 Anja Bus1,2
Anja Bus1,2 Niklas Körber1,2
Niklas Körber1,2 Isobel A. P. Parkin3
Isobel A. P. Parkin3 Birgit Samans4
Birgit Samans4 Rod J. Snowdon4
Rod J. Snowdon4 Jinquan Li1Benjamin Stich1*
Jinquan Li1Benjamin Stich1*- 1Quantitative Crop Genetics, Max Planck Institute for Plant Breeding Research, Cologne, Germany
- 2Crop Genetics and Biotechnology Unit, Institute of Crop Science and Resource Conservation, University of Bonn, Bonn, Germany
- 3Saskatoon Research Centre, Agriculture and Agri-Food Canada, Saskatoon, SK, Canada
- 4Department of Plant Breeding, Research Centre for Biosystems, Land Use and Nutrition, Justus Liebig University, Giessen, Germany
Knowing the genetic basis of the plant ionome is essential for understanding the control of nutrient transport and accumulation. The aim of this research was to (i) study mineral nutrient concentrations in a large and diverse set of Brassica napus, (ii) describe the relationships between the shoot ionome and seedling development, and (iii) identify genetic regions associated with variation of the shoot ionome. The plant material under study was a germplasm set consisting of 509 inbred lines that was genotyped by a 6K single nucleotide polymorphism (SNP) array and phenotyped by analyzing the concentrations of eleven mineral nutrients in the shoots of 30 days old seedlings. Among mineral concentrations, positive correlations were found, whereas mineral concentrations were mainly negatively correlated with seedling development traits from earlier studies. In a genome-wide association mapping approach, altogether 29 significantly associated loci were identified across seven traits after correcting for multiple testing. The associations included a locus with effects on the concentrations of Cu, Mn, and Zn on chromosome C3, and a genetic region with multiple associations for Na concentration on chromosome A9. This region was situated within an association hotspot close to SOS1, a key gene for Na tolerance in plants.
1. Introduction
Plant cells depend on the presence of nutritional elements, which fulfill a variety of functions, where elements act, amongst others, as regulators, cofactors, or structural components (Baxter, 2009). The composition of plant mineral nutrients and trace elements is referred to as the ionome (Salt et al., 2008). Its studies, so-called ionomics, aim to reveal knowledge about these functions and the networks controlling uptake, transport, and accumulation of elements. Ionomics have become of great interest in research due to its relevance in plant development and performance, where each nutrient plays a specific role. However, some elements have similar chemical properties and therefore compete for uptake or carriers (Marschner, 1995). Ionomics approaches therefore do not only address the question of quantifying nutrient concentrations but also how ions interact with each other, which factors influence their uptake and accumulation, and how the ionome can serve as indicator for associated traits.
Previous studies have characterized either partly, or as a whole, the plant ionome and used this knowledge to gain further insights into the genetic or physiologic architecture. Lahner et al. (2003) quantified 18 elements in shoots of 6000 mutagenized plants of Arabidopsis thaliana and analyzed the mutation frequency based on the elemental profiles. Their findings led to the conclusion that about 2–4% of the genome is involved in the regulation of the nutrient and trace element content in the species. Another extensive study of the ionome of A. thaliana under different iron and phosphorus conditions showed through logistic regression modeling that there is a relationship between the ionome and specific physiological responses, e.g., to Fe or P deficiency (Baxter et al., 2008). Although most of the knowledge comes from model species, ionomics approaches have recently also been performed in crops such as maize (Baxter et al., 2013), soybean (Ziegler et al., 2013), and barley (Wu et al., 2013). For the genus Brassica there is limited knowledge on the genetic basis of ionomic variation. A QTL analysis on loci controlling mineral concentrations in B. napus under normal and deficient boron conditions (Liu et al., 2009) revealed specific correlations among seven different elements and several QTLs as well as epistatic interaction pairs for mineral concentrations. These were specific for each boron regime, implying that mineral homoeostasis is controlled by genetic factors and that ion transport under different regimes is driven by multiple genes. In B. rapa, the species that forms the B. napus A genome, ten QTLs were detected for concentrations of 11 minerals under different Zn regimes (Wu et al., 2008). In B. oleracea, which in turn represents the C genome of B. napus, there are a number of known QTLs for the highly heritable Ca and Mg concentrations in shoots, of which the most significant QTLs are localized on chromosome C9 (Broadley et al., 2008). Moreover, it is known from a study on B. napus seeds that, on the one hand, multiple loci control mineral concentrations and, on the other hand, physiological and molecular mechanisms driving mineral accumulation are likely to be shared between elements (Ding et al., 2010). However, no study has provided a species- and genome-wide analysis of the B. napus ionome.
A healthy and robust development of the seedling is the prerequisite for a vigorous, high-yielding crop. Understanding the genetic basis of the shoot ionome of young plants will give deeper insights into how nutrient transport and accumulation are controlled. Furthermore, studying the relationship between ionomics and seedling development will allow an understanding of so far unknown interactions.
With regard to the aforementioned studies, association mapping (AM) based on linkage disequilibrium (LD) is a complementary approach for the detection of associations between quantitative traits and variation on the molecular level in crops. AM allows to discover the genetic bases for traits under certain scenarios, including when contributing alleles are at moderate frequencies within the population (Oraguzie et al., 2007). The decreasing costs of single nucleotide polymorphism (SNP) arrays and the advent of next-generation sequencing technologies allow the development of thousands of SNPs even in the allotetraploid B. napus (Trick et al., 2009; Bus et al., 2012; Snowdon and Iniguez Luy, 2012; Edwards et al., 2013). Therefore, SNPs have become the marker type of choice for genome-wide AM. As whole-genome resequencing is still laborious and expensive, mostly transcriptomics techniques have been chosen as high-throughput SNP detection method for B. napus. These approaches have been carried out on relatively small SNP and plant sets. Here we present an AM study on a large-size worldwide germplasm set that extensively represents the species B. napus, using a 6K SNP array to allow high resolution mapping. The objectives of our study were to (i) examine mineral nutrient concentrations and their interactions in a large and diverse set of B. napus, (ii) study the relationships between the shoot ionome and seedling development traits, and (iii) identify genetic regions associated with variation of the shoot ionome.
2. Materials and Methods
We investigated a species-wide diversity set of B. napus, consisting of 509 inbreds, of which the genetic diversity and population structure had been characterized with 89 simple sequence repeat (SSR) markers as described by Bus et al. (2011). The diversity set consists of 183 winter oilseed rape (OSR), 22 winter fodder, 73 swede (B. napus ssp. napobrassica), 7 semi-winter OSR, 204 spring OSR, 4 spring fodder, 10 vegetable, and 6 unspecified inbred lines.
