 Baohua Li
Baohua Li Daniel J. Kliebenstein
Daniel J. Kliebenstein- Department of Plant Sciences, University of California, Davis, CA, USA
Regulation of primary metabolism is a central mechanism by which plants coordinate their various responses to biotic and abiotic challenge. To identify genes responsible for natural variation in primary metabolism, we focused on cloning a locus from Arabidopsis thaliana that influences the level of TCA cycle metabolites in planta. We found that the Met.V.67 locus was controlled by natural variation in METABOLIC NETWORK MODULATOR 1 (MNM1), which encoded an AT-hook motif-containing protein that was unique to the Brassicales lineage. MNM1 had wide ranging effects on plant metabolism and displayed a tissue expression pattern that was suggestive of a function in sink tissues. Natural variation within MNM1 had differential effects during a diurnal time course, and this temporal dependency was supported by analysis of T-DNA insertion and over-expression lines for MNM1. Thus, the cloning of a natural variation locus specifically associated with primary metabolism allowed us to identify MNM1 as a lineage-specific modulator of primary metabolism, suggesting that the regulation of primary metabolism can change during evolution.
Introduction
An organism's growth and fitness within an environment is largely determined by its ability to efficiently obtain and utilize energy and nutrients. Central to this process is primary metabolism, which determines the use of inputs from the environment to produce all of the necessary building blocks for cells and the resulting biomass. To optimize fitness, primary metabolism must be precisely tuned and coordinated to make the most efficient use of available resources. This basic supposition is central to a wide range of biological fields, from the study of organismal growth to the study of organismal/environment interactions (Karban and Baldwin, 1997; Smith and Stitt, 2007). The best understood regulatory mechanism for primary metabolism involves allosteric control, in which substrate availability and product levels directly influence enzyme activities. Allosteric mechanisms allow rapid optimization of metabolic fluxes, and, theoretically, optimize energy utilization for biomass production; however, this regulatory mechanism is largely limited to internal rebalancing of primary metabolism.
Recent work has revealed other key regulatory mechanisms coordinating primary metabolism and growth. These include regulatory links whereby the circadian clock controls key components of central metabolism and coordinates daily growth with energy availability (Nozue et al., 2007; Covington et al., 2008; Gutierez et al., 2008; Fukushima et al., 2009; Harmer, 2009; Graf et al., 2010; Pracharoenwattana et al., 2010). This coordination occurs via direct molecular links between the circadian clock and primary metabolism. In addition, transcriptional analysis of circadian-responsive transcripts shows that primary metabolism likely has more regulatory inputs than simply the circadian clock (Harmer et al., 2000; Harmer and Kay, 2005).
Regulation of central metabolic pathways like the TCA cycle appears to be distributed across a number of enzymes providing distinct and independent regulatory inputs (Araujo et al., 2012). The existence of numerous regulatory inputs into a single pathway may provide the organism with a highly intricate system to modulate the flux into and out of central metabolism. Accumulating evidence indicates that organisms have developed numerous mechanisms enabling primary metabolites to self-regulate, including via transcriptional regulation of their own biosynthetic pathways (Larkin et al., 2003; Xiao et al., 2012; Finkemeier et al., 2013). Given the central importance of primary metabolism to an organism, it is likely that these initial observations on the regulation of and by primary metabolism are only a hint at the vast underlying regulatory networks. Thus, there are likely a large number of unidentified regulatory components that can influence primary metabolism.
In the past decade, the study of natural variation has become a key tool to elucidate metabolic networks and their regulation (Keurentjes et al., 2006, 2008; Schauer et al., 2006, 2008; Sulpice et al., 2009; Chan et al., 2010; Matsuda et al., 2012; Riedelsheimer et al., 2012; Li et al., 2013). Most natural alleles affecting primary metabolism are quantitative in nature and not lethal, whereas most induced mutants are either lethal or produce dwarfed plants. Thus, natural variation provides the ability to genetically perturb primary metabolism and identify the consequences without killing the plant. Analysis using natural variation has suggested a direct link between metabolomic variation and the growth of Arabidopsis thaliana accessions under a variety of conditions (Sulpice et al., 2009). Naturally variable loci that mediate differences in primary metabolism form epistatic networks in larger populations and their effects on primary metabolism depend upon the time of day (Rowe et al., 2008; Chan et al., 2010). Thus, identifying the genes underlying natural variation in primary metabolism may uncover components of the regulatory network influencing primary metabolism.
Analysis of metabolomic variation in the Bay × Sha recombinant inbred line (RIL) population of Arabidopsis thaliana identified a number of QTLs for natural variation in the plant metabolome (Rowe et al., 2008). Previous efforts identified ELF3 and AOP2 as key genes controlling natural variation underlying QTLs for both primary and secondary metabolism (Jimenez-Gomez et al., 2011a,b; Kerwin et al., 2011). These QTLs were not specific to the TCA cycle, whereas variation at the Met.V.67 locus was causing significant alteration in steady-state levels of the primary metabolite succinate (Rowe et al., 2008). Transcriptomic analysis of the same RIL population harvested at the same time of day showed that there was strong concordance in the location of QTLs controlling the expression of the circadian clock output networks with these metabolomic QTLs (West et al., 2007; Kerwin et al., 2011). This concordance was due to variation at known circadian loci likely altering metabolic networks, i.e., ELF3, and known metabolic loci altering the circadian oscillator, i.e., AOP (Jimenez-Gomez et al., 2011a,b; Kerwin et al., 2011). We have previously shown that genes with cis-eQTLs (natural variation in gene expression that map to their physical position) often identify causal loci for QTLs (Kliebenstein, 2008, 2009). Thus, we theorized that we could identify candidate genes for the remaining unknown metabolomic loci by searching for genes that are affiliated with the circadian clock and contain a cis-eQTL.
In this work we report the identification and initial characterization of METABOLIC NETWORK MODULATOR 1 (MNM1) encoding an AT-hook motif-containing protein that underlies the Met.V.67 QTL affecting the differential accumulation of TCA cycle intermediates within Arabidopsis thaliana. Analysis of T-DNA insertion lines and over-expression lines showed that they had opposite effects on flowering and growth, but highly similar effects on metabolism. Quantitative complementation from the Bay and Sha alleles in the Col-0 T-DNA insertion line backgrounds showed that the natural alleles in the AT-hook gene led to broad metabolic differences that focused on the TCA cycle with a time-of-day dependency in allelic effect. Translational fusions suggested that whereas MNM1 had broad physiological and metabolomic effects, it was largely limited to sink tissues. Together, these data show that MNM1 is causal for the Met.V.67 QTL and may act to coordinate central metabolism and physiology across the whole plant.
Materials and Methods
Plant Material and Experimental Conditions
For the metabolomics analysis of the T-DNA insertion lines and OE lines, Arabidopsis plants were grown in controlled-environment chamber at 20°C with 10 h light (noon to 10 p.m.) at 100–120 mE light intensity with two complete independent replicates of the whole experiment. The short-day conditions used were identical to those used to identify the Met.V.67 QTL allowing us to better compare results. Briefly, seeds were imbibed in water at 4°C for 3 days, and planted into Sunshine Mix 1 soil in a randomized complete block design. Seedlings were thinned to one plant per pot at 7 days after planting, for a total of 48 plants per block. At 6 weeks post germination, the chamber was set to continuous light conditions at relative dawn (actually noon), and the plant samples were harvested for metabolite analysis every 4 h across 2 days for T-DNA insertion lines and OE lines. In each experiment, four individual plants per genotype were harvested at each time point for sampling. This provides eight total samples per genotype per time point across the whole dataset.
For complementation analysis of the Bay and Sha alleles in the mnm1-1 and mnm1-2 backgrounds, homozygous T3 lines were used for each allele in each background. Plants were grown in controlled-environment chamber at 20°C with 10 h light (noon to 10 p.m.) at 100–120 mE light intensity with two complete independent replicates of the whole experiment. Briefly, seeds were imbibed in water at 4°C for 3 days, and planted into Sunshine Mix 1 soil in a randomized complete block design. Seedlings were thinned to one plant per pot at 7 days after planting. At 6 weeks after germination, the chamber was set at continuous light condition at relative dawn, and the plant samples were harvested for metabolite analysis at 4 h and 24 h post dawn. This provided 32 total samples per genotype per time point across the whole dataset.
T-DNA Lines and Generation of Transgenic Plants
T-DNA lines of SALK_085140 and SALK_128695 were ordered from ABRC (Sussman et al., 2000; Alonso et al., 2003), and lines with homozygous insertions were identified by PCR-based genotyping (Table S1).
All constructs described below were confirmed by sequencing, and transgenic Arabidopsis plants were generated by the flower-dipping method using Agrobacterium GV3101 containing different constructs (Clough and Bent, 1998). All T1 plants were selected by Basta and allowed to self-pollinate to generate T2 lines. Individual T2 plants for each independent T1 were allowed to self-pollinate and tested for homozygosity in the T3 generation. T3 homozygous transgenic plants were confirmed by PCR-based genotyping and used for this study.
To generate the overexpression lines, the MNM1 coding region was amplified from cDNA of Col-0 by primers of AKORF5 and AKORF3 (Table S1), cloned into pENTR/D-TOPO vector (Invitrogen), and subcloned to pB2GW7.0 destination vector by Gateway LR Clonase (Invitrogen). This leads to MNM1 expression being driven by the 35S promoter. Lines showing dramatic morphological differences were kept as seeds but not used for molecular analysis.
To generate the complementation transgenes, the genomic regions of MNM1 from Bay and Sha genomic DNA were amplified using the AKP5 and AK3Full primers (Table S1). These were then cloned into pENTR/D-TOPO vector, and subcloned into pMDC123 destination vector by Gateway LR Clonase. These constructs were transformed into both the mnm1-1 and mnm1-2 backgrounds to test for quantitative complementation.
