 Akira Mine
Akira Mine Masanao Sato
Masanao Sato Kenichi Tsuda
Kenichi Tsuda- 1Department of Plant Microbe Interactions, Max Planck Institute for Plant Breeding Research, Cologne, Germany
- 2Okazaki Institute for Integrative Bioscience, National Institute for Basic Biology, National Institutes of Natural Sciences, Okazaki, Japan
Plants are closely associated with microorganisms including pathogens and mutualists that influence plant fitness. Molecular genetic approaches have uncovered a number of signaling components from both plants and microbes and their mode of actions. However, signaling pathways are highly interconnected and influenced by diverse sets of environmental factors. Therefore, it is important to have systems views in order to understand the true nature of plant–microbe interactions. Indeed, systems biology approaches have revealed previously overlooked or misinterpreted properties of the plant immune signaling network. Experimental reconstruction of biological networks using exhaustive combinatorial perturbations is particularly powerful to elucidate network structure and properties and relationships among network components. Recent advances in metagenomics of microbial communities associated with plants further point to the importance of systems approaches and open a research area of microbial community reconstruction. In this review, we highlight the importance of a systems understanding of plant–microbe interactions, with a special emphasis on reconstruction strategies.
Introduction
Systems biology is an area of biology that aims at a comprehensive and mechanistic understanding of complex and dynamic biological processes and phenomena. Systems biology approaches begin with system identification, where the (almost) whole components of a biological system are identified through functional and comparative genomics. This is followed by or coupled with systems analysis, where activities of individual components and their interactions are measured, and mathematical and computational models are built to describe and predict relationships of the system’s components and to explain intrinsic properties of the system. A generated model is used to build new hypotheses to be experimentally verified, and the experimentally verified information increases resolution of a next round of modeling. These repeated processes would advance a mechanistic understanding of how properties and traits of the biological system emerge (systems understanding), and eventually provide a basis for controlling the biological system and designing new biological traits (Kitano, 2002; Ukai and Ueda, 2010).
Due to immobile lifestyles, plants need to optimize their fitness within their living environments. Pathogenic and mutualistic microbes are major factors that influence plant fitness and use host plants for proliferation. Consistent with this, host plants and microbes have coevolved and acquired a number of mechanisms that modulate outcomes of their interactions (Jones and Dangl, 2006; Oldroyd, 2013). In addition, plant responses to microbes are affected by diverse abiotic environmental factors such as temperature and light (Hua, 2013). Thus, plant–microbe interactions are very complex and dynamic biological processes. Therefore, systems biology approaches are required to understand the true nature of plant–microbe interactions.
Plants rely on their innate immune system to resist pathogenic microorganisms. Pattern-triggered immunity (PTI) and effector-triggered immunity (ETI) are two defined modes of plant innate immunity against microbial pathogens (Jones and Dangl, 2006; Tsuda and Katagiri, 2010). PTI is triggered via recognition of conserved microbial molecules known as microbe-associated molecular patterns (MAMPs) by plasma membrane-localized pattern recognition receptors (PRRs; Boller and Felix, 2009; Monaghan and Zipfel, 2012). Well-characterized Arabidopsis PRRs include the flagellin sensing 2 (FLS2) for flg22 (a 22 amino acid peptide from the bacterial protein flagellin), the elongation factor-Tu (EF-Tu) receptor (EFR) for elf18 (a 18 amino acid peptide from the bacterial protein EF-Tu), and the chitin elicitor receptor kinase 1 (CERK1) for chitin (a part of fungal cell walls; Chinchilla et al., 2006; Zipfel et al., 2006; Miya et al., 2007; Wan et al., 2008). PTI is effective against most cases of microbial invasions. However, virulent pathogens have acquired diverse mechanisms to suppress PTI during coevolution (Jones and Dangl, 2006; Boller and He, 2009). For example, a set of effector proteins are delivered into plant cells to manipulate PTI signaling (Dou and Zhou, 2012). To counteract pathogen virulence, plants have evolved resistance (R) proteins, which are often nucleotide-binding leucine-rich repeat proteins (NLRs) as intracellular receptors that specifically recognize pathogen effectors directly or indirectly and that in turn activate ETI (Jones and Dangl, 2006; Jacob et al., 2013). For instance, Arabidopsis NLRs, resistance to Pseudomonas syringae 2 (RPS2) and resistance to P. syringae pv. maculicola 1 (RPM1), recognize actions of the bacterial effectors AvrRpt2 and AvrRpm1, respectively (Jones and Dangl, 2006).
Salicylic acid (SA), jasmonic acid (JA), and ethylene (ET) are immune-related phytohormones. It is generally accepted that SA signaling plays a major role in immunity against biotrophs and hemibiotrophs which require living hosts for multiplication, such as Hyaloperonospora arabidopsidis (Hpa) and P. syringae, respectively (Glazebrook, 2005). In contrast, JA and ET signaling are major contributors of immunity against necrotrophs which actively kill hosts during infection (Glazebrook, 2005). JA, ET, and SA are produced in some cases of PTI and ETI (Spoel et al., 2003; Liu and Zhang, 2004; Mur et al., 2008; Tsuda et al., 2008, 2013; Halim et al., 2009; Tintor et al., 2013). Since JA, ET, and SA signaling pathways intimately interact with synergism and antagonism (Pieterse et al., 2009), these signaling pathways form a complex network to regulate plant immunity.
In this review, we describe how a systems understanding of plant–microbe interactions can be achieved using functional genomics such as protein–protein interactomics, transcriptomics, and metagenomics. We will then focus on experimental reconstruction of complex biological networks, such as the phytohormone signaling network, as a powerful approach to elucidate mechanisms underlying complex and dynamic properties of plant–microbe interactions.
A Physical Interaction Network Between Plant Proteins and Pathogen Effectors
Microbial pathogens range from viruses, bacteria, oomycetes to fungi but often have common hosts. For example, the hemibiotrophic bacterium, P. syringae, and the obligate biotrophic oomycete, Hpa, can colonize the model plant Arabidopsis, raising a question whether there is a common mechanism employed by these evolutionally distant pathogens to suppress Arabidopsis immunity. This question was addressed by generating protein–protein interaction networks between Arabidopsis proteins and effectors from these pathogens based on genome-wide yeast two-hybrid analysis (Figure 1A; Mukhtar et al., 2011). This approach identified 165 putative effector targets, of which 18 were targeted by effectors from both pathogens. Most of the common effector targets were experimentally proved to be important for immunity by genetic analysis. Remarkably, they were enriched in proteins that have more than 50 interactors and were therefore considered as the hubs of a highly interconnected Arabidopsis protein–protein interaction network. For instance, one of such hub proteins, CSN5a, a component of COP9 signalosome, interacted with 29 distinct effectors from Hpa and P. syringae and was a negative regulator of immunity against Hpa. These results suggest that irrespective of their lifestyles, the two pathogens from different kingdoms have independently evolved effectors that converge onto cellular hub proteins to attack the plant immune system (Mukhtar et al., 2011). This is further supported by a study showing that most Hpa effectors promoted bacterial growth in Arabidopsis (Fabro et al., 2011). As similar observations were obtained in other systems such as plant–virus, human–virus, and human–bacteria interactions (Calderwood et al., 2007; De Chassey et al., 2008; Dyer et al., 2008; Elena and Rodrigo, 2012), targeting hub components of host immune systems is likely a common and effective strategy of distinct pathogens.