For ionome and seedling development analysis, the plant material was grown in a greenhouse experiment that comprised six replicates which were performed in a row within a period of 6 months, where each replicate lasted 30 days (Körber et al., 2012). The experimental design was an alpha lattice with 24 blocks, each of 24 pots. The temperature was set to 24°C during 16 h of light exposure and to 18°C during 8 h of dark exposure. Seeds were sown in 10 × 10 cm pots filled with soil (Einheitserde, Balster Einheitserdewerk, Fröndenberg, Germany). During cultivation, the plants were treated twice with liquid fertilizer Wuxal Super 8-8-6 (Haug, Ammerbuch, Germany). From 8 to 16 days after sowing, digital infrared images of the seedlings were taken. A mask of each plant was created using a color threshold, and the projected leaf areas (LA08–LA16) of the seedlings were calculated using the digital image processing software ImageJ 1.42q (http://rsb.info.nih.gov/ij/) (Table 1). Other seedling development traits were derived from the plant masks at 10 days after sowing (Table 1). Growth parameters a (intercept, PRA) and k (growth factor, PRK) were calculated from LA08–LA16 by non-linear regression. Furthermore, data on the relative leaf chlorophyll content as measured by Minolta company-defined SPAD (Soil Plant Analysis Development) values (SPD), fresh mass (FHM), dry mass (DYM), and H2O content (H2O) of the plants were collected after a growing period of 30 days after sowing. Plant material for ionome analysis was harvested from 30 days old seedlings, oven-dried, and lyophilized.
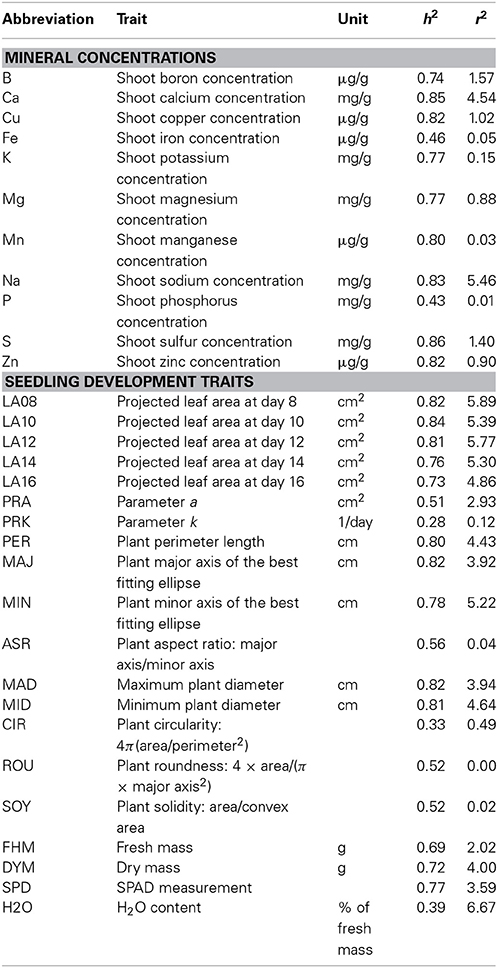
Table 1. Abbreviations of eleven mineral concentrations and twenty seedling development traits, broad sense heritability on an entry mean basis (h2), and the percentage of phenotypic variation explained by population structure (r2) measured in a B. napus germplasm set.
The entirety of all investigated mineral concentrations is herein referred to as the shoot ionome. The concentrations of altogether eleven plant nutritional elements, B, Ca, Cu, Fe, K, Mg, Mn, Na, P, S, and Zn (Table 1), were analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES, Vista Pro Radial, Varian, Palo Alto, CA, USA) at Landesanstalt für Landwirtschaftliche Chemie at the University of Hohenheim (Stuttgart, Germany).
Plant genomic DNA for SNP genotyping was extracted from fresh or lyophilized leaf material using the BioSprint 96 DNA Plant Kit and the BioSprint 96 robotic workstation (Qiagen), following the manufacturer's protocol. Genotyping of the SNPs was performed at Agriculture and Agri-Food Canada using a custom Illumina Infinium 6K array (http://aafc-aac.usask.ca/ASSYST/). Only SNPs with a minor allele frequency larger than 5% were included in the association analysis. Of all of the SNPs from the array, altogether 3910 SNPs were used, and SNP data were available for 505 inbreds of the germplasm set.
2.1. Statistical Analyses
Adjusted entry means (AEM) were calculated for each genotype-mineral concentration combination using the following mixed model:
where yikm was the observation of the ith genotype in the mth block of the kth replicate, μ the general mean, gi the effect of the ith genotype, rk the effect of the kth replicate, bkm the effect of the mth block in the kth replicate, and eikm the residual error. Outliers in residual plots were discarded. For the calculation of AEM, we considered g as fixed and all other effects as random. For estimating the genotypic variance (σ2g) and the error variance (σ2e), all effects were considered as random. The heritabilities h2 of each mineral concentration on an entry mean basis were calculated as follows:
where b was the number of replicates. Variance components and AEM were determined by the restricted maximum likelihood (REML) method. The mixed model analyses were performed with ASREML release 2.0 (Gilmour et al., 2006).
The Pearson partial correlation coefficient (Fisher, 1924) was calculated for all pairs of traits, with significance levels corrected according to Holm's method (Holm, 1979). Associations among mineral concentrations as well as between mineral concentrations and the seedling development traits described by Körber et al. (2012) were visualized by principal component analysis (PCA). To do so, phenotype data were processed by using the R prcomp() function. All of the 509 genotypes of this study had been assigned to three clusters based on SSR marker data (Bus et al., 2011). Whenever population structure needed to be accounted for in our statistical analyses, the germplasm set was separated into these clusters, referred to as MCLUST clusters 1, 2, and 3. Multiple stepwise regression of seedling development traits was run on mineral concentrations. To identify the optimum linear model which predicts the best representation of the seedling development traits, mineral concentrations were used as independent variables. The latter were selected based on the Bayesian information criterion (BIC) (Schwarz, 1978):
where yi was the observation of the ith genotype, for which the best set of predictors (mineral concentrations) was to be identified, μ the intercept term, v the number of selected variables, bp the regression coefficient of the pth mineral nutrient, xpi the AEM of the pth mineral nutrient for the ith genotype, and ei the residual.
2.2. Association Analysis and Assessment of Linkage Disequilibrium
The mixed model according to the PK method (Stich et al., 2008) was used to analyze associations between polymorphic sites and the mineral concentrations: The PK-mixed model was
where Mip was the entry mean of the ith entry carrying allele p, ap the effect of allele p, vu the effect of the uth column of the population structure matrix D, g*i the residual genetic effect of the ith entry, and eip the residual (Yu et al., 2006; Stich et al., 2008). The first and second principal coordinates from the SSR-based principal coordinate analysis in Bus et al. (2011) were used as D matrix for the association analysis. The kinship coefficient Kij between inbreds i and j were calculated according to Bernardo (1993). For the series of T values (describing the average probability that a variant from one parent of inbred i and a variant from one parent of inbred j are alike in state, given that they are not identical by descent) 0, 0.025, …, 0.975, K matrices between all inbreds were calculated. Negative kinship values between inbreds were set to 0. The optimum T value (showing the minimum likelihood) was calculated according to Stich et al. (2008). All statistical analyses, if not stated otherwise, were performed with statistical software R (R Development Core Team, 2011). For assuming an association, an adjusted P value (Bonferroni correction) of less than 0.05 was required. The association analysis of the mineral concentrations with the polymorphisms was carried out using the R package EMMA (Kang et al., 2008). The percentage of phenotypic variation explained by the significant SNPs was calculated according to Magee (1990). The threshold for significant LD (Hill and Robertson, 1968) was chosen to be a squared correlation of allele frequencies higher than 0.8. Linked SNPs in significant LD were assigned to genetic regions associated with the traits under study. Linked loci were defined as loci on the same chromosome and unlinked loci were defined as loci on different chromosomes.
