals to extremely high concentrations in their aboveground biomass. In order to provide additional molecular resources for this model metal hyperaccumulator species to study and understand the mechanism of adaptation to heavy metal exposure, we aimed to provide a comprehensive database of transcript sequences for N. caerulescens. In this study, 23,830 transcript sequences (isotigs) with an average length of 1025 bp were determined for roots, shoots and inflorescences of N. caerulescens accession “Ganges” by Roche GS-FLEX 454 pyrosequencing. These isotigs were grouped into 20,378 isogroups, representing potential genes. This is a large expansion of the existing N. caerulescens transcriptome set consisting of 3705 unigenes. When translated and compared to a Brassicaceae proteome set, 22,232 (93.2%) of the N. caerulescens isotigs (corresponding to 19,191 isogroups) had a significant match and could be annotated accordingly. Of the remaining sequences, 98 isotigs resembled non-plant sequences and 1386 had no significant similarity to any sequence in the GenBank database. Among the annotated set there were many isotigs with similarity to metal homeostasis genes or genes for glucosinolate biosynthesis. Only for transcripts similar to Metallothionein3 (MT3), clear evidence for an additional copy was found. This comprehensive set of transcripts is expected to further contribute to the discovery of mechanisms used by N. caerulescens to adapt to heavy metal exposure.
Introduction
Noccaea caerulescens (J. & C. Presl) F. K. Mey., formerly named Thlaspi caerulescens, is an outstanding model plant species to study heavy metal hyperaccumulation (Assunção et al., 2003; Peer et al., 2003; Milner and Kochian, 2008). It is one of the few plant species of which genotypes are known that are adapted to grow on soil containing high levels of zinc (Zn), cadmium (Cd), nickel (Ni), and/or lead (Pb) (Mohtadi et al., 2012). Not only are these genotypes extremely tolerant to the different heavy metals they are exposed to, but N. caerulescens can also hyperaccumulate these metals to high concentrations in shoots. Accumulations of Zn to 30,000 μg g−1, Cd to 2700 μg g−1, and Ni to 4000 μg g−1 levels are reported on a shoot dry weight (dw) base. These are two orders of magnitude higher than other, non-accumulating, species generally accumulate (Reeves and Brooks, 1983; McGrath et al., 1993; Brown et al., 1995; Lombi et al., 2000). Next to N. caerulescens, Arabidopsis halleri is developed as model metal hyperaccumulator (Meyer and Verbruggen, 2012). This species is also hypertolerant to Zn and Cd, and a strong Zn hyperaccumulator, but less of a Cd hyperaccumulator and not known to be adapted to Ni. Unlike N. caerulescens it is self-incompatible, which complicates genetic analysis, but it is much closer to the general plant model species Arabidopsis thaliana, with which is shares more sequence synteny. Adaptation to heavy metals seems to be more commonly occurring in the Brassicaceae family than in other families, with other Noccaea species known to hyperaccumulate Ni and Zn, and one, N. praecox, also hyperaccumulating Cd, as recently reviewed by Koch and German (2013). Other genera with metal hyperaccumulating species are Alyssum and Strepthanthus (both mostly Ni adapted). Thus, it is fortunate that the first plant species for which genomic data became available, A. thaliana, belongs to the Brassicaceae family, as this triggered further interest in generating genome sequence information from several members of this family (http://www.brassica.info/resource/sequencing/bmap.php). For neither A. halleri nor N. caerulescens the genome sequence has been determined, which is why most of the gene expression research on these species so far has relied on heterologous micro-array analysis using the available A. thaliana micro-arrays (Becher et al., 2004; Weber et al., 2004; Hammond et al., 2006; Talke et al., 2006; Van De Mortel et al., 2006, 2008). This revealed that both species seem to have evolved similar strategies for dealing with the high metal exposure, typically by modifying the expression of several genes normally involved in Zn and Fe mineral homeostasis. A striking example is the copy number expansion of the HMA4 gene, observed in both species, which increased expression of the gene when compared to non-accumulating species (Hanikenne et al., 2008; Ó Lochlainn et al., 2011; Craciun et al., 2012).
One reason to investigate the remarkable metal adaptation properties of N. caerulescens is that these extremophile plants are interesting target species to develop for metal phytoextraction purposes, in which plants are used to remediate soils contaminated with toxic metals (Peer et al., 2006; Anjum, 2012). Another reason is that the rare extremophile nature of metal hyperaccumulators, to have adapted to otherwise hostile environments, makes them interesting models for plant evolutionary genomics studies (Hanikenne and Nouet, 2011). For both purposes, unraveling the evolutionary and physiological mechanisms that allowed their adaptation, at the molecular level, will be needed. The main approaches that have been followed so far in identifying genes involved in metal adaptation involve comparisons of hyperaccumulating and related non-hyperaccumulating plant species, either by using genetic crosses, or by transcriptomics or proteomics comparisons (reviewed by Krämer et al., 2007; Hassan and Aarts, 2011; Lin and Aarts, 2012). However, so far, a comprehensive set of genome sequences of a metal hyperaccumulator species is not available, which is seriously limiting the progress in molecular analysis of metal adaptation.
Molecular analysis of N. caerulescens has been largely performed based on its close relationship to the well-known model species A. thaliana, allowing the use of molecular tools and genomic databases developed for this species. Although the lineages of both species separated probably some 20 Mya (Clauss and Koch, 2006), there still is substantial genome sequence similarity between these species, estimated at 87–88% sequence identity in intergenic transcribed spacer regions and 88.5% sequence identity in transcribed regions (Peer et al., 2003; Rigola et al., 2006). This high level of conservation was sufficient to use heterologous, A. thaliana, micro-arrays for comparative transcriptome analyses (Hammond et al., 2006; Van De Mortel et al., 2006, 2008). The first attempt to obtain sequence information of N. caerulescens was performed by Rigola et al. (2006), who generated an Expressed Sequence Tag (EST) database of little over 3700 transcript sequences. This resource has been used to generate a cDNA-based micro-array, which has been used for transcript profiling of N. caerulescens, but due to the limited number of probes on the array, the information that could be gathered was limited (Plessl et al., 2010). Since then, the rapid development of high-throughput sequencing techniques has made whole genome and transcriptome sequencing a lot more efficient and affordable (Morozova et al., 2009). Recently, SOLiD technology has been applied for comparative root transcriptomics of three different N. caerulescens accessions (Halimaa et al., 2014). The 454/Roche pyrosequencing method takes advantages of long read lengths and a fast running time (Metzker, 2010; Niedringhaus et al., 2011) and is a suitable method for whole transcriptome sequencing, when the main purpose is to obtain a comprehensive set of transcript sequences of substantial length to be used as a reference database for future transcriptome profiling studies.
Here we present such a comprehensive set of N. caerulescens transcripts, which largely exceeds the previous dataset in the number of identified genes (Rigola et al., 2006). We identified many transcripts involved in mineral accumulation and homeostasis, which may be relevant for metal hyperaccumulation and hypertolerance. In addition we listed genes involved in the biosynthesis of glucosinolates. These are secondary metabolites conferring resistance to herbivores, especially prominent in Brassicaceae (Bones and Rossiter, 1996). This new transcript sequence information will facilitate the further analysis of the extremophile traits of N. caerulescens and is expected to contribute to similar studies in related metal hyperaccumulators, such as A. halleri, and also less studied species like the Zn/Cd hyperaccumulator Noccaea praecox or the Ni-hyperaccumulator Noccaea goesingense (Koch and German, 2013). It will also contribute to more efficient functional studies of heavy metal related genes, to establish their role in metal adaptation or for possible applications in metal phytoextraction, or genes related to synthesis of glucosinolates.