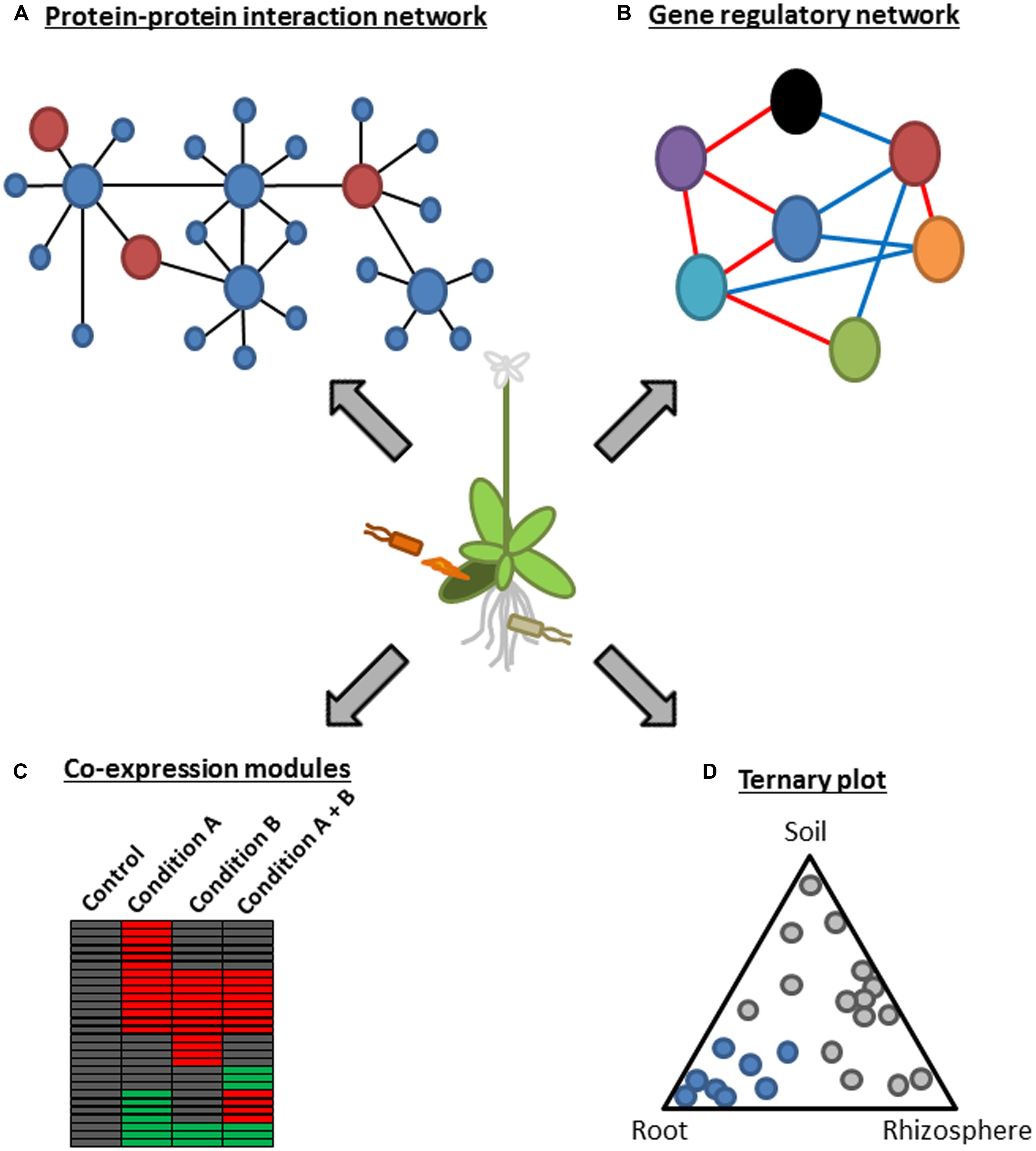
FIGURE 1. A schematic representation of systems approaches using functional genomics. (A) A protein–protein interaction network represents a global picture of a system at the protein level. In this example, several plant hub proteins (large blue circles) that interact with many plant proteins (small blue circles) are targeted by microbial effectors (large red circles). This approach was used in Mukhtar et al. (2011). (B) Gene regulatory network modeling infers regulatory relationships among components of a system. As an example, a network consisting of seven components (circles with different colors) with positive and negative regulatory relationships (red and blue lines, respectively) is depicted. This approach was used in Sato et al. (2010). (C) Co-expression module analysis is useful to visualize behavior of a system under certain conditions. As an example, co-regulated genes under different conditions are visualized in the heatmap. Red and green boxes indicate up-regulated and down-regulated genes in a certain condition, respectively. This approach was used in Zou et al. (2011), Atkinson et al. (2013), Prasch and Sonnewald (2013), and Rasmussen et al. (2013). (D) A ternary plot is used to show influence by three variables on composition. This example depicts bacterial OTUs whose relative abundance changes according to the three compartments, soil, rhizosphere, and root. Blue circles mark OTUs enriched in the root compartment. This approach was used in Bulgarelli et al. (2012) and Lundberg et al. (2012).
Network Modeling Using Expression Profiles of Genetic Mutants
Transcriptome analysis in combination with genetics and a network modeling algorithm was applied to generate a signaling network during immunity against P. syringae carrying the effector AvrRpt2 (Sato et al., 2010). The modified version of locally linear embedding (LLE) enabled detection of weak regulatory relationships by subjecting the residual from the first round of LLE to another round of LLE (Sato et al., 2010). Regulatory relationships among 22 known immune signaling components representing different signaling sectors such as the MAP kinase (MAPK) sector (MPK3 and MPK6; Liu and Zhang, 2004; Wang et al., 2007), the nitric oxide (NO) sectors (NOA1 and NIA2; Wilkinson and Crawford, 1991; Guo et al., 2003) the reactive oxygen species (ROS) sector (RBOHD and RBOHF; Torres et al., 1998), the callose sector (PMR4; Nishimura et al., 2003) and phytohormones were modeled based on similarities in mRNA expression profiles of Arabidopsis mutants deficient in one of the network components (Figure 1B; Sato et al., 2010). The resulting static model inferred extensive negative regulatory relationships among signaling sectors during AvrRpt2-ETI. For example, the JA sector was negatively linked to most of other signaling sectors including the SA sector. The model also inferred a novel negative link between the SA sector and the early MAMP-triggered signaling sectors such as the MAPK, callose, and ET sectors. Importantly, this prediction was experimentally validated, as mutual inhibition between flg22- and SA-induced marker gene inductions was observed in a dose dependent manner, highlighting the significance of the modeling approach based on expression profiles of multiple genetic mutants to generate testable hypotheses (Sato et al., 2010). The negative regulatory relationships among the signaling sectors may make the ETI signaling network robust against perturbation by, for example, pathogen effectors, as perturbation of one sector can be compensated by the other sectors through a switch-like mechanism: one sector(s) gets activated due to the loss of negative effects from the other sector(s) perturbed by pathogens (Sato et al., 2010).
Integration of Multiple Inputs Into Gene Regulation
Environmental factors such as temperature and light are known to affect plant immunity (Hua, 2013), suggesting that plants integrate complex information coming from environments and microbes to optimize their response. Yet, how plants integrate complex information into response is not well understood. Several recent studies tackled this question by comparing transcriptional responses of Arabidopsis to single and multiple stresses (Figure 1C; Atkinson et al., 2013; Prasch and Sonnewald, 2013; Rasmussen et al., 2013). All these studies found that transcriptional responses to multiple stresses are not easily predictable from the responses to single stresses. For example, effects of multiple stresses on gene expression were not simply additive. Consistent with this unpredictability, transcriptional response to the viral pathogen, turnip mosaic virus, was significantly altered when combined with additional abiotic stresses, such as heat and drought (Prasch and Sonnewald, 2013). This was associated with enhanced viral susceptibility under these abiotic stress conditions. Moreover, transcriptional response to the parasitic nematode, Heterodera schachtii, in combination with drought stress was significantly different from those to the single stresses (Atkinson et al., 2013). Rapid alkalinization factor-like 8, methionine gamma lyase, and azelaic acid induced 1 are genes that showed unpredictable expression patterns under the combined stress condition. Overexpression or loss of function of these genes resulted in altered responses to drought stress as well as to nematode infection. These results suggest that specific transcriptional responses to combinatorial stresses are crucial parts of stress tolerance mechanisms in plants.
