 Semidán Robaina Estévez
Semidán Robaina Estévez Zoran Nikoloski
Zoran Nikoloski- Systems Biology and Mathematical Modeling Group, Max Planck Institute of Molecular Plant Physiology, Potsdam-Golm, Germany
Genome-scale metabolic models (GEMs) are increasingly applied to investigate the physiology not only of simple prokaryotes, but also eukaryotes, such as plants, characterized with compartmentalized cells of multiple types. While genome-scale models aim at including the entirety of known metabolic reactions, mounting evidence has indicated that only a subset of these reactions is active in a given context, including: developmental stage, cell type, or environment. As a result, several methods have been proposed to reconstruct context-specific models from existing genome-scale models by integrating various types of high-throughput data. Here we present a mathematical framework that puts all existing methods under one umbrella and provides the means to better understand their functioning, highlight similarities and differences, and to help users in selecting a most suitable method for an application.
Introduction
Genome-scale metabolic models (GEMs) have become a useful tool to investigate metabolism. They present numerous applications, from basic research on metabolic functioning and cell physiology (Bordbar et al., 2014) to the design of novel strains for improving biotechnological processes to the analysis of metabolic diseases and the quest for novel drug targets (Milne et al., 2009; Garcia-Albornoz and Nielsen, 2013; Agren et al., 2014). Although historically biased toward microorganisms, a number of GEMs have recently been reconstructed for several plant species, including: Arabidopsis thaliana (Poolman et al., 2009; De Oliveira Dal'Molin et al., 2010; Saha et al., 2011; Arnold and Nikoloski, 2014), maize (Saha et al., 2011), maize and other C4 plants (Dal'Molin et al., 2010), rice (Dharmawardhana et al., 2013; Poolman et al., 2013) and algae (Chang et al., 2011; Gomes de Oliveira Dal'Molin et al., 2011). This late development of plant GEMs is largely due to the particular challenges of modeling plant metabolism, (in general more complex and characterized by cellular compartmentalization and an extensive secondary metabolism) and a lower coverage of annotated metabolic genes in plants in comparison with, much simpler and more experimentally accessible, microorganisms. The development plant GEMs and particular challenges are summarized in De Oliveira Dal'Molin and Nielsen (2013) and Sweetlove and Ratcliffe (2011).
The success of GEMs is largely due to their integrative nature, representing the whole known network of biochemical reactions of a given organism, and the possibility to readily use them in a mathematical model. This mathematical model can be further interrogated with powerful methods from constraint-based analysis (Lewis et al., 2012), whereby a system of mass balance equations at steady state, with additional thermodynamic and capacity constraints, define a solution space of feasible metabolic flux values. The imposed constraints may also lead to inconsistencies in the original metabolic model; for instance, by enforcing blocked reactions, i.e., reactions incapable of carrying nonzero flux at steady state. Flux balance analysis (Orth et al., 2010) represents a prominent method within constraint-based analysis, and has been widely applied to explore cell physiology. It assumes that cells adapt metabolic fluxes to optimize a certain objective function (i.e., a linear combination of metabolic fluxes). Although GEMs and constraint-based methods are convenient when modeling the entirety of known metabolism, mainly due to the smaller number of parameters to be measured (e.g., external fluxes), other available methods, such as stochastic (Wilkinson, 2009; Ullah and Wolkenhauer, 2010) or deterministic (Link et al., 2014), kinetic models may offer an alternative strategy, particularly for modeling smaller cellular subsystems. The latter is particularly the case when the focus is modeling of the dynamics of metabolite concentrations and/or of regulatory mechanisms. However, due to the dependence on a large number of (not readily measurable) parameters and the computational demand, these methods usually are not scalable. Interestingly, some hybrid approaches have been proposed merging constraint-based and kinetic methods, which may overcome individual limitations of both methods, ultimately resulting in better predictions (Jamshidi and Palsson, 2010; Soh et al., 2012; Chakrabarti et al., 2013; Chowdhury et al., 2014).
The recent advent of high-throughput technologies has propelled the GEM community to develop new methods for integrating high-throughput data into existing metabolic models. In general, these methods employ data to (1) improve flux predictions through further constraining of the solution space (Colijn et al., 2009; Chandrasekaran and Price, 2010; Jensen and Papin, 2011; Collins et al., 2012; Lee et al., 2012), and/or (2) extract context-specific metabolic models, which are a subset of the original GEM (Becker and Palsson, 2008; Shlomi et al., 2008; Jerby et al., 2010; Agren et al., 2012; Wang et al., 2012; Schmidt et al., 2013; Vlassis et al., 2014). In the first case, the metabolic model serves as a scaffold to analyze complex data sets from different sources, e.g., transcript, protein or metabolite profiles. The second case is motivated by the mounting evidence suggesting that the structure of a given metabolic network changes across different conditions, e.g., environmental changes, developmental stages as well as different cell-types or tissues. Therefore, in context-specific metabolic models only a subset of the reactions from the original GEM carry flux, and are considered active. This is of particular importance when tackling multicellular organisms, like plants, where multiple cell types with specialized metabolic functions coexist and cooperate. Following this line, a number of tissue-specific models have been reconstructed in Mintz-Oron et al. (2012) using one of such methods (the MBA, discussed below) together with a genome-scale model of Arabidopsis and publicly available tissue-specific expression profiles. However, other, manual, approaches have been used to take into account cell and tissue type in plant GEMs; for instance, in C4GEM, two cell types are modeled: bundle sheath and mesophyll cells, to capture the typical C4 carbon fixation physiology (Dal'Molin et al., 2010). In Grafahrend-Belau et al. (2013) authors go further in scope to model the metabolism of a whole barley plant, using four organ-specific models (leaf, stem, seed, and root) that are interconnected through two exchange compartments (the phloem and external environment). Here, we will use the generic term context for any of the particular conditions that may occur.