Materials and Methods
Plant Materials and RNA Preparation
An inbred line of N. caerulescens accession “Ganges” (kindly obtained from Dr. Henk Schat, Free University, Amsterdam, The Netherlands) was used. Roots and shoots were collected separately from 5-week-old plants, grown in half-strength Hoagland solutions (Assunção et al., 2001) containing 10 μM ZnSO4, in a climate controlled growth chamber (set at 20/15°C day/night temperature; 70% relative humidity; 12 h day time). Inflorescences were collected from 22-week-old plants grown in soil. RNA of these three plant parts was extracted separately by using the RNeasy® Plant Mini kit (Qiagen, cat. no. 74904) with on-column RNase-Free DNase set digestions (Qiagen, cat. no. 79254). The RNA concentrations were quantified using the highly selective Qubit™ RNA BR Assay kit (Invitrogen™, cat. no. Q10210) with a Qubit® 2.0 Fluorometer.
cDNA Library Construction
Similar amounts of RNA from roots, shoots, and flowers were pooled and used for preparation of a normalized and random-primed cDNA library for Roche/454 sequencing (Vertis Biotechnologie AG). From the total RNA sample, poly(A)+ RNA was isolated, and used for cDNA synthesis. First strand cDNA synthesis was primed using random hexamer primers. To the 5′ and 3′ ends of the cDNA, Roche/454 adapters A and B, as provided by the manufacturer, were ligated and cDNA was finally amplified using 12 PCR cycles and proofreading DNA polymerase. Normalization was performed by denaturation and re-association of cDNA. Re-associated double-stranded cDNA was separated from remaining (normalized) single-stranded cDNA (ss-cDNA) over a hydroxylapatite column. After separation, ss-DNA was PCR-amplified using six PCR cycles. Finally cDNA in the size range of 500–850 bp was eluted from a preparative agarose gel. The final normalized cDNA library contains double stranded fragments of between 500 and 850 bp, consisting of the following sequence structure: 5′-454-Adapter A (CCA-TCT-CAT-CCC-TGC-GTG-TCT-CCG-ACT-CAG), 5′- barcode (CACACG), 5′ adapter (GAC-CTT-GGC-TGT-CAC-TCA-GTT), cDNA insert (400–750 bp), 3′ adapter (TCG-CAG-TGA-GTG-ACA-GGC-CA), 3′-454-Adapter B (CTG-AGA-CTG-CCA-AGG-CAC-ACA-GGG-GAT-AGG).
De novo Sequencing and Assembly of N. caerulescens Transcript Sequences
Prior to Roche/454 sequencing, cDNA library molecules were clonally amplified using a “two copies per bead” ratio for one Large Volume Emulsion PCR, following the manufacturer's protocol (Roche, Genome Sequencer FLX Titanium Series). Two million DNA carrying beads (from a 23% enrichment) were loaded on half a picotiter plate equivalent, divided over two regions. Sequencing was done on a 454 GS FLX Titanium instrument using XLR70 chemistry and 200 flow cycles. The raw reads were assembled after adaptor trimming using Newbler (454 life Sciences Corporation) version v 2.6.
Sequence Annotation and Characterization
The predicted proteomes and genome assemblies of A. thaliana, Arabidopsis lyrata, Brassica rapa, Capsella rubella, and Thellungiella halophila, recently suggested to be named Eutrema halophila (Koch and German, 2013), were downloaded from the phytozome repository version 9.1 (Goodstein et al., 2012). The protein function annotation of A. thaliana version TAIR10.0 was obtained from the TAIR website (www.arabidopsis.org). The gene ontology (GO) annotation for A. thaliana was downloaded from the GO webpage (www.geneontology.org). The protein annotations (including GO-terms) of the four other species were downloaded from the phytozome repository. Pathway and Enzyme Code annotations for A. thaliana and B. rapa were downloaded from the PlantCyc database (www.plantcyc.org) version 8.0. The annotated GO terms were summarized using the plant GOSlim Set of the GOSlim viewer at the AgBase website (http://www.agbase.msstate.edu/cgi-bin/tools/goslimviewer_select.pl) (McCarthy et al., 2006).
The predicted Brassicaceae proteins were clustered into groups of orthologous proteins by first performing all 25 possible pairwise similarity searches between the five predicted proteomes using BlastP version 2.26 (Altschul et al., 1990). Pairwise orthologs were determined for all 10 possible species-pairs using Inparanoid version 4.1 (Remm et al., 2001). Finally, multi-species-ortholog clusters were built by feeding the Inparanoid output files to the multiparanoid Perl-script (Alexeyenko et al., 2006). Whenever possible, the annotation of orthologous protein clusters was inherited from A. thaliana or B. rapa members (in that order). Clusters without A. thaliana or B. rapa members remained un-annotated.
The transcriptome of N. caerulescens was first searched against the predicted proteomes using BlastX with an e-value cut-off of 10−5. Sequences for which the best hit was a member of an annotated cluster inherited the annotation from that cluster. The remaining sequences inherited their annotation from their best hit. Sequences without a significant match against the predicted Brassica proteomes were searched (BlastX with e-value cut-off 10−5) against the non-redundant protein database (NR) downloaded from the NCBI ftp-site (ftp://ftp.ncbi.nlm.nih.gov/). Proteins without BlastX hits were searched against the genomes of the Brassica species using BLAT (Kent, 2002) version 35. BLAT hits were filtered by requiring 90% of the N. caerulescens sequence to be aligned with an average identity of at least 85%. Finally sequences were searched against the NR nucleotide database, downloaded from the NCBI ftp-site (BlastN with e-value cut-off 10−5). Blast hits were only accepted if the corresponding HSPs had ≥85% sequence identity and covered ≥90% of the query isotigs.
Protein alignments were constructed using ClustalW2 (Larkin et al., 2007) and regions of low alignment quality were removed by hand using JalView (Waterhouse et al., 2009). Maximum likelihood trees were constructed using PhyML (the following command line parameters were used: -c 4 -m LG -o lr -v e -a e -f e -b 100) (Guindon et al., 2010). In brief, the trees were constructed using the LG amino acid substitution model (Le and Gascuel, 2008) with four relative substitution rate categories. The proportion of invariable sites and equilibrium amino-acid frequencies were estimated from the data. The alpha parameter was estimated by maximizing the likelihood of the phylogeny. Branching patterns were validated using bootstrap-analysis; 100 bootstrap-samples were generated for the ML-trees and 1000 for the NJ trees. Trees were displayed using the ETE2 python package (Huerta-Cepas et al., 2010) and dendroscope (Huson and Scornavacca, 2012). To investigate the presence of gene duplications of metal homeostasis related genes, sequence sets were created for each gene family implicated in metal homeostasis by performing blast searches against the proteomes of the five Brassicaceae reference species (with e-value cut-off 10−5). The exonerate program was used to rapidly compare transcript sequences to genome sequences (Slater and Birney, 2005). Neighbor-joining trees were subsequently constructed for all alignments using ClustalW2. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GASZ00000000. The version described in this paper is the first version, GASZ01000000. Raw sequence data have been deposited in the NCBI short read archive under the accession SRX456668.
Results
De novo Sequencing and Assembly of N. caerulescens Isotigs
To maximize the transcript diversity, a pool of RNA from roots, shoots, and inflorescences of N. caerulescens was made. A cDNA library synthesized from this RNA pool was used for sequencing. In total 834,911 raw reads with an average length of 401 nucleotides (nt) were obtained and subsequently assembled using Newbler. During the assembly process, two initial contigs can be joined together into an isotig when raw reads are found that map to the ends of both contigs. The resulting transcriptome assembly (Table 1) consisted of 26,785 contigs the vast majority of which were further assembled into a set of 23,836 putative transcripts (isotigs) with an average sequence length of 1025 base pairs (bp). Isotigs sharing at least one contig are grouped into isogroups. Isotigs are likely to be transcript isoforms from the same gene (Isogroup). The isotigs in this study were grouped into 20,378 isogroups. In total 2326 isogroups (11.4%) consisted of more than one isotig. The distributions of the isotigs sizes and the number of isotigs per isogroup are provided in Supplemental Figure S1.
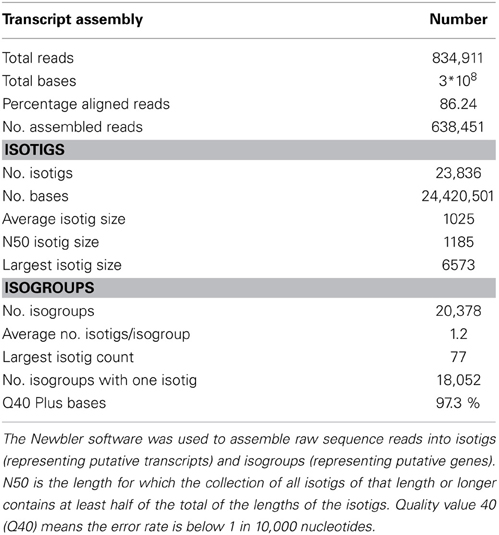
Table 1. Summary of the N. caerulescens transcriptome analysis.
Annotation and Functional Characterization of N. caerulescens Genes
N. caerulescens is a member of the Brassicaceae family, which is why we used the annotation of five other Brassicaceae species of which full genome sequence is available (A. thaliana, A. lyrata, B. rapa, C. rubella, and T. halophila) as a reference for annotation. The predicted proteomes of these five reference species were first clustered in 24,172 groups of orthologous proteins, which we called the Brassicaceae proteome (see Materials and Methods). In total 23,691 of these clusters were annotated following the A. thaliana or B. rapa annotation. We then compared the N. caerulescens isotig DNA sequences against the Brassicaceae proteome and listed the best match (Summarized in Figure 1A; full list in Supplemental Table S1). A total of 22,232 (93.2%) N. caerulescens isotigs (19191 isogroups) had a significant match with the Brassicaceae proteome and we annotated these sequences according to their best match. We also determined the best match for each N. caerulescens sequence in each of the five reference species (Figure 1B). Based on this analysis, we estimate that between 40 and 60% of all N. caerulescens protein encoding genes are represented in this transcriptome set.