To generate the GUS fusion lines, the promoters and coding regions of MNM1 from Bay and Sha without stop codons were amplified by AKP5 and AKORF3A (Table S1), cloned into pENTR/D-TOPO vector, and subcloned to pBGWFS7 destination vector by Gateway LR Clonase (Table S1). All constructs were transformed into Col-0, mnm1-1, and mnm1-2 backgrounds to allow for potential identification of differences in these backgrounds upon MNM1 expression. The transgenic plants are confirmed by PCR-based genotyping.
Flowering Time
To measure flowering time in the lines with different MNM1 genotypes, plants were arranged in a randomized complete block design within both short- and long-day conditions. For the long-day mnm1-1 and mnm1-2 comparison, the experiment was replicated five times with an average of 30 individuals per genotype per experiment such that there were 152 Col-0 individuals, 149 mnm1-1 individuals and 157 mnm1-2 individuals measured across the five experiments. For the short-day mnm1-1 and mnm1-2 plants, the experiment was replicated three independent times with a total of 49 Col-0 individuals, 48 mnm1-1 individuals, and 47 mnm1-2individuals measured. The flowering of the MNM1 OE genotypes was measured in long-day conditions with three replicated experiments providing measurements for a total of 54 Col-0 individuals, 52 OE7 individuals, and 54 OE8 individuals. Flowering time was recorded for all genotypes when the first flower opened. A secondary metric of flowering time, leaf number at flowering, was recorded only for the mnm1-1 and mnm1-2 lines because of the abnormal leaf phenotypes of overexpression lines.
Histochemical GUS Assay
Three independent transgenic lines containing either the Bay or Sha allele of the translational GUS fusion within the WT Col-0, mnm1-1, or mnm1-2 backgrounds were stained every week for 6 weeks and pictures were taken each time. GUS staining of whole plants was done as previously described (Weigel and Glazebrook, 2002).
RNA Extraction, RT-PCR, and Quantitative PCR
For qPCR and RT-PCR study, Arabidopsis plants were grown in a controlled-environment chamber at 20°C with 16 h light (6 a.m. to 10 p.m.), and 3-week-old plants were harvested at relative noon. One sample of each genotype was harvested and the entire experiment was replicated twice. All harvested samples were frozen in liquid nitrogen immediately and stored at −80°C until extraction. Total RNA was extracted using Trizol (Invitrogen), and 1 μg total RNA was treated with DNase I (Invitrogen). First strand cDNA was generated using the Reverse Transcription System (Promega) and used for RT-PCR and qPCR. Preliminary RT-PCR was performed using four pairs of primers for MNM1 with TUBULIN2 (AT5G62690) as a control (Table S1). Quantitative PCR was performed using the SYBR Green Kit in a 7300 Real Time PCR System (Applied Biosystems) with UBQ10 (At4g05320), and ACTIN2F (At3g18780), as controls (Table S1).
Metabolomics Analysis
For each metabolomics sample, one leaf disk from each of two leaves per plant was harvested, providing two leaf disks of approximately 20 mg total weight. Unless otherwise noted, all harvesting started at subjective mid-day, finishing within 15 min. Each plant was independently harvested and extracted using previously published protocols (Weckwerth et al., 2004a,b; Meyer et al., 2007). The samples were stored dry at −80°C until automated derivatization and GC-TOF-MS analysis at the UC Davis Genome Center Metabolomics Facility (http://metabolomics-core.ucdavis.edu/; Fiehn et al., 2005). Metabolite identity was determined by comparing retention time and mass to the UC Davis Genome Center Metabolomics Facility metabolites database (http://fiehnlab.ucdavis.edu/Metabolite-Library-2007/; Fiehn et al., 2005). At the time of analysis, this library contained reference spectra for 1013 known metabolites, generated by the analysis of purified reference compounds. Metabolites not contained within this library are listed as unknown or unidentified metabolites using a unique database identifier (http://fiehnlab.ucdavis.edu/Metabolite-Library-2007/). For the metabolomics measurements in these experiments, this identified 439 putative metabolites within the entire experiment (Table S2). Post analysis, samples were subject to quality control and normalization as previously described using a pooled reference standard to control for technical variance (Fiehn et al., 2008; Fernie et al., 2011).
Statistics
To test the effect of MNM1 upon metabolite accumulation at the mid-day time point, all metabolites were analyzed via ANOVA using a general linear model within (R Development Core Team, 2014). First, a primary model was run to test the effect of general genotypes (i.e., WT v mnm1-1/2 v OE7/8). In this model yrgc denotes the metabolite accumulation in each plant with Genotype g, from Replicate r. The model for the metabolite accumulation is
where εrg represents the error term and is assumed to be normally distributed with mean 0 and variance σ2ε. All parameters are reported in Table S3. Another model was also used in which the individual mnm1-1/2 and OE7/8 genotypes were nested within the broader genotype term. These two models were then compared for significant difference and this is reported in Table S3 for each metabolite. Means and standard error for each genotype class were obtained using (R Development Core Team, 2014). All presented p values are adjusted to FDR of 0.2. We have broadly used this FDR level in the past with a high level of validation (West et al., 2007; Rowe et al., 2008; Chan et al., 2011; Kerwin et al., 2011). Further, this is a similar level of significance to that used in the original QTL mapping.
To test the connection between genotype and time within the diurnal/circadian time course the same model approach was utilized to test for genotype × time interactions to obtain a precursory impression of time-specific genotype shifts. Given that this short time course has both transition and stable circadian time points because of the inclusion of the first day in the transition to constant light, this specific time testing is considered appropriate for the purpose of identifying genotypic effects upon plant metabolism. In this model yrgc denotes the metabolite accumulation in each plant with Genotype g, from Time t, from Replicate r. The model for the metabolite accumulation is
where εrgt represents the error and is assumed to be normally distributed with mean 0 and variance σ2ε. Means and standard error for each genotype class were obtained using (R Development Core Team, 2014). All presented p values are adjusted to FDR of 0.2 (Tables S2, S4). We have broadly used this FDR level in the past with a high level of validation (West et al., 2007; Rowe et al., 2008; Chan et al., 2011; Kerwin et al., 2011). Further, this is a similar level of significance to that used in the original QTL mapping.
To test the effect of Bay and Sha allelic complementation of the mnm1-1 and mnm1-2 genotypes, the following model was used:
where εrgt represents the error and is assumed to be normally distributed with mean 0 and variance σ2ε. In this model, yrgc denotes the metabolite accumulation in each plant with Allele a, Genetic background g, from Time t, from Replicate r. Allele represents the Bay and Sha alleles introduced into the mnm1-1 and mnm1-2 genetic background and is nested in genetic background. Means and standard error for each genotype class were obtained using (R Development Core Team, 2014). All presented P-values are adjusted to FDR of 0.2 (Tables S5, S6). We have broadly used this FDR level in the past with a high level of validation (West et al., 2007; Rowe et al., 2008; Chan et al., 2011; Kerwin et al., 2011). Further, this is a similar level of significance to that used in the original QTL mapping.
Results
Identification of a Candidate Gene for the Met.V.67 QTL
To test whether we could identify genes underlying metabolomic QTLs by searching for genes that are affiliated with the circadian clock and contain a cis-eQTL, we focused on the Met.V.67 QTL that is linked to variation in both metabolite accumulation (Figure 1A) and circadian network output (Figure 1B). In the previous analysis the metabolic effect had been ascribed solely due to the Bay-0 allele but this is complicated by the inability of natural variation studies to ascribe an appropriate baseline. Instead, this requires cloning of the underlying gene. To accomplish this, we first defined the physical boundaries of Met.V.67 as the complete 3-LOD interval for all metabolites with a QTL peak at this position, as previously found (Rowe et al., 2008). Within this region, we surveyed all six genes for either a cis-eQTL or circadian oscillation using previous datasets (Figure 1C) (Harmer and Kay, 2005; Covington and Harmer, 2007; West et al., 2007; Covington et al., 2008). This analysis showed that only one gene, At5g54930, had both a cis-eQTL and a circadian expression pattern in agreement with the overlapping circadian network eQTL. This gene also tightly co-expressed with TOC1 and PRR5, central components of the circadian oscillator (Figure S1) (Obayashi et al., 2007, 2009; Harmer, 2009). Thus, we hypothesized that At5g54930 could be causative for the Met.V.67 metabolomic and circadian network QTL and renamed this gene METABOLIC NETWORK MODULATOR 1, MNM1.
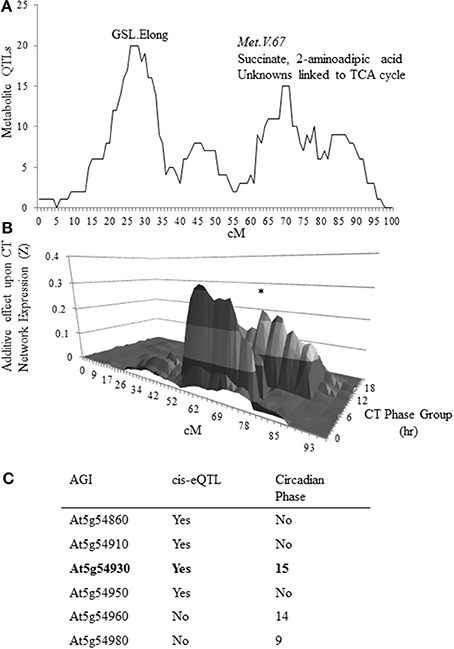
Figure 1. Candidate identification for metabolomics QTL MET.V.67. (A) The number of metabolites for which a QTL was detected in the Bay × Sha RIL population on chromosome V within a 10 cM sliding window is shown. The permuted threshold (P = 0.05) for detection of a significant metabolite hotspot within this analysis is 12 metabolite QTLs. The X axis shows the chromosomal position in cM. The known metabolomics QTLs on chromosome V are labeled as GSL.Elong and Met.V.67 with the known chemicals listed above the peak. (B) Shown is the impact of natural variation on chromosome V in the Bay × Sha RIL population upon expression of circadian CT phase group networks from previously published data. Each CT phase group consists of the genes that have a peak of circadian expression within 30 min of the time for that group. The graph presents the predicted additive effects across chromosome V for each CT phase group's expression from CT0 to CT23. The asterisk marks the CT phase group that is most impacted at the Met.V.67 locus (CT15).The bottom shows the position on the chromosome in cM. (C) Shown is the list of candidate genes in the 3-LOD interval for the metabolomics QTL Met.V.67 (At5g54830 to At5g55040) that have either a cis-eQTL or are regulated in a circadian manner with the phase reported.