2.3. Analysis of Genes Linked to Associated SNPs
The 6K SNP array positions were derived from the reference genome sequences of B. rapa (Wang et al., 2011) and B. oleracea (Yu et al., 2013). To define the positions in the B. napus genome, BLAST searches (Altschul et al., 1990) with flanking regions adjacent to the SNPs were done against a pre-publication draft (version 4) of the B. napus “Darmor-Bzh” reference genome sequence assembly, which was kindly made available prior to public release by INRA, France, Unité de Recherche en Génomique Végétale (Boulos Chaloub, INRA-URGV, Evry, France, unpublished data). For each SNP, the BLAST hit with the highest score was selected. If the two highest scores were identical or very similar, both positions were selected. SNPs that could not be assigned to a B. napus chromosome were excluded from further searches. For each locus an interval of 700 kb upstream and downstream of the defined position was screened for annotated genes. Predicted genes' sequences were annotated using Blast2GO (Conesa et al., 2005). A Gene Ontology (GO) enrichment analysis was performed using the bioconductor package GOstat (Beissbarth and Speed, 2004). The enriched GO terms of each trait and across all traits were mapped to plant GO slim terms using GOSlimViewer (McCarthy et al., 2006).
3. Results
We determined the mineral concentrations of eleven elements in a germplasm set of 509 inbred lines. The AEM of mineral concentrations were approximately normally distributed (Figure 1). Heritabilities h2 of mineral concentrations represented a range of 0.43 (P) to 0.86 (S) with a mean of 0.74 (Table 1). The phenotypic variation explained by population structure r2 ranged from 0.01 (P) to 5.46% (Na), with a mean of 1.46% (Table 1). In the PCA representing the 509 B. napus inbreds as well as the mineral concentrations and the seedling development traits, the first two principal components explained 45.0 and 10.8% of the variance (Figure 2). With respect to these two principal components, all inbreds of the germplasm types winter OSR, winter fodder, semi-winter OSR, spring OSR, and swedes were assigned to overlapping clusters, whereas spring fodder, vegetable, and unspecified lines did not show specific clustering patterns. The seedling development traits H2O, PRK, MAJ, FHM, DYM, MAD, PER, LA16, LA14, LA08, LA12, LA10, PRA, MID, MIN, and SPD and the concentrations of elements K, Na, and Ca had loadings mainly on PC 1, whereas Mn, Cu, Fe, Zn, S, B, and P primarily showed loadings on PC 2. ASR, CIR, SOY, ROU, and Mg contributed to both PC 1 and PC 2.
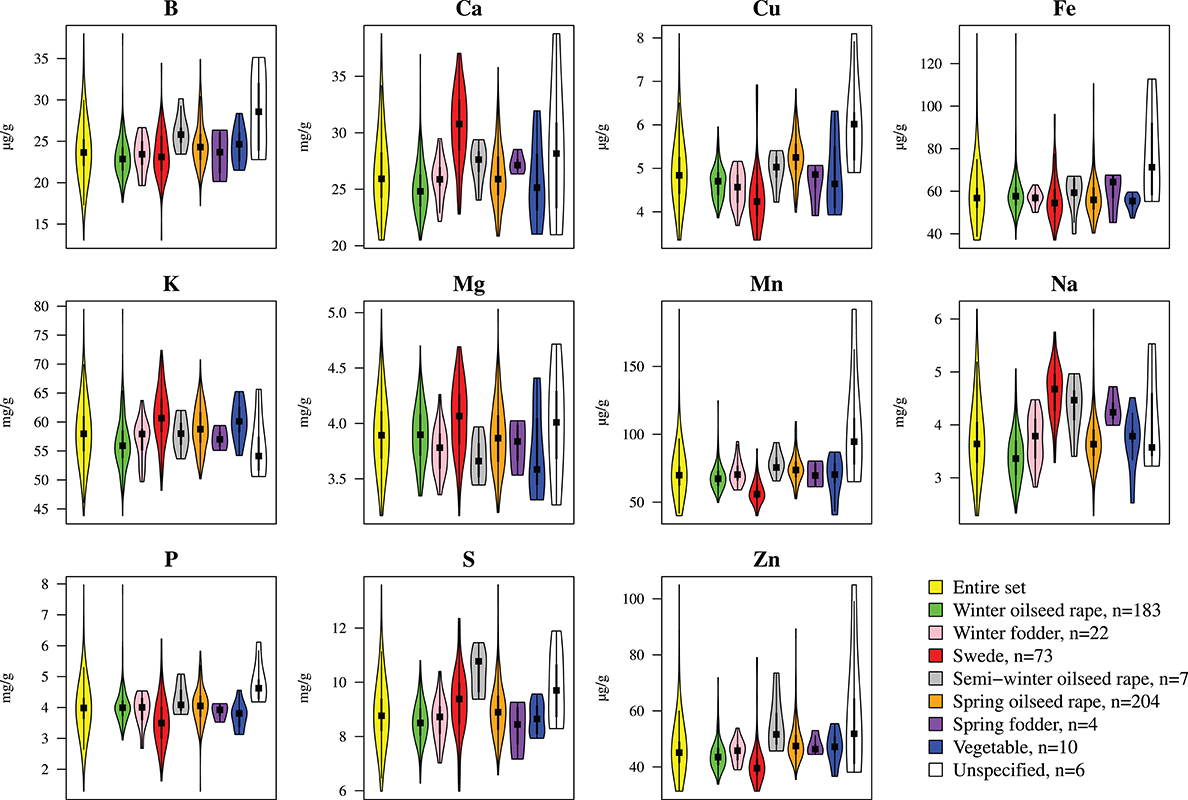
Figure 1. Frequency distribution of adjusted entry means determined for eleven different mineral concentrations in a germplasm set of B. napus inbreds. Colors represent different germplasm types. Yellow plots represent the entire germplasm set of 509 inbreds. The number of genotypes for each germplasm type is given in the legend. In each plot, a marker denotes the median of the data, a box indicates the interquartile range, and spikes extend to the upper and lower adjacent values, overlaid is the density.
