 Andriy Bilichak
Andriy Bilichak Yaroslav Ilnytskyy2
Yaroslav Ilnytskyy2 Rafal Wóycicki
Rafal Wóycicki Dawson Fogen
Dawson Fogen Igor Kovalchuk
Igor Kovalchuk- 1Lethbridge Research Centre, Agriculture and Agri-Food Canada, Lethbridge, AB, Canada
- 2Department of Biological Sciences, University of Lethbridge, Lethbridge, AB, Canada
Plants are able to maintain the memory of stress exposure throughout their ontogenesis and faithfully propagate it into the next generation. Recent evidence argues for the epigenetic nature of this phenomenon. Small RNAs (smRNAs) are one of the vital epigenetic factors because they can both affect gene expression at the place of their generation and maintain non-cell-autonomous gene regulation. Here, we have made an attempt to decipher the contribution of smRNAs to the heat-shock-induced transgenerational inheritance in Brassica rapa plants using sequencing technology. To do this, we have generated comprehensive profiles of a transcriptome and a small RNAome (smRNAome) from somatic and reproductive tissues of stressed plants and their untreated progeny. We have demonstrated that the highest tissue-specific alterations in the transcriptome and smRNAome profile are detected in tissues that were not directly exposed to stress, namely, in the endosperm and pollen. Importantly, we have revealed that the progeny of stressed plants exhibit the highest fluctuations at the smRNAome level but not at the transcriptome level. Additionally, we have uncovered the existence of heat-inducible and transgenerationally transmitted tRNA-derived small RNA fragments in plants. Finally, we suggest that miR168 and braAGO1 are involved in the stress-induced transgenerational inheritance in plants.
Introduction
Plants constantly interact with environmental factors that can either benefit or jeopardize their homeostasis depending on the intensity and nature of factors encountered. Significant environmental perturbations that exceed the optimum range of plant development can cause stress and trigger the onset of gene expression changes in plants. Stress-induced alterations in the transcriptome profile have been shown to be both stress- and tissue-specific, although the general stress response (GSR) that has been extensively studied in yeast and animals is also present in plants (Kreps et al., 2002; Kultz, 2005; Dinneny et al., 2008; Walley and Dehesh, 2010; Iyer-Pascuzzi et al., 2011). Previously, we demonstrated that the progeny of plants exposed to salt stress and Tobacco mosaic virus (TMV) exhibit a higher tolerance not only to the same stressor but also to methyl methane sulfonate, a genotoxic agent that induces DNA methylation, as compared to control progeny (Boyko et al., 2010; Kathiria et al., 2010). Hence, although plants may trigger immediate specific gene expression changes to cope with a particular stressor, transgenerational inheritance and responses to stress seem to involve non-specific priming of stress-responsive genes. Distinct epigenetic mediators of multigenerational inheritance of stress memory have been recently identified in two animal models—Drosophila (Seong et al., 2011) and C. elegans (Buckley et al., 2012), which makes it tempting to argue for the existence of similar transgenerational mediators in plants.
Plants belonging to the genus Brassica are predominantly annual cool-season economically important crops whose cultivation is acutely affected by elevated temperatures and drought (Hall, 2001; Yu et al., 2012). Heat shock (HS) stress can severely influence reproductive tissues of plants, which contributes to poor seed set yield (Zinn et al., 2010). On the other hand, the pre-treatment of plants under moderate HS conditions can protect them from an acute heat stress and provide a better stress tolerance, a phenomenon known as induced or acquired thermotolerance (Gurley, 2000). The expression of heat-shock proteins regulated by heat stress transcription factors is believed to mediate the heat stress response and acquired thermotolerance in plants (Kotak et al., 2007; Yu et al., 2012).
Curiously, the acclimation to abiotic factors and induced resistance to pathogens (Van Loon, 1997) can be propagated into the next generation, a phenomenon known as transgenerational acquired tolerance (Boyko et al., 2010; Holeski et al., 2012). Given a practical value of such response for its implementation in plant biotechnology and agriculture, there has been a remarkable interest in unraveling pathways involved in transgenerational acquired tolerance. Currently, two different mechanisms are proposed to account for the aforementioned phenomenon: maternal effects on seed size (Agrawal, 2001) and epigenetic alterations that allow a vertical propagation of acquired traits without changing the underlying genomic DNA sequence (Jablonka and Raz, 2009). Whereas the former mechanism has little support from experiments on plants subjected to multigenerational stress, the evidence favoring the second mechanism has been provided at the molecular level (Jablonka and Raz, 2009; Boyko et al., 2010; Bilichak et al., 2012; Luna et al., 2012; Rasmann et al., 2012; Slaughter et al., 2012).
Heritable fluctuations in DNA methylation, chromatin composition and smRNA metabolism are among the primary causes of transgenerational epigenetic inheritance (Holeski et al., 2012). Being reversible in their nature, the acquired components of the epigenetic machinery are usually prone to significant alterations during sexual plant development. In angiosperms, epigenetic factors have to survive the multifaceted process of gametogenesis and early embryogenesis that encompasses a significant restructuring of both cells and chromatin (Ibarra et al., 2012). Although methylation at CpG sequences found in vegetative cells is largely retained in sperm cells, embryo and conceivably in egg cells, methylation at the asymmetric CpHpH (where H = A, C, or T) sequences is largely lost as compared to vegetative cells (Gehring et al., 2009; Hsieh et al., 2009b; Ibarra et al., 2012; Jullien et al., 2012). Curiously, in all three cases (in sperm, egg cell and embryo), asymmetric DNA methylation is proposed to be restored through the RNA-dependent DNA methylation (RdDM) pathway guided by smRNAs generated by companion cells or tissues that do not contribute genetic material to the progeny. In turn, smRNAs have been shown to be vital environmental sensors, the expression of which is acutely affected by abiotic and biotic stressors (Khraiwesh et al., 2012). Therefore, we hypothesized that perturbations in the expression of smRNAs in somatic tissues caused by environmental fluctuations would eventually be imprinted in the transcriptome patterns in gametes and progeny.
Materials and Methods
Plant Cultivation
In the current work, R-o-18 a rapid-cycling self-compatible inbred line of Brassica rapa var. trilocularis (Roxb.) Hanelt (yellow sarson) was used (Williams and Hill, 1986; Amoah et al., 2012). Seeds were originally obtained from Dr. Joan E. Krochko, the NRC Plant Biotechnology Institute (Saskatoon, Saskatchewan, Canada).
