 Allison L. Creason1,2†
Allison L. Creason1,2† Edward W. Davis II1,2†Melodie L. Putnam1
Edward W. Davis II1,2†Melodie L. Putnam1 Olivier M. Vandeputte3
Olivier M. Vandeputte3 Jeff H. Chang1,2,4*
Jeff H. Chang1,2,4*- 1Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR, USA
- 2Molecular and Cellular Biology Program, Oregon State University, Corvallis, OR, USA
- 3Laboratoire de Biotechnologie Vegetale, Universite Libre de Bruxelles, Gosselies, Belgium
- 4Center for Genome Research and Biocomputing, Oregon State University, Corvallis, OR, USA
The accurate diagnosis of diseases caused by pathogenic bacteria requires a stable species classification. Rhodococcus fascians is the only documented member of its ill-defined genus that is capable of causing disease on a wide range of agriculturally important plants. Comparisons of genome sequences generated from isolates of Rhodococcus associated with diseased plants revealed a level of genetic diversity consistent with them representing multiple species. To test this, we generated a tree based on more than 1700 homologous sequences from plant-associated isolates of Rhodococcus, and obtained support from additional approaches that measure and cluster based on genome similarities. Results were consistent in supporting the definition of new Rhodococcus species within clades containing phytopathogenic members. We also used the genome sequences, along with other rhodococcal genome sequences to construct a molecular phylogenetic tree as a framework for resolving the Rhodococcus genus. Results indicated that Rhodococcus has the potential for having 20 species and also confirmed a need to revisit the taxonomic groupings within Rhodococcus.
Introduction
Defining bacteria into stable and coherent genetically similar species has many practical implications. However, multiple factors including effective population size, horizontal gene transfer and bacterial recombination, and their barriers, affect cohesiveness of different groups of bacteria to varying degrees (Doolittle and Zhaxybayeva, 2009). As a consequence, a unifying concept for bacterial species has yet to be adopted, which has made it difficult to develop criteria and thresholds that can be generally applied for defining bacterial species.
Traditional polyphasic approaches define bacterial species as a monophyletic group with at least one discriminative phenotypic trait. Though pragmatic and widely adopted, the traditional approaches are weighted toward phenotypic traits and cannot keep pace with the rate in which new genotypes are being discovered and sequenced. With major advances in contemporary methods in sequencing, operational criteria based on whole genome sequences have been developed and adopted to assist in resolving bacterial phylogeny (Konstantinidis et al., 2006). Multi-locus sequence analysis (MLSA) and trees based on whole genome sequences are powerful methods for inferring evolutionary relationships (Staley, 2009). Alternative criteria based on the degree of similarities in genome signatures have also been developed (Konstantinidis et al., 2006; Bohlin et al., 2008; Richter and Rosselló-Móra, 2009). Average nucleotide identity (ANI), for example, is a simple measure of genetic relatedness based on sequences conserved among compared genomes and has gained acceptance as a method for defining bacterial species (Konstantinidis et al., 2006; Chan et al., 2012; Kim et al., 2014). ANI has also been developed as a method for codifying bacteria based on genome similarity (Marakeby et al., 2014).
Genome-enabled comparisons and the recognition of environmental niches in structuring gene flow have revealed a diversity of population structures for groups of plant-associated bacteria. Pseudomonas fluorescens, for example, occupies multiple niches and has a level of heterogeneity consistent with limited gene flow between sub-clades that challenge their taxonomy (Loper et al., 2012). Likewise, a change to the taxonomy of Agrobacterium tumefaciens has been proposed to reflect genome-enabled discovery of clade-specific traits (Lassalle et al., 2011). The ANI method has been used to assign newly discovered isolates to known plant-associated species and discover new species of plant pathogens (Dudnik et al., 2014; Durán et al., 2014; van der Wolf et al., 2014).
The Gram-positive Rhodococcus genus is a member of the Nocardiaceae family and forms a distinct group with 30 valid species published (Jones and Goodfellow, 2012). The genus has diverse members that inhabit a wide range of terrestrial as well as aquatic habitats and are renowned for their catabolic functions and ability to degrade a large number of organic compounds (Larkin et al., 2005). Additionally, members of Rhodococcus have been recovered from extreme environments such as the deep-sea, oil-contaminated soils, and freeze-thaw tundra on glacial margins (Sheng et al., 2011; Shevtsov et al., 2013; Konishi et al., 2014). Because of their biotechnological applications and potential in bioremediation, there has been a dramatic increase in the number of sequenced Rhodococcus genomes. Their genomes are high in GC content and range in size from 4.3 megabases (Mb) to over 10 Mb. Most genomes are larger than 5 Mb and their large sizes have been attributed to both horizontal gene transfer and gene duplication (Letek et al., 2010). Partly due to the historical reliance on phenotypic traits and use of 16S rDNA sequence information, the Rhodococcus phylogeny still remains poorly resolved (Gürtler et al., 2004).
To date, Rhodococcus fascians and Rhodococcus equi are the only two members of the genus that are well documented as being pathogenic (von Bargen and Haas, 2009; Stes et al., 2011). R. fascians can infect a broad range of plants. After breaching the plant cuticle, the pathogen collapses the epidermal layer, and forms ingression sites beneath epiphytic colonies (Cornelis et al., 2001). R. fascians then grows inside the host tissue and provokes cell differentiation and de novo organogenesis, resulting in proliferations and abnormal growths called witches' brooms or leafy galls (Putnam and Miller, 2007). To gain insights into the mechanisms and evolution of virulence, we determined the genome sequences for 20 isolates of Rhodococcus (Creason et al., 2014). Like R. equi, R. fascians has few horizontally-acquired virulence genes, which are predicted to be augmented by co-option of core genes, that contribute to the ability of the bacterium to infect and cause disease (Crespi et al., 1992; Letek et al., 2008, 2010; Creason et al., 2014). Because of this mechanism of virulence evolution, phytopathogenicity is not expected to be a distinguishing trait suitable for classifying these Rhodococcus isolates.
In this study, we tested the hypothesis that leafy gall disease is caused by members of multiple species of Rhodococcus. Results from four independent methods were consistent and supported the hypothesis. Analysis of the 20 genome sequences showed the isolates formed two well supported clades, with one consisting of 16 isolates and having complex substructure indicative of multiple species. Analysis of the Rhodococcus genus associated four isolates collected from extreme environments or found in association with healthy plants to the two clades of Rhodococcus with plant-pathogenic members. Lastly, the need for revision of taxonomic grouping in Rhodococcus is suggested, as determined based on ANI distances calculated for all possible pairwise comparisons between members with available genome sequences.
Materials and Methods
Isolation of Phytopathogenic Rhodococcus
Symptomatic tissue of Leucanthemum × superbum “Becky,” received by the Oregon State University Plant Clinic, was washed, macerated in sterile saline, and incubated at room temperature for 30 min. Rhodococcus cells were selected for by culturing on semi-selective D2 media (Kado and Heskett, 1970). Isolate A22b was selected and verified as phytopathogenic based on its ability to cause leafy gall disease on pea seedlings and positive amplification for the fasA gene.
Nucleic Acid Preparations
A22b was grown in LB at 28°C with shaking (Bertani, 1951). Genomic DNA from A22b was extracted from cells grown directly from stocks. The Wizard Genomic DNA Purification Kit was used, according to the instructions of the manufacturer, to extract genomic DNA (Promega Corporation, Madison, WI, USA).