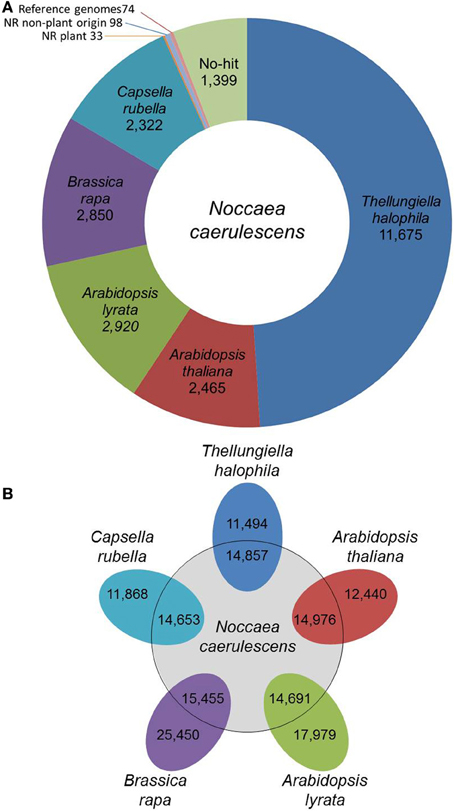
Figure 1. Classification of Noccaea caerulescens isotigs according to best-hits. (A) The number of best-hits of N. caerulescens isotigs to the Brassicaceae proteome (comprising those of Thellungiella (Eutrema) halophila, Arabidopsis thaliana, Arabidopsis lyrata, Brassica rapa, and Capsella rubella) are listed. The 1604 isotigs without any significant hit to Brassicaceae proteome were further compared to the NCBI NR protein database, identifying best-hit similarities to 33 proteins of plant origin and 98 of non-plant origin. The non-hit isotigs were further matched to the available genome sequences of the listed Brassicaceae reference species, identifying another 74 best-hit similarities. Finally, a set of 1399 isotigs remained for which no protein similarity could be found. (B) Venn diagram showing the number of genes in each of the five reference species that correspond to the best-hit in that species for each N. caerulescens isotig sequence.
Even though five closely related species were used as reference, still 1604 isotigs were found without any significant hit in the Brassicaceae proteome data set. These isotigs were therefore compared with the NCBI NR protein database. The search revealed that, although not detected in our initial search, 33 isotigs did have a significant match to a plant protein, including 16 isotigs that strongly resembled an A. thaliana protein and seven that resembled proteins from other Brassicaceae (Supplemental Table S2). Thirteen of the 33 isotigs corresponded to (retro)transposons and nearly all of the remaining ones encoded for hypothetical or unknown proteins. Next to these, an additional 98 isotigs showed only significant similarity to non-plant sequences (Supplemental Table S3). Based on the high similarity to genes of organisms often found to be associated with plants (fungi, bacteria, viruses, etc.), these are most likely reflecting such associations, rather than actual N. caerulescens genes. The remaining sequences with no hit when compared to the NR protein database, were compared against the genome sequences of the five Brassicaceae reference species. This revealed an additional 74 sequences with significant DNA matches to one or more of the reference genomes. Still, there are 1399 isotigs that had no hit to Brassica proteins and proteins in the NR database (Supplemental Table S4). When these were compared to the NR DNA database, an additional 13 isotigs were found to be similar to DNA entries in the database. Only two of these were plant genes. Five isotigs showed similarity to A. thaliana genomic sequences, one to plant mitochondrial DNA and the remaining five appeared to correspond to microbial sequences.
The list of isotigs with best-hit matches to the Brassicaceae proteome (Supplemental Table S1) has been used to obtain an estimate of transcript length coverage of the N. caerulescens isotigs (Supplemental Figure S2). This shows that around half of the transcripts cover over 80% of the protein sequence of their Brassicaceae best-hit match. In addition, we compared the full list of N. caerulescens isotigs to the previously obtained set of N. caerulescens ESTs (Rigola et al., 2006) using BlastN (≥95% identity and ≥60% coverage of shorter sequences). This retrieved 3773 of the 4289 sequences (88%). Sequence similarities between both datasets were generally around 98%, suggesting SNP frequencies of around 2 per 100 bp.
Gene Ontology (GO) Annotation of N. caerulescens Isogroups
Subsequently, we used the N. caerulescens isogroup sequences to categorize putative genes according to GO, by assigning them to three categories of GO terms: Biological Process, Cellular Compartment, and Molecular Function (see Materials and Methods) (Supplemental Table S5). The annotated GO terms were further classified with the plant set of the GO Slim Viewer (McCarthy et al., 2006) (summarized in Supplemental Figure S3, full list in Supplemental Table S6).
Within Biological Process, two major categories are cellular process (25.1%) and metabolic process (19.3%), followed by developmental process (12.3%), and response to stress/stimulus (10.4%). Genes involved in cellular ion homeostasis (involving “cations” and “metal,” but also specifically zinc, copper, calcium, iron, manganese, potassium, and phosphate) (0.1%), are likely to be crucial for the regulation of the heavy metal balance in N. caerulescens (Supplemental Figure S3), in addition to genes involved in transport (2.7%), especially ion transport (0.5%), and signal transduction (1.6%). Within Cellular Compartment, the largest category is cytoplasmic component (50.7%), followed by membrane apparatus (12%), and then intracellular components (10.7%), extracellular components (2.1%), and cell wall (0.9%). Genes encoding proteins localized to the plasma membrane (5%) and the vacuolar membrane (0.6%) will comprise metal transporter genes that are expected to be involved in metal uptake and sequestration (Supplemental Figure S3). Within Molecular Function, the largest category is binding activity (37.1%) comprising ion binding (4%) as an important sub-category for metal hyperaccumulation traits. Furthermore, the genes categorized for transporter activity (8.1%) are expected to play an important role in metal uptake and metal transportation. To see if this is different from other species, we also compared N. caerulescens and A. thaliana regarding the GO terms with the highest percentages of N. caerulescens isogroup counts (Supplemental Figure S3). This made clear that there are only minor differences between both species.
A total of 3051 isotigs (2610 isogroups) could be assigned to 352 PlantCyc pathways (Supplemental Table S7). The 26 most represented pathways (around 20 or more isogroups) are shown in Supplemental Figure S4. The top six pathways are triacylglycerol degradation, homogalacturonan degradation, glycolysis II (from fructose-6P), tRNA charging, aerobic respiration (alternative oxidase pathway), and betanidin degradation, which have more than 50 isogroups found in the pathway.
Transcripts Related to the Response to Heavy Metal Exposure
The metal hyperaccumulation and hypertolerance properties of N. caerulescens partly rely on a number of genes known in other species, mainly A. thaliana, to be involved in mineral homeostasis. A list of such genes, including 87 isogroups, is shown in Table 1. The list includes metal transporter genes belonging to the ZRT/IRT-like Protein (ZIP) gene family (9 isogroups), the Natural Resistance-Associated Macrophage Protein (NRAMP) family (8 isogroups), the Heavy Metal ATPase (HMA) family (9 isogroups), the Metal Tolerance Protein (MTP) family (7 isogroups), and the Calcium exchanger (CAX) family (9 isogroups). In addition, we listed genes belonging to the Plant Cadmium Resistance (PCR) gene family (2 isogroups), the Pleiotropic Drug Resistance protein (PDR) family (14 isogroups), and Plant Defensin (PDF) family (8 isogroups). Furthermore, transcripts related to metal chelator and metal chelator transporter functions were listed, such as genes of the Nicotianamine Synthase (NAS) family (4 isogroups), the Phytochelatin Synthase (PCS) family, (2 isogroups), the Metallothionein (MT) family (4 isogroups), the Yellow Stripe Like protein (YSL) family, (6 isogroups), the Zinc Induced Facilitator (ZIF) family (5 isogroups), and the Multidrug Resistance-associated Protein/ ATP-binding cassette transporter ABCC type (MRP/ABCC) family (26 isogroups). Nota bene we did not include the large families of genes encoding heavy metal-associated isoprenylated plant proteins (HIPP) or heavy metal-associated plant proteins (HPP), of which some members are recently suggested to be relevant for response to Cd (De Abreu-Neto et al., 2013).