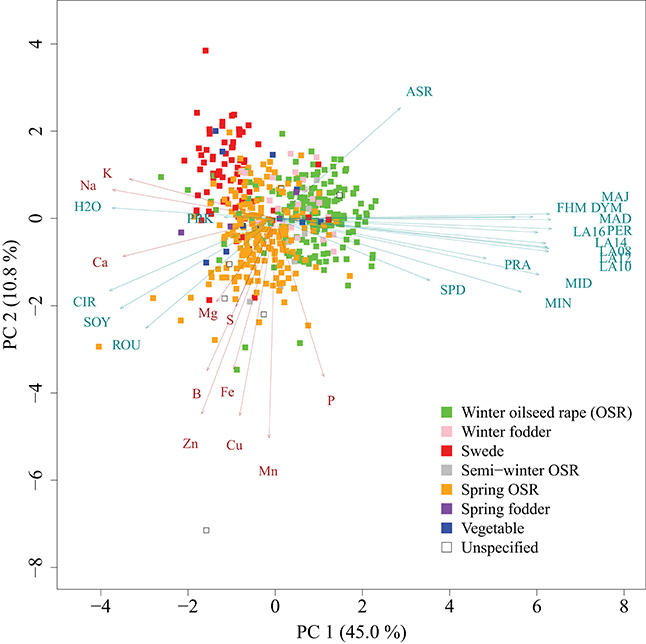
Figure 2. Principal component analysis of 509 B. napus inbreds (points), seedling development traits (turquoise arrows), and mineral concentrations (brown arrows). Germplasm types are represented by different colors. PC 1 and PC 2 are the first and second principal components, respectively. The proportion of variance explained by the principal components is given in parentheses.
Patterns of correlations among mineral concentrations described strong relationships between B/Mn, Ca/Mg, Ca/Na, Cu/Mn, Cu/Zn, Fe/Zn, and Mn/Zn (Figure 3A), which were all positively correlated with each other. Between mineral concentrations and seedling development traits there were high negative correlations between each of Ca, K, and Na and all seedling development traits except PRK, ASR, CIR, ROU, SOY, and H2O. The latter, however, was tightly positively correlated with Ca, K, and Na (Figure 3B).
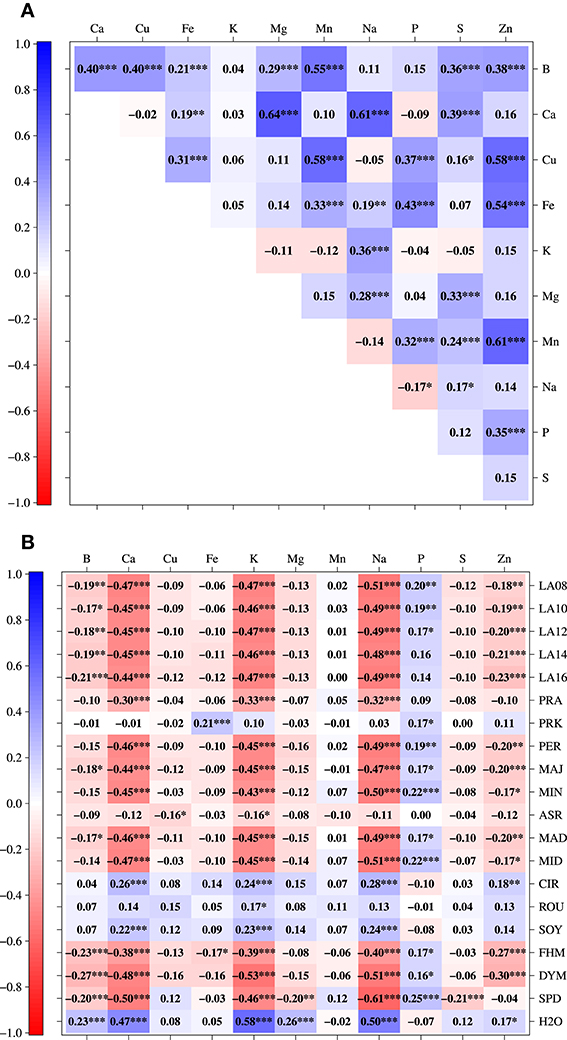
Figure 3. Pearson correlation coefficients (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, corrected using Holm's method) between pairs of eleven different mineral concentrations in 509 B. napus inbreds (A) and between pairs of mineral concentrations and seedling development traits (B).
The proportions of the phenotypic variation (adjusted R2) of the seedling development traits that could be explained by a linear combination of a selected set of the mineral concentrations in the germplasm set ranged from 0.042 (PRK) to 0.570 (DYM) (Table 2). The mean adj. R2 observed for the entire set was 0.356.

Table 2. Optimum linear combinations of eleven mineral concentrations describing seedling development traits in 509 B. napus inbreds, and the proportion of phenotypic variation explained by the selected independent variables (adjusted R2).
In the association analysis, we found altogether 29 significant (α = 0.05, Bonferroni correction) associations of 27 unique SNPs with Ca (3 associations), Cu (4 associations), Mg (2 associations), Mn (2 associations), Na (8 associations), S (2 associations), and Zn (8 associations) (Figure 4). One locus (Bn-ctg7180014703649-p1976) was associated with the three traits Cu, Mn, and Zn (Table 3). None of the SNPs was significantly associated with B, Fe, K, and P (Supplementary Figure S1). The phenotypic variation explained by single associated markers ranged from 3.02% (Bn-ctg7180014761247-p14206 and Cu) to 9.73% (p6_1705_snp15 and Mg), and the variation explained by all associated markers, determined in a simultaneous fit, ranged from 6.32% (S) to 22.89% (Zn) (Table 3).
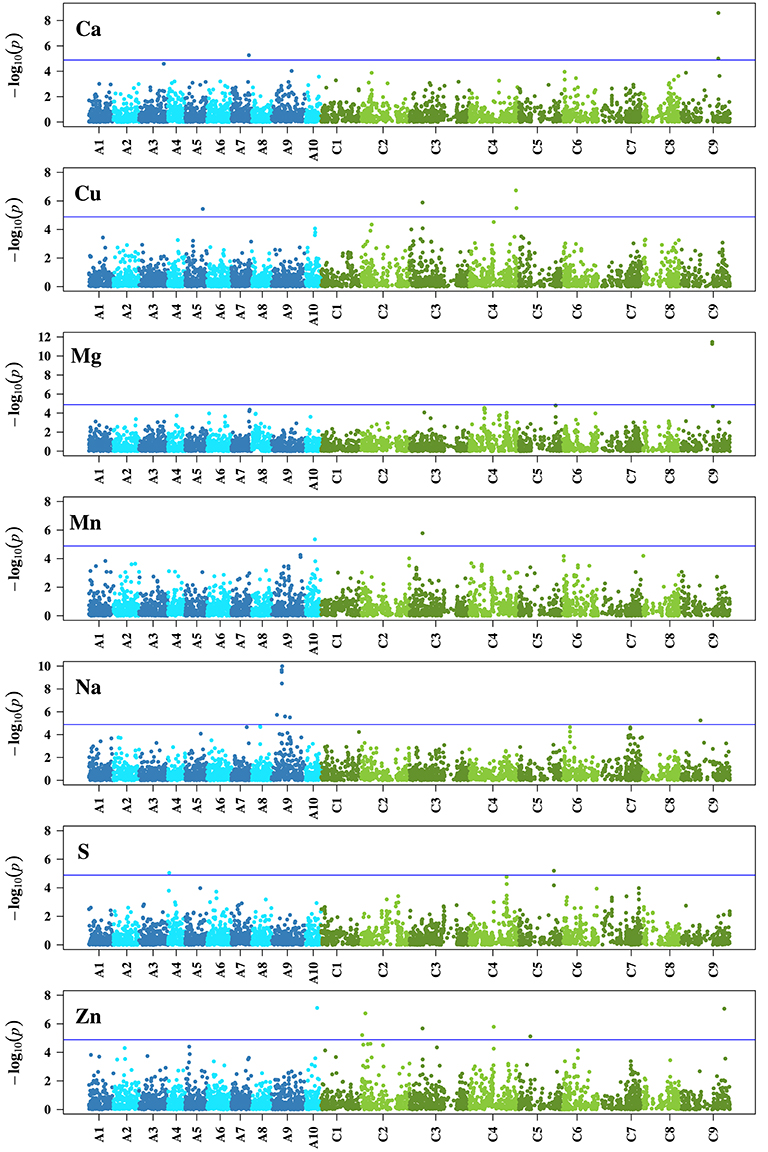
Figure 4. Genome-wide P-values for association analysis of seven mineral concentrations for which significant (α = 0.05 prior to Bonferroni correction) associations were identified in a B. napus diversity set using a 6 K single nucleotide polymorphism array after correction for multiple testing. Chromosomes of the B. napus A genome are colored blue, chromosomes of the B. napus C genome are colored green. The blue line shows the significance threshold after Bonferroni correction.