High-throughput data sets can be divided in hierarchical categories that correspond to different cellular processes. On one hand, transcript profiles capture the instantaneous expression state of a given genome under a particular condition. They have the greatest coverage, since usually all known genes are considered. They are also the most accessible in terms of experimental tractability, due to the availability of classical technologies (e.g., microarray) as well as modern developments (i.e., RNAseq). However, gene expression is also at the top of the hierarchical chain of events that govern metabolic fluxes, which may explain the relatively low correlation values between these two quantities, as reported in previous works (Yang et al., 2002; Rossell et al., 2006; Daran-Lapujade et al., 2007; Moxley et al., 2009). Protein levels may be more concordant to metabolic fluxes, and hence several methods have aimed to incorporate this source of evidence (Jerby et al., 2010; Agren et al., 2012, 2014; Bordbar et al., 2012). However, existing measurement techniques, mainly based on the combination of chromatography and mass spectrometry (Schulze and Usadel, 2010), do not offer an extensive coverage of the proteome. Finally, metabolites directly relate to metabolic fluxes, since they play the role of substrates and products of metabolic reactions. Therefore, metabolite levels may better reflect the actual state of a metabolic network. Unfortunately, current measurement methods do not permit full coverage of the metabolome to describe the metabolic state of the entire network (Fernie, 2007). Despite this shortcoming, integration of metabolite levels can substantially improve flux predictions or the extraction of context-specific models, especially when they are combined with protein and/or gene expression levels (Yizhak et al., 2010; Kleessen et al., 2012).
Several recent comprehensive reviews provide extensive coverage of computational methods for integrating high-throughput data in GEMs (Joyce and Palsson, 2006; Blazier and Papin, 2012; Lewis et al., 2012; Hyduke et al., 2013), with a recent study offering a critical systematic evaluation and performance comparison (Machado and Herrgård, 2014). Here we propose a mathematical framework that groups existing methods for context-specific model extraction in three families. This framework provides not only a mere classification but also the means to better understand the rationale behind methods and highlight their common principles and differences. We also propose a flowchart to guide interested users in selecting a method to apply in a particular setting. In the following, for each family of methods, we present its general functioning and mathematical objective, discuss its advantages and disadvantages, and we also highlight particularities of each method.
Generalization of Methods for Extraction of Context-Specific Models
Our framework for classification of the existing methods for extraction of context-specific models simultaneously offers a generalization of the mathematical and algorithmic formulation. With respect to the employed objective, these methods can be divided into three main families, namely: GIMME-, iMAT-, and MBA-like families, termed after the first representative method in each class (Figure 1). The objective employed by the GIMME-like family corresponds to the similarity of the flux phenotype to data, which is to be maximized while guaranteeing a given Required Metabolic Functionality (RMF), such as: growth or ATP production. In contrast, the iMAT-like family of methods aims at maximizing the similarity of the flux phenotype to data without imposing any RMF. Finally, the MBA-like family uses model consistency as objective, which refers to a final context-specific model without any blocked reaction. The mathematical generalization of each family of methods captures these principles, highlights the similarities, and serves as a scaffold to frame particularities of each method.
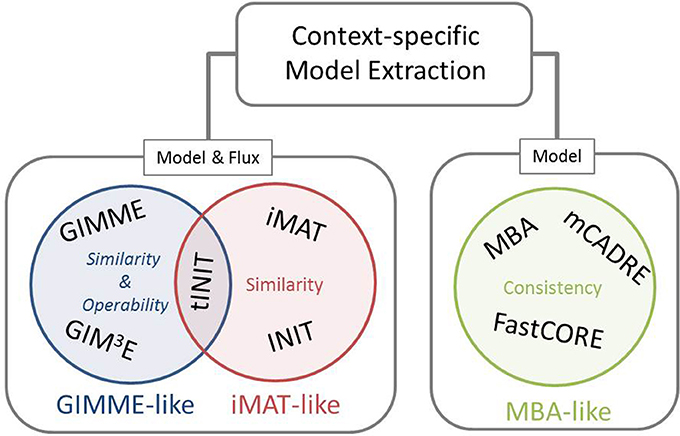
Figure 1. Existing methods for context-specific model extraction can be classified in three families: GIMME-, iMAT-, and MBA-like. This classification is based on the mathematical objective followed by the methods, i.e., similarity and operability, similarity or consistency. Moreover, methods can perform a model extraction and a flux prediction (GIMME- and iMAT-like families) or only a model extraction (MBA-like family). Although similar in formulation to INIT, the metabolic task constraint imposed by tINIT renders this algorithm close to the objective of the GIMME-like family (see main text).
GIMME-Like Family
The GIMME-like family encompasses the GIMME method (Becker and Palsson, 2008) and GIM3E, as an extension (Schmidt et al., 2013). This family reconstructs a context-specific model in two steps: First, it optimizes an objective function, the RMF, by using the classical linear programming (LP) formulation of flux balance analysis which imposes mass balance and thermodynamic constraints. This objective function is assumed to be the main cellular task in the investigated condition. It then involves solving a second LP that minimizes a penalty function, corresponding to the discrepancies between flux values and the respective transcript levels, with the additional constraint that the flux through the previous RMF must be above a given lower bound (e.g., a fraction of the optimum value found by flux balance analysis). The methods included in this family mainly differ in the way the discrepancies are minimized in the second step, the type of high-throughput data used, and in the treatment of reversible reactions, as detailed below. Box 1 displays the formulations and the generalization of this family (consult Table 1 for a glossary of used symbols).
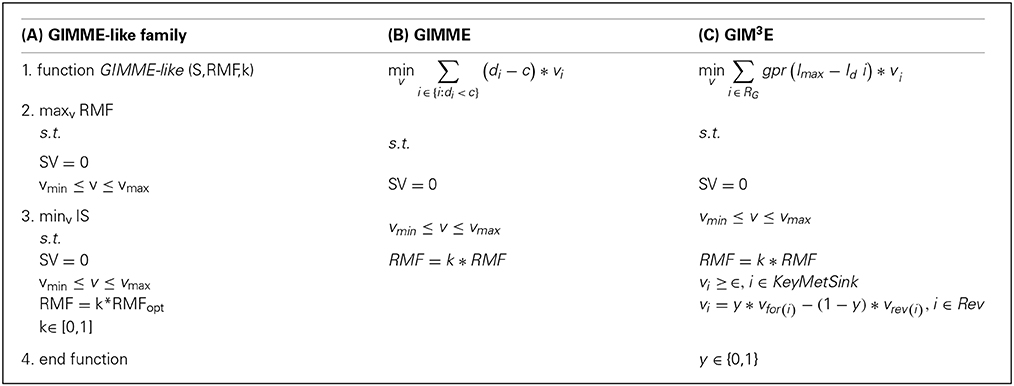
Box 1. Mathematical formulations of the GIMME-like family: (A) generalization of the family, (B) GIMME and (C) GIM3E. In (B,C) only the second LP is represented (see main text). In (C) individual reaction-associated gene penalties are first calculated and then mapped to the corresponding reaction to obtain the reaction penalty, here represented by the function: gpr(Imax−Id i) (see main text). Consult the Glossary of symbols for notation.