For two larger gene families, the ZIP and MTP families, we performed a phylogenetic analysis (Figures 3A,B) to confirm that the Blast analysis (Table 2) indeed identified the most likely A. thaliana orthologs. Of the ZIP family, no transcripts were found for orthologs of AtIRT2, AtZIP3, AtZIP5, AtZIP7, AtZIP8, and AtZIP12. For the MTP family, we did not find N. caerulescens orthologs of the AtMTP2, AtMTP3, AtMTP4, AtMTP9, and AtMTP10 genes. Next we searched for N. caerulescens specific gene duplications for all gene classes in Table 2. To this end we created for each class multiple sequence alignments with sequences from the five reference species that were similar to the N. caerulescens isogroup sequences (see Materials and Methods). Neighbor-joining trees were constructed from these alignments and inspected manually for putative N. caerulescens specific duplications. Only in the tree of the MT3 class we identified a putative N. caerulescens specific duplication. In order to obtain additional support for a recent gene duplication we identified the MT3 orthologs of the five Brassicaceae reference species and constructed a phylogenetic tree of them (Figure 3C). This confirmed that all inspected genomes had only one copy of the MT3 gene.
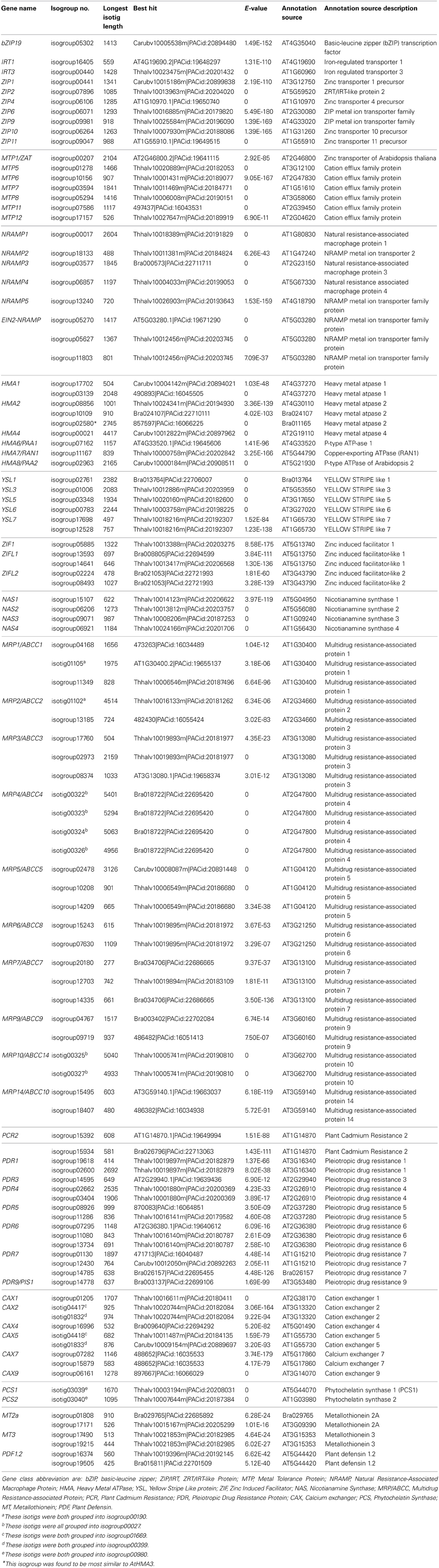
Table 2. Noccaea caerulescens isogroups corresponding to A. thaliana genes involved in mineral homeostasis and heavy metal stress response.
Glucosinolate Biosynthesis Transcripts in N. caerulescens
Next to the mineral homeostasis genes, we also examined the occurrence of transcripts representing genes in the glucosinolate biosynthesis pathway. This pathway is generally prominent in Brassicaceae as it generates glucosinolates that upon interaction with myrosinase enzymes release toxic isothiocyanates, thiocyanates, or nitrils, which display strong anti-feeding properties against herbivores (Bones and Rossiter, 1996). Most of the genes in this pathway and their roles have been identified (Sønderby et al., 2010b; Wang et al., 2011). Important genes are a series of MYB and MYC genes encoding the transcription factors controlling aliphatic and indolic glucosinolate biosynthesis (Gigolashvili et al., 2007; Sønderby et al., 2010a; Schweizer et al., 2013), next to genes involved in core structure formation, side-chain elongation, secondary modification and co-substrate pathways (as summarized by Sønderby et al., 2010b) and more recently identified genes involved in secondary modification of indolic glucosinolates and genes encoding glucosinolate transporters (Pfalz et al., 2011; Nour-Eldin et al., 2012). N. caerulescens plants originating from metalliferous soils were found to contain lower levels of glucosinolates than plants originating from non-metalliferous soils (Noret et al., 2007), which is why we were interested to see if the glucosinolate genes known from A. thaliana were also present in the N. caerulescens transcriptome. Supplemental Table S8 shows the list of genes involved in the glucosinolate biosynthesis pathway, which we classified based on their presumed function as transcription factor genes, genes involved in side-chain elongation, core structure formation and secondary modification or genes of a co-substrate pathway (according to Wang et al., 2011). Of the 61 genes in the list, 40 were found to be expressed in the N. caerulescens transcriptome.
Discussion
N. caerulescens is one of the two heavy metal hyperaccumulator plant model species, together with A. halleri, that are studied in detail to understand their adaptation to growing on soil containing extremely high concentrations of Zn and Cd (Verbruggen et al., 2013). As part of this adaptation, they not only tolerate exposure to normally lethal concentrations of both metals, but also hyperaccumulate them in their shoots, probably as protection against herbivory and microbial infection (Hoerger et al., 2013). N. caerulescens is the only species that also hyperaccumulates Ni and Pb.
The development of new and cost-effective gene expression analysis methods such as RNA-Seq prompted us to generate a comprehensive reference transcriptome dataset for N. caerulescens that will facilitate and support such gene expression analyses, without having to rely on heterologous comparisons. This data set replaces a previous data set we made, comprising only 4289 transcript sequences, representing 3709 unigenes, of accession “La Calamine” (Rigola et al., 2006). We now assembled 23,836 isotigs, further condensed into 20,378 isogroups of accession “Ganges,” which is a better Cd-hyperaccumulator than “La Calamine.” When assuming that isogroups will best represent genes, we have expanded the available transcriptome sequence information, in terms of sequenced genes, by almost 5.5-fold.
The use of isotigs, rather than contigs for listing transcripts, will account for small sequence differences between contigs corresponding to the same transcript. Such can be caused by sequence errors, allelic differences, or differences between recently duplicated gene copies. The accession “Ganges” was used for transcriptome sequencing. This accession had been inbred for at least seven generations, and as N. caerulescens is a self-compatible, readily self-fertilizing species, we expect that only few sequence differences between contigs will be due to allelic variation and that most of the differences between contigs and isotigs will be due to sequencing errors. The difference between isotigs and isogroups is mainly accounted for by differential or alternative splicing or generation of alternative transcripts from the same gene. The 454 sequence technology that was used generates relatively large reads and facilitates easier assembly compared to that of the much shorter reads created by the Illumina sequencing technology. However, despite the long reads, for about 10–15% of the isogroups more than one isotig was assembled, reflecting the occurrence of alternative transcripts (Supplemental Table S1).
When comparing the N. caerulescens transcriptome to that of other diploid Brassicaceae species for which transcriptome sequence is available, such as Thlaspi arvense (Dorn et al., 2013) or Thellungiella salsuginea (Eutrema salsugineum) (Lee et al., 2013), the N. caerulescens transcriptome set is relatively small. The draft transcriptome of T. arvense consists of 33,873 contigs, representing 25,232 Brassicaceae genes, and that of T. salsuginea comprises 42,810 unigenes, corresponding to 24,457 A. thaliana peptides. The 20,378 N. caerulescens isogroups corresponded to 19,191 protein matches in the Brassicaceae reference proteome set. The main reason for these differences will be the representation of more tissues or organs and conditions in the other sequenced libraries. For N. caerulescens, only roots, leaves and flowers of plants grown under normal Zn supply conditions were sampled, while material from more organs or conditions were sampled for the other two species. The different sequencing approaches may also account for the differences. T. arvense was sequenced using Illumina technology, which generates many more, but shorter, reads, while for T. salsuginea both a normalized and non-normalized library were sequenced, using 454 technology, and in the final assembly, previously generated EST sequences present in the NCBI GenBank database were included.