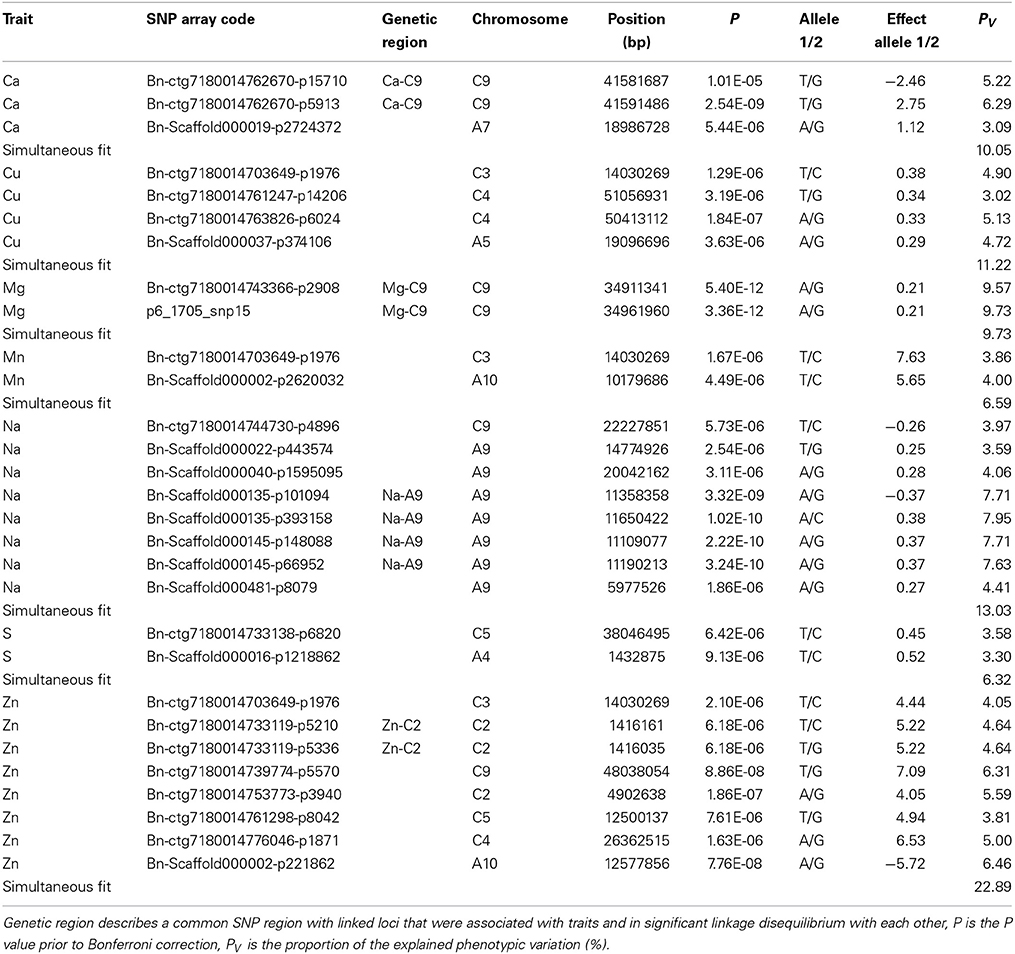
Table 3. Twenty-nine significant (α = 0.05) associations of 27 unique single nucleotide (SNP) markers with mineral concentrations after Bonferroni correction in a B. napus diversity set.
Linked SNPs from marker-trait associations that were in significant LD with each other were combined to represent genetic regions associated with the respective traits. For Ca, Mg, Na, and Zn, two (Ca, Mg, Zn) and four (Na) linked loci associated with the respective trait were in significant LD (Supplementary Table S1) and assigned to the genetic regions Ca-C9, Mg-C9, Na-A9, and Zn-C2 (Table 3). We therefore identified genetic regions on chromosomes A9, C2, and C9. None of the loci in significant LD were associated with different traits.
Based on the highest scores of the BLAST results, the chromosome information derived from the B. rapa and B. oleracea references was in accordance with the information derived from the B. napus reference assembly for 23 of all 27 unique SNPs with significant associations (Supplementary Table S2). In case there were two hits with the same or very similar top score, the information could be verified by one of them. We detected within the 700 kb interval each upstream and downstream the significant associations between 105 (Bn-Scaffold000002-p221862) and 524 (Bn-Scaffold000481-p8079) candidate genes. The GO enrichment analysis done on each trait revealed between 62 (Mg) and 207 (Na) enriched terms for the category biological process, between 6 (Ca, S) and 33 (Na) for cellular component, and between 27 (Mg) and 135 (Na) for molecular function (P ≤ 0.05) (Supplementary Table S3). Across all traits, the analysis resulted in 190 (biological process), 42 (cellular component), and 121 (molecular function) enriched terms. The enriched GO terms showed unequal representations of plant GO slim terms (Supplementary Figure S2). About 540 kb away from SNP Bn-Scaffold000022-p443574, which is located within the association hotspot for Na on chromosome A9 (Figure 4), we found the gene SOS1 (Salt Overly Sensitive 1).
4. Discussion
4.1. Correlation between Population Structure and the Shoot Ionome
A serious issue with AM is the identification of spurious associations caused by the presence of population structure (Flint-Garcia et al., 2003). In the germplasm set under study, we had previously detected population structure, which became apparent through MCLUST clusters which were represented mainly by winter types, spring types, and swedes (Bus et al., 2011). The proportion of shoot ionome variation explained by population structure r2 was nevertheless low across the shoot ionome (Table 1), and mineral concentrations varied moderately across germplasm types (Figure 1), indicating that population structure had little influence on our phenotypic observations of the shoot ionome. Based on this information, we decided against a separate analysis for each MCLUST cluster.