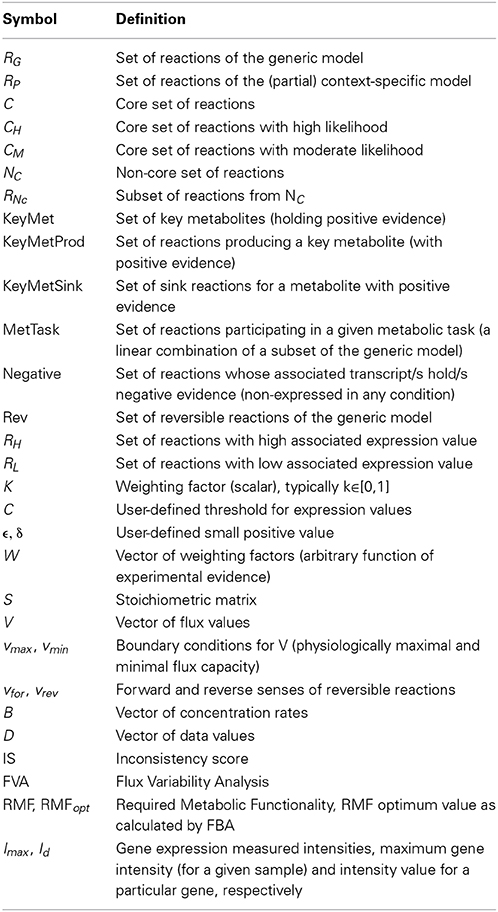
Table 1. Glossary of symbols.
GIMME
In GIMME, the penalty function is termed inconsistency score. This function penalizes flux values of reactions whose associated expression levels are below a user-defined cut-off (i.e., threshold). More specifically, the inconsistency score is given by the dot product of the flux distribution and the reaction penalty, defined as the vector difference of the associated expression values from the threshold. The reaction associated expression level is obtained following the standard GPR rules (Becker and Palsson, 2008), which take into account the presence of isoenzymes and protein complexes. Although transcript profiles were used in the original formulation, a variant called GIMMEp allows for the integration of proteomic data (Bordbar et al., 2012). The result of applying this algorithm is a flux distribution which ensures that a given RMF can be carried out and is as consistent as possible to the employed data.
GIM3E
GIM3E introduces several modifications to the original GIMME. First, it allows integration of metabolomics data, imposing a nonzero flux value to reactions involving a metabolite for which there is evidence of being synthesized in an investigated condition. Second, it modifies the definition of the reaction penalty; here, the penalties for all reaction-associated genes are determined separately and are then mapped to the reaction following the GPR rules. Moreover, the penalties are calculated as the distance between each transcript and the maximum expression level of the set. Consequently, after mapping transcript penalties all reactions obtain a penalty value, rather than only the set below the threshold which is the case in GIMME. Finally, GIM3E takes into account directionality of reversible reactions by constraining them to operate in only one direction, which is modeled by introducing a binary variable for the direction of choice. As a result, GIM3E is formulated as a mixed integer linear program (MILP), which is more computationally challenging than the LP formulation of GIMME.
Advantages and disadvantages of the GIMME-like family
When a given RMF operates in different contexts, the operability constraint may lead to more accurate context-specific model reconstructions and flux distributions. This issue has been evaluated in a recent review (Machado and Herrgård, 2014), demonstrating that methods which do not impose network operability were incapable of predicting growth using a yeast metabolic model. Furthermore, the total sum of the inconsistency score also quantifies the correspondence of the RMF to the set of expression data, which may provide further insights into cellular functionality.
Nevertheless, while the selection of a RMF can be a relatively easy task for prokaryotes, whereby experimental evidence supports the choice of cellular growth or biomass maximization as a plausible RMF, this task is much more challenging for eukaryotic organisms, especially the multicellular. In this case, choosing a RMF for a given tissue or cellular type is a complicated task, as each cell type is specialized in certain biochemical functions, modulated on the level of the entire organism. Therefore, methods that do not require a RMF may be applied easier to models of multicellular organisms.
There are existing implementations for both methods: GIMME can be executed using the createTissueSpecificModel function built in the COBRA toolbox within the Matlab environment (Schellenberger et al., 2011). The GIM3E implementation is however built under Python (“The OpenCOBRA Project1,” n.d.).
iMAT-Like Family
The iMAT-like family comprises three methods, iMAT (Shlomi et al., 2008), INIT (Agren et al., 2012) and its extension, tINIT (Agren et al., 2014), which also aim at extracting a context-specific model compatible with a given data set. However, in contrast to the GIMME-like family of methods, the iMAT-like family does not assume a RMF achieved by the cell. More specifically, these methods maximize the number of matches between reaction states (i.e., active or inactive) and corresponding data states (i.e., expressed or not non-expressed). The mathematical formulation results in a MILP, in which the value of the binary variable denotes the most concordant reaction state for a given (data) context. Although sharing the general strategy, iMAT, INIT and tINIT differ considerably respecting to how they deal with data: iMAT integrates data in the constraints, INIT and tINIT do so directly in the objective function. See Box 2 for mathematical formulations and generalization of the family, and Table 1 for a glossary of used symbols.

Box 2. Mathematical formulations of the iMAT-like family: (A) generalization of the family, (B) iMAT, and (C) INIT. In (C), the tINIT extension is displayed in blue.. Consult the Glossary of symbols for notation.
iMAT
The algorithm first classifies reactions into two groups based on a previously defined threshold for the corresponding expression data; this results in the groups of reactions with a high and low associated expression values. It then maximizes the number of matches between a reaction state, defined through a minimum flux value, and the group to which the reaction belongs. Thus, if a reaction is included in the highly expressed group, the aim is to obtain a flux value over the minimum, which is performed by solving the MILP in Box 2.
Several network states can yield the same overall similarity to expression data, i.e., multiple flux distributions may yield the same objective function value. iMAT tackles this issue through an adapted flux variability analysis (FVA): First, it forces each reaction to be active and evaluates the similarity, and then repeats the process in a similar way by forcing each reaction to be inactive. The final outcome is computed by comparing the two obtained similarities. A reaction is termed active if its inclusion results in higher similarity to data, and it is termed as inactive, if its inclusion decreases this similarity. In the case that both similarities are equal, iMAT categorizes the reaction as undetermined.