Next-Generation Sequencing, Assembly, and Annotation
Library construction and sequencing on an Illumina MiSeq were done in the Center for Genome Research and Biocomputing at Oregon State University. The A22b genome was assembled using Velvet (v1.2.08), with a hash length of 125 (Zerbino and Birney, 2008). The insert size was determined based on the estimated fragment size of the library preparation. Multiple assemblies were done, in which coverage cutoff, expected coverage, and hash length parameters were changed (Creason et al., 2014). The highest quality assembly was identified based on the number of contigs and having a sum total size between 5 and 6 Mb. Contigs were reordered using the genome sequence of R. fascians A44a as a reference and the Mauve Contig Mover (Rissman et al., 2009). The genome was annotated using Prokka (Seemann, 2014). As part of the Prokka pipeline, CDSs were annotated in part, based on BLASTP analysis and a database of genomes core to the Rhodococcus genus, including whole-genome assemblies from Rhodococcus jostii RHA1, Rhodococcus opacus B4, Rhodococcus erythropolis PR4, R. equi 103S, and the R. fascians linear plasmid, pFiD188 (Na et al., 2005; McLeod et al., 2006; Sekine et al., 2006; Letek et al., 2010; Francis et al., 2012). The whole genome shotgun project for A22b has been deposited at DDJB/EMBL/GenBank under the accession JOKB00000000 (BioProject PRJNA252927, BioSample SAMN02864791). The version described in this paper is version JOKB01000000. The A22b short reads and annotated genome sequences are available for download (SRS641819, http://dx.doi.org/10.7267/N9PN93H8). In order to be consistent, publicly available wgs sequences used in this study were similarly annotated. Their genome annotations are available upon request.
Phylogenetic Analyses
We used Hal (−a muscle and −y 100 settings) to construct the multi-gene tree of the 20 isolates and the Nocardia farcinica type strain as the outgroup (Ishikawa et al., 2004; Robbertse et al., 2011).
Sequences for the maximum-likelihood MLSA tree were gathered from the NCBI nt and wgs databases, using FtsY, InfB, RpoB, RsmA, SecY, TsaD, and YchF from Rhodococcus jostii RHA1 and Bifidobacterium longum subsp. infantis ATCC 15697 as queries in TBLASTN+ (v2.2.29) searches (with default settings; Adékambi et al., 2011). The query sequences were selected to provide coverage of the Actinobacteria phylum. Duplicate results from the two TBLASTN+ (v2.2.29) results, and strains in which all seven translated sequences were not detected, were filtered out. A total of 1316 strains passed filter.
The filtered sequences were aligned using the L-INS-i algorithm in MAFFT (v7.149b) with the –legacygappenalty flag. Gblocks (v0.91b) was used to trim the alignments prior to concatenation with half gapped positions allowed (−b5 = h setting; Castresana, 2000). Concatenated sequences with 100% identity, excluding those in the Rhodococcus genus, were collapsed into one entry, resulting in 961 sequences as input for tree generation. The most appropriate models of substitution for each gene were selected using the ProteinModelSelection.pl script provided with RAxML (v8; Katoh and Standley, 2013; Stamatakis, 2014). Trees were generated using RAxML (v8), based on the guidelines provided in the users manual (Stamatakis, 2014). Briefly, five starting parsimony trees were generated using the −y option; fixed initial arrangements were run on the five trees separately with the −i 10 setting. Automatic initial arrangements were also run on the five trees. The best log likelihood scores were used to choose the proper initial arrangement setting for further tree generation (−i 10 was the best for the dataset). A total of 500 rapid bootstraps (−x setting with −N 500) were performed on this dataset, and 10 distinct (−f d setting with −N 10) trees were generated. Bootstrap values were mapped on the best of the 10 distinct trees using the −f b setting. See Supplemental Data 1 for the full tree, accession values, and duplicated sequences that were removed.
Alignments were visualized using Belvu (Sonnhammer and Hollich, 2005). Images were generated using the iTOL (Letunic and Bork, 2011).
Bioinformatic Analyses
The progressiveMauve (v2.3.1) alignment was produced using default settings and as input, the chromosomal sequences for isolates D188, A21d2, 05-339-1, and A44a (Darling et al., 2010).
JSpecies with whole genome FASTA sequences as input, was used to calculate average nucleotide identities (BLAST; ANIb) and do pairwise comparisons of tetranucleotide frequencies (TETRA; Richter and Rosselló-Móra, 2009). Codon usage tables were constructed using EMBOSS cusp and sum difference statistics were calculated using EMBOSS codcmp (default settings; http://emboss.sourceforge.net/apps/cvs/emboss/apps/cusp.html). Reciprocal best BLASTP analysis was done according to methods previously reported (Creason et al., 2014).
The ANI values used to generate the distance dendrogram were calculated using published methods (Konstantinidis and Tiedje, 2005). The following were automated using ad-hoc scripts. Genome sequences were split into 1020-nucleotide long segments. The genome segments were used as queries in BLASTN+ (v2.2.27) searches against all other complete genomes in an all-by-all pairwise analysis. BLASTN+ (v2.2.27) was used, with the extra settings, “blastn –task blastn –dust no –xdrop_gap 150 –penalty −1 –reward 1 –gapopen 5 –gapextend 2,” for the searches (Camacho et al., 2009). Sequences with less than 70% coverage and 30% identity were filtered out, the number of results above the cutoffs were counted, and the average nucleotide identity of the resulting sequences were calculated. Results were comparable to those calculated using jSpecies and were better at handling the larger number of samples (Richter and Rosselló-Móra, 2009).
The distance dendrogram was generated using the all-by-all pairwise ANI divergence values as input, which is defined as 100%—ANI (Chan et al., 2012). The hcluster Python package was installed along with all dependencies (http://scipy-cluster.googlecode.com/). IPython, in interactive mode, was used to generate the dendogram (Perez and Granger, 2007). The matplotlib library was also required (ipython –matplotlib; Hunter, 2007; http://matplotlib.org). The pdist() function from hcluster was used to calculate the Euclidean distance between the ANI divergence values, and the complete linkage on the distance matrix was calculated using the complete() function. The dendrogram was generated using the dendrogram() function of hcluster. Input data and the resulting script from the interactive IPython session can be found in Supplemental Data 2.
Graphs were generated in R (R Core Team, 2013). The 3-D scatter plot was generated using plot3d{rgl} and quads3d{rgl}. Heatmaps were generated using heatmap.2{gplots}.
Results
Whole Genome-Based Phylogeny Supports Multiple Lineages of Plant Pathogenic Rhodococcus
In our first sequencing effort, we used hybrid approaches to generate high quality assemblies for isolates D188 and A44a (Creason et al., 2014). Unexpectedly, initial attempts to align the genome sequences were challenging, leading us to hypothesize that the two isolates represented different species of Rhodococcus. In order to test this hypothesis, we determined the genome sequences for 18 additional isolates of Rhodococcus identified from diseased plants or initially typed as R. fascians (Table 1; Miteva et al., 2004). The alignment of four genome sequences shows conservation of collinear blocks, with A44a being the most disparate in respect to the level of conservation and number and size of gaps between blocks (Figure S1).
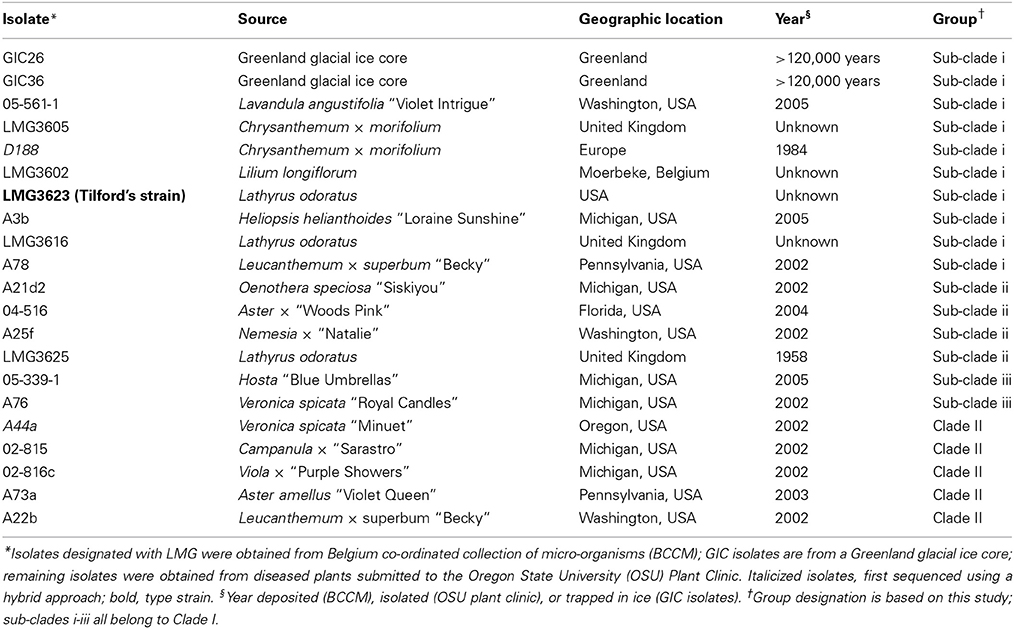
Table 1. Isolates of Rhodococcus selected for whole genome sequencing.