It is an interesting question whether signal integration for gene regulation is achieved at a transcription factor which is regulated by multiple signaling pathways or at a gene promoter where multiple transcription factors recognize distinct cis-regulatory elements (CREs). Although a number of CREs have been experimentally identified in promoters of stress-responsive genes, this information is not sufficient to correctly predict stress-responsive gene expression. Based on co-expression patterns of Arabidopsis genes under many different biotic and abiotic stresses, a number of putative CREs with characteristics of authentic cis-elements were computationally identified (Figure 1C; Zou et al., 2011). Strikingly, prediction of gene up-regulation by salt, ultraviolet or flg22 was markedly improved by considering binary combinations of known or putative CREs compared to that based on the presence or absence of single CREs. Thus, stress-responsive gene expression is apparently governed by multiple CREs which are signal integration sites for gene regulation, and further systems analysis will be required to untangle the complexity.
Structure Identification of Plant Root-Inhabiting Bacterial Microbiota
Plant–microbe interaction studies have been mostly focused on binary interactions, typically consisting of a single host plant and either pathogenic or mutualistic microbes (Jones and Dangl, 2006; Oldroyd, 2013). However, plants are surrounded by numerous microbes in natural environments, especially in soil, where one of the richest bacterial diversities exists (Gans et al., 2005). Recently, two independent research groups developed similar experimental pipelines for classifying bacterial 16S ribosomal DNA sequences into operational taxonomic unit (OTU) based on high-resolution pyrosequencing, and defined bacterial communities associated with Arabidopsis roots (Bulgarelli et al., 2012; Lundberg et al., 2012). In accordance with prior studies, plant-associated bacterial OTUs that are present inside or firmly attached to roots were significantly different from OTUs found in rhizosphere (defined as soil particles associated with roots) and in bulk soil (Figure 1D), suggesting a selection mechanism(s) that shapes the structure of root-inhabiting bacterial microbiota. Strikingly, by comparing bacterial community profiles associated with different Arabidopsis accessions and in soils from different locations, both studies independently identified a similar profile of root-inhabiting microbiota and reached the same conclusion that the soil type had a greater influence than the host genotype on the bacterial community composition (Bulgarelli et al., 2012; Lundberg et al., 2012). This suggests that the selection process may be facilitated by enrichment and/or exclusion of particular bacteria through interactions among different bacterial species present in a soil. Overall, these studies demonstrated the power of metagenomics for characterization of interactions between plants and microbial communities.
A Stepwise Reconstruction of a Plant Immune Signaling Network From a Ground Level State
One of the longstanding questions in plant–pathogen interactions is why ETI response is very robust compared to PTI, which is overcome by virulent pathogens. A systems analysis using an Arabidopsis quadruple mutant and combinatorial mutants regarding the core immune signaling components, JA, ET, SA, and phytoalexin deficient 4 (PAD4), revealed differential properties of PTI and ETI signaling networks, which provided an answer for this question (Figure 2; Tsuda et al., 2009). Delayed-dehiscence 2 (DDE2), ethylene insensitive 2 (EIN2), and salicylic acid induction-deficient 2 (SID2) are essential components of JA, ET, and SA signaling, respectively (Alonso et al., 1999; Wildermuth et al., 2001; Park et al., 2002). PAD4 is required for pathogen-induced SA accumulation and other immune responses (Jirage et al., 1999), and the latter is considered as the PAD4 signaling sector. Thus, the JA, ET, PAD4, and SA signaling sectors are all compromised in the dde2/ein2/pad4/sid2-quadruple mutant. Growth measurement of P. syringae showed that flg22-triggered immunity (flg22-PTI) and AvrRpt2-triggered immunity (AvrRpt2-ETI) are largely (∼80%) lost in the quadruple mutant. Hence, we consider the quadruple mutant as a (almost) ground level state of the plant immune signaling network accounting for flg22-PTI and AvrRpt2-ETI. The stepwise reconstruction (triple, double, and single mutants to the wild-type) of the plant immune signaling network from the ground level state (the quadruple mutant) followed by signaling allocation analysis revealed that contrary to previous ideas, the JA, ET, PAD4, and SA signaling sectors can all positively contribute to both flg22-PTI and AvrRpt2-ETI. The analysis also illustrated differential relationships among the signaling sectors in PTI and ETI: partly synergistic and partly compensatory in PTI but almost exclusively compensatory in ETI. For example, the interaction between the PAD4 and SA sectors is synergistic in PTI but compensatory in ETI. Thus, reconstructing a network from a ground level state is a powerful approach to understand the true contribution of a signaling sector in and properties of a highly interconnected network.
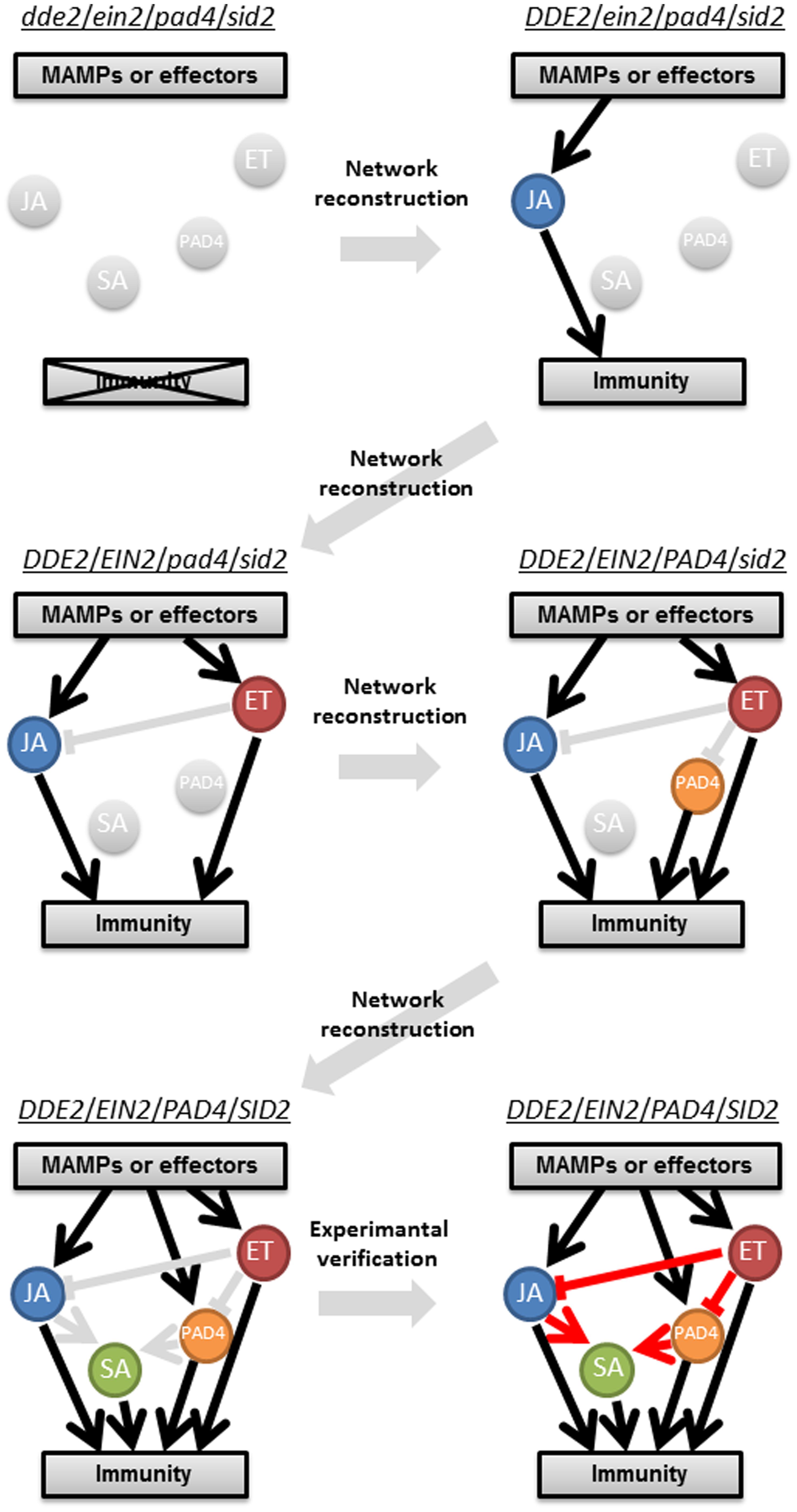
FIGURE 2. A schematic representation of a stepwise reconstruction of a plant immune signaling network from a ground level state. The Arabidopsis dde2/ein2/pad4/sid2-quadruple mutant is considered a ground level state of the plant immune signaling network consisting of the JA, ET, PAD4, and SA signaling sectors. Network reconstruction from the ground level state to the wild-type state via all combinatorial mutants is conducted to quantitatively measure contribution of the individual sectors to immunity (black lines) and activities (circles) and predict their regulatory relationships (gray lines). These predictions are experimentally verified (red lines). For simplicity, only one example is shown for triple, double, and single mutants. This approach was used in Tsuda et al. (2009) and Kim et al. (2014).