INIT
INIT was optimized to integrate evidences from the Human Protein Atlas, although expression data are integrated when proteomic evidences are missing. In this case, INIT does not group reactions in categories in contrast to iMAT. Instead, it adopts experimental data to weight the binary variable of the corresponding reaction, whereby the weight is a function of experimental data (e.g., gene expression profiles) or a set of arbitrary numbers that quantify the color code of the entries of the Human Protein Atlas. In addition, INIT imposes a positive net production of metabolites for which there is experimental support for that context or tissue. Hence, when a metabolite is experimentally determined to be present, its net production is forced to comply with a given lower bound. As a result, INIT allows the integration of metabolomics data in a qualitative way. This method has been applied to generate a human metabolic reaction database (“Human Metabolic Atlas2,” n.d.) where several tissue-specific model reconstructions can be examined.
tINIT
tINIT, an extension of INIT, has been recently proposed (Agren et al., 2014). Here, the main innovation comes with the definition of a set of metabolic tasks that the final context-specific model must perform. These tasks can represent production or consumption of a certain metabolite or the activation of entire pathways that are known to occur in a given context. Furthermore, reversible reactions are constrained to operate in only one direction, which introduces an extra binary variable. The user can choose between establishing a net production of certain metabolites, as in INIT, or maintaining the steady state. Finally, the task-driven strategy of tINIT renders this algorithm close to the principles of the GIMME-like family, since it aims to obtain operational context-specific models in coherence with experimental data.
Advantages and disadvantages of the iMAT-like family
The main advantage of this family of methods is the independence of a RMF; therefore, these methods are convenient for extracting context-specific models when no specific RMF is known to dominate the context, which is often the case for tissue-specific models of multicellular organisms. However, MILP problems are computationally more challenging in comparison to LP problems, and may, in general, require longer computation time. This is particularly the case of iMAT, in which two MILPs have to be solved in the modified FVA per reaction. iMAT can be easily implemented using the createTissueSpecificModel function of the COBRA toolbox, although only one MILP is solved in this implementation, ultimately reducing the computation time at the expense of neglecting the exhaustive search through the space of possible multiple optima. The INIT and tINIT methods are integrated within the RAVEN toolbox (Agren et al., 2013) for Matlab, and the user can define a set of metabolic tasks to be performed (tINIT) or run the algorithm without any (INIT). Note that selection of direction in reversible reactions is disabled by default.
MBA-Like Family
The MBA-like family is composed of MBA (Jerby et al., 2010), mCADRE (Wang et al., 2012) and FastCORE (Vlassis et al., 2014). While previous methods perform both a flux prediction and a context-specific model reconstruction, MBA-like methods only return a context-specific model as output. This family a priori categorizes reactions in two sets, the core and the non-core. The core set includes those reactions with positive evidences (e.g., high-throughput data and/or well-curated biochemical knowledge) of being active in a certain context. Once these sets are defined, the MBA-like methods prune the GEM by eliminating non-core reactions that are unnecessary to ensure consistency in the core set, i.e., no blocked reaction is allowed in the final model. Thereby, all reactions must carry non-zero flux in at least one feasible solution. As a result, checking model consistency is a crucial part of these methods and also the main difference in comparison to the other methods. FVA have been used to pinpoint blocked reactions, but it is computationally expensive since it requires solving two optimization problems per reaction (Mahadevan and Schilling, 2003). Thus, the major changes in formulation are due to finding faster alternatives to perform the same task. However, other differences arise when defining the core set and during the pruning process. Box 3 shows the three MBA-like algorithms in pseudocode, as well as the generalization of the family (consult Table 1 for a glossary of used symbols).

Box 3. Pseudocode describing algorithms of the MBA-like family corresponding to: (A) the generalization of the family, (B) MBA, (C) FastCORE, (D) mCADRE. The CheckModelConsistency function (E) of MBA and FindSparseMode (F) of FastCORE are presented separately. Consult the Glossary of symbols for notation.
MBA
MBA divides the core set in two subcores: a set with high likelihood to be present in the context-specific model (CH), if evidence comes from well-curated biochemical knowledge in that particular context, and a set with moderate likelihood (CM) if evidence comes from context-specific high-throughput data. The algorithm performs the pruning iteratively and randomly by selecting a non-core (NC) reaction to be eliminated, and checking consistency at each step: if CH and a user-defined fraction of CM remain unblocked, MBA removes the reaction out of the model along with CM and NC corresponding blocked reactions. This routine is repeated until no reaction is left in NC. The topology of the final model clearly depends on the order in which non-core reactions are eliminated. Therefore, to remove artifacts due to the order, the algorithm is repeated a number of times (1000 in Jerby et al., 2010) to obtain a population of context-specific models. Later, reactions are ranked according to their occurrence in the population and added up to CH until a consistent model is obtained. MBA proposes an alternative to FVA to check consistency in a more efficient way: First, it solves a LP problem which maximizes the total sum of fluxes. It then removes active reactions (i.e., carrying non-zero flux) and repeats the LP over the remaining set of reactions. If no reaction is found to be active, FVA is applied to each reaction to determine whether it is blocked. The process is repeated until all reactions have been classified either as blocked or unblocked.
mCADRE
A prominent characteristic of mCADRE lies in ranking reactions of the genome-scale reconstruction according to three scores: expression-, connectivity-, and confidence-level-based. In addition, this ranking determines the core set of reactions as well as the order by which non-core reactions are eliminated. The core is determined by fixing a threshold value to the expression-based score; therefore, reactions whose values are above the threshold are included in the core, and the rest constitute the non-core reactions. Unlike other methods, the expression-based score does not directly consider the levels of expression. Instead, it calculates the frequency of expressed states over a battery of transcript profiles in the same context, and, thus, requires a previous binarization of the expression data. Reactions outside the core are then ranked according to the connectivity-based score, which assesses the connectedness of adjacent reactions, and the confidence level-based score, which accounts for the type of evidences supporting a reaction in the genome-scale reconstruction.