Genetic Analysis of MNM1 Variation
MNM1 encodes a putative 286-amino-acid protein that contains an AT-Hook motif (RGRP; Figure 2). At-Hook motifs are often involved in binding DNA minor-grooves (Aravind and Landsman, 1998; Fujimoto et al., 2004) and can play roles in plant disease resistance, hormone metabolism, development, and other complex phenotypes (Matsushita et al., 2007; Ng et al., 2009; Gallavotti et al., 2011; Jin et al., 2011; Yadeta et al., 2011). The AT-hook motif was found close to a Degradation box and a nuclear localization sequence, but no other identifiable motifs were present in the protein sequence (Figure 2). Interestingly, MNM1 is not a member of the previously identified family of Arabidopsis AT-hook motif-containing proteins (Fujimoto et al., 2004). Instead, MNM1 is a single copy gene within Arabidopsis thaliana and has a full-length homolog only in the Brassicales genomes of Arabidopsis lyrata, Capsella rubella, and E. salsugineum. All other tested eudicot genomes, such as Brassica rapa, Vitis vinifera, Populus trichocarpa, and Ricinus communis, have only partial paralogs that are related solely by the 80 amino acids surrounding the AT-hook motif (Figures S2, S3). This leads to an average identity of only 33% to the closest paralogs solely centered around the AT-hook motif (Figure S2 and Table S7). Thus, MNM1 appears to be unique to the Brassicales lineage and it was apparently lost in the Brassica species (Figure S2).
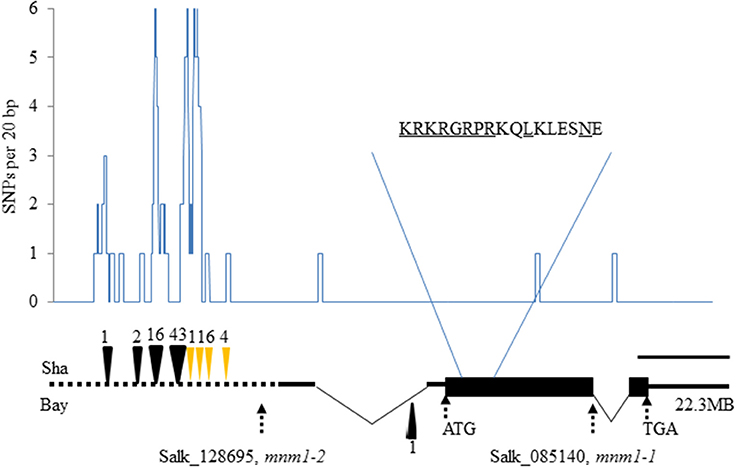
Figure 2. Natural and induced variation in MNM1. The gene structure of MNM1 is shown with the wide black rectangle showing the open reading frame. The 5′ and 3′ untranslated regions are shown as solid black lines with introns shown by the thin triangle. The promoter analyzed is shown by the dotted black line. The graph shows the number of single nucleotide polymorphisms per 20 basepairs between the Bay and Sha genomic sequences. The deletions with respect to the Col-0 sequence are shown as black triangles at the respective positions, while insertions are shown as yellow triangles. The position of mnm1-2 (SALK_128695) and mnm1-1 (SALK_085140) are presented. The scale represents a distance of 400 bp. The amino acid sequence shows the portion of the protein containing the AT-hook motif (RGRP), the potential nuclear localization sequence (KRKR) and the putative D-box (RxxLxxxxN). The start codon, stop codon, and chromosomal position are also labeled.
To test if MNM1 had the potential to be a causal basis of the Met.V.67 QTL we sequenced the gene in both Bay-0 and Sha parental accessions to look for genetic variation. This identified numerous polymorphisms within the MNM1 promoter, including 40 SNPs and nine separate insertion/deletion events (Figure 2 and Figure S4). There were three SNPs within the predicted body of the RNA but these did not affect any key splicing sites and only a single Leu to Ile change was introduced into the protein. Thus, while it is more likely that the MNM1 QTL is caused by the variation between the Bay and Sha promoters the amino acid change could potentially affect the protein function or the sequence change potentially alter RNA processing.
Genetic Analysis of MNM1 Effect on Arabidopsis Physiology
Two T-DNA insertion lines from the Arabidopsis Biological Resource Center were obtained to further analyze the potential role of MNM1 in Arabidopsis metabolism. SALK_085140, hereafter mnm1-1, contained a T-DNA in the open reading frame, whereas SALK_128695, hereafter mnm1-2, contained a T-DNA very near the transcription start site (Figure 2; Alonso et al., 2003). qRT-PCR of these alleles showed that mnm1-1 leads to a 5-fold reduction in transcript abundance with complete ablation of the last exon (Figure S5). By contrast, mnm1-2 had a 2-fold increase in transcript abundance above WT. Neither mnm1-2 nor mnm1-1 displayed altered expression of the neighboring At5g54920 and At5g54940 genes (Figure S6). We created MNM1 overexpression constructs by fusing the coding sequence to a 35S promoter. Two independent T3 lines, OE7 and OE8, from independent T1 events that had homozygous insertions with minimal gross morphological effects were chosen for further analysis. These lines both exhibited a >10× increase in MNM1 transcript levels (Figure S5).
Using a nested ANOVA allowed us to test for differences between MNM1 and mnm1 as well as between the mnm1 alleles. This showed that both mnm1-1 and mnm1-2 displayed slightly earlier flowering under both short and long-day conditions, suggesting that the T-DNA insertions similarly affect the flowering function of MNM1 even with their different effects on expression of the transcript (Figure 3 and Figure S7, Table S8). This idea is supported by the finding that both OE lines had a slight delay in flowering (Figure 3), suggesting that the effects of high-level overexpression are opposite to those of slight overexpression or down-regulation of MNM1 (Figure 3 and Figure S8). Approximately half of all obtained MNM1 OE lines showed even more dramatic developmental consequences but these lines were not further analyzed because of the difficulty of separating the morphological and metabolic consequences of MNM1 (Figure S8). Thus, the mnm1-1 and mnm1-2 alleles have similar physiological consequences that are the opposite of those observed in the OE lines, suggesting that MNM1 plays a role in plant development. There are several possible explanations about why the mnm1-1 and 1-2 alleles have similar physiological phenotypes while divergent transcript abundances. mnm1-2 may have altered transcript processing, stability or translation which would disconnect RNA abundance from function. Additional work is required to understand the precise basis of the phenotypic cause in mnm1-2.
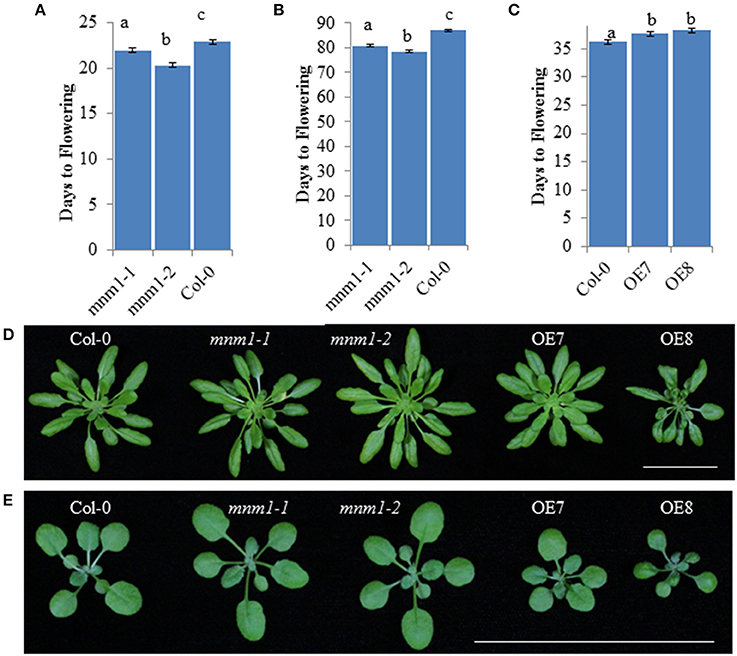
Figure 3. Morphological analysis of MNM1 Genotypes. Letters show statistically different groupings within a graph as determined by ANOVA at a 0.001 level. All ANOVA models included genotype and the interaction of genotype and replicate. In all instances the interaction term was non-significant. All values are least squared means with standard error. For (A,B), all genotypes were tested together in four independent experiments per day length with an average of 200 plants in total across the experiments for each genotype. For C, Col-0 and the two MNM1 OE genotypes were tested in three replicates with 50–54 plants per genotype. (A) Number of days to flowering under long-day conditions for WT and the mnm1-1 and mnm1-2 mutants. (B) Number of days to flowering under short-day conditions for WT and the mnm1-1 and mnm1-2 mutants. (C) Number of days to flowering under long-day conditions for WT and two MNM1 overexpression genotypes. (D) Growth of 6-week old plants under short-day conditions. The bar shows a 5 cm scale. (E) Growth of 3-week old plants under long-day conditions. The bar shows a 5 cm scale.