We constructed a multi-gene phylogenetic tree for the 20 isolates of Rhodococcus based on the whole genome sequences to infer the evolutionary relationships. Homologous sequences were identified from all 20 and from the type strain of Nocardia, which we used as an outgroup (Ishikawa et al., 2004). Clusters with paralogs, but not those with sequences potentially acquired via recombination, were filtered out, leaving 1727 clusters. The corresponding sequences from each isolate were concatenated, aligned, and used to derive a neighbor-joining (NJ) tree. The isolates formed two distinct clades (Figure 1). Clade I has substructure, with the largest and deepest branching sub-clade i consisting of the type strain LMG3623, D188, the two glacial ice core isolates, three other culture collection isolates, and three isolated from diseased plants. Sub-clade ii includes three isolates obtained directly from diseased plants and one obtained from a culture collection. Sub-clade iii is represented by just two isolates, 05-339-1, and A76, collected from diseased plants. As indicated by the longer branch lengths, the two isolates are more diverged. Clade II consists of four isolates from the US that includes A44a.
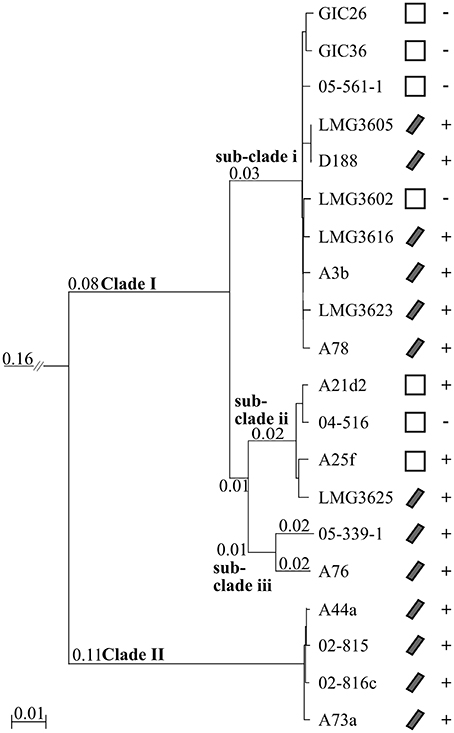
Figure 1. Neighbor-joining tree based on 1727 homologous genes. A rooted neighbor-joining tree was constructed using translated sequences from 1727 genes present in all 20 isolates and the Nocardia farcinica type strain (not shown). Clade and sub-clade designations are indicated at the corresponding node. Scale bar, number of amino acid substitutions per site; only branches with lengths greater than zero are indicated. Isolates with a linear plasmid are denoted with a diagonal bar; empty boxes, absence of linear plasmid. Phytopathogenic isolates are indicated with, “+”; non-pathogenic isolates are indicated with, “−.”
We projected the presence/absence of a linear virulence plasmid and trait of phytopathogenicity on to the phylogenetic tree (Figure 1; Creason et al., 2014). Phytopathogenicity is not exclusive to any clade or sub-clade. The five non-pathogenic isolates clustered in Clade I with four and one found in sub-clades i and ii, respectively. Sub-clade ii is the most variable in respect to virulence loci structure, as phytopathogenic A21d2 and A25f lack a linear virulence plasmid and A21d2 also lacks the entire fas operon (Creason et al., 2014). These data are consistent with our hypothesis that leafy gall disease is caused by multiple species of Rhodococcus and explain why initial attempts in aligning the whole genome sequences of D188 and A44a were difficult.
Alternative Whole-Genome Based Analyses Support the Multiple Lineages of Rhodococcus
To support the existence of distinct groups of plant pathogenic Rhodococcus, we used alternative methods to cluster the bacteria based on similarities in their genome sequence features. We determined the average nucleotide identity values (ANIb; calculated with the BLAST algorithm) and tetranucleotide usage patterns (TETRA) for all pairwise comparisons (Teeling et al., 2004; Konstantinidis and Tiedje, 2005; Richter and Rosselló-Móra, 2009). A plot of ANI vs. TETRA formed three distinct clouds (Figure 2). The plots of the comparisons of isolates within sub-clades i and ii as well as Clade II coalesced into cloud 1. These comparisons were wholly within the calibrated and strictest thresholds of 96% ANI and 0.997 TETRA values that are recommended for circumscribing prokaryotic taxa (Richter and Rosselló-Móra, 2009). Results were identical when we relaxed ANI thresholds to 94% (data not shown). The reciprocal comparisons between isolates 05-339-1 and A76 of sub-clade iii associated with Cloud 1 but fell below ANI thresholds, regardless of which strictness level was used. The failure to exceed threshold is consistent with the greater divergence between these isolates, as observed in the NJ tree. Cloud 2 represented all possible comparisons between isolates in different sub-clades of Clade I. This cloud spanned the TETRA threshold value but was well below the ANI threshold values (Richter and Rosselló-Móra, 2009). Cloud 3 contains the most dissimilar comparisons between isolates of Clades I and II. The values within this cloud are similar to those derived from a comparison between R. equi 103S and an environmental isolate of Rhodococcus (McLeod et al., 2006; Letek et al., 2010). Thus, not only was the structure of the Rhodococcus samples supported by analysis with ANI and TETRA, but the consistency in results reconfirmed the use of ANI for inferring genetic relatedness, as was previously demonstrated by others (Goris et al., 2007; Richter and Rosselló-Móra, 2009).
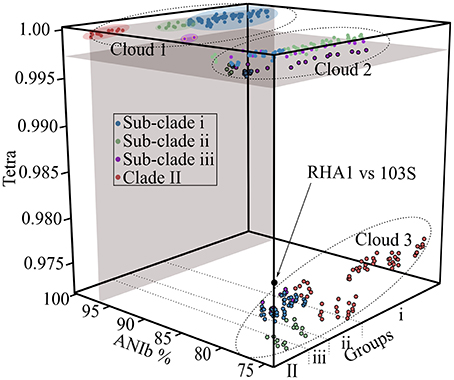
Figure 2. Cloud plot of ANIb vs. TETRA for all possible pairwise comparisons. Average nucleotide identity (ANIb; x-axis) and tetranucleotide (TETRA; y-axis) usage patterns were calculated and plotted as a factor of isolate grouping (z-axis). All pairwise comparisons, including reciprocal comparisons are presented, with colors assigned based on the clade membership of the isolate being compared to. Gray colored areas demark 96% ANI and 0.997 TETRA thresholds. Clouds are demarcated by dotted lines. Circles with black borders are below the TETRA threshold. Circles with yellow borders (comparisons between isolates of sub-clade iii; purple) exceed the TETRA threshold but not the ANIb threshold. The black circle represents a comparison between R. jostii RHA1 and R. equi 103S.
The genetic code is nearly universal but synonymous codons are not used with equal frequencies across species because of a complex balance of multiple forces (Plotkin and Kudla, 2011). Because this codon bias can be used to distinguish between dissimilar groups of organisms, we compared all possible pairwise combinations between the 20 isolates and displayed the similarity values as a heat map (Sharp and Li, 1987). Codon usage preferences clearly differentiated the isolates of Clade I from those of Clade II (Figure 3A). Codon usage preferences also revealed a pattern consistent with the substructure observed in the NJ tree and ANI vs. TETRA analysis (Figures 1, 2). The three sub-clades were easily discernable, though the relationships within sub-clade i differed slightly from those inferred from the NJ tree.