Non-core reactions are in turn sequentially removed according to the previous ranking, and consistency is evaluated. Here, mCADRE presents two other innovations: it defines a set of key metabolites, with positive evidences of appearing in the context-specific model reconstruction, and relaxes the stringent condition of including all core reactions in the final model. More specifically, a reaction can only be eliminated if it does not prevent the production of a key metabolite and if it is unnecessary to ensure core consistency. However, if evidence exists for the respective transcript to be unexpressed in any of the context-specific samples, mCADRE allows the elimination of the reaction even if it blocks some of the core reactions. To this end, two conditions have to be satisfied: (1) production of key metabolites is not impaired and (2) the relation between the number of blocked core and non-core reactions matches a predefined ratio. To check model consistency, mCADRE maintains the procedure proposed in MBA, although adapted to use FastFVA (Gudmundsson and Thiele, 2010) instead of maximizing the total sum of flux values. mCADRE has been used to create the Tissue-Specific Encyclopedia of Metabolism (“Tissue-Specific Encyclopedia of Metabolism3,” n.d.) using the Recon1 human metabolic reconstruction (Duarte et al., 2007) and data from the Gene Expression Barcode Project (McCall et al., 2014) to extract 126 tissue-specific model reconstructions.
FastCORE
While FastCORE aims also at obtaining a minimal consistent model containing all core reactions, typical for this family of methods, it differs principally from MBA and mCADRE in the algorithmic strategy. Instead of eliminating one non-core reaction followed by consistency evaluation at each step, FastCORE solves two LPs: The first LP maximizes the cardinality of the core set of reactions, computed as the number of reaction values above a small positive constant. On the other hand, the second LP minimizes the cardinality outside the core set by minimizing the L1-norm of the flux vector, under the constraint that the entire core set must remain active. These two LPs are repeatedly applied in alternating fashion until the core set is consistent, whereby activation of all core reactions is ensured while including a minimum set of non-core reactions in the final model. To deal with reversible reactions, FastCORE evaluates both directions by changing the sign of the corresponding column of the stoichiometric matrix.
Advantages and disadvantages of MBA-like methods
One of the main advantages of this family over other methods is the possibility to integrate multiple data sets of different nature together with well-curated biochemical knowledge. Defining a core set of reactions from such a diverse collection of experimental evidence may increase the confidence for a particular set of reactions to appear in a certain context (e.g., tissue), as missing information on one data set can be complemented by another. Moreover, imposing the whole core set inclusion can be highly advantageous, as reactions with overwhelming evidence would always be included in the context-specific model. Moreover, like the iMAT-like family, MBA-like methods are independent of a RMF and, hence, appropriate to be employed if no RMF is known to operate in a given context. Nevertheless, we would like to emphasize that MBA-like methods provide only a context-specific model reconstruction, in contrast to the iMAT-like methods which generate both a context-specific reconstruction and a flux distribution.
MBA-like methods follow two ways to define the core set of reactions: MBA takes into account well-curated biochemical knowledge and a variety of experimental data (e.g., transcript, protein, metabolite, and/or metabolic flux profiles). While this approach to define the core set of reactions may be more accurate, it is also time-consuming due its manual nature. On the other hand, the definition of the core set in mCADRE allows for full automation, since it relies only on determining a threshold to expression-based evidence.
In terms of computation time, FastCORE outperforms the contending alternatives. Therefore, it has advantages over other methods when computing time is the limiting resource, provided that a properly defined core set is given (note that FastCORE does not provide an operational definition of a core set). The good time-related performance of FastCORE is due to two main innovations: First, the maximization of the cardinality represents a softer objective than the maximization of the total sum of flux values (used in MBA), since fluxes are only required to be above a small positive value. Consequently, solving this optimization problem usually results in more active reactions per iteration than the MBA counterpart. Second, the computation of the L1-norm to prune non-core reactions renders the pruning step more efficient due to the possibility to remove a once more than one reaction. These modifications make FastCORE the fastest algorithm in this family of methods, as it is able to extract a context-specific model in a computational time two to three orders of magnitude smaller than that expended by mCADRE and MBA (Vlassis et al., 2014). Finally, both mCADRE and FastCORE can be run under the Matlab environment (“FastCORE in COBRA toolbox4,” n.d., “mCADRE source code5,” n.d.).
Conclusions
Here we presented a classification of the existing approaches for extracting context-specific metabolic models. We classified the methods into three families according to their mathematical formulation. Furthermore, we also proposed a mathematical generalization for each family, which summarizes the fundamental principles shared by its members.
Altogether, the classification and generalization constitutes a mathematical framework that aims to fulfill three main purposes: First, it provides a better understanding of the rationale behind methods, allowing an easy inspection of its main characteristics as well as highlighting the advantages and shortcomings. Second, such structured knowledge may facilitate the envisioning of novel approaches to extract context-specific models. Third, it may help users in choosing a best suited method for their particular problem, since the classification outlines the differences in the data and knowledge requirements as input to the particular methods.
The flow-chart on Figure 2 demonstrates that an optimal choice with respect to the parameters and available data (Table 2) may be executed in a simple and concise manner by answering few questions. Initially, one may select between methods that perform both, a model extraction and a flux prediction (GIMME-and iMAT-like families), or methods which only provide a context-specific model (MBA-like families). To further select between the GIMME- and the iMAT-like families, one can take into account if a RMF is known to operate in the context under consideration. In that case the GIMME-like family may provide the method of choice, since the resulting model would be guaranteed to include the RMF. Selection of GIMME or GIM3E may depend on the interest to integrate metabolomics data along with transcripts profiles, the computational platform of the current implementations, or the difference in computing time. For instance, the choice is between the COBRA toolbox in Matlab, for GIMME, or its version in Python, for GIM3E (“The OpenCOBRA Project,” n.d.), or between the LP formulation of GIMME, vs. the more computationally demanding MILP of GIM3E.