Growing B. rapa Plants for Heat-Stress Experiments and Tissue Harvesting
Seeds obtained from a single unstressed plant were germinated on All purpose potting soil (Plant Etc; Lethbridge, AB, Canada) mixed in the proportion 4:1 with vermiculite (The Professional Gardener Co LTD, AB, Canada) in 4 × 4 inch square pots. The soil mixture was pre-soaked once with Miracle-Gro fertilizer (Scotts Canada Ltd., Mississauga, ON, Canada) and was maintained constantly moist with tap water. Rapid-cycling B. rapa parental plants (30 plants per group) and their progeny were cultivated in biochambers (BiofootTM, model GC-20, Winnipeg, MB, Canada) under continuous high-intensity cold light illumination (200 μmol m−2 s−1) provided by fluorescent lamps and a 60% relative humidity at 22°C as described previously (Daugherty and Musgrave, 1994; Tel-Zur and Goldman, 2007). The cultivation of plants under constant light did not affect their phenotypic appearance as compared to plants grown under a long-day photoperiod (16 h day, 8 h night). Two weeks post germination, before the appearance of apical inflorescence shoots, plants from the treated group were exposed to 42°C for 3 h per day for 7 days in a row (Supplementary Figure 1). Subsequently, plants from both control and treated groups and their progeny were grown in continuous light at 22°C.
In order to monitor the development of stress memory and follow its transmission to germ cells, 17 days after heat-shock treatment, middle leaves and up to 1 cm of the inflorescence meristem (32 days post germination) dissected from flower buds of control and stressed plants were harvested.
To follow the occurrence of epigenetic marks in reproductive tissues, the pollen and unfertilized and fertilized ovules from both groups were harvested separately. The inflorescences were covered with plastic bags to prevent cross-pollination between treatment groups. The pollen from control and heat-shock treated plants was harvested separately using a vacuum manifold method (Johnson-Brousseau and McCormick, 2004).
The unfertilized ovules containing mature embryo sacs were synchronized developmentally by emasculating flowers at stage 13 (Smyth et al., 1990). Twenty four hours later, the ovules were collected from hand-dissected pistils (Le et al., 2010). The fertilized ovules containing zygotes were harvested from siliques 24 h after hand-pollination of emasculated flowers (Le et al., 2010). Finally, after 21–28 days-after-pollination, mature-green embryos and endosperms were hand dissected using fine forceps from seed coat and harvested separately.
In order to track the transmission of epigenetic marks to the next unstressed generation, the control and treated plants were propagated, and tissue samples from 2-week-old seedlings were harvested. Each developmental stage was represented by two biological replicates (15 plants per repeat) that were harvested independently.
Molecular Techniques and Methods Used in this Study
Total RNA isolation and purification
Total RNA isolation was done using the Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Total RNA was further purified and concentrated using the RNA Clean-Up and Concentration Kit (Norgen Biotek Corp., Ontario, Canada).
Both, the quality and concentration of every sample were quantified using the NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific Inc.). Agarose gel electrophoresis was performed to verify RNA sample integrity.
mRNA deep sequencing
The mRNA libraries were prepared from 16 samples of total RNA in 2 biological replicates for most of the tissues according to the TruSeq RNA sample Prep v2 LS protocol (Illumina, San Diego, CA, U.S.A.). Briefly, mRNA was purified from the total RNA samples using poly-T oligo-attached magnetic beads followed by mRNA fragmentation, first- and second-strand cDNA synthesis. Later, the overhangs resulting from the fragmentation of double-stranded (ds) cDNA were repaired to form blunt ends. A single “A” nucleotide was added to the 3′ ends of the blunt fragments to prevent them from ligating to one another during the adapter ligation reaction. Multiple indexing adapters were ligated to the ends of ds cDNA to prepare them for hybridization onto a flow cell followed by a PCR amplification step. The libraries were quantified using the qPCR technique and analyzed on a Bioanalyzer 2100 (Agilent Technologies) using a DNA specific chip. Subsequently, the libraries were normalized and pooled together followed by flow-cell cluster generation using a cBot fully automated clonal cluster generation system for Illumina sequencing. Single-end multiplexed sequencing was done using the Illumina GAIIx platform with the total of 100 cycles.
Deep sequencing of small non-coding RNAs
Small non-coding RNA libraries were generated from the aforementioned tissues using the TruSeq small RNA library construction kit according to the manufacturer's protocol (Illumina, San Diego, CA, U.S.A.). Briefly, the 3′ and 5′ adapters were ligated to small RNAs from the total RNA sample followed by reverse-transcription PCR amplification. PCR was performed with two primers that annealed to the ends of adapters and contained indexes. Subsequently, the libraries with unique indexes were pooled together; the cDNA was gel-purified using a TBE PAGE gel and then concentrated by ethanol. Following a successful library quality control by qPCR, flow cell cluster generation was performed using a cBot. Single-end multiplexed sequencing was done using the Illumina GAIIx platform with the total of 36 cycles.
Northern blot analysis of small non-coding RNAs
The confirmation of small RNA sequencing data was performed using a non-radioactive northern blot method as described previously (Kim et al., 2010). Briefly, 3 ug of total RNA was separated on a 15% urea PAGE (National diagnostics, USA) and transferred to a positively charged nylon membrane (Roche). The pre-hybridization was performed with ULTRAhyb Ultrasensitive Hybridization Buffer (Ambion) at 37°C for at least 30 min in the hybridization oven followed by hybridization step with the DIG-labeled probe in ULTRAhyb Ultrasensitive Hybridization Buffer at 37°C overnight with a slow rotation. The DIG-labeled miR168 probe—AB492 (Supplementary Table 9) was synthesized by Eurofins MWG Operon (Huntsville, AL, USA). Subsequently, the membrane was washed and incubated with Anti-Digoxigenin-AP Fab fragments (Roche) followed by the detection using CDP-Star (Roche). The membrane was photographed using the FluorChem HD2 MultiImage™ Light Cabinet (Cell Biosciences Pty Ltd, Heidelberg, Australia), and the bands were quantified using the Image J program (NIH, http://rsbweb.nih.gov/ij/).
cDNA synthesis and qPCR gene expression analysis
500 ng of total RNA from every tissue in 2 biological replicates was treated with DNase I, purified, converted into cDNA and quantified with qPCR. The real-time quantitative PCR was performed using SsoFast EvaGreen Supermix (Bio-Rad). cDNAs were amplified under the following conditions: (1) 98°C for 2 min for one cycle; 98°C for 5 s, 48°C for 5 s, 65°C to 95°C for 5 s; for 40 cycles; (2) melt-curve analysis −65°C to 95°C for 5 s, with a 0.5°C increment. Primers for the real-time quantitative PCR were designed using the Beacon Designer7 program (Supplementary Table 9). The optimization of the annealing temperature, melt-curve analysis, and gel analysis of amplicons were performed for each set of primers. To evaluate the PCR efficiency, the standard curve was established using a series of cDNA dilutions. Gene expression was confirmed for four differentially expressed genes in the progeny: Bra029235, Bra031065, Bra029719, Bra040903, and Bra032254 (AtAGO1 homolog). The normalization was done against four B. rapa housekeeping genes: GAPDH, TUBULIN, EF1α, and UBC (Qi et al., 2010).