Although a synergistic interaction in PTI can be easily attenuated by a pathogen effector that disrupts one of the synergistically interacting signaling sectors, compensatory interactions such as in ETI would make the signaling network highly robust to perturbations. An interesting question is if robust immunity is correlated with robust gene expression. A genome-wide expression profile showed that SA is not essential during AvrRpt2-ETI for regulation of the majority of SA-responsive genes which are regulated in an SA-dependent manner in PTI (Figure 1C; Tsuda et al., 2013), demonstrating extensive compensation by other signaling mechanism(s) for the loss of SA. This seems reasonable because hemibiotrophic and biotrophic pathogens have diverse mechanisms to perturb SA signaling (Zheng et al., 2012; Caillaud et al., 2013; Jiang et al., 2013; Gimenez-Ibanez et al., 2014). The two immune-related MAPKs, MPK3, and MPK6, are activated with different kinetics during PTI and ETI: transient during flg22-PTI but sustained during AvrRpt2-ETI (Tsuda et al., 2013). The SA compensation in ETI seems to be regulated by, in part, sustained activation of MPK3 and MPK6. Thus, although the MAPKs are shared by PTI and ETI, different activation kinetics leads to different downstream events (Tsuda et al., 2013). ROS production and Ca2+ flux are also known to be more sustained during ETI than PTI (Shapiro and Zhang, 2001; Torres et al., 2006; Gao et al., 2013). Thus, time-resolved analysis is needed to fully understand downstream signaling mechanisms.
It should be noted that contribution of the four signaling sectors to immunity differs depending on trigger of immunity and/or pathogens (Tsuda et al., 2009; Maekawa et al., 2012). Therefore, further elucidation of network components is needed to understand the complete structure of the plant immune signaling network. MAPKs (Meng and Zhang, 2013; Tsuda et al., 2013), calcium-dependent protein kinases (Tena et al., 2011; Gao et al., 2013), NO (Zeidler et al., 2004; Zeier et al., 2004), ROS (Torres et al., 2002, 2006), and other phytohormones such as abscisic acid, auxin, and gibberellin (Asselbergh et al., 2008; Robert-Seilaniantz et al., 2011; Mang et al., 2012) are important players in plant immunity and should be integrated in further network analysis.
A Stepwise Reconstruction of a Pathogen Effector Repertoire
Although individual functions of pathogen effectors have been studied, how these effectors function in a coordinated manner has been rarely investigated (Lindeberg et al., 2012). Therefore, a stepwise reconstruction of effectors was used to understand a pathogen virulence strategy (Cunnac et al., 2011). P. syringae pv. tomato DC3000 (Pto) is a model pathogen for systems analysis because virtually all effector candidates that are injected by the type III secretion system (T3SS) were identified (Lindeberg et al., 2006). The Pto mutant (DC3000D28E), which is deficient in the 28 effectors, was found to grow less than the T3SS-deficient mutant in Nicotiana benthamiana. Therefore, this functionally effector less mutant can be considered as a ground level state (Cunnac et al., 2011). A stepwise reconstruction of the effector repertoire to restore virulence of DC3000D28E identified a minimal set of eight effectors with hierarchical functions (AvrPtoB, HopM1, AvrE, HopE1, HopG1, HopAM1, HopAA1, and HopN1; Figure 3). AvrPtoB blocks initiation of PTI signaling by targeting PRRs such as FLS2 (Göhre et al., 2008; Gimenez-Ibanez et al., 2009) and the co-receptor BRI1-associated kinase 1 (BAK1; Shan et al., 2008; Monaghan and Zipfel, 2012). AvrPtoB alone was sufficient to promote growth of DC3000D28E (Cunnac et al., 2011). Bacterial growth was further promoted when HopM1, which targets a PTI component involved in vesicle trafficking (Nomura et al., 2006), was introduced together with AvrPtoB, suggesting that attenuation of receptor functions is a prerequisite for the function of HopM1 (Cunnac et al., 2011). The remaining six effectors supported bacterial growth to near the wild-type level when introduced into DC3000D28E harboring AvrPtoB and HopM1. HopE1, HopG1, HopAM1, HopAA1, and HopN1 seem less important for the bacterial virulence in N. benthamiana, because these effectors are lacking in the genome of P. syringae pv. syringae B728a and P. syringae pv. tabaci 11528, both of which cause disease in N. benthamiana (Vinatzer et al., 2006; Studholme et al., 2009), suggesting that these five effectors can be replaced with other effectors. It would be interesting to investigate whether these two P. syringae strains use effectors with functions similar to or distinct from the five effectors to promote bacterial growth. In summary, reconstruction of the functional effector repertoire from the ground level state together with the comparative genomics of P. syringae strains revealed the virulence strategy by which the bacterial pathogen manipulates plant immunity by deploying a few core effectors and many interchangeable effectors (Lindeberg et al., 2012).