4.2. Relationships between Traits
We characterized the shoot ionome in a diverse panel of B. napus. The correlation coefficients among mineral concentrations were largely positive (Figure 3A). This observation suggests either common ion uptake and transport mechanisms shared between the respective elements or pleiotropy of the causal variants responsible for correlated mineral concentrations. The tightest positive correlation among mineral concentrations was found between Ca and Mg (0.64). The phenomenon of strong positive correlations between Ca and Mg accumulation in plants has been reviewed earlier (Baxter, 2009; White and Broadley, 2009). Similar to our results, Broadley et al. (2008) observed a high correlation (0.97) between the mean shoot Ca and Mg across ten B. oleracea subtaxa in an investigation of genetic variation of these two elements in 355 diverse accessions, 74 modern F1 cultivars, and a mapping population of B. oleracea. They pointed out that Ca and Mg homoeostasis is very likely to be controlled by common regulatory networks because they are chemically very similar. This is potentially also the reason for the tight correlation we found. The relationship between Ca and Mg in plants was furthermore described in a large study by Watanabe et al. (2007), who investigated 42 elements in more than 2000 leaf samples from 670 plant species and determined a correlation of 0.36 between Ca and Mg. The relationship between Ca and Mg we observed was therefore in line with previous studies.
Ding et al. (2010) measured the seed contents of Ca, Cu, Fe, Mg, Mn, P, and Zn in in 124 recombinant inbreds of B. napus under low and normal P conditions in two consecutive years. The correlations measured under the normal P regime across 2 years showed similar trends when compared with our data. However, major differences were found between Ca and P (−0.09 in our study vs. 0.49 in year 1 and 0.38 in year 2) as well as Mg and P (0.04 vs. 0.86 in year 1 and 0.39 in year 2), and Mn and Zn, although differences were only obvious for year 1 (0.61 vs. −0.08 in year 1 and 0.53 in year 2). These differences might be due to the measurement of elements in different tissues, as the ion balance is partly tissue-specific (Baxter, 2009). Nevertheless, the discrepancies with regard to the correlation between Mn and Zn need to be validated as they are only meaningful when regarding the results of year 1 in the work of Ding et al. (2010). Moreover, Wu et al. (2008) also found a significant positive correlation between Mn and Zn (0.61 in our study vs. 0.25 in Wu et al., 2008) when measuring 11 minerals (9 of which were identical with those from our research) in leaves of 183 B. rapa ssp. pekinensis DH lines grown in an open field. The relationship between Mn and Zn that we observed might be due to a locus with pleiotropic effects on Mn and Zn on the C genome that was so far unknown in B. napus. However, it cannot be verified by the results of Wu et al. (2008) as B. rapa represents the A genome of B. napus.
Not only relationships among traits representing the shoot ionome were evaluated but also those between mineral concentrations and traits characterizing seedling development. The relationship between K and H2O as well as Na and H2O (Figure 3B) is in all probability partly caused by the osmotic function of the two elements. But the similar chemical properties of alkali metals in general will also greatly influence the correlation between the two. The correlation between Ca and CIR, SOY, and ROU (Figures 2, 3B) hints at the cell wall stabilizing properties of Ca (Demarty et al., 1984). On the other hand, there is a tight negative correlation between Ca, K, and Na with most of the seedling development traits (except for PRK, ASR, CIR, ROU, SOY, and, as described before, H2O). Many of these ions are leaf growth-related. It is therefore conceivable that strong growth causes a dilution of these ions.
The identified relationships between the shoot ionome and seedling development raised the question whether one trait category might be predicted by the other. Multiple linear regression models were used to assess mineral concentrations as predictors for seedling development traits. The mean adj. R2 for the entire set was 35.6%. Körber et al. (2012) fitted linear models to predict agronomic and seed quality traits through the same set of seedling development trait data used in here. In 217 lines of a winter trial (subdivided into two subgroups) and 188 lines of a spring trial (all from the B. napus diversity set), the mean adj. R2 was 13%. We can therefore conclude that mineral concentrations are more suitable as predictors for seedling development traits than seedling development traits as predictors for agronomic and seed quality traits. This also implies that ionome data from field trials have the potential to predict agronomic data, but this will require further research.
4.3. Identified Marker-Trait Associations
Heritabilities were moderate for the two traits Fe (0.46) and P (0.43) and high for all other traits (between 0.74 for B and 0.86 for S, Table 1), which decreased the power to detect marker-trait associations for the former traits. This might explain why no significant associations were found for the traits Fe and P (Supplementary Figure S1). Broadley et al. (2008) described a high heritability of Ca and Mg in B. oleracea, which was confirmed also for B. napus by the results from our study (0.85 for Ca, 0.77 for Mg, Table 1).
Owing to the manifold networks, pathways and transporters that influence the presence of minerals in plants, mineral concentrations are complex traits which are likely to be affected by a number of loci with small effects—a phenomenon that has been described before for different traits like yield (Shi et al., 2009). We observed on chromosome C9 two significant SNPs for Ca and another two SNPs for Mg, next to one significant association for Na and Zn, respectively (Table 3, Figure 4). The detection of association peaks for Ca and Mg are in line with the earlier findings of significant QTLs on C9 for Ca and Mg in B. oleracea (Broadley et al., 2008), although further studies are required to determine whether any of the loci from our and the previous research are in close proximity to each other, or even identical. The most pronounced association peak in this study was found for Na on A9, comprising seven SNPs of which four were in significant LD (Table 3, Figure 4, Supplementary Table S1). To search for annotated genes close to the significantly associated SNPs, an interval of 700 kb upstream and downstream of each SNP position was chosen as it corresponds to the extent of significant LD in B. napus (Bus et al., 2011), assuming a linear transformation with a rate of 0.674 Mb/cM according to Bancroft et al. (2011). The gene SOS1 that was found in the proximity (about 540 kb) to the association hotspot is known to be an Na+/H+ antiporter that controls the uptake and efflux of Na, a mechanism which is crucial for plants under Na-rich conditions (Fraile-Escanciano et al., 2010). SOS1 was first identified in A. thaliana (Wu et al., 1996), and its orthologs in Brassica species are also known (Chakraborty et al., 2012; Ford et al., 2012). Our identification of a closely situated gene that plays a key role in Na tolerance underlines the importance of the hotspot, which includes the genetic region Na-A9, in B. napus. The region might be particularly interesting in the framework of breeding for Na tolerance. However, it requires further research whether the hotspot is due to genotypic variation in SOS1.
Associated with the candidate genes within 700 kb upstream or downstream the significant SNPs, we identified a number of enriched GO terms that are functionally associated with the traits under study (Supplementary Table S3). When considering enriched GO terms across all traits it becomes obvious that these are mainly associated with general biological, cellular, and metabolic processes as well as enzyme and transporter activity and many cellular components involved in photosynthesis (Supplementary Table S3, Supplementary Figure S2). According to the detection of SOS1, the smallest P value (2.65 × 10−7) in the category biological process for Na was found for the term “sodium ion transport.” Furthermore, the second smallest P value (6.46 × 10−6) in the category molecular function for Na was found for “sodium ion transmembrane transporter activity,” and the GO slim term “transporter activity” was overrepresented for Na (Supplementary Figure S2). These findings support the assumption that the association hotspot is involved in Na uptake and transport. Many enriched terms hint at the known biological processes the examined traits are typically involved in, such as many photosynthesis-related terms for Cu, Mg, and Mn; ions that are essential for photosynthesis (Marschner, 1995). Similarly, phototropism was significantly enriched (P value 3.37 × 10−4) for biological process in Ca. A relationship between Ca and phototropism is expected because Ca controls the auxin-induced elongation of cells, and it has been described before in maize seedlings (Gehring et al., 1990). Hence, the enriched GO terms provide insights into what might be the roles of genes linked to the significantly associated SNPs we found.