The statistical significance between treatment groups was evaluated using the two-tailed paired Student's t-test (α = 0.05) and performed using JMP 10.0 software (SAS Institute Inc.).
The Bioinformatic Treatment of Deep Sequencing Data
mRNA deep sequencing data analysis
Base calling and demultiplexing of transcriptome sequencing reads were performed using the Consensus Assessment of Sequence and Variance (CASAVA) v 1.6 and Novobarcode software (http://www.novocraft.com/). FastQC v 0.10.1 software was used for the preliminary quality check. The reads were mapped to the genome, and de novo splice site prediction was performed using TopHat v 2.0.4 beta software (Trapnell et al., 2009). The de novo predicted splice-sites obtained were used to perform transcript assembly for each sample separately using Cufflinks v 2.0.2 (Trapnell et al., 2010). The assemblies were merged using the cuffmerge tool in Cufflinks software with the reference file containing B. rapa predicted genes (Wang et al., 2011; Trapnell et al., 2012). The aim of this analysis was to compare our de novo assembly with the in silico predicted transcriptome (Wang et al., 2011).
The merged transcript assembly was used to assess differentially expressed features between treatments with the cuffdiff tool in Cufflinks software (Trapnell et al., 2012). The q-value Benjamini-Hochberg method (Benjamini and Hochberg, 1995) below 0.05 was considered as a significant difference of gene expression between treatment groups.
Deep sequencing data analysis of small non-coding RNAs
Base calling and demultiplexing of sequencing reads generated by the Illumina GAIIx platform were performed using the CASAVA v 1.8.1. software (http://support.illumina.com/downloads/casava_181.ilmn). Then, the sequencing reads were processed using adapter trimming Cutadapt v 1.1 software (Martin, 2011) with options specified to search for adapter sequences anywhere within the sequencing read and to retain only the sequences that were longer than 17 nucleotides; quality trimming was performed with a Sanger quality cutoff score of 20. Summary statistics and run quality data were collected from the adapter trimmed libraries using FastQC v 0.10.1 software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Samples that passed quality control tests were aligned to the B. rapa genome (accession number AENI01000000) (Wang et al., 2011) using Bowtie v 2 2.0.0—beta2 aligner run (Langmead et al., 2009). Bowtie command was: bowtie -v 2 --best -m 50 -p 4 -S <brassica_index> <trimmed_reads> Normalization was performed using DESeq (Anders and Huber, 2010). The reads that could be aligned to the genome were further classified based on feature classes. The alignment was performed in a stepwise fashion. The reads that could be aligned to features of a certain class were counted and excluded from subsequent alignments. In this way, the pool of sequences was gradually depleted. The mapping process continued until the remaining reads could not be assigned to any of the known mapping categories and were labeled as “unclassified.” Both unique reads and reads that matched multiple loci (cutoff ≥ 50 loci for reads mapped to multiple loci) were considered for this analysis.
The alignment to B. rapa mature and passenger strand miRNA sequences was performed using conservative and predicted miRNA sequences described before (Yu et al., 2012). A novel miRNA prediction was done by using MiRDeep-P software (Yang and Li, 2011) followed by its alignment to both the predicted trans-acting siRNAs (ta-siRNAs) by using the UEA sRNA workbench (Stocks et al., 2012) and non-coding RNA genes, structured cis-regulatory elements and self-splicing RNAs from Rfam database, v 10.01 (Burge et al., 2013). The remaining reads were aligned to repeats and predicted genes (B. rapa gene database v 1.2). As a result, all small RNAs were sorted into 7 groups: miRNA candidates, gene-aligned small RNAs, conserved miRNAs, Rfam v 10.01, ta-siRNA candidates, transposon-aligned and unclassified small RNAs.
To perform statistical comparisons, the sequence reads were collapsed to unique tags after adapter trimming using a fastx_collapser program from the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Raw read counts assigned to unique tags were compared between treatments and tissues. Normalization and statistical tests were performed using DESeq bioconductor package as described in the user's manual (Anders and Huber, 2010). Reads with the sum raw counts ≤ 5 across all libraries that participated in a particular comparison were excluded from the analysis. The cutoff value for significance was q < 0.2 (the Benjamini-Hochberg method) (Benjamini and Hochberg, 1995).
The precise mapping of all assorted small RNAs was done using MicroRazerS v 1.0 software with default settings (i.e., the first 16-nt-long ones were matched, no mismatch allowed) (Emde et al., 2010). Only tags that were considered to be significantly changed (q < 0.2) were annotated. Due to the repetitive nature of some tag sequences, some single tag sequences had multiple annotations.
The prediction of putative gene targets for miRNAs
The psRNATarget software was used with default settings to predict and retrieve gene IDs of putative miRNA gene targets from the B. rapa CDS library v 1.1 (Dai and Zhao, 2011). Subsequently, putative gene-target IDs were identified in the differentially expressed gene dataset of the corresponding tissue and annotated using the SWISS-PROT database (Bairoch and Apweiler, 2000).
Results
The Analysis of Gene Expression in B. rapa Parental Leaves, Inflorescence Meristem, Pollen, Unfertilized Ovules, 24-h Post-Fertilization Ovules, Embryo, Endosperm and Leaf Tissues of Progeny Plants after Heat Shock Treatment
The transcriptome libraries were generated and analyzed from the total RNA of somatic and reproductive tissues of control and exposed parental B. rapa plants as well as from the untreated progeny (Supplementary Table 1). In the experiment, the genome-matched reads comprised of on average 67.70% of the total reads. Unfortunately, the whole transcriptome sequencing data for B. rapa species that could be used for the comparison with our data are not available yet.