Genetic Analysis of MNM1 in Arabidopsis Metabolism
To test the link between MNM1 and the Met.V.67 QTL, we measured metabolite accumulation in mnm1-1 and mnm1-2 and MNM1 overexpression genotypes as well as the wild type Col-0 accession. The samples were harvested at mid-day in short-day conditions, which is the same time and light regime as the original metabolomic and expression QTL experiments used to identify the candidate gene (West et al., 2007; Rowe et al., 2008; Kerwin et al., 2011). 459 metabolites were detected and all were subjected to nested ANOVA at an FDR adjusted level of 0.2 (Table 1 and Table S3) (West et al., 2007; Rowe et al., 2008; Kerwin et al., 2011). We have broadly used this FDR level in the past with a high level of validation (West et al., 2007; Rowe et al., 2008; Chan et al., 2011; Kerwin et al., 2011). Further, this is a similar level of significance to that used in the original QTL mapping. Supporting the hypothesis that MNM1 variation causes the Met.V.67 QTL, succinate was significantly altered in the OE lines in comparison to Col-0. Nested ANOVA again showed that the two mnm1 alleles behaved identically with respect to each other and that the two OE lines were identical to one another (Table 1 and Table S3), this allows us to combine data from the recessive mnm1 and dominant OE lines in further analysis.
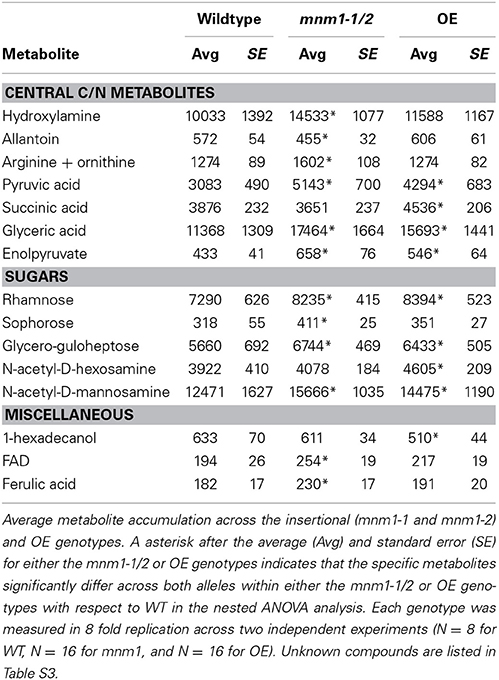
Table 1. Known metabolites altered at mid-day by MNM1 genotypes.
Comparison of Col-0 WT to the OE lines mimicked the QTL pattern of additive effects between the Bay and Sha alleles of Met.V.67. Specifically, the OE lines displayed higher accumulation of all compounds associated with the Met.V.67 QTL in comparison to Col-0, just as the Sha allele led to higher accumulation of these compounds in comparison to the Bay allele (Table S3). This is in accordance with previous observations that the Col-0 and Bay-0 alleles of MNM1 are expressed at similar levels whereas the Sha allele displays higher expression (Kliebenstein et al., 2006; Van Leeuwen et al., 2007; West et al., 2007), and our current result that the genomic sequence of MNM1 in Bay is more similar to Col-0 than Sha. Therefore, the comparison of MNM1 OE lines with Col-0 better recreates the natural variation originally studied in the Bay-0 × Sha RIL population. Further supporting MNM1 as the causative locus of the Met.V.67 QTL, none of the metabolites linked to QTLs flanking Met.V.67 (±10 cM) were altered by the various MNM1 genotypes (Table S3).
The specific manipulation of MNM1 within an isogenic background provides us more statistical power and genetic precision to fully interrogate the effects of MNM1 upon plant metabolism in comparison to the previous Bay-0 by Sha QTL analysis. Therefore, we analyzed the full metabolomics data to identify any additional metabolites that might be linked to alterations in MNM1. ANOVA of all 459 detected metabolites showed that 49 metabolites were significantly altered by the differences in the MNM1 genotypes (Table S3). Of these, 38 metabolites did not show a significant nesting term when comparing mnm1-1 and mnm1-2 to each other as well as the OE alleles to each other, indicating that mnm1-1 and mnm1-2 as well as OE7 and OE8 behaved similarly to each other in terms of those metabolites (Table S3). Surprisingly, levels of only six of the known metabolites were statistically altered in both the OE and mnm1-1/2 lines and in these cases the directionality of the change was always the same in both groups of lines (Table 1). This directional agreement between the mnm1-1/2 and OE genotypes was also true for unidentified metabolites in this study (Table S3, Figure S9). This similarity in effect of both OE and mnm1-1/2 upon steady-state metabolite accumulation suggests that MNM1 may play a role in determining metabolic balance rather than any specific directional effect. The discrepancy wherein the OE and mnm1-1/2 lines display opposite flowering time phenotypes despite similar metabolic alterations suggests that these metabolic and physiological outcomes may reflect independent functions of MNM1. Alternatively, there may be an as-yet unmeasured aspect of metabolism, such as flux, that is more directly linked to the physiological consequences of MNM1 activity. Future experiments are necessary to resolve this interesting observation.
Genetic Analysis of MNM1 in Arabidopsis Metabolic Balance
To further examine the effect of MNM1 on metabolic homeostasis within Arabidopsis, we analyzed metabolite accumulation in the mnm1-1/2 and OE lines across a 48 h time course. Given that MNM1 was linked to circadian clock output (Figure 1), we left the plants in constant light for the entire time course to get a preliminary view of how the genetic polymorphisms within MNM1 affect metabolic homeostasis. 459 metabolites were detected and all were subjected to nested ANOVA (Tables S2, S4). This identified 122 metabolites that accumulated to significantly different levels in both mnm1-1/2 or both OE lines in comparison to the WT genotype. Of the metabolites significantly different between the MNM1 mutant alleles and the WT genotype, 96 showed no significant difference when comparing mnm1-1 to mnm1-2 or OE7 to OE8, suggesting that these metabolites show identical shifts upon any perturbation of MNM1 (Tables S2, S4). While the vast majority of these metabolites showed significant variation across the time course, the genotypic influence was the same across all timepoints as indicated by the Genotype:Time interaction (Tables S2, S4). The known metabolites that were affected by the mnm1-1/2 or OE genotypes at all timepoints are shown in a map of central metabolism (Figure 4). These metabolic effects were spread across the TCA cycle and general amino acid metabolism, but had no apparent effect on lipid metabolism (Figure 4). In both the mnm1-1/2 and OE genotypes there seemed to be a rebalancing of metabolite accumulation away from TCA and sugars toward specific amino acids.
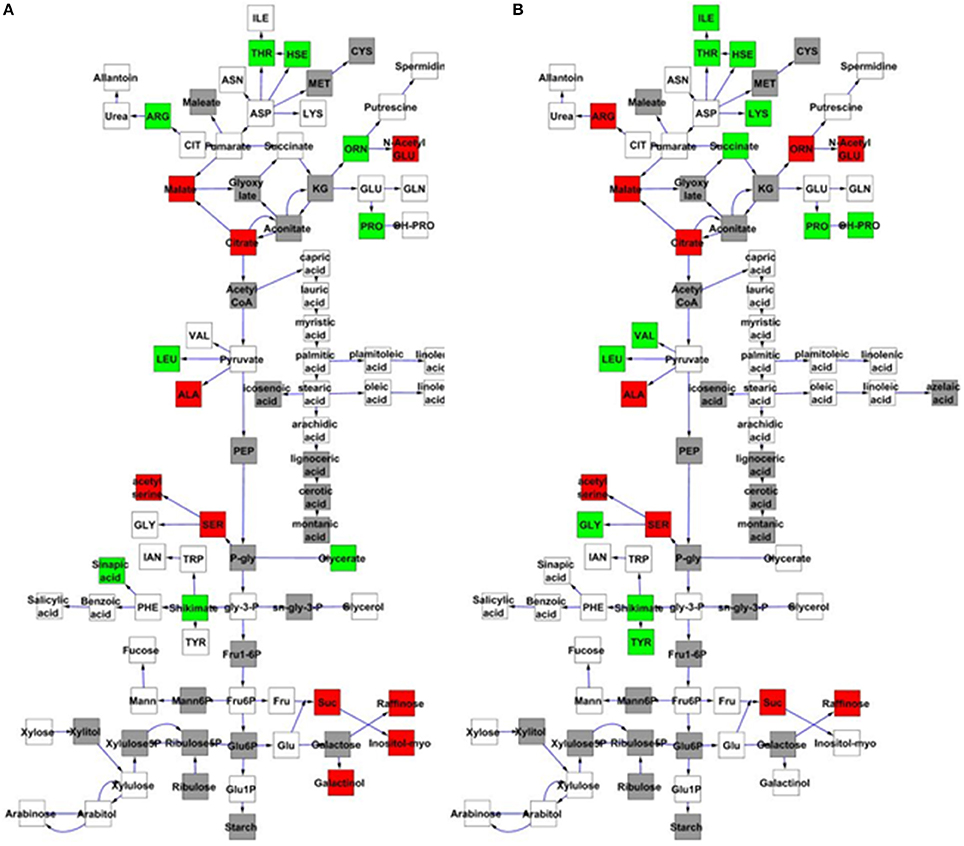
Figure 4. MNM1 effects upon the metabolic network. Shown are the known primary metabolites that are altered at all timepoints by the mnm1-1 and mnm1-2 and overexpression genotypes in comparison to wildtype controls within the free-run experiment. The gray boxes show metabolites not measured in these experiments, white boxes are metabolites that had no significant difference, green boxes were up regulated in the respective MNM1 genotypes and red boxes were down-regulated in the respective MNM1 genotypes. Only metabolites showing a significant effect across the mnm1-1 and mnm1-2 and OE genotypes are presented. The central metabolites are presented in a stylized representation of central metabolism with common abbreviations for each chemical. Arrows represent enzymatic linkages between detected compounds but are not necessarily showing single enzymatic steps. All detected intermediates such as shikimate are included in the network. (A) Metabolites altered in both the mnm1-1 and mnm1-2 genotypes. (B) Metabolites altered in both MNM1 OE genotypes.