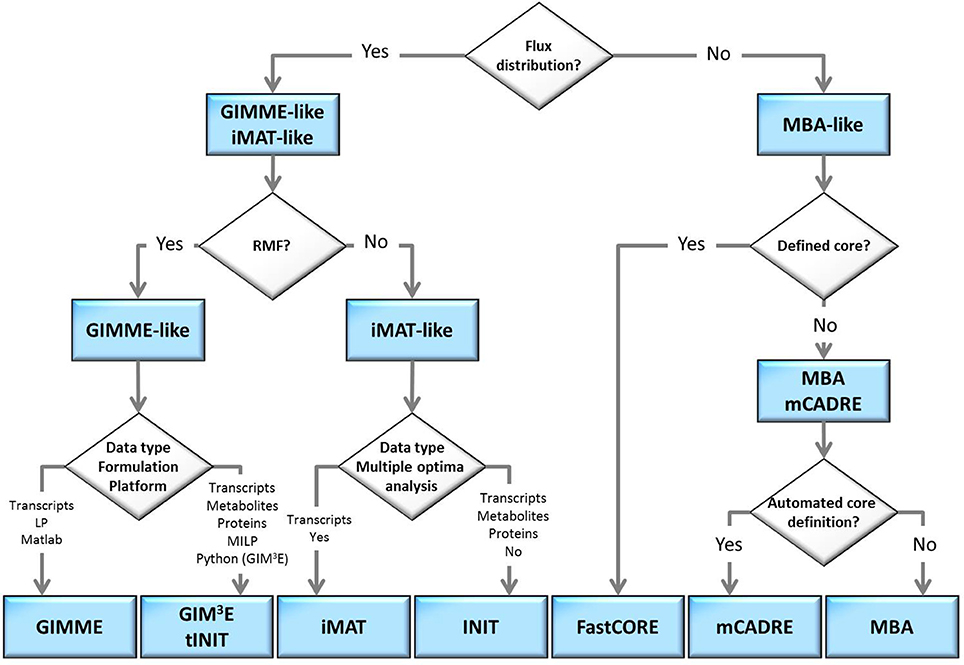
Figure 2. Optimal choice of methodologies when tackling a context-specific reconstruction problem. The choice can be made by answering a few questions, in a flowchart manner, related to: demand of model extraction and flux prediction, knowledge on a required metabolic functionality, the type of experimental data available or the computational platform.
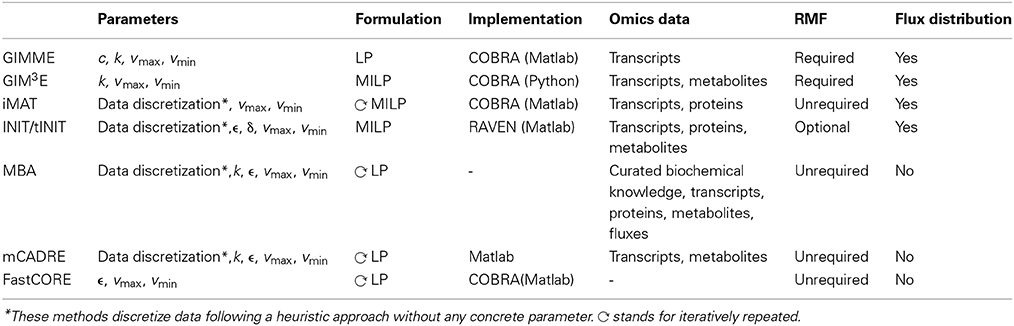
Table 2. Summary of methods for context-specific metabolic model extraction.
Without the information about the operability of a particular RMF in a given context, the iMAT-like family may provide the method of choice. To select between iMAT and INIT one could take into account the flexibility on integrating different types of experimental data, since iMAT was developed to integrate transcript profiles, whereas INIT can integrate semi-quantitative proteomic data, transcript profiles and metabolic evidences. In addition, one could consider the possibility of the method to discriminate between multiple optima with same similarity score, together with the computational cost for performing this task.
In contrast, if only a context-specific model extraction is required, one may opt for any of the presented method. However, the methods in the MBA-like family have some advantageous properties, namely, the integration of a variety of experimental data sources and the inclusion of reactions for which there is strong experimental evidence in the context-specific reconstruction. One may then choose based on the core set definition of each method as well as on the total computational time required. The MBA-like family proposes two ways to define the core: the MBA semi-automated procedure, whereby reactions are included in the core set if there is sufficient positive evidence across different databases, and the mCADRE automated procedure, whereby reactions are included if the expression value of the respective transcript is larger than a given threshold. Thus, if an appropriate number of databases contain experiments about the context of interest and the computation time is not a primary limitation, the MBA core definition may be a suitable alternative. As previously commented, this procedure can cross-validate the confidence on a reaction to belong to a certain context, due to the simultaneous usage of several databases. Subsequently, one can readily employ MBA to extract the context-specific model, or can opt for FastCORE, which can perform the extraction, using the previously defined core, in a more efficient way. On the other hand, mCADRE could be preferentially applied when an automated core definition is preferred. Moreover, the mCADRE relaxation of whole core inclusion can improve accuracy when a core reaction diminishes the overall coherence with respect to the data, through the inclusion of non-core reactions with negative evidences to ensure consistency. Finally, one can also apply FastCORE to a core set defined in an automated way to benefit of its rapid computation. However, neglecting the characteristic core relaxation and ranking of non-core reactions of mCADRE.
Development of new approaches for extraction of context-specific metabolic models can further expand on the advantages of the existing methods, while facilitating efficient computation accounting for the shortcomings. This will allow rapid devising of context-specific models and their interconnection in larger multilevel models, typical for complex eukaryotes, to allow for more realistic simulation scenarios.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge the Max Planck Society and the International Max Planck Research School in Primary Metabolism and Plant Growth (IMPRS-PMPG) for funding support.