Unique tissue-specific alterations in mRNA accumulation in heat-shock treated B. rapa plants was observed, with a nearly even representation of the number of up- and down-regulated genes (Figure 1). Whereas in the leaves of parental plants that were directly exposed to stress, we detected 562 differentially expressed genes as compared to the untreated controls (the Benjamini-Hochberg method, q < 0.05, Figure 1), in the inflorescence meristem derived from the exposed shoot apical meristem, there were only 79 differentially expressed genes as compared to the untreated controls (Figure 1). Both paternal and maternal reproductive tissues responded to HS with little changes in gene expression (3, 78, and 24 differentially expressed genes in pollen, unfertilized ovules and fertilized ovules, respectively, Figure 1).
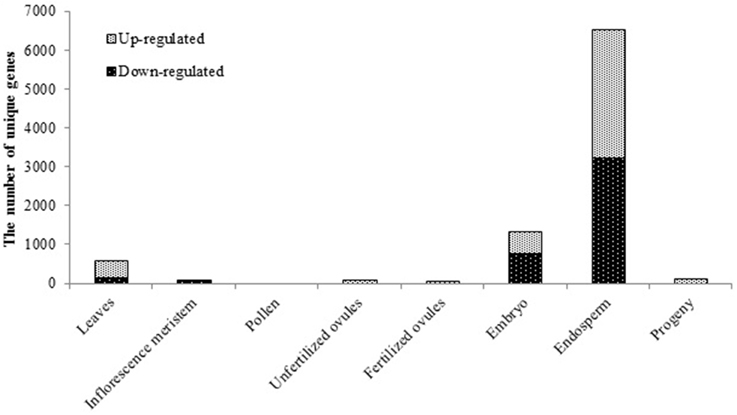
Figure 1. The effect of exposure to the elevated temperature on gene expression in tissues of parental plants and leaves of untreated progeny plants of Brassica rapa. The bars represent the number of unique differentially expressed genes in response to HS in parental tissues and progeny, the Benjamini-Hochberg method, q < 0.05.
Enigmatically, the highest oscillations in gene expression were observed in the tissue that was not directly exposed to HS—the endosperm (6365 differentially expressed genes, q < 0.05, Figure 1) as compared to control tissue. Such behavior of the endosperm could possibly be attributed to global genome demethylation and a decrease in the expression of silencing-related genes as compared to other tissues, as it was previously reported to occur in the Arabidopsis endosperm under normal conditions (Hsieh et al., 2009b; Jullien et al., 2012). Considerable oscillations of gene expression were also observed in the embryo, albeit to a lesser extent than that in the endosperm (1311 differentially expressed genes, q < 0.05, Figure 1).
The analysis showed that despite the fact that drastic changes in gene expression were observed in the embryo and endosperm tissues, few changes were passed on to the leaf tissue of progeny when examined at 2 weeks post germination stage (116 differentially expressed genes, q < 0.05, Figure 1). This suggests that the reversal of changes in gene expression takes place either during the final steps of seed maturation or later throughout seed germination and plantlet development. At the same time we did not examine the seed tissues from the progeny of stressed plants, hence we cannot rule out the possibility of re-occurrence of similar pattern in embryo and endosperm. Additionally, unfertilized and fertilized ovules collected 24 h post-fertilization also responded to HS treatment with moderate changes in the gene expression profile (Figure 1). Therefore, it may be suggested that alterations in the transcriptional activity of genes occur during stages of cell division/expansion of seed development and proceeds into green embryo and endosperm tissues (Le et al., 2010). Noteworthy, since we did not control the actual fertilization of ovules in the emasculated flowers that were manually pollinated, we were unable to rule out the possibility that fertilized ovule tissue samples could have been contaminated with unfertilized embryo sacs. This could eventually affect the sequencing outcome in the ovule tissue samples collected at 24 h postfertilization. Nevertheless, a comparison of unfertilized and fertilized ovule transcriptomes from control plants revealed 791 significantly differentially expressed genes (q < 0.05, the Benjamini-Hochberg method), with 603 being up-regulated in fertilized ovules (data not shown), which suggests that in our experiment, fertilized and unfertilized ovule samples were indeed different. However, it does not completely rule out the presence of unfertilized ovules in fertilized ovule samples.
A unique tissue-specific pattern of transcriptome fluctuations following HS stress was further observed in the comparison analysis (Figure 2). The most pronounced overlap of differentially expressed genes was seen between embryo and endosperm (1240 commonly changed genes, Figures 2C,D) and then between embryo and leaves (51 commonly changed genes, Figure 2E). The untreated progeny of plants stressed by HS had the highest overlap of differentially expressed genes with the endosperm (32 commonly changed genes, Figure 2D) followed by the inflorescence meristem (15 commonly changed genes, Figure 2E) and embryo (13 commonly changed genes, Figure 2E). Surprisingly, the embryo and unfertilized and fertilized ovules of stressed parental plants had the lowest number of common differentially expressed genes, which can conceivably be attributed to the overall quiescent response of maternal reproductive organs to stress (Figures 2B,C).
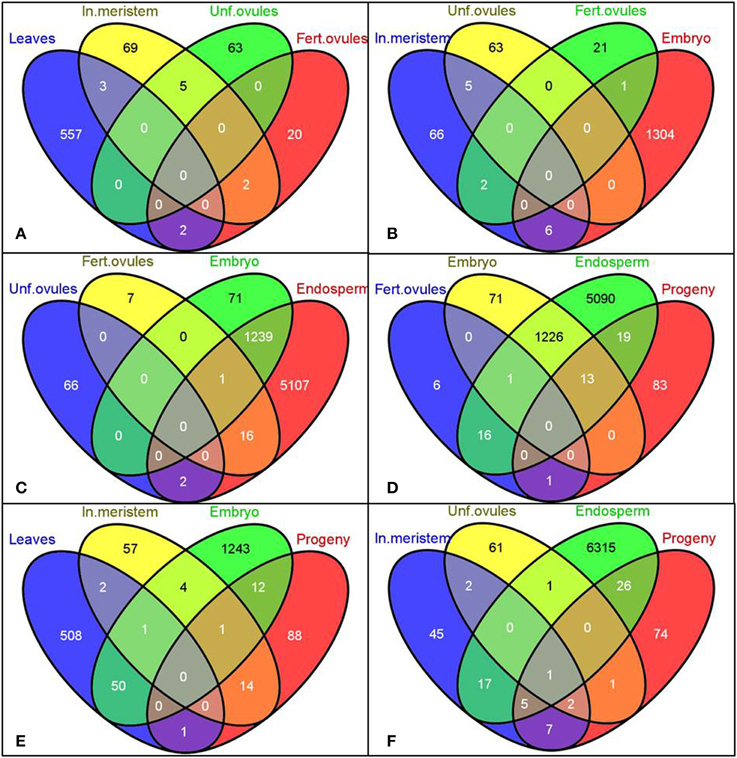
Figure 2. Venn diagrams representing common genes the expression of which was significantly changed in the selected tissues. Gene IDs of differentially expressed genes (both up- and down-regulated) were used to create the Venn diagrams using the Venny program (Oliveros). Labels: In. meristem—Inflorescence meristem, Unf. Ovules—Unfertilized ovules, Fert. ovules—Fertilized ovules.