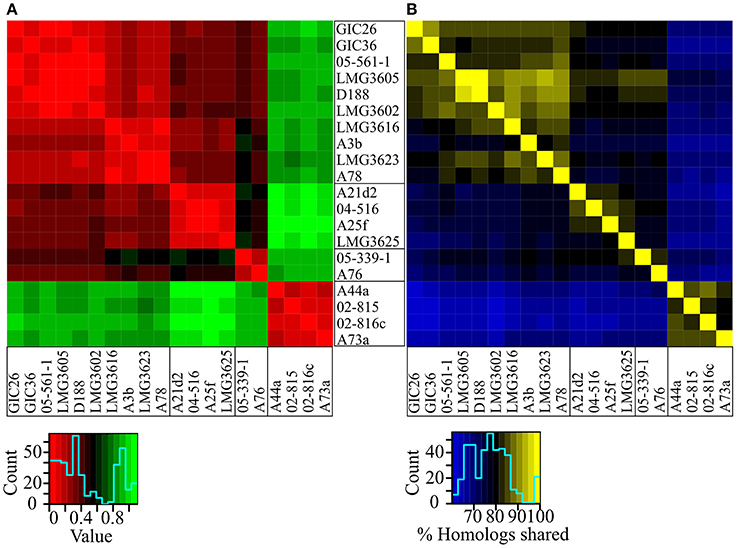
Figure 3. Heatmaps of codon usage preference and homology. (A) Codon usage preference similarities were calculated for all possible pairwise comparisons. Lower values indicate fewer differences. Isolates were ordered according to their phylogenetic relationships. (B) Reciprocal BLASTP analysis was used to determine percent homology for all possible pairwise comparisons. Larger values indicate greater similarities. Isolates were ordered according to their phylogenetic relationships.
The core genome hypothesis suggests that coherent clusters of bacteria have a core set of functions, and each member augments the core with a variable accessory genome that contributes functions for niche adaptation (Lan and Reeves, 1996). The 20 sequenced isolates share a core of 3063 genes. However, sample size indubitably affects estimation of core genome identities and sizes. Given the small sample size of our collection and the imbalance in numbers of isolates between clades, we elected to cluster based on the percent of shared homologs rather than on core genomes (Figure 3B). The members of Clades I and II separated into distinct clusters, which could be taken as evidence for limited gene flow between clades. Relative to results from other approaches, the relationship of the isolates within Clade I were noticeably different, as sub-clade ii and sub-clade iii were not clearly demarcated. Regardless of these subtle differences, the results were entirely consistent in separating the 20 isolates into two groups of phytopathogenic Rhodococcus, with one also having evidence for substructure.
A Molecular Phylogeny Based on Whole Genome Sequences Provides a Framework for Resolving the Rhodococcus Genus
To develop a framework for resolving the Rhodococcus phylogeny, we constructed a multi-locus sequence analysis (MLSA) maximum likelihood (ML) tree based on 961 concatenated sequences representing 1316 members of the Actinobacteria phylum (Figure 4). We used seven marker genes that were previously identified as conserved and informative for the subclass Actinobacteridae (Adékambi et al., 2011). Rhodococcus and Nocardia formed sister groups and the members of the Rhodococcus genus formed a well-supported phylogenetically coherent cluster (bootstrap percentage of 98%). We were able to identify two relatively defined groups and one small, less defined, group within the Rhodococcus genus (Figure 4; marked as a, b, and c). The two larger groups were consistent with the two clades previously described in a phylogeny based on 16S rDNA sequences (Jones and Goodfellow, 2012). In contrast, in the MLSA ML tree, the smaller R. equi clade is within the larger Rhodococcus clade (bootstrap percentage of 93%), unlike previous studies, which associated the R. equi clade with Nocardia.
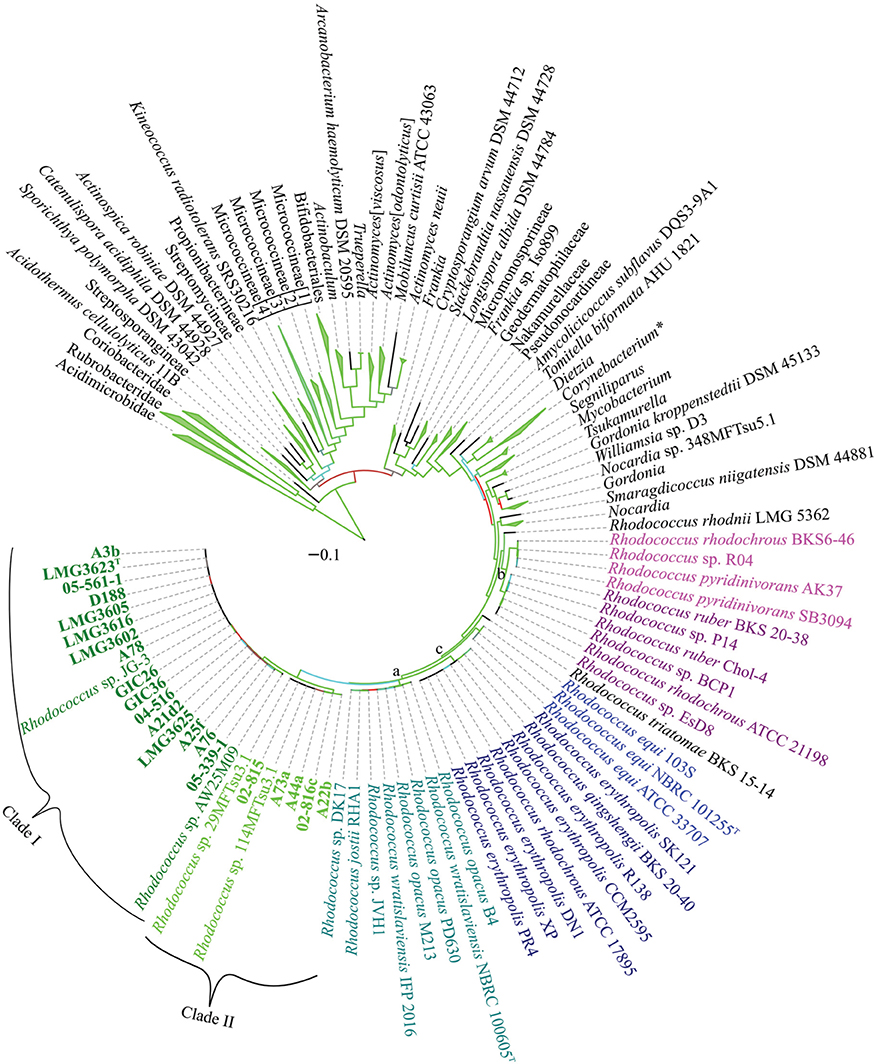
Figure 4. Multi-locus sequence analysis maximum likelihood tree of the Actinobacterium phylum. Translated sequences for ftsY, infB, rpoB, rsmA, secY, tsaD, and ychF from 1316 members were identified using TBLASTN, aligned, and used to generate a multi-locus maximum likelihood tree. A total of 961 sequences were used as input for tree generation. The 21 Rhodococcus isolates sequenced by our group are shown in bold and the two clades that include phytopathogenic Rhodococcus spp. are indicated. The R. erythropolis, R. rhodochrous, and R. equi clades, previously identified based on a 16S rDNA phylogeny of Rhodococcus are labeled with a, b, and c, respectively. Type strains are indicated with a superscript “T.” Branches outside of the Rhodococcus genus were collapsed at the genus, family, suborder, order, and subclass level, as appropriate, with the corners of the triangle indicating the shortest and longest total branch lengths for the members of the collapsed clade. A total of 500 rapid bootstraps were performed on this dataset, and branches are colored on a gradient to indicate bootstrap percentage (Green-Cyan-Red, with cutoffs of 100-75-50 and below, respectively). Scale bar, mean number of amino acid substitutions per site.