T. salsuginea is predicted to contain 26,521 protein encoding genes (Yang et al., 2013), of which 18,970 were found to be represented in the 42,810 unigene set (Lee et al., 2013). Similar gene numbers are also found for Leavenworthia alabamica (30,343 genes) and Sisymbrium irio (28,917 genes) (Haudry et al., 2013). These three species have a genome size comparable to the genome size of N. caerulescens, which is expected to be 310–330 Mb as based on 2C DNA content (Peer et al., 2003). Assuming that N. caerulescens has around 26,000–30,000 genes, this means around 60% of its transcriptome is covered in the current dataset.
N. caerulescens belongs to the Coluteocarpeae tribe of the Brassicaceae that consists mainly of Noccaea species, which, when tested, are all found to be metal hyperaccumulators (Koch and German, 2013). Some of the other species in this tribe, e.g., Raparia bulbosa and Thlaspiceras oxyceras, are also known to accumulate metals. This tribe belongs to the expanded lineage II of the Brassicaceae, to which also the Brassica and Thellungiella (Eutrema) genera belong. This fits well with the observation that most N. caerulescens transcript sequences find their best BLASTX hit with T. halophila proteins (Supplemental Table S1). Only 98 sequences appeared to be originating from non-plant organisms, potentially corresponding to organisms living in association with the N. caerulescens plants we used to generate the cDNA libraries. Considering that for the T. arvense transcriptome set, 424 out of 33,873 contigs showed similarity to fungal genes, the contribution of such genes to the N. caerulescens dataset is modest. Upon comparing the N. caerulescens isotigs to existing sequence information available in the NCBI GenBank, 1386 isotigs (5.8%) were found to have no significant similarity to any other sequence in database (Supplemental Table S4). Potentially these could be genes unique to N. caerulescens; however, they may also represent miRNA precursors that are much less conserved and hard to detect using Blast analysis. Of course it is possible that these sequences represent genomic DNA sequences (not very likely considering the use of DNAses in the cDNA library construction), or transcripts of plant-associated organisms that have not been sequenced yet. In the previous N. caerulescens transcriptome dataset we generated, we found 8% of the unigenes to show no similarity to any other entry in the NCBI database (Rigola et al., 2006). Comparison of the “no-hit” sequences to the N. caerulescens whole genome sequence, when available, will be needed to clarify if these are from N. caerulescens.
Isogroups rather than isotigs were used for GO annotation and analysis to avoid overrepresentation of certain GO terms due to alternative transcripts generating many isotigs for the same gene. Sometimes the isogrouping appears to be too strict, which has forced transcripts from different paralogs into one isogroup, as was found for some of the mineral homeostasis related genes (Table 2). The GO analysis (Figure 2, Supplemental Figure S3 and Supplemental Tables S5 and S6) showed that N. caerulescens isogroups were largely GO-annotated in comparable GO-class representations as for A. thaliana.
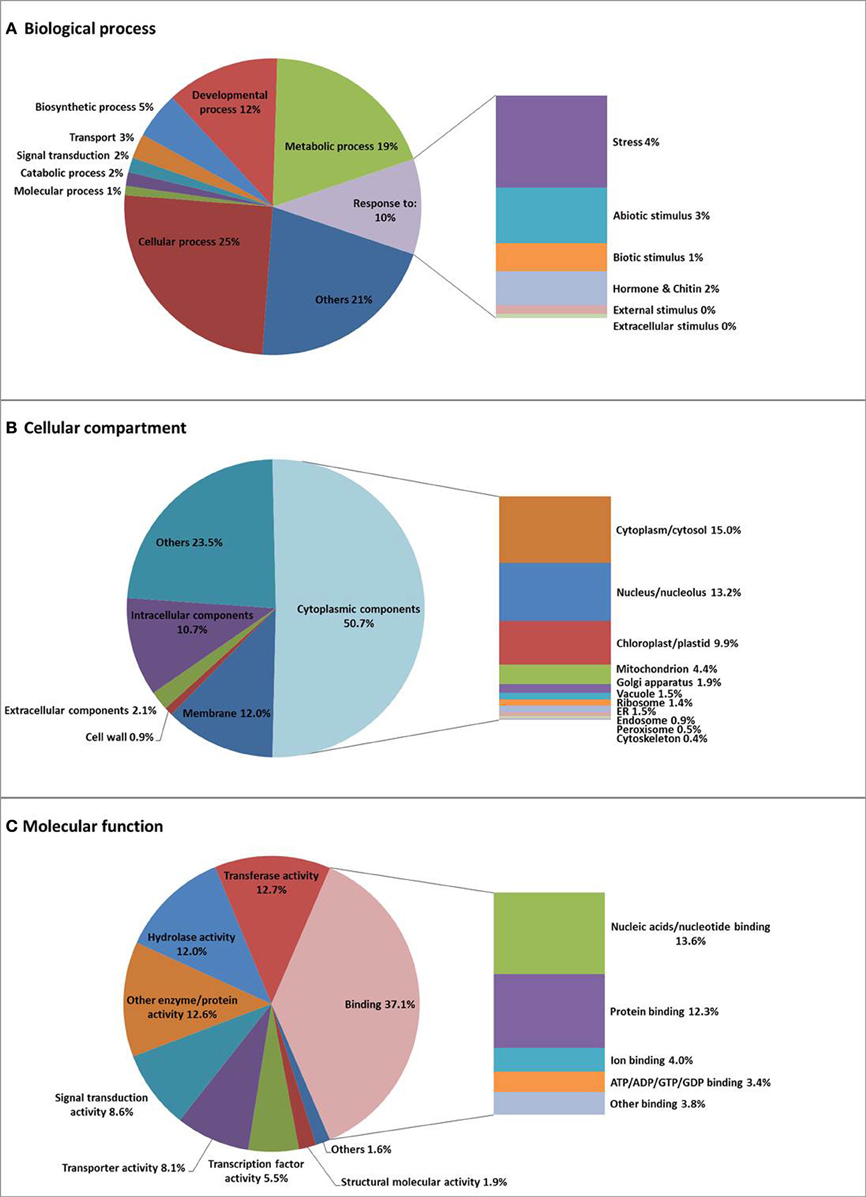
Figure 2. Gene Ontology (GO) classification of N. caerulescens isogroups. N. caerulescens isogroups were classified into three major functional GO groups: Biological Process (A), Cellular Component (B), and Molecular Function (C) and subsequently sub-classified as indicated.
Many mineral homeostasis genes were identified among the N. caerulescens isotigs (Table 2). Although we identified genes for all relevant gene families, not all genes for each family were found to have N. caerulescens counterparts, representing their potential orthologs. For instance, for the ZIP gene family of plasma membrane metal importers, no N. caerulescens transcript was found for IRT2 and ZIP3, 5, 7, 8, and 12. Transcripts for these genes have been found by Halimaa et al. (2014), but except for the ZIP5 ortholog, these are expressed at low levels in GA roots. Not finding a potential ortholog for ZIP5 is remarkable, since transcripts for these genes were also found previously (Rigola et al., 2006) and at least the ortholog of ZIP5 was expressed in roots and shoots (Wu et al., 2009). Also for the MTP family of vacuolar metal importers we did not detect transcripts similar to all A. thaliana MTP genes, but most of them have been found by Halimaa et al. (2014). Phylogenetic analysis of the ZIP and MTP isotigs showed that those were classified correctly (Figure 3). For the HMA family of plasma membrane metal exporter genes, we initially did not find a potential ortholog of the NcHMA3 gene (Ueno et al., 2011). However, upon re-examining the isogroups showing similarity to HMA proteins, isogroup02580, which was found to be very similar to a B. rapa protein annotated as HMA2, was actually highly similar (~90% DNA identity) to A. halleri and A. thaliana HMA3 genes.