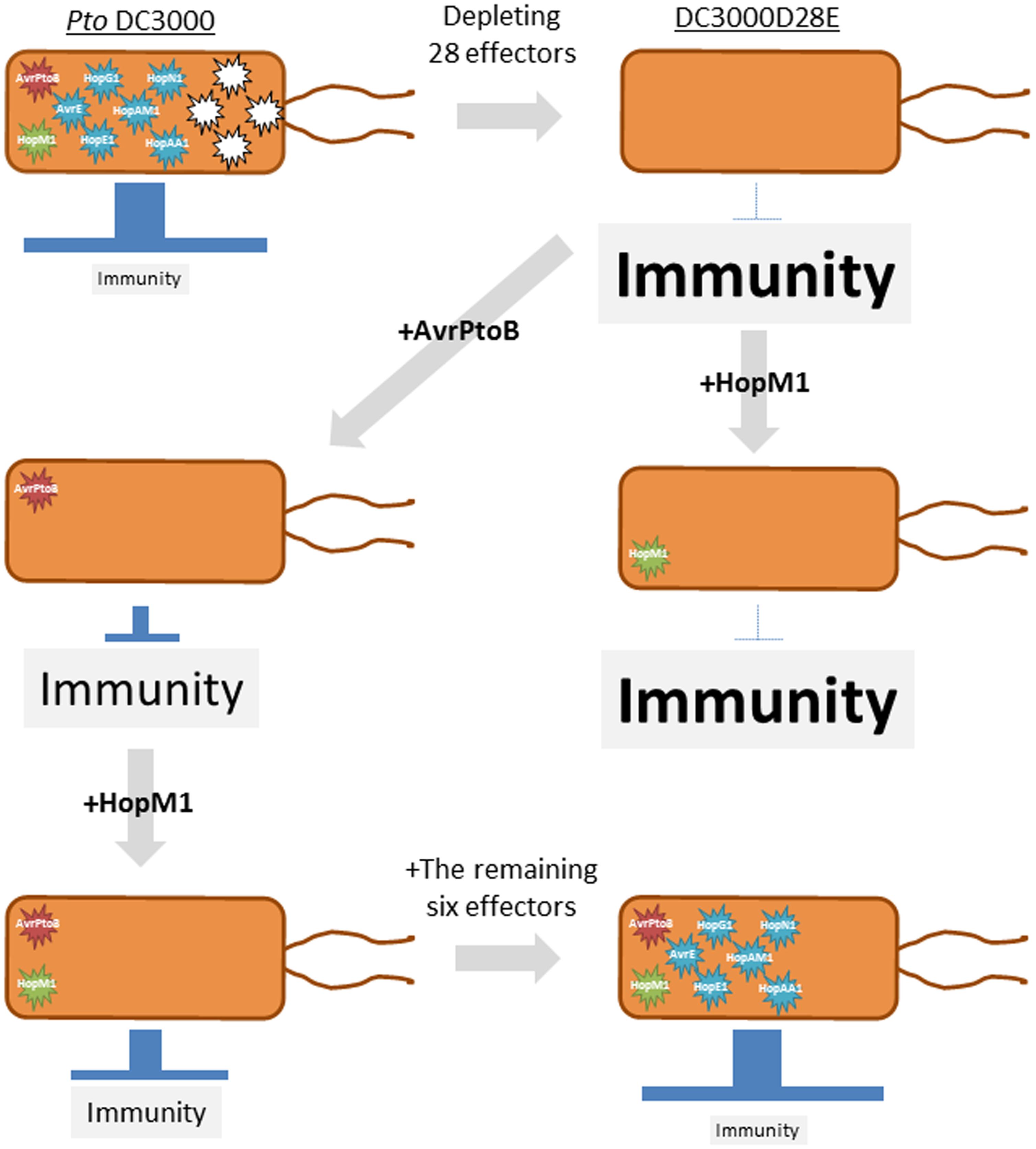
FIGURE 3. A schematic representation of a stepwise reconstruction of a functional effector repertoire of Pseudomonas syringae. The P. syringae pv. tomato (Pto) DC3000 mutant lacking 28 effectors (DC3000D28E) is considered a ground level state for virulence. AvrPtoB but not HopM1 is sufficient to promote in planta bacterial growth. HopM1 requires AvrPtoB to promote bacterial growth. The remaining six effectors (AvrE, HopE1, HopG1, HopAM1, HopAA1, and HopN1) support bacterial growth to near the wild-type level in the presence of AvrPtoB and HopM1. This approach was used in Cunnac et al. (2011).
Modeling Signal Flows Using Experimental Network Reconstruction with Multiple Inputs and Outputs
The plant immune signaling network needs to be robust against pathogen attack and at the same time tunable to achieve optimal fitness since plants combat a diverse range of microbial pathogens including biotrophs and necrotrophs and since unnecessary immune responses have a negative impact on plant fitness (Tsuda and Katagiri, 2010; Alcázar et al., 2011; Mengiste, 2012). The PTI signaling network has some level of robust property although the level of robustness is lower compared to that in ETI (Tsuda et al., 2009). To elucidate the mechanisms underlying intrinsic properties of the PTI signaling network, such as robustness and tunability, the stepwise network reconstruction (Tsuda et al., 2009) was used to generate a dynamic model that describes signal flows and their contributions to immunity in the network consisting of the JA, ET, PAD4, and SA sectors (Kim et al., 2014). Expression levels of marker genes for each of the four sectors at two different time points and growth inhibition of two different P. syringae strains were measured as proxies of signaling sector activities and immune outputs, respectively, after the treatment with three different MAMPs, flg22, elf18 and a modified form of chitin, chitosan (Silipo et al., 2010) as well as mock treatment (Figure 2). Using this multifactorial quantitative data set, a highly predictable dynamic PTI network model was generated. We describe four key findings below.
First, the model predicted that the ET sector suppresses the JA and PAD4 sectors, which is a source of the robustness in the PTI signaling network. Second, although it is often thought that JA signaling inhibits SA signaling (Vlot et al., 2009), the JA sector was predicted to activate the SA sector. This prediction was surprising but experimentally verified as introducing the dde2 mutation into the genotypes containing the pad4 mutation abolished the flg22-induced SA accumulation to the same level as in sid2, indicating that the positive effect of the JA sector on the SA sector is evident only when the PAD4 sector is missing (Kim et al., 2014). This clearly points to the significance of combinatorial perturbations: simultaneous perturbations of multiple components are necessary to truly understand component’s functions in a highly interconnected network. Third, the model revealed that different MAMPs activate the four signaling sectors with different strength, resulting in different immune outputs. For example, flg22 strongly activates the JA, ET, and PAD4 sectors, which leads to strong contributions of the PAD4 and SA sectors to immunity against P. syringae. In contrast, elf18 and chitosan predominantly activate the JA and ET sectors, resulting in weaker immunity against the bacterial pathogens. Since chitosan is a MAMP from fungal pathogens including necrotrophs, it may have been selected to prioritize the JA and ET sectors over the other sectors to mount immunity effective against this type of pathogens (Glazebrook, 2005). Given that a microbe likely presents multiple MAMPs, it is tempting to speculate that plants tune sector activities of the PTI signaling network by sensing different compositions of MAMPs from different microbes to tailor appropriate immune responses for a specific condition. Last, the contribution of the PAD4 sector to immunity against the bacterial pathogens was clearly larger than that of the SA sector. Although a primary function of PAD4 is often thought to be the amplification of SA signaling (Vlot et al., 2009), this result suggests it may be the other way around: a major function of the SA sector is to activate the PAD4 sector rather than its direct contribution to immunity against biotrophic and hemibiotrophic pathogens. It will be necessary to characterize activities of the PAD4 and SA sectors independently of each other in order to re-define the relationship between the two sectors in plant immunity.