The 20 isolates of interest in this study formed a distinct subgroup (bootstrap percentage of 100%) within the MLSA ML tree and also included five other isolates (Figure 4). Phytopathogenic isolate A22b, which was identified from a diseased plant and sequenced independently from the 20 isolates, clustered in Clade II. Two of the isolates that clustered with the 20 of interest in this study, were identified independent of plants and in extreme environments. Rhodococcus sp. JG-3 was isolated from permafrost (GenBank BioProject PRJNA195882) and Rhodococcus sp. AW25M09 was isolated from the stomach of an Atlantic Hagfish (Hjerde et al., 2013). Two others, Rhodococcus spp. 29MFTsu3.1 and 114MTsu3.1 were found associated with the rhizosphere or endosphere of Arabidopsis thaliana (GenBank BioProject PRJNA201196).
The topology of the tree outside of the Rhodococcus genus was similar to previously reported trees and revealed inconsistencies in currently defined taxonomic groups, as previously observed (Adékambi et al., 2011; Gao and Gupta, 2012; Jones and Goodfellow, 2012; Verma et al., 2013). The Micrococcineae formed a polyphyletic group, with three to four distinct clades, depending on the tree and tree generation method (bootstrap percentages of 88–100%). We also had difficulties in accurately placing the Frankineae into a discrete group, as they were found throughout the phylogeny. The Actinomyces neuii species formed a separate, but somewhat poorly supported (bootstrap percentage of 63%) clade with Mobiluncus curtisii, as was the case in a phylogeny based on 16S rDNA sequences (Jones and Goodfellow, 2012). We observed a cryptic relationship with Actinopolysporineae included within the Pseudonocardineae clade. However, some of the branches within the Pseudonocardineae were poorly supported (bootstrap percentages <50%), indicating poor resolution with the clade as a whole. One key addition of the MLSA ML tree was that the branching of Actinomyces was well supported and is consistent with there being two large clades (bootstrap percentages of 90 and 100%).
ANI Provides a Framework for Resolving the Rhodococcus Genus
Because of the relatively few informative sites, bootstrap values at the tips of the MLSA ML tree were often low and insufficient for resolving the Rhodococcus genus; compare for example, the two clades of Rhodococcus with plant pathogenic members (Figures 1, 4). Therefore, to develop a molecular framework for the Rhodococcus genus, we used ANIb as a tool for inferring similarity. The values were calculated for 3422 pairwise comparisons of the 59 Rhodococcus isolates, compiled into a distance matrix, and used to generate a divergence dendogram (Figure 5; Chan et al., 2012). Seven distinct clusters formed with inter-group comparisons that exceeded ANI values of 70–75%, a range that is typically found between members of the same genus.
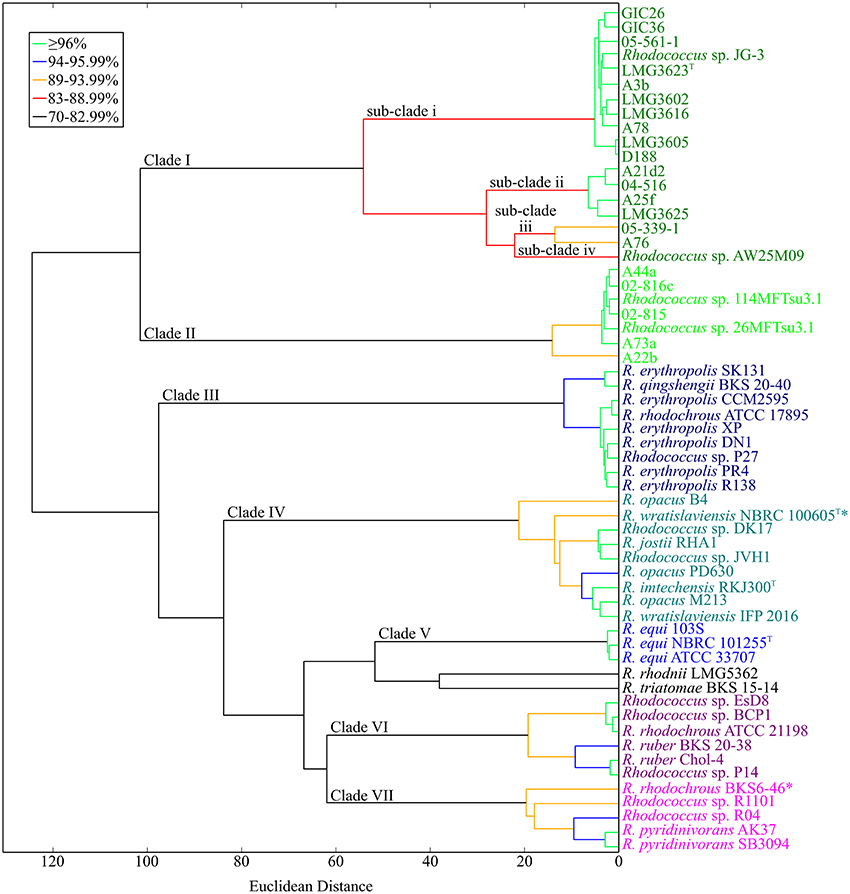
Figure 5. Average nucleotide identity dendogram for 59 isolates of Rhodococcus. Complete genome sequences for 59 isolates of Rhodococcus were used to generate an ANI matrix. The matrix was used to calculate an ANI divergence dendrogram. Groups are color coded according to groups represented in the MLSA ML tree. Branches are colored using cutoffs for pairwise comparisons of all taxa after the nodal point. The 21 Rhodococcus isolates sequenced by our group are shown in bold; type strains are designated with a superscript “T.” Clades and sub-clades of the phytopathogenic Rhodococcus isolates are labeled at the corresponding node. *Indicates conflict between placement within the dendogram and calculated ANI values (see corresponding text for details).
The two clades of phytopathogenic Rhodococcus spp. represent seven different species, when using ANI and a 94% threshold. As previously observed, Clade I has complex substructure and represents four different species (Figures 1–3, 5). Like conclusions described above, subclade i represents the originally named R. fascians species, as it includes the type strain LMG3623, along with the most sequenced isolates. Isolates in subclade iii were not as closely grouped, which was also consistent with results from the NJ tree and analysis of codon usage. In fact, based on the single criterion of ANI, 05-339-1, and A76 should be considered as separate species. Subclade iv is represented by a single isolate, Rhodococcus sp. AW25M09. In Clade II, isolate A22b is just below the 94% ANI cutoff and may represent a separate subspecies from those that formed this second major clade in previously described analyses (Figures 1–3, 5).
The other isolates of Rhodococcus, which are not known as plant-associated, formed five additional clades. Within these clades, and including single isolates (singletons), there were 13 smaller groups defined by ANI values of 94% ANI or greater. These relationships can be used to infer species groups.
Clade III consists mostly of isolates named as Rhodococcus erythropolis. The group also included Rhodococcus qinshengii BKS 20-40, Rhodococcus rhodochrous ATCC 17895, and Rhodococcus sp. P27, which based on their association to the clade, could be considered members of the R. erythropolis species.
Clade IV has two subgroups and two singletons. The first subgroup is represented by Rhodococcus jostii RHA1 and also included Rhodococcus spp. DK17 and JVH1. The second subgroup varied in terms of named species and consisted of two isolates of Rhodococcus opacus, the type strain of Rhodococcus imtechensis and Rhodococcus wratislaviensis IFP 2016. Because the latter strain did not cluster with the type strain of R. wratislaviensis, NBRC 100605, we suggest that IFP 2016 is not a member of the R. wratislaviensis species and it belongs to a different species of Rhodococcus. The other singleton isolate was R. opacus B4. A whole genome sequence of the R. opacus type strain is unavailable. Thus, we cannot suggest whether the subgroup or the singleton should be designated as the R. opacus species. Furthermore, 16S rDNA sequences of the three named R. opacus isolates are too similar for resolving this issue. Interestingly, R. opacus PD630 is most similar to R. wratislaviensis NBRC 100605 in respect to ANI values, but the two did not associate with one another in the divergence dendogram.
Clade V contains a tightly clustered group with members of the Rhodococcus equi species. Its placement within Rhodococcus was consistent with the MLSA ML tree.