With the association of the SNP Bn-ctg7180014703649-p1976 with Cu, Mn, and Zn (Table 3, Figure 4), we identified a locus with effects on three different heavy metals. This observation and the tight correlations between the three elements (Figures 2, 3A) suggest a pleiotropic effect of this SNP. On the other hand, the significant association with the three traits may be caused by linkage between the underlying genes. This hypothesis is supported by the extent of LD over distances of about 1 cM in the germplasm set under study (Bus et al., 2011). From the detected associated genes (data not shown), it is currently not obvious which one might be the causal gene or genes underlying this association. Additional approaches like RNA-seq analysis will be needed to reveal them.
Using SNPs from arrays in AM may cause ascertainment bias which brings forward an oversampling of mutations at intermediate frequencies, these again cause amounts of LD that are lower than those present in completely randomly selected SNP sets (Ingvarsson and Street, 2011). How strongly ascertainment bias affects the power of AM, however, is hard to say because it depends on various factors such as whether low- or intermediate-frequency SNPs are assumed to have larger effects on the trait under consideration (Clark et al., 2005; Manolio et al., 2009).
The significant associations found in this study explained between 3.02 and 9.73% of the phenotypic variation in single marker analyses (Table 3), and jointly up to 22.89%. The fact that none of the associations in single marker analyses explained more than 10% of the variation underlines the complexity of the examined traits, but it might also be due to imbalanced allele frequencies (Wricke and Weber, 1986). By way of example, for Ca, the SNPs Bn-ctg7180014762670-p15710 and Bn-Scaffold000019-p2724372 explain a comparable proportion of the variance (5.22 and 3.09%, respectively Table 3), but the minor allele frequency of the former is much lower (0.09) than that of the latter (0.47). These two SNPs could be studied further in a biparental population where allele frequencies would be balanced, and the SNP that explains a higher variance could be identified. Phenotypic differences in mineral concentrations, however, are not only due to the associations reported here but most likely also to further small effects of many undetected loci (Figure 4, Supplementary Figure S1).
Author Contributions
Anja Bus analyzed the data. Niklas Körber contributed the data on seedling development. Isobel A. P. Parkin provided the 6K array data. Birgit Samans and Rod J. Snowdon carried out the candidate gene and GO term enrichment analysis on the pre-published B. napus reference sequence. Jinquan Li supported the AM analysis. Anja Bus and Benjamin Stich wrote the manuscript. Benjamin Stich designed and supervised the study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the Landesanstalt für Landwirtschaftliche Chemie at the University of Hohenheim (Stuttgart, Germany) for carrying out the ICP emission spectrometry. We are grateful to Isabell Scheibert for the technical assistance. We thank Boulos Chaloub (INRA-URGV, Èvry, France) for providing the pre-publication draft of the B. napus Darmor-Bzh reference sequence to Rod Snowdon and Birgit Samans. We acknowledge the funding by the Deutsche Forschungsgemeinschaft (DFG) and the Max Planck Society for this work, which was part of the ERA-NET PG project “ASSYST.” Finally, we thank the associate editor Stewart Gillmor and the two reviewers for helpful discussions on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00485/abstract
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bancroft, I., Morgan, C., Fraser, F., Higgins, J., Wells, R., Clissold, L., et al. (2011). Dissecting the genome of the polyploid crop oilseed rape by transcriptome sequencing. Nat. Biotechnol. 29, 762–766. doi: 10.1038/nbt.1926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baxter, I. (2009). Ionomics: studying the social network of mineral nutrients. Curr. Opin. Plant Biol. 12, 381–386. doi: 10.1016/j.pbi.2009.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baxter, I. R., Gustin, J. L., Settles, A. M., and Hoekenga, O. A. (2013). Ionomic characterization of maize kernels in the intermated B73 x Mo17 population. Crop Sci. 53, 208–220. doi: 10.2135/cropsci2012.02.0135
Baxter, I. R., Vitek, O., Lahner, B., Muthukumar, B., Borghi, M., Morrissey, J., et al. (2008). The leaf ionome as a multivariable system to detect a plant's physiological status. Proc. Natl. Acad. Sci. U.S.A. 105, 12081–12086. doi: 10.1073/pnas.0804175105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beissbarth, T., and Speed, T. P. (2004). GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics 20, 1464–1465. doi: 10.1093/bioinformatics/bth088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bernardo, R. (1993). Estimation of coefficient of coancestry using molecular markers in maize. Theor. Appl. Genet. 85, 1055–1062. doi: 10.1007/BF00215047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Broadley, M. R., Hammond, J. P., King, G. J., Astley, D., Bowen, H. C., Meacham, M. C., et al. (2008). Shoot calcium and magnesium concentrations differ between subtaxa, are highly heritable, and associate with potentially pleiotropic loci in Brassica oleracea. Plant Physiol. 146, 1707–1720. doi: 10.1104/pp.107.114645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bus, A., Hecht, J., Huettel, B., Reinhardt, R., and Stich, B. (2012). High-throughput polymorphism detection and genotyping in Brassica napus using next-generation RAD sequencing. BMC Genomics 13:281. doi: 10.1186/1471-2164-13-281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bus, A., Körber, N., Snowdon, R. J., and Stich, B. (2011). Patterns of molecular variation in a species-wide germplasm set of Brassica napus. Theor. Appl. Genet. 123, 1413–1423. doi: 10.1007/s00122-011-1676-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chakraborty, K., Sairam, R. K., and Bhattacharya, R. C. (2012). Differential expression of salt overly sensitive pathway genes determines salinity stress tolerance in Brassica genotypes. Plant Physiol. Biochem. 51, 90–101. doi: 10.1016/j.plaphy.2011.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clark, A. G., Hubisz, M. J., Bustamante, C. D., Williamson, S. H., and Nielsen, R. (2005). Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 15, 1496–1502. doi: 10.1101/gr.4107905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demarty, M., Morvan, C., and Thellier, M. (1984). Calcium and the cell wall. Plant Cell Environ. 7, 441–448. doi: 10.1111/j.1365-3040.1984.tb01434.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ding, G., Yang, M., Hu, Y., Liao, Y., Shi, L., Xu, F., et al. (2010). Quantitative trait loci affecting seed mineral concentrations in Brassica napus grown with contrasting phosphorus supplies. Ann. Bot. 105, 1221–1234. doi: 10.1093/aob/mcq050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edwards, D., Batley, J., and Snowdon, R. J. (2013). Accessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 126, 1–11. doi: 10.1007/s00122-012-1964-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flint-Garcia, S. A., Thornsberry, J. M., and Buckler, E. S. (2003). Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54, 357–374. doi: 10.1146/annurev.arplant.54.031902.134907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ford, B. A., Ernest, J. R., and Gendall, A. R. (2012). Identification and characterization of orthologs of AtNHX5 and AtNHX6 in Brassica napus. Front. Plant Sci. 3:208. doi: 10.3389/fpls.2012.00208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fraile-Escanciano, A., Kamisugi, Y., Cuming, A. C., Rodríguez-Navarro, A., and Benito, B. (2010). The SOS1 transporter of Physcomitrella patens mediates sodium efflux in planta. New Phytol. 188, 750–761. doi: 10.1111/j.1469-8137.2010.03405.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gehring, C. A., Williams, D. A., Cody, S. H., and Parish, R. W. (1990). Phototropism and geotropism in maize coleoptiles are spatially correlated with increases in cytosolic free calcium. Nature 345, 528–530. doi: 10.1038/345528a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilmour, A. R., Gogel, B. J., Cullis, B. R., Welham, S. J., and Thompson, R. (2006). ASReml User Guide Release 2.0. Hemel Hempstead: VSN International Ltd.