The transcriptome sequencing data were further confirmed in the progeny of stressed and non-stressed plants for four genes with the most significant change in gene expression (log2FC) using the qPCR method (Supplementary Table 2, q < 0.05, the Benjamini-Hochberg method).
Gene Ontology Annotation of Differentially Expressed Genes in Tissues of B. rapa Plants Subjected to Heat Shock
Differentially expressed genes in all tissues were further grouped into gene ontology categories according to the biological process they participate in Conesa et al. (2005). As expected, the majority of genes in all tissues fall into the gene ontology (GO) category “response to stress” (Supplementary Figures 2, 3), with the only exception of unfertilized ovules (Supplementary Figure 3C) where the GO category “response to stress” was absent. Instead, genes coding for the proteins involved in the overall cell structure development were predominant.
A Small RNA Transcriptome Analysis of B. rapa Parental Leaves, Inflorescence Meristem, Pollen, Unfertilized Ovules, 24-h Post-Fertilization Ovules, Embryo, Endosperm and Leaf Tissues of Progeny Plants after Heat Shock Treatment
Illumina small RNA sequencing was performed to compare alterations in smRNA transcriptomes in somatic and reproductive tissues of B. rapa plants and in their progeny in response to HS treatment. We hypothesized that it would allow us to reveal possible messengers of transgenerational stress memory inheritance in plants. For smRNA sequencing, we used the same tissue samples as for mRNA sequencing. An average of 4,873,042 reads per library was achieved. Genome-matched reads comprised on average 52.68% of the total reads (Supplementary Table 3), and it was comparable with the previously published report on Chinese cabbage (56.96% of genome-mapped smRNA reads) (Wang et al., 2012). We observed drastic fluctuations in the percentage of genome-mapped sequencing reads: the highest percentage was observed in the inflorescence meristem (79.57% mapped reads on average) and the lowest one—in leaves (25.71% mapped reads on average). Lower mapping rate in some libraries may be attributed to tissue-specific factors, for instance, in tissues containing chlorophyll significant fraction may map to plastid genome. Quick examination of several unaligned reads from progeny of control 1 library, which originates from the leave tissue, showed that they indeed originate from chloroplast genomes.
A general compositional analysis of smRNA libraries revealed substantial variations in the relative smRNA enrichment among tissues, whereas differences between control and exposed groups within the same tissue were not so pronounced (Figure 3). The overwhelming majority of aligned sequencing reads were mapped to gDNA gene regions (30.29% of library reads on average) followed by those ones mapped to transposons (15.88% of library reads on average) and miRNAs (4.76% of library reads on average). The remaining classified reads comprised of Rfam database (v 10.01) mapped to smRNAs (1.38% of library reads on average), ta-siRNA candidates (0.23% of library reads on average) and miRNA candidates (0.14% of library reads on average). We observed considerable differences in the relative smRNA library composition between parental somatic tissues such as leaves and the inflorescence meristem and between paternal and maternal reproductive tissues.
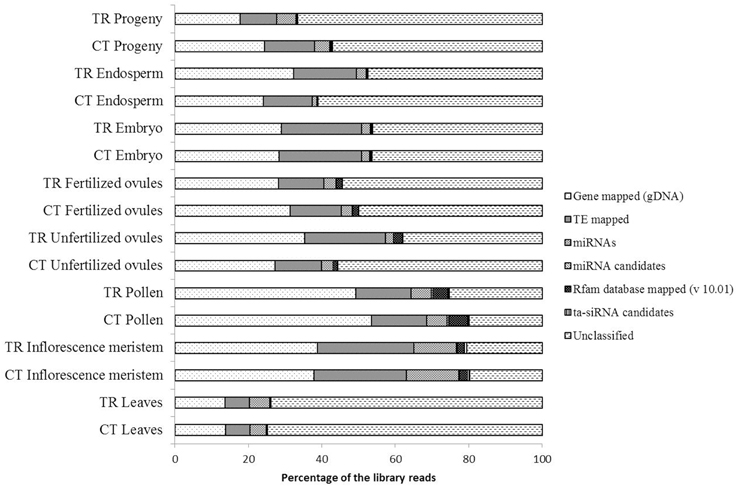
Figure 3. A general compositional analysis of small RNA libraries sequenced from the total RNA of corresponding tissues. The length of stacked bars represents the percentage of smRNA fraction occupied by a specific smRNA type in the corresponding library.
The most pronounced alterations in the library composition after HS stress were observed in unfertilized ovule tissues with a 9.27 and 8.15% increase in the number of transposon- and gene-mapped smRNA reads as compared to control, respectively (Figure 3). Changes in the endosperm were less pronounced. The progeny of stressed plants had a detectable decrease in the number of smRNA reads mapped to genes (6.66% as compared to control) and to transposons (3.61% as compared to control, Figure 3), which was in accordance with the overall up-regulation of gene expression observed in the progeny of stressed as compared to control plants (Figure 1). The leaf tissues that were directly subjected to stress suffered only minor oscillations in the smRNA library composition as compared to control, albeit both the control and treated smRNA libraries had the highest relative amount of unclassified reads among all tissues that conceivably could contain responsive smRNAs (Figure 3).