Clade VI consists of two smaller subgroups. The first subgroup has R. rhodochrous ATCC 21198 and two undesignated isolates (EsD8 and BCP1). The low ANI of 72% placed R. rhodochrous ATCC 21198 in a clade separate from isolate R. rhodochrous BKS6-46. To further investigate this discrepancy, we used the 16S rDNA sequence from R. rhodochrous ATCC 21198 as a query in a BLASTN+ search. The sequence identified corresponding sequences from Rhodococcus aetherivorans (100% identity, 100% subject coverage), including the type strain DSM 44752. When we used the 16S rDNA sequence from the type strain of R. rhodochrous as a query, it showed greater similarity to the corresponding sequence of R. rhodochrous BKS6-46 rather than ATCC 21198. In all, these data suggest the identity of ATCC 211983 should be revisited. The second subgroup of Clade VI consists of two Rhodococcus ruber isolates (BKS 20-38 and Chol-4) and another undesignated isolate (P14). Thus, we suggest that P14 is a member of the R. ruber species.
Clade VII has one cluster of defined species and two singletons. Two isolates of Rhodococcus pyridinivorans (AK37 and SB3094) and Rhodococcus sp. R04 clustered, which we suggest represents the R. pyridinivorans species. R. rhodochrous BKS6-46 is a singleton in this clade but its precise placement within this clade of the dendrogram is somewhat misleading. The ANI values for the pairwise comparisons of R. rhodochrous BKS6-46 to the two named R. pyridinivorans, but not Rhodococcus sp. R04, exceeded the 94% threshold used to define a species relationship. The values derived from comparison with Rhodococcus sp. R04 likely caused R. rhodochrous BKS6-46 to form its own branch. The last singleton is Rhodococcus sp. R1101, which had ANI values around 90% relative to the other isolates of Clade VII.
There were two outliers in the dendogram. Rhodococcus rhodnii and Rhodococcus triatomae were identified as singletons and placed in a clade closest to Clade V (R. equi). These isolates had low ANI values (between 71 and 75%) in comparison to all of the sequenced Rhodococcus isolates.
Discussion
Leafy gall disease is a substantial economic problem for the horticultural industry. The pathogen has an extensive host range that includes most plants important to the industry (Putnam and Miller, 2007). Current management strategies rely on visual inspection and the only method of control is the destruction of infected plant material. While visual inspection of plants for disease is superficially trivial, the absence of fundamental information on its epidemiology and the lack of robust, on-site diagnostics contribute to make disease management challenging. Whole genome sequencing is a cost-effective and facile approach for studying bacterial species and has important practical implications for diagnosis of disease. In this study, we used whole genome sequences to analyze the genetic diversity of plant pathogenic Rhodococcus as a first step toward the development of better management strategies for this pathogen.
Twenty isolates, many of which were identified from diseased plants, were previously selected for whole genome sequencing (Creason et al., 2014). Based on a tree derived from 1727 homologous genes, we demonstrated that the 20 isolates separated into distinct clades and sub-clades (Figure 1). Similarities in genome features, including ANI, TETRA, codon usage preference, and degree of genome homology, were all consistent in clustering the isolates into distinct and coherent groups (Figures 2, 3). The similarity in results between the tree and ANI was encouraging and gave us confidence in using the latter for inferring evolutionary relationships of isolates within a larger dataset (Figure 5).
The phytopathogenic isolates formed a subgroup distinct from other members of the Rhodococcus genus, which could be taken as evidence for cohesion (Figures 4, 5). However, within this subgroup, phytopathogenicity is not a discriminative trait (Figures 1, 4, 5). Although, we speculate that members of this subgroup are potentiated toward phytopathogenicity. Virulence evolution in Rhodococcus has been modeled according to a mechanism of gene co-option whereby limited, but key horizontally acquired virulence genes, trigger the co-option of core genes for virulence (Letek et al., 2010; Creason et al., 2014). As few as four functions, most often conferred by a cluster of genes vectored by a conjugative virulence plasmid, are hypothesized to be sufficient for phytopathogenicity for members of these genetically diverse clades of Rhodococcus.
The non-pathogenic isolates recovered from a Greenland glacier ice core, GIC26, and GIC36, as well as Rhodococcus sp. JG-3 from permafrost and AW25M09 from the stomach of the Atlantic Hagfish, clustered with phytopathogenic isolates in Clade I (Miteva et al., 2004; Hjerde et al., 2013). Rhodococcus spp. 29MFTsu3.1 and 114MTsu3.1, both of which associate with plants, clustered in Clade II. Inspection of the genome sequences of isolates JG-3, AW25M09, 29MFTsu3.1, and 114MTsu3.1, failed to reveal any of the virulence genes known to be necessary for Rhodococcus to cause leafy gall disease. We did detect a linear plasmid-like sequence in the draft genome of Rhodococcus sp. AW25M09, but it lacks genes known to be necessary for virulence toward plants (Hjerde et al., 2013). It would be interesting to test whether these isolates, upon acquisition of genes that confer the four key virulence functions, gain the ability to infect and cause disease to plants.
Our genome sequencing effort of plant-associated isolates contributed to increasing the number of sequenced Rhodococcus isolates by 50%. Not since a decade ago, have genomic features been used to address the Rhodococcus genus (Gürtler et al., 2004). We therefore used these genome sequences, an additional sequence we generated for phytopathogenic isolate A22b, along with most currently available rhodococcal genome sequences, to construct a molecular phylogeny to help resolve the genus and shed light on the phylum Actinobacteria as a whole. MLSA provided a framework for defining genus level relationships that can be further explored (Figure 4). In this study, the R. equi species was placed within the Rhodococcus genus, consistent with previously reported results derived from whole-genome based approaches, and in contrast to phylogenetic analysis based on 16S rDNA sequences (Letek et al., 2010; Jones and Goodfellow, 2012). Use of MLSA to infer phylogeny revealed inconsistencies in the placement of Frankineae, in contrast to a 16S rDNA-based phylogeny, which formed a Frankineae cluster near the root of the phylum (Jones and Goodfellow, 2012). However, MLSA was not sufficient for resolving some of the branches within the Pseudonocardineae (bootstrap percentages <50%). More genome sequences, or informative marker sequences for members of Actinopolysporineae might help discern the finer details within the suborder. Other minor discrepancies in single isolate naming were also noted (Supplemental Data 1).
With ANI, we were able to place the 59 Rhodococcus isolates with sequenced genomes into seven large groups. Results were consistent with the MLSA ML tree but ANI provided greater resolution of their relationships (Figure 5). We were also able to infer at least 20 different species from these 59 isolates. Some conflicts between clustering and species naming were noted and suggest a need to revisit their taxonomical groupings. However, we recognize that the use of ANI to infer taxonomical groupings merely provides a framework and is not sufficient, by itself, for defining bacterial species. At present, in order to validly designate a species, it needs to be further characterized for at least one discriminative trait. Moreover, the designation of a species would also require inclusion of their corresponding type strain.
The methods that were used were congruent in supporting the existence of multiple species of Rhodococcus. However, each of the methods has limitations. The tree developed based on whole genome sequences was computationally and time intensive and precluded us from using methods with stronger statistical frameworks for phylogenetic reconstruction and also became too time-intensive for larger datasets. MLSA is limited by the need for a sufficient set of generalizable and informative marker genes. Despite the reduced number of sequences, the MLSA approach was still computationally and time intensive with larger datasets. ANIb can be limiting in projecting relationships in an evolutionary context (Figure 2). Though the dendrogram constructed based on Euclidean distances was a convenient way for visualizing dissimilarities, there were nevertheless some discrepancies between placement of isolates and their measured ANI values (Figure 5). For example, R. opacus PD630 and R. wratislaviensis NBRC 100605 have an ANI value that warranted consideration as a species, but their dissimilarities to other isolates prevented the two from clustering in the dendogram (Figure 5). Lastly, similarities based on codon usage preferences and genome inventories were inadequate for resolving relationships between isolates with highly similar genome signatures (Figure 3). Nevertheless, when multiple methods were coupled, we were able to make strong inferences regarding taxonomical relationships.