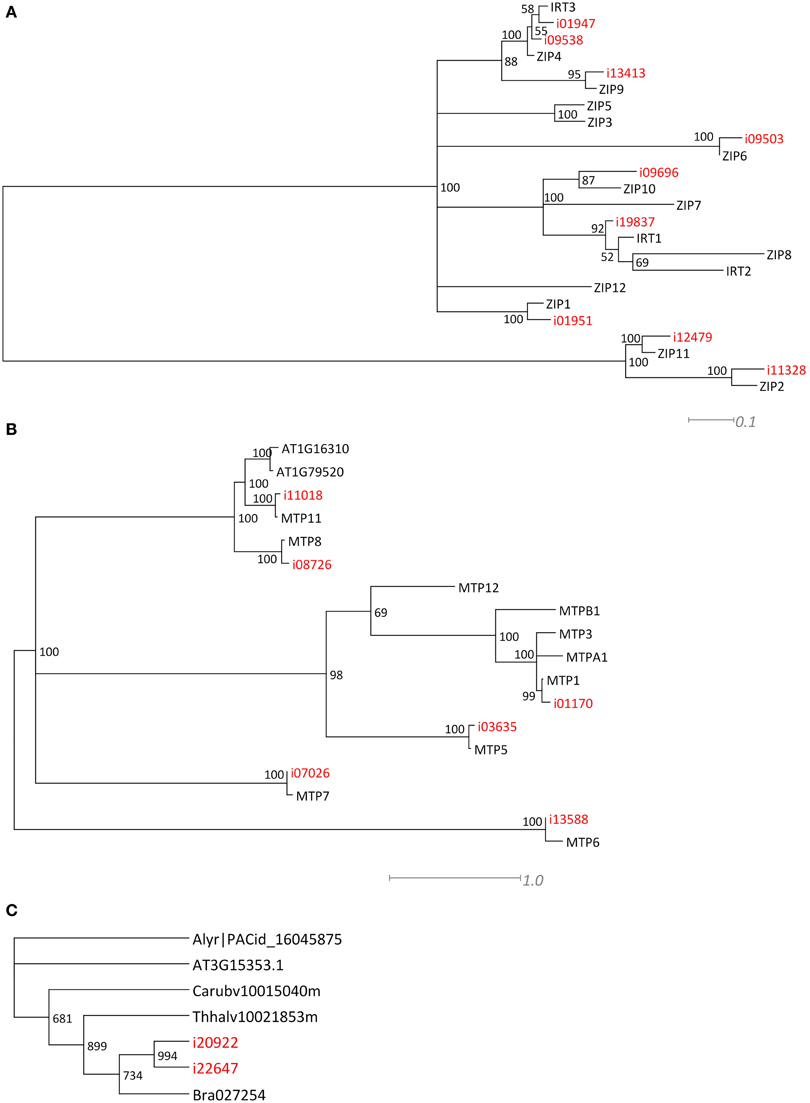
Figure 3. Phylogenetic comparison of ZIP, MTP, and MT related N. caerulescens isotigs. Maximum Likelihood trees, showing the longest isotigs (indicated in red) belonging to different isogroups of the ZRT/IRT-like Protein (ZIP) (A) and Metal Tolerance Protein (MTP) gene families (B) as identified in Table 2, and compared at amino acid sequence level to A. thaliana genes to indicate the most likely orthologs. When all proteins listed in Table 2 were manually examined for N. caerulescens specific gene duplications, this appeared to be the case only for Metallothionein 3 (MT3). The Neighbor Joining tree is shown in (C), displaying the comparison of nucleotide sequences of MT3 isotigs to most similar genes of the previously used five Brassicaceae reference gene sets, indicates that N. caerulescens expresses an additional gene copy. Note that all branches in the Maximum Likelihood and Neighbor Joining trees with 50% or less bootstrap support were collapsed.
Only for the MT3 gene we could identify an additional copy in N. caerulescens compared to the single copies found in related species (Figure 3C). This gene has been implicated in Cu homeostasis, and expression in a southern France accession (“St. Felix,” which is close to the origin of “Ganges”) is much higher than in the two other tested accessions and appears to be constitutive, rather than induced by Cu (Roosens et al., 2004). Such could well be the consequence of an additional copy, which would mean the additional copy is accession, rather than species specific, and is worth further investigations. Not finding copy number expansion for other genes is remarkable, as for several of the other genes listed in Table 2, such as HMA3 and HMA4, multiple copies have already been reported in other accessions, including “Ganges” (Ó Lochlainn et al., 2011; Ueno et al., 2011; Craciun et al., 2012; Iqbal et al., 2013). However, for HMA4, for which cDNA sequences of “Ganges” are available, the different cDNA copies are very similar in sequence (Iqbal et al., 2013), only differing in a few bp and three InDels, which will not be distinguished from sequence differences due to sequencing errors by the assembly software we used. This is likely to be the case for more recently duplicated gene copies. Detailed analysis of copy number variation will be much less ambiguous upon availability of a whole genome sequence, where non-encoding sequence can be taken into account to distinguish duplicated copies.
When examining genes involved in glucosinolate biosynthesis, we identified isotigs corresponding to all transcription factor genes involved in aliphatic glucosinolates, but not for two of the MYB genes involved in indolic glucosinolates (MYB34 and MYB122) (Supplemental Table S8). In contrast, hardly any of the aliphatic side-chain elongation genes as well as the two CYP79F genes involved in aliphatic core structure formation was represented in the N. caerulescens isogroup list. Of course, not finding them in the isotig list does not mean these genes are not expressed, but it is remarkable that expression of several of the structural genes involved in aliphatic glucosinolate biosynthesis is so low that apparently these genes are more likely to missed by 454 sequencing than the other genes. Low expression of glucosinolate genes would be in line with a previous report that especially metallicolous accessions from the south of France, where also “Ganges” is originating from, are low in total glucosinolate levels (Noret et al., 2007).
The main reason for studying N. caerulescens is to learn more on its extraordinary capacities to tolerate exposure to high concentrations of heavy metals and accumulate these to extremely high levels in the leaves. The transcriptome sequence as such will not tell us that much on which genes will be relevant for these traits, but it can be used as an excellent reference for RNA-Seq studies to determine gene expression in organs of different accessions, exposed to a range of metal concentrations. There are many different N. caerulescens populations in Europe, which can differ substantially in their ability to tolerate and accumulate metals, as well as differ in their metal preferences. Many populations grow on non-metallicolous soils, which are not enriched in metals. At those sites, they are likely to accumulate mainly Zn. When exposed to Zn, Ni, and Cd they can hyperaccumulate them, but as they are generally not metal tolerant, plants will rapidly die upon exposure. Other natural populations grow on (ultramafic) serpentine outcrops, which are generally rich in metals, but not Zn. At those sites, they often hyperaccumulate Ni, and when exposed to Zn or Cd, will also hyperaccumulate these metals in their shoot. However, they are often only tolerant to Ni and will suffer or die from exposure to Cd. Finally, there are several local populations spread over different sites in Europe that have been heavily contaminated by Zn and Cd, and often also Pb (Mohtadi et al., 2012). These calamine populations are Zn and often Cd hyperaccumulating and are also tolerant to these metals. When exposed to Ni, they will also hyperaccumulate it, but they are less tolerant to it than populations from serpentine sites (Assunção et al., 2003). Especially populations found on metal contaminated sites in the Cevennes region, around the town of Ganges in the south of France, are particularly good at hyperaccumulating Cd. Most of the phenotypic differences related to metal tolerance and accumulation between populations are due to genetic differences, often reflected in differences in gene expression (Van De Mortel et al., 2008). With the N. caerulescens transcriptome dataset we generated, it will be much easier to study such differences. Also, when the whole genome sequence of N. caerulescens will be determined, the transcriptome data can be used to annotate predicted genes and delineate potential transcripts of such genes. This will be useful to determine the function of these genes. Finally, since the previous transcriptome dataset was obtained from accession “La Calamine,” and the new one from “Ganges,” the comparison of transcripts from the same genes revealed a SNP frequency of around 2 per 100 bp. Although the presence of sequence errors in both sets will have inflated this number, it will be straightforward to convert these differences into genetic markers that can be used in mapping quantitative trait loci in segregating populations of N. caerulescens (Assunção et al., 2006; Deniau et al., 2006).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the Dutch Graduate School for Experimental Plant Sciences and the Zenith-scheme of the Dutch Organization for Scientific Research (NWO) for financially supporting this research. Corrie Hanhart is gratefully acknowledged for providing the N. caerulescens inflorescences used for RNA extraction.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00261/abstract
References
Alexeyenko, A., Tamas, I., Liu, G., and Sonnhammer, E. L. L. (2006). Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics 22, e9–e15. doi: 10.1093/bioinformatics/btl213
Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman, D. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Anjum, N. A. (2012). The Plant Family Brassicaceae Contribution Towards Phytoremediation [Online]. Dordrecht; New York: Springer. doi: 10.1007/978-94-007-3913-0
Assunção, A. G. L., Da Costa Martins, P., De Folter, S., Vooijs, R., Schat, H., and Aarts, M. G. M. (2001). Elevated expression of metal transporter genes in three accessions of the metal hyperaccumulator Thlaspi caerulescens. Plant Cell Environ. 24, 217–226. doi: 10.1111/j.1365-3040.2001.00666.x
Assunção, A. G. L., Pieper, B., Vromans, J., Lindhout, P., Aarts, M. G. M., and Schat, H. (2006). Construction of a genetic linkage map of Thlaspi caerulescens and quantitative trait loci analysis of zinc accumulation. New Phytol. 170, 21–32. doi: 10.1111/j.1469-8137.2005.01631.x
Assunção, A. G. L., Schat, H., and Aarts, M. G. M. (2003). Thlaspi caerulescens, an attractive model species to study heavy metal hyperaccumulation in plants. New Phytol. 159, 351–360. doi: 10.1046/j.1469-8137.2003.00820.x
Becher, M., Talke, I. N., Krall, L., and Krämer, U. (2004). Cross-species microarray transcript profiling reveals high constitutive expression of metal homeostasis genes in shoots of the zinc hyperaccumulator Arabidopsis halleri. Plant J. 37, 251–268. doi: 10.1046/j.1365-313X.2003.01959.x
Bones, A. M., and Rossiter, J. T. (1996). The myrosinase-glucosinolate system, its organisation and biochemistry. Physiol. Plant. 97, 194–208. doi: 10.1111/j.1399-3054.1996.tb00497.x
Brown, S., Angle, J., Chaney, R., and Baker, A. (1995). Zinc and cadmium uptake by hyperaccumulator Thlaspi caerulescens grown in nutrient solution. Soil Sci. Soc. Am. J. 59, 125–133.