In central metabolism, ornithine was the sole exception to the finding that any perturbation of MNM1 gave rise to identical metabolic consequences. For ornithine, the mnm1-1/2 genotypes led to higher accumulation whereas the OE genotypes had lower accumulation in comparison to WT. Outside of central metabolism, nine unknown metabolites and methionine sulfoxide, the position of which within the metabolic network is unknown, showed a similar pattern to ornithine, while glycerate had the same trend but was not significantly different in the OE background (Figure 4 and Tables S2, S4). By contrast, only three unknown metabolites showed the opposite pattern, with significantly higher accumulation in the mnm1-1/2 lines and the reduced accumulation in the OE lines (Tables S2, S4). Thus, genetic alterations in MNM1 lead to linear changes in accumulation for some metabolites, while for the other metabolites MNM1 behaves more as a homeostasis controller whereby any change in MNM1 function leads to identical fluctuations in these metabolites (Figure 4). These data suggest that the flowering time phenotype in the MNM1 lines may be more linked to ornithine, methionine sulfoxide or any of the unknown compounds that are linearly related to MNM1 function.
There was a small collection of metabolites for which the genotype: time interaction terms had more significance than the genotype main effect term (Tables S2, S4). This was most pronounced for a set of sugars (maltose, trehalose, raffinose, cellobiotol, and sophorose) (Figure 5). Both the OE and mnm1-1/2 insertion lines show a diminished and shifted oscillation in free raffinose accumulation with a concomitant shift toward higher accumulation of the other sugars. These sugars are connected by the conversion of raffinose to glucose via sucrose, and the other sugars in this pathway (sucrose, galactinol, and myo-inositol) are all also decreased in the OE and T-DNA insertion lines. This suggests that MNM1 plays a role in mediating the temporal partitioning of sugars within the Arabidopsis leaf. Further experiments will be required to test which is the most immediate effect on the metabolomics network of polymorphisms within MNM1.
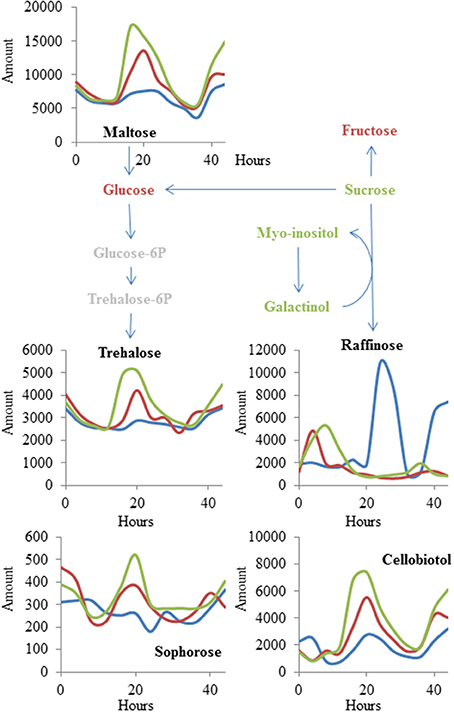
Figure 5. MNM1 effects upon cycling within the metabolic network. Shown are the known metabolites that were altered in a time-dependent fashion by the mnm1-1, mnm1-2, and MNM1 OE genotypes in comparison to wildtype controls within the free-run experiment. The metabolites are placed within a sugar network for illustration. Red metabolites had no significant effect of MNM1 function, green metabolites had a significant effect of MNM1 function but no interaction with time and are shown in Figure 4. Metabolites labeled in black had a significant interaction of MNM1 function and time within the analysis. Their levels are shown across the 44 h of the experiment in relative abundance levels obtained from the GC-TOF analysis. Blue lines in the graph represent WT Col-0, Red lines are the effect of mnm1-1 and mnm1-2, while green is the effect of the OE7 and OE8 genotypes.
Quantitative Complementation of MNM1
The above experiments demonstrated that MNM1 alters metabolism and physiology of Arabidopsis in a manner similar to phenotypes that are linked to variation at the Met.V.67 metabolomic QTL. To validate that variation in the Bay and Sha alleles of MNM1 were potentially causal for the Met.V.67 QTL, we cloned the full length-genomic sequences of MNM1 from both the Bay and Sha genotypes and independently transformed these into both the mnm1-1 and mnm1-2 backgrounds. Comparison of two opposing alleles in a common genetic background is the definitive test for establishing that the alleles differ in their function (Mackay, 2001). Given that the mnm1-1 and mnm1-2 lines had similar phenotypes in terms of both the metabolite and flowering time data, we obtained independent homozygous transgenic lines containing either the Bay or Sha genomic allele in both backgrounds. We then measured metabolites in these complementation lines at 6 weeks of age at either 4 or 24 h past dawn. Four hours is the time point equivalent to that used for the QTL mapping sample, whereas 24 h was the time point that showed the most differences in the metabolomics time course (Figures 4, 5). Of the 11 metabolites originally detected as being altered by the Met.V.67 QTL, nine were detected in this experiment, succinate, 2-aminoadipic acid and seven unknowns (211891, 214529, 204348, 200450, 208664, 212373, and 213330). Only two unknown compounds were not found in this analysis. To test for quantitative complementation differences between the Bay and Sha MNM1 alleles, we used an ANOVA that specifically tested all metabolites for differences between the transgenic plants containing the Bay and Sha MNM1 alleles while controlling for any potential difference between the mnm1-1 and mnm1-2 backgrounds. This showed that the accumulation of eight of the nine metabolites linked to the Met.V.67 QTL was statistically different between the quantitative complementation lines containing the Bay or Sha allele (Tables S5, S6). In all instances, the direction of effect agreed with the previously published analysis from Met.V.67 (Tables S5, S6) (Rowe et al., 2008). The only exception was the unknown 212373. Thus, variation between the Bay and Sha alleles of MNM1 is responsible for the vast majority of metabolomic variation at the Met.V.67 QTL in the Bay × Sha RIL population.
Most of these metabolites showed a genotype by time interaction, i.e., the Bay and Sha alleles had time-dependent effects on metabolite levels. This was particularly striking in the TCA cycle where succinate, fumarate, and malate shifted from the Bay allele conferring higher accumulation at the 4 h time point to the Sha allele leading to higher accumulation at the 24 h time point (Figure 6 and Tables S5, S6). Glycine, serine, phenylalanine, threonine and salicylic acid also displayed a time-dependent allelic effect. By contrast, other metabolites like glutamate and glutamine display more consistent behavior, with plants harboring the Bay and Sha alleles showing the same relationship at both time points. This suggests that the MNM1 allelic variation underlying the Met.V.67 QTL has different temporal effects upon the metabolomics network.
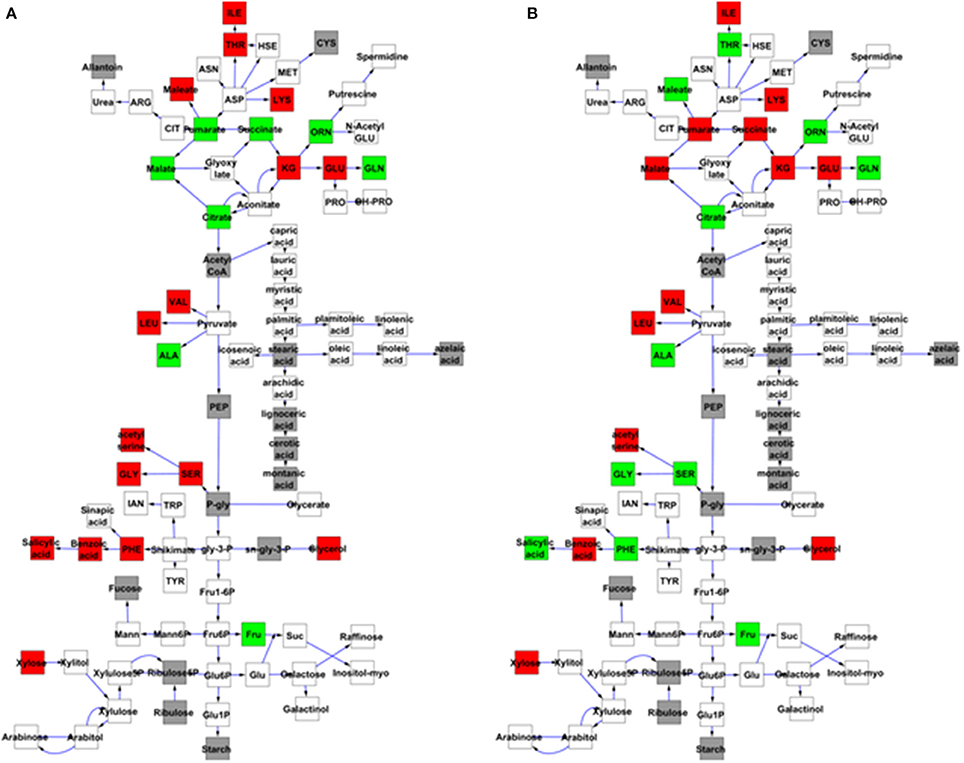
Figure 6. Quantitative complementation to validate MNM1 as the Met.V.67 QTL. Full genomic Bay or Sha alleles of MNM1 were transgenically reintroduced into both the mnm1-1 and mnm1-2 background genotypes. Homozygous T2 lines were analyzed for metabolomics at either 4 or 24 h in a free-run experiment as identified as key timepoints for MNM1 function. The 4 h timepoint is the same that was sampled to identify the Met.V.67 QTL. The effect on only central metabolites showing a significant difference between the Bay and Sha allele when introduced into both the mnm1-1/2 genotypes presented at the specific timepoint shown. The gray boxes show metabolites not measured in these experiments, white boxes are metabolites that had no significant difference, green boxes are metabolites higher in Bay allele complementation lines and red boxes are metabolites higher in the Sha allele complementation lines. The metabolites in central metabolism are presented in a stylized representation of central metabolism with common abbreviations for each chemical. Arrows represent enzymatic linkages between detected compounds but are not necessarily showing single enzymatic steps. All detected intermediates such as shikimate are included in the network. (A) Metabolites that significantly differ between the Bay and Sha MNM1 alleles at the 4 h timepoint. (B) Metabolites that significantly differ between the Bay and Sha MNM1 alelles at the 24 h timepoint in constant light.