Conclusion and Perspectives
The first step of systems biology, system identification, has been achieved in many studies of plant–microbe interactions by means of functional and comparative genomics using model organisms. However, only some of these studies reached the second step, systems analysis, which is the most critical step to understand complex and dynamic properties of plant–microbe interactions (Tsuda et al., 2009, 2013; Sato et al., 2010; Cunnac et al., 2011; Mukhtar et al., 2011; Bulgarelli et al., 2012; Lundberg et al., 2012; Kim et al., 2014). Indeed, systems analysis using reconstruction approaches revealed how a plant immune signaling network is structured to be robust and tunable and how pathogen effectors manipulate plant immunity (Tsuda et al., 2009; Cunnac et al., 2011; Kim et al., 2014). Furthermore, reconstruction of microbial communities would undoubtedly facilitate our understanding of selection mechanisms and functions of plant root-inhabiting bacterial microbiota (Bulgarelli et al., 2012; Lundberg et al., 2012), as shown in studies of phyllosphere and gut microbiota (Atarashi et al., 2013; Bodenhausen et al., 2014). All these studies provide bases for improving future agricultural productivity and food security. For instance, we will, in principle, be able to develop chemical compounds that target core effectors to control pathogens effectively (Cunnac et al., 2011). Another idea will be potentiation of plant immunity by, for example, conferring ETI-like robustness to PTI (Tsuda et al., 2009, 2013; Kim et al., 2014). Notably, transgenic potato plants that trigger ETI-like sustained MAPK activation through recognition of MAMPs were generated (Yamamizo et al., 2006). This transgenic plants showed no developmental abnormalities but exhibited high resistance to both the necrotrophic fungus, Alternaria solani, and the biotrophic oomycete,Phytophthora infestans (Yamamizo et al., 2006). Conventional molecular genetic approaches for rigorous testing of hypotheses emerging from systems analysis will further elucidate molecular mechanisms underlying complexity and dynamics of plant–microbe interactions, which will again provide a basis for controlling the outcomes of plant–microbe interactions.
So far, most systems biology studies have utilized data sets obtained under controlled laboratory conditions. However, complex and dynamic environmental conditions, which plants actually face in nature, should be taken into account to model plant–microbe interactions. Given the significant impact of environmental factors on not only plants but also microbes, it will be a key future challenge to directly incorporate relevant information from a complex environment with changing conditions in order to understand the true nature of plant–microbe interactions. Considering recent technological advances and lowering cost for quantitative measurement such as RNA-seq, we propose that it is time to tackle the grand challenge with the power of systems biology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by The Max Planck Society (Kenichi Tsuda) and a Max Planck fellowship (Akira Mine). Akira Mine was a recipient of a Post-doctoral Fellowship for Research Abroad from Japan Society for the Promotion of Science.
References
Alcázar, R., Reymond, M., Schmitz, G., and de Meaux, J. (2011). Genetic and evolutionary perspectives on the interplay between plant immunity and development. Curr. Opin. Plant Biol. 14, 378–384. doi: 10.1016/j.pbi.2011.04.001
Alonso, J. M., Hirayama, T., Roman, G., Nourizadeh, S., and Ecker, J. R. (1999). EIN2 a bifunctional transducer of ethylene and stress responses in Arabidopsis. Science 284, 2148–2152. doi: 10.1126/science.284.5423.2148
Asselbergh, B., De Vleesschauwer, D., and Hofte, M. (2008). Global switches and fine-tuning-ABA modulates plant pathogen defense. Mol. Plant Microbe Interact. 21, 709–719. doi: 10.1094/MPMI-21-6-0709
Atarashi, K., Tanoue, T., Oshima, K., Suda, W., Nagano, Y., Nishikawa, H.,et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. doi: 10.1038/nature12331
Atkinson, N. J., Lilley, C. J., and Urwin, P. E. (2013). Identification of genes involved in the response of Arabidopsis to simultaneous biotic and abiotic stresses. Plant Physiol. 162, 2028–2041. doi: 10.1104/pp.113.222372
Bodenhausen, N., Bortfeld-Miller, M., Ackermann, M., and Vorholt, J. A. (2014). A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 10:e1004283. doi: 10.1371/journal.pgen.1004283
Boller, T., and Felix, G. (2009). A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 60, 379–406. doi: 10.1146/annurev.arplant.57.032905.105346
Boller, T., and He, S. Y. (2009). Innate immunity in plants: an arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science 324, 742–744. doi: 10.1126/science.1171647
Bulgarelli, D., Rott, M., Schlaeppi, K., Ver Loren Van Themaat, E., Ahmadinejad, N., Assenza, F.,et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Caillaud, M. C., Asai, S., Rallapalli, G., Piquerez, S., Fabro, G., and Jones, J. D. (2013). A downy mildew effector attenuates salicylic acid-triggered immunity in Arabidopsis by interacting with the host mediator complex. PLoS Biol. 11:e1001732. doi: 10.1371/journal.pbio.1001732
Calderwood, M. A., Venkatesan, K., Xing, L., Chase, M. R., Vazquez, A., Holthaus, A. M.,et al. (2007). Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. U.S.A. 104, 7606–7611. doi: 10.1073/pnas.0702332104
Chinchilla, D., Bauer, Z., Regenass, M., Boller, T., and Felix, G. (2006). The Arabidopsis receptor kinase FLS2 binds flg22 and determines the specificity of flagellin perception. Plant Cell 18, 465–476. doi: 10.1105/tpc.105.036574
Cunnac, S., Chakravarthy, S., Kvitko, B. H., Russell, A. B., Martin, G. B., and Collmer, A. (2011). Genetic disassembly and combinatorial reassembly identify a minimal functional repertoire of type III effectors in Pseudomonas syringae. Proc. Natl. Acad. Sci. U.S.A. 108, 2975–2980. doi: 10.1073/pnas.1013031108
De Chassey, B., Navratil, V., Tafforeau, L., Hiet, M. S., Aublin-Gex, A., Agaugue, S.,et al. (2008). Hepatitis C virus infection protein network. Mol. Syst. Biol. 4, 230. doi: 10.1038/msb.2008.66
Dou, D., and Zhou, J. M. (2012). Phytopathogen effectors subverting host immunity: different foes, similar battleground. Cell Host Microbe 12, 484–495. doi: 10.1016/j.chom.2012.09.003
Dyer, M. D., Murali, T. M., and Sobral, B. W. (2008). The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 4:e32. doi: 10.1371/journal.ppat.0040032
Elena, S. F., and Rodrigo, G. (2012). Towards an integrated molecular model of plant-virus interactions. Curr. Opin. Virol. 2, 719–724. doi: 10.1016/j.coviro.2012.09.004
Fabro, G., Steinbrenner, J., Coates, M., Ishaque, N., Baxter, L., Studholme, D. J.,et al. (2011). Multiple candidate effectors from the oomycete pathogen Hyaloperonospora arabidopsidis suppress host plant immunity. PLoS Pathog. 7:e1002348. doi: 10.1371/journal.ppat.1002348
Gans, J., Wolinsky, M., and Dunbar, J. (2005). Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309, 1387–1390. doi: 10.1126/science.1112665
Gao, X., Chen, X., Lin, W., Chen, S., Lu, D., Niu, Y.,et al. (2013). Bifurcation of Arabidopsis NLR immune signaling via Ca2+-dependent protein kinases. PLoS Pathog. 9:e1003127. doi: 10.1371/journal.ppat.1003127
Gimenez-Ibanez, S., Boter, M., Fernandez-Barbero, G., Chini, A., Rathjen, J. P., and Solano, R. (2014). The bacterial effector HopX1 targets JAZ transcriptional repressors to activate jasmonate signaling and promote infection in Arabidopsis. PLoS Biol. 12:e1001792. doi: 10.1371/journal.pbio.1001792
Gimenez-Ibanez, S., Hann, D. R., Ntoukakis, V., Petutschnig, E., Lipka, V., and Rathjen, J. P. (2009). AvrPtoB targets the LysM receptor kinase CERK1 to promote bacterial virulence on plants. Curr. Biol. 19, 423–429. doi: 10.1016/j.cub.2009.01.054
Glazebrook, J. (2005). Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227. doi: 10.1146/annurev.phyto.43.040204.135923
Göhre, V., Spallek, T., Haweker, H., Mersmann, S., Mentzel, T., Boller, T.,et al. (2008). Plant pattern-recognition receptor FLS2 is directed for degradation by the bacterial ubiquitin ligase AvrPtoB. Curr. Biol. 18, 1824–1832. doi: 10.1016/j.cub.2008.10.063
Guo, F. Q., Okamoto, M., and Crawford, N. M. (2003). Identification of a plant nitric oxide synthase gene involved in hormonal signaling. Science 302, 100–103. doi: 10.1126/science.1086770
Halim, V. A., Altmann, S., Ellinger, D., Eschen-Lippold, L., Miersch, O., Scheel, D.,et al. (2009). PAMP-induced defense responses in potato require both salicylic acid and jasmonic acid. Plant J. 57, 230–242. doi: 10.1111/j.1365-313X.2008.03688.x
Hua, J. (2013). Modulation of plant immunity by light, circadian rhythm, and temperature. Curr. Opin. Plant Biol. 16, 406–413. doi: 10.1016/j.pbi.2013.06.017
Jacob, F., Vernaldi, S., and Maekawa, T. (2013). Evolution and conservation of plant NLR functions. Front. Immunol. 4:297. doi: 10.3389/fimmu.2013.00297
Jiang, S., Yao, J., Ma, K. W., Zhou, H., Song, J., He, S. Y.,et al. (2013). Bacterial effector activates jasmonate signaling by directly targeting JAZ transcriptional repressors. PLoS Pathog. 9:e1003715. doi: 10.1371/journal.ppat.1003715
Jirage, D., Tootle, T. L., Reuber, T. L., Frost, L. N., Feys, B. J., Parker, J. E.,et al. (1999). Arabidopsis thaliana PAD4 encodes a lipase-like gene that is important for salicylic acid signaling. Proc. Natl. Acad. Sci. U.S.A. 96, 13583–13588. doi: 10.1073/pnas.96.23.13583
Jones, J. D., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286.