The Length Distribution of Small RNA Libraries
Previous reports have shown a conservative pattern of the smRNA length distribution in plants which is compatible with DICER-dependent transcriptome processing (Rajagopalan et al., 2006; Fahlgren et al., 2007; Moxon et al., 2008; Szittya et al., 2008; Song et al., 2010; Chi et al., 2011). We observed the overall bimodal length distribution of smRNA sequencing reads in all tissues (Supplementary Figure 4) with a major peak at 24 nucleotides (40.19% of all reads on average) and a smaller shoulder at 21-nt (18.30% of all reads on average), which was consistent with the published data on B. rapa plants (He et al., 2008; Wang et al., 2012). Patterns for the 21- and 24-mers distribution were dissimilar between tissues. In a stark contrast to other tissues, the 21-nt smRNA fraction was predominant in leaves (28.06% on average) followed by 24-nt-long smRNAs (22.20% on average, Supplementary Figure 4A). Also, an equivalent accumulation of 21- and 24-nt-long reads was observed in leaves of 2-week-old progenies (28.42 and 29.55% for 21- and 24-nt-long reads, respectively, Supplementary Figure 4H) that is in agreement with the previous report (He et al., 2008). Whereas the paternal (Supplementary Figure 4C) and maternal (Supplementary Figures 4D,E) reproductive tissues had a different distinguishable pattern of smRNA library length distribution, the length of smRNA reads derived from the inflorescence meristem (Supplementary Figure 4B), embryo (Supplementary Figure 4F) and endosperm (Supplementary Figure 4G) tissues was highly similar. None of examined tissues, except for pollen, responded to HS with detectable fluctuations in the relative smRNA length distribution (Supplementary Figure 4C). The most prominent alterations were observed in the 24-nt-long smRNA fraction (a 1.4 fold increase as compared to control, Student's t-test, α = 0.05), which usually corresponds to the smRNA fraction deriving from heterochromatic genomic regions in angiosperms (Axtell, 2013).
The length distribution analysis of sequencing reads mapped to the prominent genomic sequence categories revealed that the majority of gene- (Supplementary Figure 5) and transposon-mapped (Supplementary Figure 6) smRNAs fell mostly into the 24- and 21-nt-long categories in all tissues except for pollen. In male reproductive tissues, a vast amount of low range small RNAs (19-mers and smaller) observed in the total smRNA library were mapped to gene regions in gDNA (17.09 and 16.97% of the relative fraction of 17-nt-long reads in the total smRNA and gene-mapped smRNA libraries, respectively, Supplementary Figure 5C), which suggests that a tissue-specific transcriptome degradation process takes place in pollen. Similar results were previously reported in mature Arabidopsis pollen where 16-nt-long reads were prevailing in the smRNA sequencing library. Unfortunately, the authors did not explain the origin of small size smRNAs in pollen (Grant-Downton et al., 2009b).
smRNAs mapped to transposable elements (TEs) in male tissues were distributed between a major peak of 24-nt long reads (3.96% of the total reads on average) and an unusually broad but minor peak stretching from 17 to 22-nt in length (Supplementary Figure 6C). Previous reports on smRNA metabolism in Arabidopsis pollen (Slotkin et al., 2009) give us a reason to speculate that low range smRNAs can apparently be non-cell autonomous silencing signals generated in the vegetative nucleus to suppress the TE activity in sperm cells.
Pollen and Endosperm of Parental Plants and Leaf Tissues of Progeny Plants Demonstrate Significant Alterations in the Small RNAome Profile in Response to Heat Shock
Despite the fact that we did not detect significant fluctuations in the sequence read length distribution in response to HS, considerable changes in the differential expression of smRNAs were observed in all tissues except unfertilized ovules (Figure 4). Surprisingly, the most striking alterations were detected in systemic tissues that were not directly exposed to stress such as pollen (621 differentially expressed smRNAs), the endosperm (385 differentially expressed smRNAs) and more importantly, in leaf tissues of the progeny (376 differentially expressed smRNAs). Minor changes were recorded in leaves (12 differentially expressed smRNAs), the inflorescence meristem (15 differentially expressed smRNAs), fertilized ovules (31 differentially expressed smRNAs) and the embryo (eight differentially expressed smRNAs). Strikingly, none of smRNAs responded to HS in unfertilized ovules (Figure 4, the Benjamini-Hochberg method, q < 0.2).
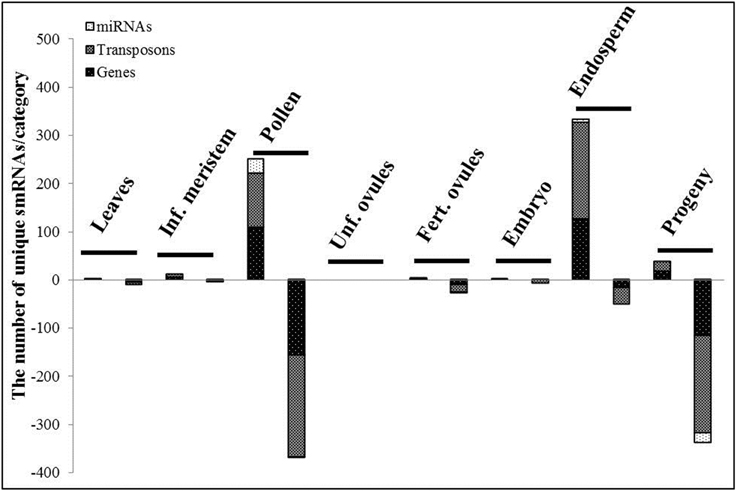
Figure 4. The differentially expressed small RNAs, TR vs. CT. The stacked bars represent the number of the unique differentially expressed smRNAs per category in the corresponding tissue. SmRNAs were divided into 3 categories regarding the genome regions they were mapped to: miRNAs, transposon and gene regions. The first and second stacked bars show the number of smRNAs with the positive and negative log2fold change values (TR vs. CT) in the corresponding tissue, respectively (the Benjamini-Hochberg method, q < 0.2).
The detailed mapping and analysis of differentially expressed smRNAs showed a lower representation of gene-mapped siRNAs as compared to those ones derived from transposable elements in all tissues (566 and 823 in total for genes and transposons, respectively). Whereas in the endosperm, 86.75% of all differentially expressed smRNAs were up-regulated (Figure 4), in pollen and leaves of the progeny, 59.58 and 89.89% of all differentially expressed smRNAs were down-regulated, respectively. MiRNAs comprised a minor fraction with only 59 of them being differentially expressed in toto.
Differentially Expressed siRNAs are Mapped to Genes that are Unique for Every Tissue
To reveal the commonalities in action of the altered siRNAs mapped to gene regions, we extracted their gene IDs and used them for inter-tissue comparisons. Since TEs are known to get activated in response to environmental perturbations (Grandbastien, 1998), we hypothesized that the levels of TE- and gene-derived siRNAs would be affected by stress exposure, and moreover this response is expected to be tissue-specific.