Clauss, M. J., and Koch, M. A. (2006). Poorly known relatives of Arabidopsis thaliana. Trends Plant Sci. 11, 449–459. doi: 10.1016/j.tplants.2006.07.005
Craciun, A. R., Meyer, C.-L., Chen, J., Roosens, N., De Groodt, R., Hilson, P., et al. (2012). Variation in HMA4 gene copy number and expression among Noccaea caerulescens populations presenting different levels of Cd tolerance and accumulation. J. Exp. Bot. 63, 4179–4189. doi: 10.1093/jxb/ers104
De Abreu-Neto, J. B., Turchetto-Zolet, A. C., De Oliveira, L. F. V., Bodanese Zanettini, M. H., and Margis-Pinheiro, M. (2013). Heavy metal-associated isoprenylated plant protein (HIPP): characterization of a family of proteins exclusive to plants. FEBS J. 280, 1604–1616. doi: 10.1111/febs.12159
Deniau, A., Pieper, B., Ten Bookum, W., Lindhout, P., Aarts, M., and Schat, H. (2006). QTL analysis of cadmium and zinc accumulation in the heavy metal hyperaccumulator Thlaspi caerulescens. Theor. Appl. Gene. 113, 907–920. doi: 10.1007/s00122-006-0350-y
Dorn, K. M., Fankhauser, J. D., Wyse, D. L., and Marks, M. D. (2013). De novo assembly of the pennycress (Thlaspi arvense) transcriptome provides tools for the development of a winter cover crop and biodiesel feedstock. Plant J. 75, 1028–1038. doi: 10.1111/tpj.12267
Gigolashvili, T., Berger, B., Mock, H.-P., Müller, C., Weisshaar, B., and Flügge, U.-I. (2007). The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 50, 886–901. doi: 10.1111/j.1365-313X.2007.03099.x
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Halimaa, P., Lin, Y.-F., Ahonen, V. H., Blande, D., Clemens, S., Gyenesei, A., et al. (2014). Gene expression differences between Noccaea caerulescens ecotypes help to identify candidate genes for metal phytoremediation. Environ. Sci. Technol. 48, 3344–3353. doi: 10.1021/es4042995
Hammond, J. P., Bowen, H. C., White, P. J., Mills, V., Pyke, K. A., Baker, A. J. M., et al. (2006). A comparison of the Thlaspi caerulescens and Thlaspi arvense shoot transcriptomes. New Phytol. 170, 239–260. doi: 10.1111/j.1469-8137.2006.01662.x
Hanikenne, M., and Nouet, C. (2011). Metal hyperaccumulation and hypertolerance: a model for plant evolutionary genomics. Curr. Opin. Plant Biol. 14, 252–259. doi: 10.1016/j.pbi.2011.04.003
Hanikenne, M., Talke, I. N., Haydon, M. J., Lanz, C., Nolte, A., Motte, P., et al. (2008). Evolution of metal hyperaccumulation required cis-regulatory changes and triplication of HMA4. Nature 453, 391–395. doi: 10.1038/nature06877
Hassan, Z., and Aarts, M. G. M. (2011). Opportunities and feasibilities for biotechnological improvement of Zn, Cd or Ni tolerance and accumulation in plants. Environ. Exp. Bot. 72, 53–63. doi: 10.1016/j.envexpbot.2010.04.003
Haudry, A., Platts, A. E., Vello, E., Hoen, D. R., Leclercq, M., Williamson, R. J., et al. (2013). An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat. Genet. 45, 891–898. doi: 10.1038/ng.2684
Hoerger, A. C., Fones, H. N., and Preston, G. (2013). The current status of the elemental defense hypothesis in relation to pathogens. Front. Plant Sci. 4:395. doi: 10.3389/fpls.2013.00395
Huerta-Cepas, J., Dopazo, J., and Gabaldón, T. (2010). ETE: a python environment for tree exploration. BMC Bioinformatics 11:24. doi: 10.1186/1471-2105-11-24
Huson, D. H., and Scornavacca, C. (2012). Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61, 1061–1067. doi: 10.1093/sysbio/sys062
Iqbal, M., Nawaz, I., Hassan, Z., Hakvoort, H. W. J., Bliek, M., Aarts, M. G. M., et al. (2013). Expression of HMA4 cDNAs of the zinc hyperaccumulator Noccaea caerulescens from endogenous NcHMA4 promoters does not complement the zinc-deficiency phenotype of the Arabidopsis thaliana hma2hma4 double mutant. Front. Plant Sci. 4:404. doi: 10.3389/fpls.2013.00404
Kent, W. J. (2002). BLAT—the blast-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Koch, M. A., and German, D. (2013). Taxonomy and systematics are key to biological information: Arabidopsis, Eutrema (Thellungiella), Noccaea and Schrenkiella (Brassicaceae) as examples. Front. Plant Sci. 4:267. doi: 10.3389/fpls.2013.00267
Krämer, U., Talke, I. N., and Hanikenne, M. (2007). Transition metal transport. FEBS Lett. 581, 2263–2272. doi: 10.1016/j.febslet.2007.04.010
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Lee, Y., Giorgi, F., Lohse, M., Kvederaviciute, K., Klages, S., Usadel, B., et al. (2013). Transcriptome sequencing and microarray design for functional genomics in the extremophile Arabidopsis relative Thellungiella salsuginea (Eutrema salsugineum). BMC Genomics 14:793. doi: 10.1186/1471-2164-14-793
Lin, Y.-F., and Aarts, M. G. M. (2012). The molecular mechanism of zinc and cadmium stress response in plants. Cell. Mol. Life Sci. 69, 3187–3206. doi: 10.1007/s00018-012-1089-z
Lombi, E., Zhao, F. J., Dunham, S. J., and McGrath, S. P. (2000). Cadmium accumulation in populations of Thlaspi caerulescens and Thlaspi goesingense. New Phytol. 145, 11–20. doi: 10.1046/j.1469-8137.2000.00560.x
McCarthy, F., Wang, N., Magee, G. B., Nanduri, B., Lawrence, M., Camon, E., et al. (2006). AgBase: a functional genomics resource for agriculture. BMC Genomics 7:229. doi: 10.1186/1471-2164-7-229
McGrath, S. P., Sidoli, C. M. D., Baker, A. J. M., and Reeves, R. D. (1993). “The potential for the use of metal-accumulating plants for the in situ decontamination of metal-polluted soils,” in Integrated Soil and Sediment Research: A Basis for Proper Protection, (Proceedings of the European Conference on Integrated Research for Soil and Sediment Protection and Remediation, EUROSOL, MECC Maastricht, The Netherlands), eds H. J. P. Eijsackers and T. Hamers (Dordrecht: Kluwer Academic Publishers), 673–676.