Kim, Y., Tsuda, K., Igarashi, D., Hillmer, R. A., Sakakibara, H., Myers, C. L.,et al. (2014). Mechanisms underlying robustness and tunability in a plant immune signaling network. Cell Host Microbe 15, 84–94. doi: 10.1016/j.chom.2013.12.002
Kitano, H. (2002). Systems biology: a brief overview. Science 295, 1662–1664. doi: 10.1126/science.1069492
Lindeberg, M., Cartinhour, S., Myers, C. R., Schechter, L. M., Schneider, D. J., and Collmer, A. (2006). Closing the circle on the discovery of genes encoding Hrp regulon members and type III secretion system effectors in the genomes of three model Pseudomonas syringae strains. Mol. Plant Microbe Interact. 19, 1151–1158. doi: 10.1094/MPMI-19-1151
Lindeberg, M., Cunnac, S., and Collmer, A. (2012). Pseudomonas syringae type III effector repertoires: last words in endless arguments. Trends Microbiol. 20, 199–208. doi: 10.1016/j.tim.2012.01.003
Liu, Y., and Zhang, S. (2004). Phosphorylation of 1-aminocyclopropane-1-carboxylic acid synthase by MPK6 a stress-responsive mitogen-activated protein kinase, induces ethylene biosynthesis in Arabidopsis. Plant Cell 16, 3386–3399. doi: 10.1105/tpc.104.026609
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S.,et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Maekawa, T., Kracher, B., Vernaldi, S., Ver Loren Van Themaat, E., and Schulze-Lefert, P. (2012). Conservation of NLR-triggered immunity across plant lineages. Proc. Natl. Acad. Sci. U.S.A. 109, 20119–20123. doi: 10.1073/pnas.1218059109
Mang, H. G., Qian, W., Zhu, Y., Qian, J., Kang, H. G., Klessig, D. F.,et al. (2012). Abscisic acid deficiency antagonizes high-temperature inhibition of disease resistance through enhancing nuclear accumulation of resistance proteins SNC1 and RPS4 in Arabidopsis. Plant Cell 24, 1271–1284. doi: 10.1105/tpc.112.096198
Meng, X., and Zhang, S. (2013). MAPK cascades in plant disease resistance signaling. Annu. Rev. Phytopathol. 51, 245–266. doi: 10.1146/annurev-phyto-082712-102314
Mengiste, T. (2012). Plant immunity to necrotrophs. Annu. Rev. Phytopathol. 50, 267–294. doi: 10.1146/annurev-phyto-081211-172955
Miya, A., Albert, P., Shinya, T., Desaki, Y., Ichimura, K., Shirasu, K.,et al. (2007). CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 104, 19613–19618. doi: 10.1073/pnas.0705147104
Monaghan, J., and Zipfel, C. (2012). Plant pattern recognition receptor complexes at the plasma membrane. Curr. Opin. Plant Biol. 15, 349–357. doi: 10.1016/j.pbi.2012.05.006
Mukhtar, M. S., Carvunis, A. R., Dreze, M., Epple, P., Steinbrenner, J., Moore, J.,et al. (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333, 596–601. doi: 10.1126/science.1203659
Mur, L. A. J., Laarhoven, L. J. J., Harren, F. J. M., Hall, M. A., and Smith, A. R. (2008). Nitric oxide interacts with salicylate to regulate biphasic ethylene production during the hypersensitive response. Plant Physiol. 148, 1537–1546. doi: 10.1104/pp.108.124404
Nishimura, M. T., Stein, M., Hou, B. H., Vogel, J. P., Edwards, H., and Somerville, S. C. (2003). Loss of a callose synthase results in salicylic acid-dependent disease resistance. Science 301, 969–972. doi: 10.1126/science.1086716
Nomura, K., Debroy, S., Lee, Y. H., Pumplin, N., Jones, J., and He, S. Y. (2006). A bacterial virulence protein suppresses host innate immunity to cause plant disease. Science 313, 220–223. doi: 10.1126/science.1129523
Oldroyd, G. E. (2013). Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 11, 252–263. doi: 10.1038/nrmicro2990
Park, J. H., Halitschke, R., Kim, H. B., Baldwin, I. T., Feldmann, K. A., and Feyereisen, R. (2002). A knock-out mutation in allene oxide synthase results in male sterility and defective wound signal transduction in Arabidopsis due to a block in jasmonic acid biosynthesis. Plant J. 31, 1–12. doi: 10.1046/j.1365-313X.2002.01328.x
Pieterse, C. M., Leon-Reyes, A., Van Der Ent, S., and Van Wees, S. C. (2009). Networking by small-molecule hormones in plant immunity. Nat. Chem. Biol. 5, 308–316. doi: 10.1038/nchembio.164
Prasch, C. M., and Sonnewald, U. (2013). Simultaneous application of heat, drought, and virus to Arabidopsis plants reveals significant shifts in signaling networks. Plant Physiol. 162, 1849–1866. doi: 10.1104/pp.113.221044
Rasmussen, S., Barah, P., Suarez-Rodriguez, M. C., Bressendorff, S., Friis, P., Costantino, P.,et al. (2013). Transcriptome responses to combinations of stresses in Arabidopsis. Plant Physiol. 161, 1783–1794. doi: 10.1104/pp.112.210773
Robert-Seilaniantz, A., Grant, M., and Jones, J. D. (2011). Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annu. Rev. Phytopathol. 49, 317–343. doi: 10.1146/annurev-phyto-073009-114447
Sato, M., Tsuda, K., Wang, L., Coller, J., Watanabe, Y., Glazebrook, J.,et al. (2010). Network modeling reveals prevalent negative regulatory relationships between signaling sectors in Arabidopsis immune signaling. PLoS Pathog. 6:e1001011. doi: 10.1371/journal.ppat.1001011
Shan, L., He, P., Li, J., Heese, A., Peck, S. C., Nurnberger, T.,et al. (2008). Bacterial effectors target the common signaling partner BAK1 to disrupt multiple MAMP receptor-signaling complexes and impede plant immunity. Cell Host Microbe 4, 17–27. doi: 10.1016/j.chom.2008.05.017
Shapiro, A. D., and Zhang, C. (2001). The role of NDR1 in avirulence gene-directed signaling and control of programmed cell death in Arabidopsis. Plant Physiol. 127, 1089–1101. doi: 10.1104/pp.010096
Silipo, A., Erbs, G., Shinya, T., Dow, J. M., Parrilli, M., Lanzetta, R.,et al. (2010). Glyco-conjugates as elicitors or suppressors of plant innate immunity. Glycobiology 20, 406–419. doi: 10.1093/glycob/cwp201
Spoel, S. H., Koornneef, A., Claessens, S. M., Korzelius, J. P., Van Pelt, J. A., Mueller, M. J.,et al. (2003). NPR1 modulates cross-talk between salicylate- and jasmonate-dependent defense pathways through a novel function in the cytosol. Plant Cell 15, 760–770. doi: 10.1105/tpc.009159