Footnotes
1. ^Retrieved from http://opencobra.sourceforge.net/openCOBRA/Welcome.html
2. ^Retrieved from http://www.metabolicatlas.com/
3. ^Retrieved from https://price.systemsbiology.net/tissue-specific-encyclopedia-metabolism-tsem
4. ^Retrieved from https://github.com/opencobra/cobratoolbox/tree/cd4368bda07ef4d63486a3683865f4d9f3bc53fe
5. ^Retrieved from https://price.systemsbiology.net/tissue-specific-encyclopedia-metabolism-tsem
References
Agren, R., Bordel, S., Mardinoglu, A., Pornputtapong, N., Nookaew, I., and Nielsen, J. (2012). Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 8:e1002518. doi: 10.1371/journal.pcbi.1002518
Agren, R., Liu, L., Shoaie, S., Vongsangnak, W., Nookaew, I., and Nielsen, J. (2013). The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 9:e1002980. doi: 10.1371/journal.pcbi.1002980
Agren, R., Mardinoglu, A., Asplund, A., Kampf, C., Uhlen, M., and Nielsen, J. (2014). Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 10:721. doi: 10.1002/msb.145122
Arnold, A., and Nikoloski, Z. (2014). Bottom-up metabolic reconstruction of arabidopsis and its application to determining the metabolic costs of enzyme production. Plant Physiol. 165, 1380–1391. doi: 10.1104/pp.114.235358
Becker, S. A., and Palsson, B. O. (2008). Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 4:e1000082. doi: 10.1371/journal.pcbi.1000082
Blazier, A. S., and Papin, J. A. (2012). Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 3:299. doi: 10.3389/fphys.2012.00299
Bordbar, A., Mo, M. L., Nakayasu, E. S., Schrimpe-Rutledge, A. C., Kim, Y.-M., Metz, T. O., et al. (2012). Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 8:558. doi: 10.1038/msb.2012.21
Bordbar, A., Monk, J. M., King, Z. A., and Palsson, B. O. (2014). Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120. doi: 10.1038/nrg3643
Chakrabarti, A., Miskovic, L., Soh, K. C., and Hatzimanikatis, V. (2013). Towards kinetic modeling of genome-scale metabolic networks without sacrificing stoichiometric, thermodynamic and physiological constraints. Biotechnol. J. 8, 1043–1057. doi: 10.1002/biot.201300091
Chandrasekaran, S., and Price, N. D. (2010). Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 107, 17845–17850. doi: 10.1073/pnas.1005139107
Chang, R. L., Ghamsari, L., Manichaikul, A., Hom, E. F. Y., Balaji, S., Fu, W., et al. (2011). Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol. Syst. Biol. 7:518. doi: 10.1038/msb.2011.52
Chowdhury, A., Zomorrodi, A. R., and Maranas, C. D. (2014). k-OptForce: integrating kinetics with flux balance analysis for strain design. PLoS Comput. Biol. 10:1003487. doi: 10.1371/journal.pcbi.1003487
Colijn, C., Brandes, A., Zucker, J., Lun, D. S., Weiner, B., Farhat, M. R., et al. (2009). Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 5:1000489. doi: 10.1371/journal.pcbi.1000489
Collins, S. B., Reznik, E., and Segrè, D. (2012). Temporal expression-based analysis of metabolism. PLoS Comput. Biol. 8:e1002781. doi: 10.1371/journal.pcbi.1002781
Dal'Molin, C. G., Quek, L.-E., Palfreyman, R. W., Brumbley, S. M., and Nielsen, L. K. (2010). C4GEM, a genome-scale metabolic model to study C4 plant metabolism. Plant Physiol. 154, 1871–1885. doi: 10.1104/pp.110.166488
Daran-Lapujade, P., Rossell, S., van Gulik, W. M., Luttik, M. A. H., de Groot, M. J. L., Slijper, M., et al. (2007). The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc. Natl. Acad. Sci. U.S.A. 104, 15753–15758. doi: 10.1073/pnas.0707476104
De Oliveira Dal'Molin, C. G., and Nielsen, L. K. (2013). Plant genome-scale metabolic reconstruction and modelling. Curr. Opin. Biotechnol. 24, 271–277. doi: 10.1016/j.copbio.2012.08.007
De Oliveira Dal'Molin, C. G., Quek, L.-E., Palfreyman, R. W., Brumbley, S. M., and Nielsen, L. K. (2010). AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 152, 579–589. doi: 10.1104/pp.109.148817
Dharmawardhana, P., Ren, L., Amarasinghe, V., Monaco, M., Thomason, J., Ravenscroft, D., et al. (2013). A genome scale metabolic network for rice and accompanying analysis of tryptophan, auxin and serotonin biosynthesis regulation under biotic stress. Rice (N.Y.) 6:15. doi: 10.1186/1939-8433-6-15
Duarte, N. C., Becker, S. A., Jamshidi, N., Thiele, I., Mo, M. L., Vo, T. D., et al. (2007). Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. U.S.A. 104, 1777–1782. doi: 10.1073/pnas.0610772104
Fernie, A. R. (2007). The future of metabolic phytochemistry: larger numbers of metabolites, higher resolution, greater understanding. Phytochemistry 68, 2861–2880. doi: 10.1016/j.phytochem.2007.07.010
Garcia-Albornoz, M. A., and Nielsen, J. (2013). Application of genome-scale metabolic models in metabolic engineering. Ind. Biotechnol. 9, 203–214. doi: 10.1089/ind.2013.0011
Gomes de Oliveira Dal'Molin, C., Quek, L.-E., Palfreyman, R. W., and Nielsen, L. K. (2011). AlgaGEM—a genome-scale metabolic reconstruction of algae based on the Chlamydomonas reinhardtii genome. BMC Genomics 2(Suppl. 4):S5 doi: 10.1186/1471-2164-12-S4-S5
Grafahrend-Belau, E., Junker, A., Eschenröder, A., Müller, J., Schreiber, F., and Junker, B. H. (2013). Multiscale metabolic modeling: dynamic flux balance analysis on a whole-plant scale. Plant Physiol. 163, 637–647. doi: 10.1104/pp.113.224006
Gudmundsson, S., and Thiele, I. (2010). Computationally efficient flux variability analysis. BMC Bioinformatics 11:489. doi: 10.1186/1471-2105-11-489
Hyduke, D. R., Lewis, N. E., and Palsson, B. Ø. (2013). Analysis of omics data with genome-scale models of metabolism. Mol. BioSyst. 9, 167–174. doi: 10.1039/c2mb25453k
Jamshidi, N., and Palsson, B. (2010). Mass action stoichiometric simulation models: incorporating kinetics and regulation into stoichiometric models. Biophys. J. 98, 175–185. doi: 10.1016/j.bpj.2009.09.064
Jensen, P. A., and Papin, J. A. (2011). Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 27, 541–547. doi: 10.1093/bioinformatics/btq702
Jerby, L., Shlomi, T., and Ruppin, E. (2010). Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol. Syst. Biol. 6:401. doi: 10.1038/msb.2010.56