In summary, we used whole genome sequences to resolve the phytopathogenic members of Rhodococcus into multiple sister species and developed a dataset that contributes to reconstructing the phylogeny of the Rhodococcus genus.
Author Contributions
These authors contributed equally. Allison L. Creason, Edward W. Davis II, Melodie L. Putnam, Olivier M. Vandeputte and Jeff H. Chang contributed to the conception and design of the work, analysis of data, and wrote and approved the manuscript. Allison L. Creason, Edward W. Davis II, Melodie L. Putnam, and Olivier M. Vandeputte also contributed to the acquisition of isolates and/or genome sequences. The authors are accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Alex Buchanan, Ethan Chang, and Tyler Horton, of the Chang lab for their assistance. We would also like to thank Joyce Loper for critical reading of this manuscript. This work is supported by a grant from the Agricultural Research Foundation awarded to Jeff H. Chang. Edward W. Davis II is supported by a Provost's Distinguished Graduate Fellowship awarded by Oregon State University. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1314109 to Edward W. Davis II. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation. This work is also supported in part by the Society of American Florists and a cooperative agreement with USDA-ARS awarded to Melodie L. Putnam. Olivier M. Vandeputte was supported by grants from FRS-FNRS and the Fonds David et Alice Van Buuren (Belgium). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00406/abstract
Figure S1. Genome alignments of four members of phytopathogenic Rhodococcus. Sequences of the chromosomes of isolates D188, A21d2, 05-339-1, and A44a were aligned using progressiveMauve. Each colored square represents a block of sequences that is collinear to a corresponding block of sequences in another genome sequence; linear collinear blocks (LCBs). The extent of homology within LCBs is represented by the height of the colored plot in each block.
Supplemental Data 1. Compressed folder with MLSA tree in Newick tree format file and its associated data. The phylogenetic tree of the entire Actinobacteria phylum is available as a Newick file. Also included are three comma separate files. (1) SD1_identical_seqs.csv: lists the names of the duplicate sequences that were removed prior to generating the final tree. The names of the species used in generating the tree are listed in the first column. The remaining columns contain the names of the species that were filtered out based on having an identical sequence. (2) SD1_names_identifiers.csv: associates genome sequence identifiers (either GI number for complete genomes, or 4 letter code for wgs sequences) to corresponding isolates in the tree. (3) SD1_taxonomy.csv: lists the taxonomy values for each of the taxa.
Supplemental Data 2. Compressed folder with Python script and input data for ANI dendrogram generation. A Python script file is included that can generate an ANI dendrogram from the two input files, one that includes the ANI divergence matrix (SD2_ani_data.txt), and one that includes the names of the genomes included in the analysis (SD2_ani_names.txt).
References
Adékambi, T., Butler, R. W., Hanrahan, F., Delcher, A. L., Drancourt, M., and Shinnick, T. M. (2011). Core gene set as the basis of multilocus sequence analysis of the subclass Actinobacteridae. PLoS ONE 6:e14792. doi: 10.1371/journal.pone.0014792
Bertani, G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300.
Bohlin, J., Skjerve, E., and Ussery, D. W. (2008). Reliability and applications of statistical methods based on oligonucleotide frequencies in bacterial and archaeal genomes. BMC Genomics 9:104. doi: 10.1186/1471-2164-9-104
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chan, J. Z.-M., Halachev, M. R., Loman, N. J., Constantinidou, C., and Pallen, M. J. (2012). Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 12:302. doi: 10.1186/1471-2180-12-302
Cornelis, K., Ritsema, T., Nijsse, J., Holsters, M., Goethals, K., and Jaziri, M. (2001). The plant pathogen Rhodococcus fascians colonizes the exterior and interior of the aerial parts of plants. Mol. Plant Microbe. Interact. 14, 599–608. doi: 10.1094/MPMI.2001.14.5.599
Creason, A. L., Vandeputte, O. M., Savory, E. A., Davis, E. W., Putnam, M. L., Hu, E., et al. (2014). Analysis of genome sequences from plant pathogenic Rhodococcus reveals genetic novelties in virulence Loci. PLoS ONE 9:e101996. doi: 10.1371/journal.pone.0101996
Crespi, M., Messens, E., Caplan, A. B., van Montagu, M., and Desomer, J. (1992). Fasciation induction by the phytopathogen Rhodococcus fascians depends upon a linear plasmid encoding a cytokinin synthase gene. EMBO J. 11, 795–804.
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Doolittle, W. F., and Zhaxybayeva, O. (2009). On the origin of prokaryotic species. Genome. Res. 19, 744–756. doi: 10.1101/gr.086645.108
Dudnik, A., Bigler, L., and Dudler, R. (2014). The Endophytic Strain Rhizobium sp. AP16 produces the proteasome inhibitor Syringolin A. Appl. Environ. Microbiol. 80, 3741–3748. doi: 10.1128/AEM.00395-14
Durán, D., Rey, L., Mayo, J., Zuñiga-Davila, D., Imperial, J., Ruiz-Argüeso, T., et al. (2014). Bradyrhizobium paxllaeri sp. nov. and Bradyrhizobium icense sp. nov., nitrogen-fixing rhizobial symbionts of Lima bean (Phaseolus lunatus L.) in Peru. Int. J. Syst. Evol. Microbiol. 64, 2072–2078. doi: 10.1099/ijs.0.060426-0
Francis, I., De Keyser, A., De Backer, P., Simón-Mateo, C., Kalkus, J., Pertry, I., et al. (2012). pFiD188, the Linear Virulence Plasmid of Rhodococcus fascians D188. Mol. Plant Microbe. Interact. 25, 637–647. doi: 10.1094/MPMI-08-11-0215
Gao, B., and Gupta, R. S. (2012). Phylogenetic framework and molecular signatures for the main clades of the phylum Actinobacteria. Microbiol. Mol. Biol. Rev. 76, 66–112. doi: 10.1128/MMBR.05011-11
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Gürtler, V., Mayall, B. C., and Seviour, R. (2004). Can whole genome analysis refine the taxonomy of the genus Rhodococcus? FEMS Microbiol. Rev. 28, 377–403. doi: 10.1016/j.femsre.2004.01.001
Hjerde, E., Pierechod, M. M., Williamson, A. K., Bjerga, G. E. K., Willassen, N. P., Smalås, A. O., et al. (2013). Draft genome sequence of the Actinomycete Rhodococcus sp. Strain AW25M09, Isolated from the Hadsel Fjord, Northern Norway. Genome Announc. 1:e0005513. doi: 10.1128/genomeA.00055-13
Hunter, J. D. (2007). Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95. doi: 10.1109/MCSE.2007.55
Ishikawa, J., Yamashita, A., Mikami, Y., Hoshino, Y., Kurita, H., Hotta, K., et al. (2004). The complete genomic sequence of Nocardia farcinica IFM 10152. Proc. Natl. Acad. Sci. U.S.A. 101, 14925–14930. doi: 10.1073/pnas.0406410101
Jones, A. L., and Goodfellow, M. L. (2012). “Genus IV. Rhodococcus (Zopf 1891) emend Goodfellow, Alderson and Chun 1998a,” in Bergey's Manual of Systematic Bacteriology 2nd Edn. Vol. 5 The Actinobacteria, Part A, eds M. Goodfellow, P. Kampfer, H. J. Busee, M. E. Trujillo, K. Suzuki, W. Ludwig, and W. B. Whitman (New York, NY: Springer), 437–477.