Metzker, M. L. (2010). Sequencing technologies - the next generation. Nat. Rev. Genet. 11, 31–46. doi: 10.1038/nrg2626
Meyer, C.-L., and Verbruggen, N. (2012). The use of the model species Arabidopsis halleri towards phytoextraction of cadmium polluted soils. N. Biotechnol. 30, 9–14. doi: 10.1016/j.nbt.2012.07.009
Milner, M. J., and Kochian, L. V. (2008). Investigating heavy-metal hyperaccumulation using Thlaspi caerulescens as a model system. Ann. Bot. 102, 3–13. doi: 10.1093/aob/mcn063
Mohtadi, A., Ghaderian, S., and Schat, H. (2012). A comparison of lead accumulation and tolerance among heavy metal hyperaccumulating and non-hyperaccumulating metallophytes. Plant Soil 352, 267–276. doi: 10.1007/s11104-011-0994-5
Morozova, O., Hirst, M., and Marra, M. A. (2009). Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet. 10, 135–151. doi: 10.1146/annurev-genom-082908-145957
Niedringhaus, T. P., Milanova, D., Kerby, M. B., Snyder, M. P., and Barron, A. E. (2011). Landscape of next-generation sequencing technologies. Anal. Chem. 83, 4327–4341. doi: 10.1021/ac2010857
Noret, N., Meerts, P., Vanhaelen, M., Santos, A. D., and Escarré, J. (2007). Do metal-rich plants deter herbivores? A field test of the defence hypothesis. Oecologia 152, 92–100. doi: 10.1007/s00442-006-0635-5
Nour-Eldin, H. H., Andersen, T. G., Burow, M., Madsen, S. R., Jorgensen, M. E., Olsen, C. E., et al. (2012). NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 488, 531–534. doi: 10.1038/nature11285
Ó Lochlainn, S., Bowen, H. C., Fray, R. G., Hammond, J. P., King, G. J., White, P. J., et al. (2011). Tandem quadruplication of HMA4 in the zinc (Zn) and cadmium (Cd) hyperaccumulator Noccaea caerulescens. PLoS ONE 6:e17814. doi: 10.1371/journal.pone.0017814
Peer, W. A., Mahmoudian, M., Freeman, J. L., Lahner, B., Richards, E. L., Reeves, R. D., et al. (2006). Assessment of plants from the Brassicaceae family as genetic models for the study of nickel and zinc hyperaccumulation. New Phytol. 172, 248–260. doi: 10.1111/j.1469-8137.2006.01820.x
Peer, W. A., Mamoudian, M., Lahner, B., Reeves, R. D., Murphy, A. S., and Salt, D. E. (2003). Identifying model metal hyperaccumulating plants: germplasm analysis of 20 Brassicaceae accessions from a wide geographical area. New Phytol. 159, 421–430. doi: 10.1046/j.1469-8137.2003.00822.x
Pfalz, M., Mikkelsen, M. D., Bednarek, P., Olsen, C. E., Halkier, B. A., and Kroymann, J. (2011). Metabolic engineering in Nicotiana benthamiana reveals key enzyme functions in Arabidopsis indole glucosinolate modification. Plant Cell 23, 716–729. doi: 10.1105/tpc.110.081711
Plessl, M., Rigola, D., Hassinen, V., Tervahauta, A., Kärenlampi, S., Schat, H., et al. (2010). Comparison of two ecotypes of the metal hyperaccumulator Thlaspi caerulescens (J. & C. PRESL) at the transcriptional level. Protoplasma 239, 81–93. doi: 10.1007/s00709-009-0085-0
Reeves, R. D., and Brooks, R. R. (1983). European species of Thlaspi L. (Cruciferae) as indicators of nickel and zinc. J. Geochem. Explor. 18, 275–283.
Remm, M., Storm, C. E. V., and Sonnhammer, E. L. L. (2001). Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 314, 1041–1052. doi: 10.1006/jmbi.2000.5197
Rigola, D., Fiers, M., Vurro, E., and Aarts, M. G. M. (2006). The heavy metal hyperaccumulator Thlaspi caerulescens expresses many species-specific genes, as identified by comparative expressed sequence tag analysis. New Phytol. 170, 753–766. doi: 10.1111/j.1469-8137.2006.01714.x
Roosens, N. H., Bernard, C., Leplae, R., and Verbruggen, N. (2004). Evidence for copper homeostasis function of metallothionein (MT3) in the hyperaccumulator Thlaspi caerulescens. FEBS Lett. 577, 9–16. doi: 10.1016/j.febslet.2004.08.084
Schweizer, F., Fernández-Calvo, P., Zander, M., Diez-Diaz, M., Fonseca, S., Glauser, G., et al. (2013). Arabidopsis basic Helix-Loop-Helix transcription factors MYC2, MYC3, and MYC4 regulate glucosinolate biosynthesis, insect performance, and feeding behavior. Plant Cell 25, 3117–3132. doi: 10.1105/tpc.113.115139
Slater, G., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. doi: 10.1186/1471-2105-6-31
Sønderby, I. E., Burow, M., Rowe, H. C., Kliebenstein, D. J., and Halkier, B. A. (2010a). A complex interplay of three R2R3 MYB transcription factors determines the profile of aliphatic glucosinolates in Arabidopsis. Plant Physiol. 153, 348–363. doi: 10.1104/pp.109.149286
Sønderby, I. E., Geu-Flores, F., and Halkier, B. A. (2010b). Biosynthesis of glucosinolates - gene discovery and beyond. Trends Plant Sci. 15, 283–290. doi: 10.1016/j.tplants.2010.02.005
Talke, I. N., Hanikenne, M., and Kramer, U. (2006). Zinc-dependent global transcriptional control, transcriptional deregulation, and higher gene copy number for genes in metal homeostasis of the hyperaccumulator Arabidopsis halleri. Plant Physiol. 142, 148–167. doi: 10.1104/pp.105.076232
Ueno, D., Milner, M. J., Yamaji, N., Yokosho, K., Koyama, E., Clemencia Zambrano, M., et al. (2011). Elevated expression of TcHMA3 plays a key role in the extreme Cd tolerance in a Cd-hyperaccumulating ecotype of Thlaspi caerulescens. Plant J. 66, 852–862. doi: 10.1111/j.1365-313X.2011.04548.x
Van De Mortel, J. E., Almar Villanueva, L., Schat, H., Kwekkeboom, J., Coughlan, S., Moerland, P. D., et al. (2006). Large expression differences in genes for iron and zinc homeostasis, stress response, and lignin biosynthesis distinguish roots of Arabidopsis thaliana and the related metal hyperaccumulator Thlaspi caerulescens. Plant Physiol. 142, 1127–1147. doi: 10.1104/pp.106.082073
Van De Mortel, J. E., Schat, H., Moerland, P. D., Ver Loren Van Themaat, E., Van Der Ent, S., Blankestijn, H., et al. (2008). Expression differences for genes involved in lignin, glutathione and sulphate metabolism in response to cadmium in Arabidopsis thaliana and the related Zn/Cd-hyperaccumulator Thlaspi caerulescens. Plant Cell Environ. 31, 301–324. doi: 10.1111/j.1365-3040.2007.01764.x
Verbruggen, N., Juraniec, M., Baliardini, C., and Meyer, C.-L. (2013). Tolerance to cadmium in plants: the special case of hyperaccumulators. Biometals 26, 633–638. doi: 10.1007/s10534-013-9659-6
Wang, H., Wu, J., Sun, S., Liu, B., Cheng, F., Sun, R., et al. (2011). Glucosinolate biosynthetic genes in Brassica rapa. Gene 487, 135–142. doi: 10.1016/j.gene.2011.07.021
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M., and Barton, G. J. (2009). Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Weber, M., Harada, E., Vess, C., Roepenack-Lahaye, E. V., and Clemens, S. (2004). Comparative microarray analysis of Arabidopsis thaliana and Arabidopsis halleri roots identifies nicotianamine synthase, a ZIP transporter and other genes as potential metal hyperaccumulation factors. Plant J. 37, 269–281. doi: 10.1046/j.1365-313X.2003.01960.x
Wu, J., Zhao, F.-J., Ghandilyan, A., Logoteta, B., Guzman, M., Schat, H., et al. (2009). Identification and functional analysis of two ZIP metal transporters of the hyperaccumulator Thlaspi caerulescens. Plant Soil 325, 79–95. doi: 10.1007/s11104-009-0151-6
Keywords: metal hyperaccumulation, metal hypertolerance, phytoremediation, zinc, cadmium, gene expression, Brassicaceae
Citation: Lin Y-F, Severing EI, te Lintel Hekkert B, Schijlen E and Aarts MGM (2014) A comprehensive set of transcript sequences of the heavy metal hyperaccumulator Noccaea caerulescens. Front. Plant Sci. 5:261. doi: 10.3389/fpls.2014.00261
Received: 11 December 2013; Accepted: 21 May 2014;
Published online: 20 June 2014.
Edited by:
Cécile Nouet, University of Liège, BelgiumReviewed by:
Henk Schat, Vrije Universiteit Amsterdam, NetherlandsSirpa Orvokki Kärenlampi, University of Eastern Finland, Finland
Copyright © 2014 Lin, Severing, te Lintel Hekkert, Schijlen and Aarts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark G. M. Aarts, Laboratory of Genetics, Wageningen University, Droevendaalsesteeg 1, 6708 PB Wageningen, Netherlands e-mail:bWFyay5hYXJ0c0B3dXIubmw=
†These authors have contributed equally to this work.