In our study, in accordance with our hypothesis, the differentially expressed siRNAs were mostly tissue-specific, with a small number of siRNAs overlapping between pollen and endosperm (32 common genes, Figures 5B–D), pollen and leaf tissues in the progeny (16 common genes, Figures 5C,D), and endosperm and leaf tissues in the progeny (14 common genes, Figures 5C,D). The number of differentially expressed siRNAs in fertilized ovules was very small (11 gene-mapped siRNAs) despite a substantially larger number of them found in pollen (267 gene-mapped siRNAs, Figure 5).
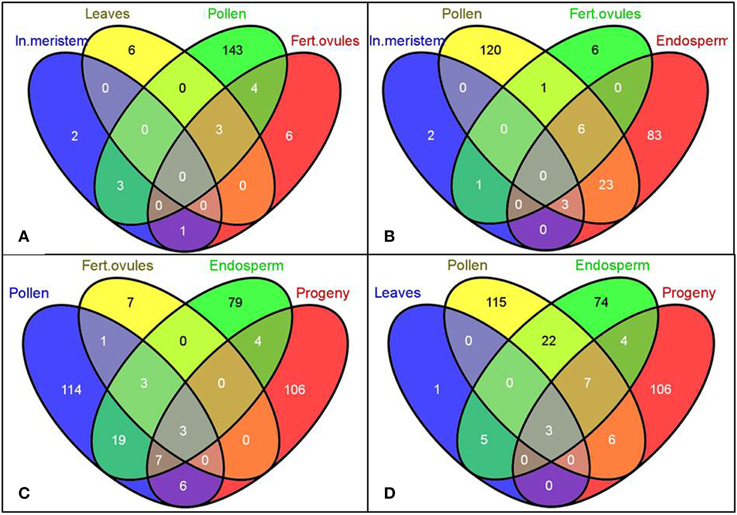
Figure 5. The Venn diagrams representing common genes mapped to the significantly altered siRNAs among tissues of B. rapa plants exposed to heat stress. Gene IDs of putative gene-mapped siRNA targets were used to create the Venn diagrams using the Venny program (Oliveros)1. Labels: In. meristem—Inflorescence meristem, Fert. ovules—Fertilized ovules. (A–D) Comparison of common genes mapped to the significantly altered siRNAs among denoted tissues.
Expression Hot Spots of Small Non-Coding RNAs Observed in B. rapa Tissues after Heat Shock Treatment
In our study, we observed hot spots of changes in the expression of smRNAs involved in stress response common for leaves, pollen, fertilized ovules, endosperm and leaves in the progeny (the Benjamini-Hochberg method, q < 0.2, Table 1). Despite the fact that the commonly regulated smRNAs were mapped to three different genes (Bra003466, Bra018314, and Bra030669), the sequences of these smRNAs were highly similar because they were produced from the predicted tRNAs which resided in the intron region of the three aforementioned genes. tRNA-derived RNA fragments (tRFs) comprise a novel class of smRNAs discovered recently in plants (Hsieh et al., 2009a; Chen et al., 2011; Hackenberg et al., 2013) and in silico predicted to have a regulatory role in gene expression through the miRNA pathway (Loss-Morais et al., 2013). The existence in plants of transgenerationally transmitted, heat-responsive tRFs is a novel finding that adds one more variable to environmentally induced epigenetic responses to stress.

Table 1. The common stress-responsive hot spots mapped to the differentially expressed siRNAs in leaves, pollen, fertilized ovules, endosperm and leaves of the progeny of heat-treated B. rapa plants.
The Functional Annotation of Gene-Mapped siRNAs
To examine common biological pathways that are presumably affected by differentially expressed smRNAs mapped to genes, their putative targets were further annotated and classified according to the biological process they were involved in. To do this, we included only the data sets of the three tissues that demonstrated the maximum smRNAome disequilibrium after HS—pollen, endosperm and leaf tissues of the progeny. Due to the repetitive nature of the B. rapa genome, a number of differentially expressed sequencing reads were mapped to multiple genomic loci. Also, a vast majority of siRNAs originated from the intron sequences apparently do not have an ability to regulate gene expression at the posttranscriptional level, excluding the alternative transcripts that rarely occur in plants.
Overall, whereas the genes involved in “response to stress” were the predominant putative targets in pollen and leaves of the progeny tissues (19.13 and 19.78%, respectively, Figures 6A,C); in endosperm, smRNAs mapped to the genes involved in “RNA metabolic process” and “transport” were the most enriched (Figure 6B).
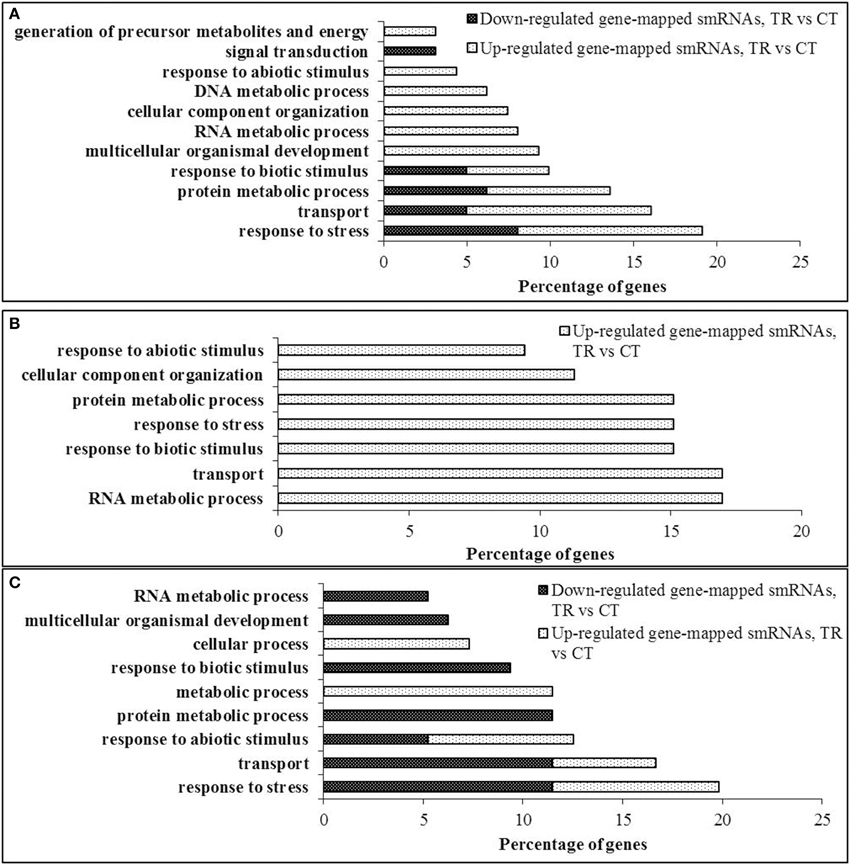
Figure 6. Gene ontology annotation of putative siRNA targets. (A) Pollen, (B) Endosperm, (C) Progeny. Coding sequences of putative targets of the differentially expressed gene-mapped siRNAs were extracted from the B. rapa transcriptome database v 1.2 and loaded as a FASTA file into the Blast2GO v 2.6.2 software for the NCBI BLAST similarity search using a blastx option (Conesa et al., 2005). Further, the recovered ontologies were annotated and grouped into gene ontology categories using default settings. Gene ontology nodes were combined into the most prominent categories using a GO-slim-TAIR tool and represented as bar graphs.