Studholme, D. J., Ibanez, S. G., Maclean, D., Dangl, J. L., Chang, J. H., and Rathjen, J. P. (2009). A draft genome sequence and functional screen reveals the repertoire of type III secreted proteins of Pseudomonas syringae pathovar tabaci 11528. BMC Genomics 10:395. doi: 10.1186/1471-2164-10-395
Tena, G., Boudsocq, M., and Sheen, J. (2011). Protein kinase signaling networks in plant innate immunity. Curr. Opin. Plant Biol. 14, 519–529. doi: 10.1016/j.pbi.2011.05.006
Tintor, N., Ross, A., Kanehara, K., Yamada, K., Fan, L., Kemmerling, B.,et al. (2013). Layered pattern receptor signaling via ethylene and endogenous elicitor peptides during Arabidopsis immunity to bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 110, 6211–6216. doi: 10.1073/pnas.1216780110
Torres, M. A., Dangl, J. L., and Jones, J. D. (2002). Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc. Natl. Acad. Sci. U.S.A. 99, 517–522. doi: 10.1073/pnas.012452499
Torres, M. A., Jones, J. D., and Dangl, J. L. (2006). Reactive oxygen species signaling in response to pathogens. Plant Physiol. 141, 373–378. doi: 10.1104/pp.106.079467
Torres, M. A., Onouchi, H., Hamada, S., Machida, C., Hammond-Kosack, K. E., and Jones, J. D. (1998). Six Arabidopsis thaliana homologues of the human respiratory burst oxidase (gp91phox). Plant J. 14, 365–370. doi: 10.1046/j.1365-313X.1998.00136.x
Tsuda, K., and Katagiri, F. (2010). Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant Biol. 13, 459–465. doi: 10.1016/j.pbi.2010.04.006
Tsuda, K., Mine, A., Bethke, G., Igarashi, D., Botanga, C. J., Tsuda, Y.,et al. (2013). Dual regulation of gene expression mediated by extended MAPK activation and salicylic acid contributes to robust innate immunity in Arabidopsis thaliana. PLoS Genet. 9:e1004015. doi: 10.1371/journal.pgen.1004015
Tsuda, K., Sato, M., Glazebrook, J., Cohen, J. D., and Katagiri, F. (2008). Interplay between MAMP-triggered and SA-mediated defense responses. Plant J. 53, 763–775. doi: 10.1111/j.1365-313X.2007.03369.x
Tsuda, K., Sato, M., Stoddard, T., Glazebrook, J., and Katagiri, F. (2009). Network properties of robust immunity in plants. PLoS Genet. 5:e1000772. doi: 10.1371/journal.pgen.1000772
Ukai, H., and Ueda, H. R. (2010). Systems biology of mammalian circadian clocks. Annu. Rev. Physiol. 72, 579–603. doi: 10.1146/annurev-physiol-073109-130051
Vinatzer, B. A., Teitzel, G. M., Lee, M. W., Jelenska, J., Hotton, S., Fairfax, K.,et al. (2006). The type III effector repertoire of Pseudomonas syringae pv. syringae B728a and its role in survival and disease on host and non-host plants. Mol. Microbiol. 62, 26–44. doi: 10.1111/j.1365-2958.2006.05350.x
Vlot, A. C., Dempsey, D. A., and Klessig, D. F. (2009). Salicylic acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 47, 177–206. doi: 10.1146/annurev.phyto.050908.135202
Wan, J., Zhang, X. C., Neece, D., Ramonell, K. M., Clough, S., Kim, S. Y.,et al. (2008). A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 20, 471–481. doi: 10.1105/tpc.107.056754
Wang, H., Ngwenyama, N., Liu, Y., Walker, J. C., and Zhang, S. (2007). Stomatal development and patterning are regulated by environmentally responsive mitogen-activated protein kinases in Arabidopsis. Plant Cell 19, 63–73. doi: 10.1105/tpc.106.048298
Wildermuth, M. C., Dewdney, J., Wu, G., and Ausubel, F. M. (2001). Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 414, 562–565. doi: 10.1038/35107108
Wilkinson, J. Q., and Crawford, N. M. (1991). Identification of the Arabidopsis CHL3 gene as the nitrate reductase structural gene NIA2. Plant Cell 3, 461–471. doi: 10.1105/tpc.3.5.461
Yamamizo, C., Kuchimura, K., Kobayashi, A., Katou, S., Kawakita, K., Jones, J. D.,et al. (2006). Rewiring mitogen-activated protein kinase cascade by positive feedback confers potato blight resistance. Plant Physiol. 140, 681–692. doi: 10.1104/pp.105.074906
Zeidler, D., Zahringer, U., Gerber, I., Dubery, I., Hartung, T., Bors, W.,et al. (2004). Innate immunity in Arabidopsis thaliana: lipopolysaccharides activate nitric oxide synthase (NOS) and induce defense genes. Proc. Natl. Acad. Sci. U.S.A. 101, 15811–15816. doi: 10.1073/pnas.0404536101
Zeier, J., Delledonne, M., Mishina, T., Severi, E., Sonoda, M., and Lamb, C. (2004). Genetic elucidation of nitric oxide signaling in incompatible plant-pathogen interactions. Plant Physiol. 136, 2875–2886. doi: 10.1104/pp.104.042499
Zheng, X. Y., Spivey, N. W., Zeng, W., Liu, P. P., Fu, Z. Q., Klessig, D. F.,et al. (2012). Coronatine promotes Pseudomonas syringae virulence in plants by activating a signaling cascade that inhibits salicylic acid accumulation. Cell Host Microbe 11, 587–596. doi: 10.1016/j.chom.2012.04.014
Zipfel, C., Kunze, G., Chinchilla, D., Caniard, A., Jones, J. D., Boller, T.,et al. (2006). Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell 125, 749–760. doi: 10.1016/j.cell.2006.03.037
Keywords: systems biology, experimental network reconstruction, plant immunity, phytohormone, robustness, tunability, pathogen effector, microbial community
Citation: Mine A, Sato M and Tsuda K (2014) Toward a systems understanding of plant–microbe interactions. Front. Plant Sci. 5:423. doi: 10.3389/fpls.2014.00423
Received: 15 May 2014; Accepted: 07 August 2014;
Published online: 25 August 2014.
Edited by:
Cris Argueso, Colorado State University, USAReviewed by:
Camila Caldana, Brazilian Bioethanol Science and Technology Laboratory – Centro Nacional de Pesquisa em Energia e Materiais/Associação Brasileira de Tecnologia de Luz Síncrotron, BrazilMehdi Kabbage, University of Wisconsin–Madison, USA
Copyright © 2014 Mine, Sato and Tsuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenichi Tsuda, Department of Plant Microbe Interactions, Max Planck Institute for Plant Breeding Research, Carl-von-Linne Weg 10, Cologne 50829, Germany e-mail:dHN1ZGFAbXBpcHoubXBnLmRl