Expression Analysis of MNM1
The temporal aspect of the effect of MNM1 allelic variation and the preponderance of natural variation in the promoter of MNM1 led us to test for expression difference of the Bay and Sha alleles of MNM1. To do this, we cloned the genomic copies of MNM1 from Bay and Sha and used these to make 3′ translational GUS-GFP fusions. Both the Bay and Sha GUS-GFP translational fusions were transformed into the Col-0, mnm1-1 and mnm1-2 backgrounds. Three to four independent T1s were chosen for each genotype/transgene combination and allowed to self-pollinate; then, three homozygous T3 plants from independent T1 lines were analyzed for developmental expression patterning of the two alleles of MNM1. GUS staining was performed once a week for 6 weeks post-germination in short-day conditions and once every week for 4 weeks post-germination in long-day conditions. There was no detectable difference between the expression patterns of the Bay and Sha alleles under any condition in any background in any of the independent transgenic lines (Figures S10–S12). At 1-week post germination, MNM1 was most highly expressed in the newly emerging leaves and in the vasculature of the root and cotyledons (Figure 7). By 2-weeks post germination, the vascular expression was largely gone while the newly emerging leaves showed the highest expression. In the maturing leaves of these plants, MNM1 was expressed at the base and excluded from the vasculature (Figure 7). During the development of short day plants, the newly emerging leaves always showed high expression of MNM1 but as these leaves matured, the expression was gradually lost in the lamina until only the margin showed any MNM1 expression (Figure 7). Finally, all detectable MNM1 expression was lost in the most mature leaves. Thus, it does not appear that the variation in the Bay and Sha promoters of MNM1 leads to altered developmental patterning of the translational fusion. Further work will be required to ascertain the mechanism by which variation in these two alleles leads to the altered metabolic networks.
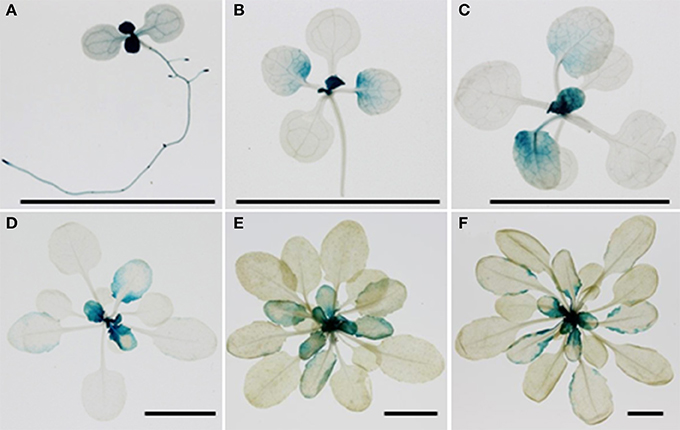
Figure 7. Developmental patterning of MNM1. Shown is histochemical GUS staining of transgenic Arabidopsis plants expressing the MNM1 translational fusion from the Sha accession in the Col-0 background. Three independent transgenic lines per construct were visualized for each time point and a representative image is shown. The black bar in each graph represents a 1 cm scale. (A) One-week-old seedling grown in MS medium in long day condition. (B) Two week-old rosette leaves grown in soil in short day condition. (C) Three week-old rosette leaves grown in soil in short day condition. (D) Four week-old rosette leaves grown in soil in short day condition. (E) Five week-old rosette leaves grown in soil in short day condition. (F) Six week-old rosette leaves grown in soil in short day condition.
Discussion
This work identifies MNM1 as a causal gene behind the Met.V.67 QTL that determines natural variation in primary metabolism. The Met.V.67 metabolomics QTL in the Bay × Sha RIL population was linked to variation in TCA cycle metabolites, such as succinate, as well as altered expression of genes that peak during a circadian cycle (Rowe and Kliebenstein, 2008; Kerwin et al., 2011). To elucidate the molecular basis of this locus, we used a systems interrogation of genes within the region of the Met.V.67 QTL and found a single gene, MNM1, which had variable expression in the Bay × Sha population and was expressed at the proper time of the circadian oscillation (Figure 1). We validated the effect of MNM1 on natural variation using quantitative complementation to compare the phenotypic effects of introducing the Bay and Sha genomic alleles of MNM1 into two independent T-DNA insertion backgrounds. The quantitative complementation showed that variation within the Bay and Sha alleles led to the genotypes containing these transgenes to have identical changes in the steady state accumulation of eight of nine metabolites linked to variation in this locus (Figure 6) (Rowe and Kliebenstein, 2008). The availability of these isogenic lines containing only MNM1 variation also allowed us to show that there were a large number of other metabolites affected by this variation (Figure 6). Thus, the Bay and Sha alleles of MNM1 confer differential accumulation of metabolites associated with the Met.V.67 QTL, demonstrating that allelic variation in MNM1 in part underlies the Met.V.67 QTL. It remains to be tested if additional genes within this region also contribute to the Met.V.67 QTL.
The most likely causal polymorphisms between the Bay and Sha alleles of MNM1 are within the promoter region. Large indels were found when comparing the two promoters (Figures 2 and Figure S4). This agrees with the gene having a cis-eQTL in the Bay and Sha population, suggesting that there is a promoter polymorphism leading to differential expression of the two alleles (West et al., 2007). However, all of our efforts to identify differential expression between the Bay and Sha translational fusions failed to reveal a dramatic difference between the two alleles (Figure 7). The cis-eQTL for this gene represents a quite modest effect of only 15% (West et al., 2007), and it is possible that a difference this small would not be apparent in the translational GUS fusion assays. Additionally, the eQTL analysis was performed using whole rosette tissue and as such, there may be tissue or even temporal aspects to the cis-eQTL that we have not yet identified (West et al., 2007). It is also possible that the non-synonymous change in the protein may also be responsible for the allelic difference possibly due to differential translation, RNA metabolism or subtle effects on protein structure. Further work is required to identify the specific difference between the Bay and Sha alleles that leads to the alterations in plant metabolism (Figures 6, 7).
MNM1 Alters Metabolic Networks both Temporally and Non-Temporally.
The allelic effect of the Bay vs. Sha alleles of MNM1 was temporally dependent, with the Bay and Sha alleles having opposite effects at the different time points (Figure 6). For example, the Bay allele had generally higher TCA metabolites at relative noon but lower levels of some amino acids compared to the Sha allele. This pattern then switched at the 24 h time point (Figure 6). Interestingly, other metabolites, such as glutamate and glutamine, were affected by alterations in MNM1, but these effects were not dependent on the sampling time. Thus, MNM1 has the capacity to regulate primary metabolism homeostasis with both temporal and non-temporal effects depending upon the specific metabolite being measured. The temporally conditional effects agree with the linkage between MNM1, Met.V.67 and the circadian clock. However, it remains an open question how manipulating a single gene can lead to both stable and temporally conditional effects within the interconnected metabolomics network.
A conundrum of the work is that the over-expresser and knockout lines have similar phenotypic consequences upon the metabolic network. Given that MNM1 is not likely an enzyme, the metabolic changes are likely not the direct result of MNM1 activity but instead a consequence of the direct target of MNM1. One could suppose that this might be a consequence of a secondary stress situation. However, none of the common stress related metabolites (Salicylate, Proline, OH-Proline, and polyamines) show changes in the MNM1 genotypes. Further, the mnm1-1 which has little to know RNA has the same effect suggesting that this is not the result of MNM1 mis-expression in the wrong place or time. Thus, it suggests that if MNM1 is high or low there is the same metabolic output which is contrary to the typical gene where the function is correlated to the output. While there are very few biological examples of this function, the behavior of MNM1 to the metabolome is very similar to electrical circuits that display a XOR or XNOR function (Bonnet et al., 2013). In both XOR and XNOR circuits, high and low levels of input (MNM1 activity) give rise to one level of output (metabolite) while middle levels of input give rise to a different output. Thus, it is possible that the MNM1 gene functions within a XOR or XNOR style genetic circuit that can influence the metabolome. It will require cloning other genes surrounding MNM1 to test this possibility.
Could MNM1 be a Controller of Sink-Source Relations?