Kado, C. I., and Heskett, M. G. (1970). Selective media for isolation of Agrobacterium, Corynebacterium, Erwinia, Pseudomonas, and Xanthomonas. Phytopathology 60, 969–976. doi: 10.1094/Phyto-60-969
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, M., Oh, H.-S., Park, S.-C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Konishi, M., Nishi, S., Fukuoka, T., Kitamoto, D., Watsuji, T.-O., Nagano, Y., et al. (2014). Deep-sea Rhodococcus sp. BS-15, Lacking the Phytopathogenic fas Genes, Produces a Novel Glucotriose Lipid Biosurfactant. Mar. Biotechnol. 16, 484–493. doi: 10.1007/s10126-014-9568-x
Konstantinidis, K. T., Ramette, A., and Tiedje, J. M. (2006). The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1929–1940. doi: 10.1098/rstb.2006.1920
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Lan, R., and Reeves, P. R. (1996). Gene transfer is a major factor in bacterial evolution. Mol. Biol. Evol. 13, 47–55. doi: 10.1093/oxfordjournals.molbev.a025569
Larkin, M. J., Kulakov, L. A., and Allen, C. C. R. (2005). Biodegradation and Rhodococcus–masters of catabolic versatility. Curr. Opin. Biotechnol. 16, 282–290. doi: 10.1016/j.copbio.2005.04.007
Lassalle, F., Campillo, T., Vial, L., Baude, J., Costechareyre, D., Chapulliot, D., et al. (2011). Genomic species are ecological species as revealed by comparative genomics in Agrobacterium tumefaciens. Genome Biol. Evol. 3, 762–781. doi: 10.1093/gbe/evr070
Letek, M., González, P., Macarthur, I., Rodríguez, H., Freeman, T. C., Valero-Rello, A., et al. (2010). The genome of a pathogenic Rhodococcus: cooptive virulence underpinned by key gene acquisitions. PLoS Genet. 6:e1001145. doi: 10.1371/journal.pgen.1001145
Letek, M., Ocampo-Sosa, A. A., Sanders, M., Fogarty, U., Buckley, T., Leadon, D. P., et al. (2008). Evolution of the Rhodococcus equi vap pathogenicity island seen through comparison of host-associated vapA and vapB virulence plasmids. J. Bacteriol. 190, 5797–5805. doi: 10.1128/JB.00468-08
Letunic, I., and Bork, P. (2011). Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478. doi: 10.1093/nar/gkr201
Loper, J. E., Hassan, K. A., Mavrodi, D. V., Davis, E. W., Lim, C. K., Shaffer, B. T., et al. (2012). Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 8:e1002784. doi: 10.1371/journal.pgen.1002784
Marakeby, H., Badr, E., Torkey, H., Song, Y., Leman, S., Monteil, C. L., et al. (2014). A system to automatically classify and name any individual genome-sequenced organism independently of current biological classification and nomenclature. PLoS ONE 9:e89142. doi: 10.1371/journal.pone.0089142
McLeod, M. P., Warren, R. L., Hsiao, W. W. L., Araki, N., Myhre, M., Fernandes, C., et al. (2006). The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. U.S.A. 103, 15582–15587. doi: 10.1073/pnas.0607048103
Miteva, V. I., Sheridan, P. P., and Brenchley, J. E. (2004). Phylogenetic and physiological diversity of microorganisms isolated from a deep Greenland glacier ice core. Appl. Environ. Microbiol. 70, 202–213. doi: 10.1128/AEM.70.1.202-213.2004
Na, K.-S., Kuroda, A., Takiguchi, N., Ikeda, T., Ohtake, H., and Kato, J. (2005). Isolation and characterization of benzene-tolerant Rhodococcusopacus strains. J. Biosci. Bioeng. 99, 378–382. doi: 10.1263/jbb.99.378
Perez, F., and Granger, B. E. (2007). IPython: a system for interactive scientific computing. Comput. Sci. Eng. 9, 21–29. doi: 10.1109/MCSE.2007.53
Plotkin, J. B., and Kudla, G. (2011). Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42. doi: 10.1038/nrg2899
Putnam, M. L., and Miller, M. L. (2007). Rhodococcusfascians in herbaceous perennials. Plant Dis. 91, 1064–1076. doi: 10.1094/PDIS-91-9-1064
R Core Team. (2013). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available online at: http://www.R-project.org
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Nat. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rissman, A. I., Mau, B., Biehl, B. S., Darling, A. E., Glasner, J. D., and Perna, N. T. (2009). Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25, 2071–2073. doi: 10.1093/bioinformatics/btp356
Robbertse, B., Yoder, R. J., Boyd, A., Reeves, J., and Spatafora, J. W. (2011). Hal: an automated pipeline for phylogenetic analyses of genomic data. PLoS Curr. 3:RRN1213. doi: 10.1371/currents.RRN1213
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sekine, M., Tanikawa, S., Omata, S., Saito, M., Fujisawa, T., Tsukatani, N., et al. (2006). Sequence analysis of three plasmids harboured in Rhodococcus erythropolis strain PR4. Environ. Microbiol. 8, 334–346. doi: 10.1111/j.1462-2920.2005.00899.x
Sharp, P. M., and Li, W. H. (1987). The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.3.1281
Sheng, H. M., Gao, H. S., Xue, L. G., Ding, S., Song, C. L., Feng, H. Y., et al. (2011). Analysis of the composition and characteristics of culturable endophytic bacteria within subnival plants of the Tianshan Mountains, northwestern China. Curr. Microbiol. 62, 923–932. doi: 10.1007/s00284-010-9800-5
Shevtsov, A., Tarlykov, P., Zholdybayeva, E., Momynkulov, D., Sarsenova, A., Moldagulova, N., et al. (2013). Draft genome sequence of Rhodococcus erythropolis DN1, a crude oil biodegrader. Genome Announc. 1:e00846-13. doi: 10.1128/genomeA.00846-13
Sonnhammer, E. L. L., and Hollich, V. (2005). Scoredist: a simple and robust protein sequence distance estimator. BMC Bioinformatics 6:108. doi: 10.1186/1471-2105-6-108
Staley, J. T. (2009). The phylogenomic species concept for Bacteria and Archaea. Microbe 4, 361–365.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stes, E., Vandeputte, O. M., El Jaziri, M., Holsters, M., and Vereecke, D. (2011). A successful bacterial coup d'état: how Rhodococcus fascians redirects plant development. Annu. Rev. Phytopathol. 49, 69–86. doi: 10.1146/annurev-phyto-072910-095217
Teeling, H., Waldmann, J., Lombardot, T., Bauer, M., and Glöckner, F. O. (2004). TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics 5:163. doi: 10.1186/1471-2105-5-163
van der Wolf, J. M., Nijhuis, E. H., Kowalewska, M. J., Saddler, G. S., Parkinson, N., Elphinstone, J. G., et al. (2014). Dickeya solani sp. nov., a pectinolytic plant-pathogenic bacterium isolated from potato (Solanum tuberosum). Int. J. Syst. Evol. Microbiol. 64, 768–774. doi: 10.1099/ijs.0.052944-0
Verma, M., Lal, D., Kaur, J., Saxena, A., Kaur, J., Anand, S., et al. (2013). Phylogenetic analyses of phylum actinobacteria based on whole genome sequences. Res. Microbiol. 164, 718–728. doi: 10.1016/j.resmic.2013.04.002
von Bargen, K., and Haas, A. (2009). Molecular and infection biology of the horse pathogen Rhodococcus equi. FEMS Microbiol. Rev. 33, 870–891. doi: 10.1111/j.1574-6976.2009.00181.x
Keywords: Gram-positive, plant pathogen, prokaryotic taxonomy, leafy gall, average nucleotide identity
Citation: Creason AL, Davis EW II, Putnam ML, Vandeputte OM and Chang JH (2014) Use of whole genome sequences to develop a molecular phylogenetic framework for Rhodococcus fascians and the Rhodococcus genus. Front. Plant Sci. 5:406. doi: 10.3389/fpls.2014.00406
Received: 20 June 2014; Accepted: 30 July 2014;
Published online: 19 August 2014.
Edited by:
Gail Preston, University of Oxford, UKReviewed by:
David John Studholme, University of Exeter, UKLeighton Pritchard, James Hutton Institute, UK
Copyright © 2014 Creason, Davis, Putnam, Vandeputte and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeff H. Chang, Department of Botany and Plant Pathology, Oregon State University, 2082 Cordley Hall, Corvallis, OR 97331, USA e-mail:Y2hhbmdqQHNjaWVuY2Uub3JlZ29uc3RhdGUuZWR1
†These authors have contributed equally to this work.