Joyce, A. R., and Palsson, B. Ø. (2006). The model organism as a system: integrating “omics” data sets. Nat. Rev. Mol. Cell Biol. 7, 198–210. doi: 10.1038/nrm1857
Kleessen, S., Araujo, W. L., Fernie, A. R., and Nikoloski, Z. (2012). Model-based confirmation of alternative substrates of mitochondrial electron transport chain. J. Biol. Chem. 287, 11122–11131. doi: 10.1074/jbc.M111.310383
Lee, D., Smallbone, K., Dunn, W. B., Murabito, E., Winder, C. L., Kell, D. B., et al. (2012). Improving metabolic flux predictions using absolute gene expression data. BMC Syst. Biol. 6:73. doi: 10.1186/1752-0509-6-73
Lewis, N. E., Nagarajan, H., and Palsson, B. O. (2012). Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305. doi: 10.1038/nrmicro2737
Link, H., Christodoulou, D., and Sauer, U. (2014). Advancing metabolic models with kinetic information. Curr. Opin. Biotechnol. 29, 8–14. doi: 10.1016/j.copbio.2014.01.015
Machado, D., and Herrgård, M. (2014). Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 10:e1003580. doi: 10.1371/journal.pcbi.1003580
Mahadevan, R., and Schilling, C. H. (2003). The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 5, 264–276. doi: 10.1016/j.ymben.2003.09.002
McCall, M. N., Jaffee, H. A., Zelisko, S. J., Sinha, N., Hooiveld, G., Irizarry, R. A., et al. (2014). The Gene Expression Barcode 3.0: improved data processing and mining tools. Nucleic Acids Res. 42, D938–D943. doi: 10.1093/nar/gkt1204
Milne, C. B., Kim, P. J., Eddy, J. A., and Price, N. D. (2009). Accomplishments in genome-scale in silico modeling for industrial and medical biotechnology. Biotechnol. J. 4, 1653–1670. doi: 10.1002/biot.200900234
Mintz-Oron, S., Meir, S., Malitsky, S., Ruppin, E., Aharoni, A., and Shlomi, T. (2012). Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 339–344. doi: 10.1073/pnas.1100358109
Moxley, J. F., Jewett, M. C., Antoniewicz, M. R., Villas-Boas, S. G., Alper, H., Wheeler, R. T., et al. (2009). Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p. Proc. Natl. Acad. Sci. U.S.A. 106, 6477–6482. doi: 10.1073/pnas.0811091106
Orth, J. D., Thiele, I., and Palsson, B. Ø. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi: 10.1038/nbt.1614
Poolman, M. G., Kundu, S., Shaw, R., and Fell, D. A. (2013). Responses to light intensity in a genome-scale model of rice metabolism. Plant Physiol. 162, 1060–1072. doi: 10.1104/pp.113.216762
Poolman, M. G., Miguet, L., Sweetlove, L. J., and Fell, D. A. (2009). A genome-scale metabolic model of Arabidopsis and some of its properties. Plant Physiol. 151, 1570–1581. doi: 10.1104/pp.109.141267
Rossell, S., van der Weijden, C. C., Lindenbergh, A., van Tuijl, A., Francke, C., Bakker, B. M., et al. (2006). Unraveling the complexity of flux regulation: a new method demonstrated for nutrient starvation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 103, 2166–2171. doi: 10.1073/pnas.0509831103
Saha, R., Suthers, P. F., and Maranas, C. D. (2011). Zea mays irs1563: a comprehensive genome-scale metabolic reconstruction of maize metabolism. PLoS ONE 6:e21784. doi: 10.1371/journal.pone.0021784
Schellenberger, J., Que, R., Fleming, R. M. T., Thiele, I., Orth, J. D., Feist, A. M., et al. (2011). Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 6, 1290–1307. doi: 10.1038/nprot.2011.308
Schmidt, B. J., Ebrahim, A., Metz, T. O., Adkins, J. N., Palsson, B., and Hyduke, D. R. (2013). GIM3E: condition-specific models of cellular metabolism developed from metabolomics and expression data. Bioinformatics 29, 2900–2908. doi: 10.1093/bioinformatics/btt493
Schulze, W. X., and Usadel, B. (2010). Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 61, 491–516. doi: 10.1146/annurev-arplant-042809-112132
Shlomi, T., Cabili, M. N., Herrgård, M. J., and Palsson, B. Ø. Ruppin, E. (2008). Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 26, 1003–1010. doi: 10.1038/nbt.1487
Soh, K. C., Miskovic, L., and Hatzimanikatis, V. (2012). From network models to network responses: integration of thermodynamic and kinetic properties of yeast genome-scale metabolic networks. FEMS Yeast Res. 12, 129–143. doi: 10.1111/j.1567-1364.2011.00771.x
Sweetlove, L. J., and Ratcliffe, R. G. (2011). Flux-balance modeling of plant metabolism. Front. Plant Sci. 2:38. doi: 10.3389/fpls.2011.00038
Ullah, M., and Wolkenhauer, O. (2010). Stochastic approaches in systems biology. WIREs Syst. Biol. Med. 2, 385–397. doi: 10.1002/wsbm.78
Vlassis, N., Pacheco, M. P., and Sauter, T. (2014). Fast reconstruction of compact context-specific metabolic network models. PLoS Comput. Biol. 10:e1003424. doi: 10.1371/journal.pcbi.1003424
Wang, Y., Eddy, J. A., and Price, N. D. (2012). Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 6:153. doi: 10.1186/1752-0509-6-153
Wilkinson, D. J. (2009). Stochastic modelling for quantitative description of heterogeneous biological systems. Nat. Rev. Genet. 10, 122–133. doi: 10.1038/nrg2509
Yang, C., Hua, Q., and Shimizu, K. (2002). Integration of the information from gene expression and metabolic fluxes for the analysis of the regulatory mechanisms in Synechocystis. Appl. Microbiol. Biotechnol. 58, 813–822. doi: 10.1007/s00253-002-0949-0
Keywords: genome-scale models, high-throughput data, data integration, context-specific models, mathematical programming
Citation: Robaina Estévez S and Nikoloski Z (2014) Generalized framework for context-specific metabolic model extraction methods. Front. Plant Sci. 5:491. doi: 10.3389/fpls.2014.00491
Received: 10 July 2014; Accepted: 03 September 2014;
Published online: 19 September 2014.
Edited by:
Lee Sweetlove, University of Oxford, UKReviewed by:
Rui Alves, Universitat de Lleida, SpainThomas Christopher Rhys Williams, Universidade Federal de Vicosa, Brazil
Copyright © 2014 Robaina Estévez and Nikoloski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zoran Nikoloski, Systems Biology and Mathematical Modeling Group, Max-Planck Institute of Molecular Plant Physiology, Am Mühlenberg 1, 14424 Potsdam, Germany e-mail:bmlrb2xvc2tpQG1waW1wLWdvbG0ubXBnLmRl