The metabolomics analysis showed that MNM1 affects steady state metabolite accumulation in fully mature leaves of 6-week-old plants (Figures 4–6). For these plants, we specifically sampled the lamina at the widest point of the leaf, avoiding the edge and vasculature. Interestingly, all translational and transcriptional GUS fusions showed that at this age, MNM1 is restricted to the young leaves and the edge of older leaves. As such, the tissue sampled for metabolomics has no evidence of expression of MNM1. This suggests that either MNM1 is transported as a protein or that there is some other mechanism for how this gene influences leaf metabolite profiles. One suggestion comes from the observation that raffinose levels across time are altered in genotypes that affect MNM1 function. Raffinose is a key sugar in phloem transport that plays a central role in moving energy between source and sink tissues in plants (Lalonde et al., 2003). The fact that altering MNM1 function leads to a dramatic reduction in raffinose in combination with the trend of MNM1 expression in what should be sink tissues raises the possibility that MNM1 plays a role in coordinating sink and source tissues with regard to the movement of energy or metabolites (Figures 5, 7).
A role in determining energy or metabolite homeostasis between source and sink tissues could be one possible explanation for how altering MNM1 affects metabolite accumulation in tissues that do not appear to express this protein or transcript. It is likely that metabolism is not cell autonomous but instead is linked throughout the plant via the transpiration stream and movement of metabolites between source and sink tissues (Zhang et al., 2010). This potential role for MNM1 in modulating metabolite flows within the plant could also explain why the different MNM1 genotypes lead to altered physiological performance as measured by flowering time and biomass accumulation (Figure 3). The tissue-specific pattern of expression for this gene means that future efforts to describe its regulatory mechanism should focus on newly emerging leaves or leaf edges, tissues that are not frequently investigated.
How does a Novel Gene Obtain the Ability to Affect Primary Metabolism?
Our results suggest that MNM1 has the capacity to regulate central metabolism homeostasis and consequently alter growth and flowering time in Arabidopsis thaliana, potentially from within sink tissues. The ability to regulate source/sink relations is a fundamental physiological process that is shared by all plants. This would then lead one to expect that genes controlling this shared process should be present in all plant species, yet all of our bioinformatics analysis suggests that MNM1 is a gene unique to the Brassicales lineage as we cannot find a close homolog within other eudicot genomes. Other genomes contain paralogs that are more closely related to different Arabidopsis genes (Figure S2). Thus, it appears that the Brassicales lineage has derived a new gene with the ability to influence a core physiological process. This raises the intriguing question of how a new gene can arise and intercalate into the regulatory network for a core physiological process without disrupting it. Similarly, if this can occur once in a plant lineage it could happen in other lineages. The modern reliance on focusing upon homologs of genes with known function leads to an ascertainment bias against studying unique genes, and as such there is no way of currently assessing how frequently conserved physiological processes may have lineage-specific factors. The fact that MNM1 was found through studying natural variation suggests that if these variable lineage-specific factors exist in other plants and crops that they could be playing an unrecognized role in domestication or adaptation. At the very least, this suggests that more effort needs to be put into understanding how lineage-specific genes may link to non-lineage specific physiology.
Conclusion
Our quantitative complementation shows that MNM1, a gene unique to the Brassicales, is likely a causal gene for the metabolomics QTL Met.V.67 in the Arabidopsis Bay × Sha RIL population. This demonstrates that genes can be identified solely through their ability to alter metabolomics variation. Analysis of gene expression and metabolomics effects lead us to hypothesize that MNM1 may modulate source/sink relations in the plant. Little is known about how plants control source/sink relations or how they control metabolic networks in a whole-plant level; however, MNM1 provides a unique tool to begin investigating these questions central to plant metabolism. Future experiments will be required to identify the specific molecular mechanism by which MNM1 generates the observed metabolomics and physiological whole plant effects. Similarly, future work is required to investigate how often lineage-specific genes may influence phenotypes previously thought to be conserved across diverse lineages.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This effort was funded by the NSF DBI grant 0820580 and by the USDA National Institute of Food and Agriculture, Hatch project number CA-D-PLS-7033-H to Daniel J. Kliebenstein.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00415/abstract
References
Alonso, J. M., Stepanova, A. N., Leisse, T. J., Kim, C. J., Chen, H. M., Shinn, P., et al. (2003). Genome-wide Insertional mutagenesis of Arabidopsis thaliana. Science, 301, 653–657. doi: 10.1126/science.1086391
Araujo, W. L., Nunes-Nesi, A., Nikoloski, Z., Sweetlove, L. J., and Fernie, A. R. (2012). Metabolic control and regulation of the tricarboxylic acid cycle in photosynthetic and heterotrophic plant tissues. Plant Cell Environ. 35, 1–21. doi: 10.1111/j.1365-3040.2011.02332.x
Aravind, L., and Landsman, D. (1998). AT-hook motifs identified in a wide variety of DNA binding proteins. Nucleic Acids Res. 26, 4413–4421. doi: 10.1093/nar/26.19.4413
Bonnet, J., Yin, P., Ortiz, M. E., Subsoontorn, P., and Endy, D. (2013). Amplifying genetic logic gates. Science 340, 599–603. doi: 10.1126/science.1232758
Chan, L E. K., Rowe, H. C., Corwin, J. A., Joseph, B., and Kliebenstein, D. J. (2011). Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana. PLoS Biol. 9:e1001125. doi: 10.1371/journal.pbio.1001125
Chan, E. K., Rowe, H. C., Hansen, B. G., and Kliebenstein, D. J. (2010). The complex genetic architecture of the metabolome. PLoS Genet. 6:e1001198. doi: 10.1371/journal.pgen.1001198
Clough, S. J., and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. doi: 10.1046/j.1365-313x.1998.00343.x
Covington, M. F., and Harmer, S. L. (2007). The circadian clock regulates auxin signaling and responses in Arabidopsis. PLoS Biol. 5:e222. doi: 10.1371/journal.pbio.0050222
Covington, M. F., Maloof, J. N., Straume, M., Kay, S. A., and Harmer, S. L. (2008). Global transcriptome analysis reveals circadian regulation of key pathways in plant growth and development. Genome Biol. 9, R130. doi: 10.1186/gb-2008-9-8-r130
Fernie, A. R., Aharoni, A., Willmitzer, L., Stitt, M., Tohge, T., Kopka, J., et al. (2011). Recommendations for reporting metabolite data. Plant Cell 23, 2477–2482. doi: 10.1105/tpc.111.086272
Fiehn, O., Wohlgemuth, G., and Scholz, M. (2005). “Setup and annotation of metabolomic experiments by integrating biological and mass spectrometric metadata,” in Data Integration In The Life Sciences, Proceedings (Berlin). doi: 10.1007/11530084_18
Fiehn, O., Wohlgemuth, G., Scholz, M., Kind, T., Lee, D. Y., Lu, Y., et al. (2008). Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J. 53, 691–704. doi: 10.1111/j.1365-313X.2007.03387.x
Finkemeier, I., Koenig, A. -C., Heard, W., Nunes-Nesi, A., Phuong Anh, P., Leister, D., et al. (2013). Transcriptomic analysis of the role of carboxylic acids in metabolite signaling in arabidopsis leaves. Plant Physiol. 162, 239–253. doi: 10.1104/pp.113.214114
Fujimoto, S., Matsunaga, S., Yonemura, M., Uchiyama, S., Azuma, T., and Fukui, K. (2004). Identification of a novel plant MAR DNA binding protein localized on chromosomal surfaces. Plant Mol. Biol. 56, 225–239. doi: 10.1007/s11103-004-3249-5
Fukushima, A., Kusano, M., Nakamichi, N., Kobayashi, M., Hayashi, N., Sakakibara, H., et al. (2009). Impact of clock-associated Arabidopsis pseudo-response regulators in metabolic coordination. Proc. Natl. Acad. Sci. U.S.A. 106, 7251–7256. doi: 10.1073/pnas.0900952106
Gallavotti, A., Malcomber, S., Gaines, C., Stanfield, S., Whipple, C., Kellogg, E., et al. (2011). BARREN STALK FASTIGIATE1 Is an AT-Hook protein required for the formation of maize ears. Plant Cell 23, 1756–1771. doi: 10.1105/tpc.111.084590
Graf, A., Schlereth, A., Stitt, M., and Smith, A. M. (2010). Circadian control of carbohydrate availability for growth in Arabidopsis plants at night. Proc. Natl. Acad. Sci. U.S.A. 107, 9458–9463. doi: 10.1073/pnas.0914299107
Gutierez, R. A., Stokes, T. L., Thum, K., Xu, X., Obertello, M., Katari, M. S., et al. (2008). Systems approach identifies an organic nitrogen-responsive gene network that is regulated by the master clock control gene CCA1. Proc. Natl. Acad. Sci. U.S.A. 105, 4939–4944. doi: 10.1073/pnas.0800211105