Hill, W. G., and Robertson, A. (1968). Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38, 226–231. doi: 10.1007/BF01245622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ingvarsson, P. K., and Street, N. R. (2011). Association genetics of complex traits in plants. New Phytol. 189, 909–922. doi: 10.1111/j.1469-8137.2010.03593.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, H. M., Zaitlen, N. A., Wade, C. M., Kirby, A., Heckerman, D., Daly, M. J., et al. (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. doi: 10.1534/genetics.107.080101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Körber, N., Wittkop, B., Bus, A., Friedt, W., Snowdon, R. J., and Stich, B. (2012). Seedling development in a Brassica napus diversity set and its relationship to agronomic performance. Theor. Appl. Genet. 125, 1275–1287. doi: 10.1007/s00122-012-1912-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lahner, B., Gong, J., Mahmoudian, M., Smith, E. L., Abid, K. B., Rogers, E. E., et al. (2003). Genomic scale profiling of nutrient and trace elements in Arabidopsis thaliana. Nat. Biotechnol. 21, 1215–1221. doi: 10.1038/nbt865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, J., Yang, J., Li, R., Shi, L., Zhang, C., Long, Y., et al. (2009). Analysis of genetic factors that control shoot mineral concentrations in rapeseed (Brassica napus) in different boron environments. Plant Soil 320, 255–266. doi: 10.1007/s11104-009-9891-6
Magee, L. (1990). R2 Measures based on Wald and likelihood ratio joint significance tests. Am. Stat. 44, 250–253.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCarthy, F. M., Wang, N., Magee, G. B., Nanduri, B., Lawrence, M. L., Camon, E. B., et al. (2006). AgBase: a functional genomics resource for agriculture. BMC Genomics 7:229. doi: 10.1186/1471-2164-7-229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oraguzie, N. C., Wilcox, P., Rikkerink, E. H., and de Silva, H. N. (2007). “Linkage Disequilibrium,” in Association Mapping in Plants, eds N. C. Oraguzie, E. H. Rikkerink, S. E. Gardiner, and H. N. de Silva (New York, NY: Springer), 11–39. doi: 10.1007/978-0-387-36011-9-2
R Development Core Team. (2011). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Salt, D. E., Baxter, I., and Lahner, B. (2008). Ionomics and the study of the plant ionome. Annu. Rev. Plant Biol. 59, 709–733. doi: 10.1146/annurev.arplant.59.032607.092942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwarz, G. (1978). Estimating the dimensions of a model. Ann. Stat. 6, 461–464. doi: 10.1214/aos/1176344136
Shi, J., Li, R., Qiu, D., Jiang, C., Long, Y., Morgan, C., et al. (2009). Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182, 851–861. doi: 10.1534/genetics.109.101642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Snowdon, R. J., and Iniguez Luy, F. L. (2012). Potential to improve oilseed rape and canola breeding in the genomics era. Plant Breed. 131, 351–360. doi: 10.1111/j.1439-0523.2012.01976.x
Stich, B., Möhring, J., Piepho, H.-P., Heckenberger, M., Buckler, E. S., and Melchinger, A. E. (2008). Comparison of mixed-model approaches for association mapping. Genetics 178, 1745–1754. doi: 10.1534/genetics.107.079707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trick, M., Long, Y., Meng, J., and Bancroft, I. (2009). Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 7, 334–346. doi: 10.1111/j.1467-7652.2008.00396.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, T., Broadley, M. R., Jansen, S., White, P. J., Takada, J., Satake, K., et al. (2007). Evolutionary control of leaf element composition in plants. New Phytol. 174, 516–523. doi: 10.1111/j.1469-8137.2007.02078.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
White, P. J., and Broadley, M. R. (2009). Biofortification of crops with seven mineral elements often lacking in human diets - iron, zinc, copper, calcium, magnesium, selenium and iodine. New Phytol. 182, 49–84. doi: 10.1111/j.1469-8137.2008.02738.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wricke, G., and Weber, W. E. (1986). Quantitative Genetics and Selection in Plant Breeding. Berlin: De Gruyter. doi: 10.1515/9783110837520
Wu, D., Shen, Q., Cai, S., Chen, Z.-H., Dai, F., and Zhang, G. (2013). Ionomic responses and correlations between elements and metabolites under salt stress in wild and cultivated barley. Plant Cell Physiol. 54, 1976–1988. doi: 10.1093/pcp/pct134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, J., Yuan, Y.-X., Zhang, X.-W., Zhao, J., Song, X., Li, Y., et al. (2008). Mapping QTLs for mineral accumulation and shoot dry biomass under different Zn nutritional conditions in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Plant Soil 310, 25–40. doi: 10.1007/s11104-008-9625-1
Wu, S. J., Ding, L., and Zhu, J. K. (1996). SOS1, a genetic locus essential for salt tolerance and potassium acquisition. Plant Cell 8, 617–627. doi: 10.1105/tpc.8.4.617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, J., Pressoir, G., Briggs, W. H., Vroh Bi, I., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, J., Zhao, M., Wang, X., Tong, C., Huang, S., Tehrim, S., et al. (2013). Bolbase: a comprehensive genomics database for Brassica oleracea. BMC Genomics 14:664. doi: 10.1186/1471-2164-14-664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: genome-wide association mapping, single nucleotide polymorphism, shoot ionome, Brassica napus, linkage disequilibrium
Citation: Bus A, Körber N, Parkin IAP, Samans B, Snowdon RJ, Li J and Stich B (2014) Species- and genome-wide dissection of the shoot ionome in Brassica napus and its relationship to seedling development. Front. Plant Sci. 5:485. doi: 10.3389/fpls.2014.00485
Received: 23 June 2014; Accepted: 02 September 2014;
Published online: 30 September 2014.
Edited by:
Stewart Gillmor, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Eduard Akhunov, Kansas State University, USALewis Lukens, University of Guelph, Canada
Copyright © 2014 Bus, Körber, Parkin, Samans, Snowdon, Li and Stich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin Stich, Max Planck Institute for Plant Breeding Research, Carl-von-Linné-Weg 10, 50829 Cologne, Germany e-mail:c3RpY2hAbXBpcHoubXBnLmRl