Harmer, S. L. (2009). The circadian system in higher plants. Annu. Rev. Plant Biol. 60, 357–377. doi: 10.1146/annurev.arplant.043008.092054
Harmer, S. L., Hogenesch, L. B., Straume, M., Chang, H. S., Han, B., Zhu, T., et al. (2000). Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290, 2110–2113. doi: 10.1126/science.290.5499.2110
Harmer, S. L., and Kay, S. A. (2005). Positive and negative factors confer phase-specific circadian regulation of transcription in Arabidopsis. Plant Cell 17, 1926–1940. doi: 10.1105/tpc.105.033035
Jimenez-Gomez, J. M., Corwin, J. A., Joseph, B., Maloof, J. N., and Kliebenstein, D. J. (2011a). Genomic analysis of QTLs and genes altering natural variation in stochastic noise. PLoS Genet. 7:e1002295. doi: 10.1371/journal.pgen.1002295
Jimenez-Gomez, J. M., Wallace, A. D., and Maloof, J. N. (2011b). Network analysis identifies ELF3 as a QTL for the shade avoidance response in Arabidopsis. PLoS Genet. 6:e1001100. doi: 10.1371/journal.pgen.1001100
Jin, Y., Luo, Q., Tong, H., Wang, A., Cheng, Z., Tang, J., et al. (2011). An AT-hook gene is required for palea formation and floral organ number control in rice. Dev. Biol. 359, 277–288. doi: 10.1016/j.ydbio.2011.08.023
Karban, R., and Baldwin, I. T. (1997). Induced Responses to Herbivory. Chicago, IL: University of Chicago Press. doi: 10.7208/chicago/9780226424972.001.0001
Kerwin, R. E., Jiménez-Gómez, J. M., Fulop, D., Harmer, S. L., Maloof, J. N., and Kliebenstein, D. J. (2011). Network quantitative trait loci mapping of circadian clock outputs identifies metabolic pathway-to-clock linkages in Arabidopsis. Plant Cell 23, 471–485. doi: 10.1105/tpc.110.082065
Keurentjes, J. J. B., Fu, J. Y., de Vos, C. H. R., Lommen, A., Hall, R. D., Bino, R. J., et al. (2006). The genetics of plant metabolism. Nat. Genet. 38, 842–849. doi: 10.1038/ng1815
Keurentjes, J. J. B., Sulpice, R., Gibon, Y., Steinhauser, M.-C., Fu, J., Koornneef, M., et al. (2008). Integrative analysis of genetic variation in enzyme activities of primary carbohydrate metabolism reveal distinct modes of regulation in Arabidopsis thaliana. Genome Biol. 9:R129. doi: 10.1186/gb-2008-9-8-r129
Kliebenstein, D. (2009). Quantitative genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTLs. Annu. Rev. Plant Biol. 60, 93–114. doi: 10.1146/annurev.arplant.043008.092114
Kliebenstein, D. J. (2008). A role for gene duplication and natural variation of gene expression in the evolution of metabolism. PLoS ONE 3:e1838. doi: 10.1371/journal.pone.0001838
Kliebenstein, D. J., West, M. A. L., Van Leeuwen, H., Kyunga, K., Doerge, R. W., Michelmore, R. W., et al. (2006). Genomic survey of gene expression diversity in Arabidopsis thaliana. Genetics 172, 1179–1189. doi: 10.1534/genetics.105.049353
Lalonde, S., Tegeder, M., Throne-Holst, M., Frommer, W. B., and Patrick, J. W. (2003). Phloem loading and unloading of sugars and amino acids. Plant Cell Environ. 26, 37–56. doi: 10.1046/j.1365-3040.2003.00847.x
Larkin, R. M., Alonso, J. M., Ecker, J. R., and Chory, J. (2003). GUN4, a regulator of chlorophyll synthesis and intracellular signaling. Science 299, 902–906. doi: 10.1126/science.1079978
Li, H., Peng, Z., Yang, X., Wang, W., Fu, J., Wang, J., et al. (2013). Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50. doi: 10.1038/ng.2484
Mackay, T. F. C. (2001). The genetic architecture of quantitative traits. Annu. Rev. Genet. 35, 303–339. doi: 10.1146/annurev.genet.35.102401.090633
Matsuda, F., Okazaki, Y., Oikawa, A., Kusano, M., Nakabayashi, R., Kikuchi, J., et al. (2012). Dissection of genotype-phenotype associations in rice grains using metabolome quantitative trait loci analysis. Plant J. 70, 624–636. doi: 10.1111/j.1365-313X.2012.04903.x
Matsushita, A., Furumoto, T., Ishida, S., and Takahashi, Y. (2007). AGF1, an AT-hook protein, is necessary for the negative feedback of AtGA3ox1 encoding GA 3-oxidase. Plant Physiol. 143, 1152–1162. doi: 10.1104/pp.106.093542
Meyer, R. C., Steinfath, M., Lisec, J., Becher, M., Witucka-Wall, H., Törjék, O., et al. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 104, 4759–4764. doi: 10.1073/pnas.0609709104
Ng, K.-H., Yu, H., and Ito, T. (2009). AGAMOUS controls GIANT KILLER, a multifunctional chromatin modifier in reproductive organ patterning and differentiation. PLoS Biol. 7:e1000251. doi: 10.1371/journal.pbio.1000251
Nozue, K., Covington, M. F., Duek, P. D., Lorrain, S., Fankhauser, C., Harmer, S. L., et al. (2007). Rhythmic growth explained by coincidence between internal and external cues. Nature, 448, 358–361. doi: 10.1038/nature05946
Obayashi, T., Hayashi, S., Saeki, M., Ohta, H., and Kinoshita, K. (2009). ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res. 37, D987–D991. doi: 10.1093/nar/gkn807
Obayashi, T., Kinoshita, K., Nakai, K., Shibaoka, M., Hayashi, S., Saeki, M., et al. (2007). ATTED-II a database of co-expressed genes and cis elements for identifying co-regulated gene groups in Arabidopsis. Nucleic Acids Res. 35, D863–D869. doi: 10.1093/nar/gkl783
Pracharoenwattana, I., Zhou, W., Keech, O., Francisco, P. B., Udomchalothorn, T., Tschoep, H., et al. (2010). Arabidopsis has a cytosolic fumarase required for the massive allocation of photosynthate into fumaric acid and for rapid plant growth on high nitrogen. Plant J. 62, 785–795. doi: 10.1111/j.1365-313X.2010.04189.x
R Development Core Team (2014). R: a Language and Environment for Statistical Computing. Vienna: R. F. f. S. Computing.
Riedelsheimer, C., Lisec, J., Czedik-Eysenberg, A., Sulpice, R., Flis, A., Grieder, C., et al. (2012). Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. U.S.A. 109, 8872–8877. doi: 10.1073/pnas.1120813109
Rowe, H. C., Hansen, B. G., Halkier, B. A., and Kliebenstein, D. J. (2008). Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell 20, 1199–1216. doi: 10.1105/tpc.108.058131
Rowe, H. C., and Kliebenstein, D. J. (2008). Complex genetics control natural variation in Arabidopsis thaliana resistance to Botrytis cinerea. Genetics 180, 2237–2250. doi: 10.1534/genetics.108.091439
Schauer, N., Semel, Y., Balbo, I., Steinfath, M., Repsilber, D., Selbig, J., et al. (2008). Mode of inheritance of primary metabolic traits in tomato. Plant Cell 20, 509–523. doi: 10.1105/tpc.107.056523
Schauer, N., Semel, Y., Roessner, U., Gur, A., Balbo, I., Carrari, F., et al. (2006). Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 24, 447–454. doi: 10.1038/nbt1192
Smith, A. M., and Stitt, M. (2007). Coordination of carbon supply and plant growth. Plant Cell Environ. 30, 1126–1149. doi: 10.1111/j.1365-3040.2007.01708.x
Sulpice, R., Pyl, E. T., Ishihara, H., Trenkamp, S., Steinfath, M., Witucka-Wall, H., et al. (2009). Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. U.S.A. 106, 10348–10353. doi: 10.1073/pnas.0903478106
Van Leeuwen, H., Kliebenstein, D. J., West, M. A. L., Kim, K. D., van Poecke, R., Katagiri, F., et al. (2007). Natural variation among Arabidopsis thaliana accessions for transcriptome response to exogenous salicylic acid. Plant Cell 19, 2099–2110. doi: 10.1105/tpc.107.050641
Weckwerth, W., Loureiro, M. E., Wenzel, K., and Fiehn, O. (2004a). Differential metabolic networks unravel the effects of silent plant phenotypes. Proc. Natl. Acad. Sci. U.S.A. 101, 7809–7814. doi: 10.1073/pnas.0303415101
Weckwerth, W., Wenzel, K., and Fiehn, O. (2004b). Process for the integrated extraction identification, and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics 4, 78–83. doi: 10.1002/pmic.200200500
Weigel, D., and Glazebrook, J. (2002). Arabidopsis: a Laboratory Manual. New York, NY: Cold Spring Harbor.
West, M. A. L., Kim, K., Kliebenstein, D. J., van Leeuwen, H., Michelmore, R. W., Doerge, R. W., et al. (2007). Global eQTL mapping reveals the complex genetic architecture of transcript level variation in Arabidopsis. Genetics 175, 1441–1450. doi: 10.1534/genetics.106.064972
Xiao, Y., Savchenko, T., Baidoo, E. E. K., Chehab, W. E., Hayden, D. M., Tolstikov, V., et al. (2012). Retrograde signaling by the plastidial metabolite MEcPP regulates expression of nuclear stress-response genes. Cell 149, 1525–1535. doi: 10.1016/j.cell.2012.04.038
Yadeta, K. A., Hanemian, M., Smit, P., Hiemstra, J. A., Pereira, A., Marco, Y., et al. (2011). The Arabidopsis thaliana DNA-binding protein AHL19 mediates verticillium wilt resistance. Mol. Plant Microbe Interact. 24, 1582–1591. doi: 10.1094/MPMI-04-11-0090
Keywords: primary metabolism, QTL, TCA, AT-hook motif, Arabidopsis
Citation: Li B and Kliebenstein DJ (2014) The AT-hook motif-encoding gene METABOLIC NETWORK MODULATOR 1 underlies natural variation in Arabidopsis primary metabolism. Front. Plant Sci. 5:415. doi: 10.3389/fpls.2014.00415
Received: 12 June 2014; Accepted: 04 August 2014;
Published online: 22 August 2014.
Edited by:
Zoran Nikoloski, Max-Planck Institute of Molecular Plant Physiology, GermanyReviewed by:
Paula Casati, Centro de Estudios Fotosinteticos-CONICET, ArgentinaAlisdair Fernie, Max Planck Institut for Plant Physiology, Germany
Roosa Anna Emilia Laitinen, Max Planck Institute of Molecular Plant Physiology, Germany
Copyright © 2014 Li and Kliebenstein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel J. Kliebenstein, Department of Plant Sciences, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA e-mail:a2xpZWJlbnN0ZWluQHVjZGF2aXMuZWR1