Common Differentially Expressed miRNAs in Examined Tissues
The analysis of siRNA fractions of sequencing reads was followed by a detailed examination of those reads mapped to conservative plant micro RNA (miRNA) sequences described in the recent paper (Yu et al., 2012) and those ones that were de novo predicted (Yang and Li, 2011). The unique sequences mapped to miRNAs constituted a minor fraction of differentially expressed smRNAs that responded to HS regardless of the tissue sample. The largest number of altered mature miRNA sequences were observed in pollen (31 miRNAs, 4.99% of all altered smRNAs, Supplementary Table 4), in the endosperm (6 miRNAs, 1.56% of all altered smRNAs, Supplementary Table 5) and in the progeny (20 miRNAs, 5.32% of all altered smRNAs, Supplementary Table 6).
Only one miRNA was differentially expressed in the inflorescence meristem and fertilized ovules as compared to controls (Supplementary Table 7), and none of altered miRNAs was observed in the unfertilized ovules, embryo and leaves of heat stressed B. rapa plants (the Benjamini-Hochberg method, q < 0.2).
Our analysis allowed us to predict one novel miRNA in the pool of differentially expressed smRNAs in pollen (miR22711, Supplementary Table 4) and two novel miRNAs in the progeny (miR31241, miR315691, Supplementary Table 6), suggesting that the potential for the sequencing-based discovery of novel miRNAs in B. rapa plants is not exhausted.
MicroRNAs are known to play an important role in plant stress response since they act as global regulators of gene expression (Kruszka et al., 2012). More importantly, recently miRNAs have been implicated in a non-cell autonomous mode of action (Carlsbecker et al., 2010; Marin et al., 2010). This raises a question whether miRNAs generated in the directly exposed tissues can be mobilized to distinct systemic organs, such as reproductive tissues, and modulate the inheritance of transgenerational stress memory. Hence, we performed the commonality analysis of miRNAs between sequencing libraries of parental plants and untreated progeny. We observed a unique pattern of tissue response to stress (Figure 7A), with only a few overlapping differentially expressed mature miRNAs among pollen, endosperm and leaf tissues of the progeny belonging to three microRNA gene families: bra-miR167, bra-miR390, and bra-miR168 (Figure 7B, Table 2). The members of miR167 and miR390 gene families have been implemented in the regulation of auxin response factors (ARFs) in Arabidopsis which are transcription factors that bind to auxin response elements in the promoters of early auxin response genes (Tiwari et al., 2003; Mallory et al., 2005; Montgomery et al., 2008). The discovery of differential expression of miR168 that regulates ARGONAUTE 1 (AGO1) level in Arabidopsis was the most intriguing (Vaucheret et al., 2004). AGO1 has been shown to be vital for plant development due to its unique and essential role in microRNA metabolism. Hence, the discovery of altered expression of miR168 in parental B. rapa plants exposed to heat stress and in the untreated progeny makes it tempting to speculate about its role in transgenerational epigenetic inheritance of stress memory. Consequently, the members of the miR168 microRNA gene family were selected for further analysis, and their expression was related to the AGO1 transcript level in both parental plants exposed to stress and the untreated progeny.
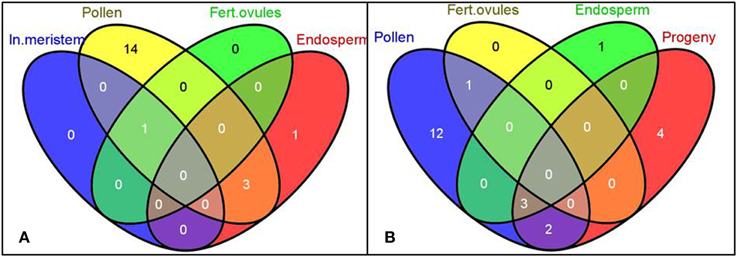
Figure 7. The Venn diagrams representing the common microRNA gene families of differentially expressed mature miRNAs among tissues in heat-stressed B. rapa plants as compared to controls. The differentially expressed smRNAs mapped to conservative miRNAs (TR vs. CT, q < 0.2, the Benjamini-Hochberg method) were grouped into microRNA gene families and used for the generation of Venn diagrams using the Venny software. Labels: In. meristem—Inflorescence meristem, Fert. ovules—Fertilized ovules. (A,B) Comparison of common microRNA gene families of differentially expressed mature miRNAs among denoted tissues.
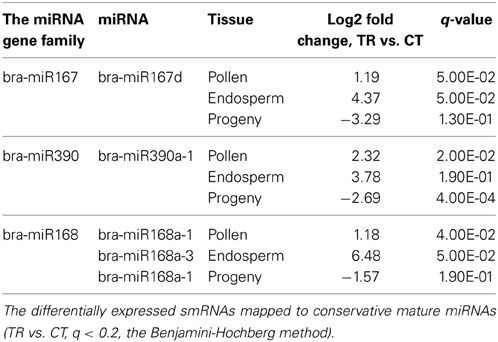
Table 2. The common significantly altered microRNA gene families in the pollen, endosperm and leaves of progeny of heat-stressed plants.
miR168 is a Putative Messenger of Transgenerational Epigenetic Inheritance
Simply detecting miRNAs in stressed parental tissues and untreated progeny does not provide evidence that they are functional as smRNAs and can guide targeting RNA and DNA substrates (Grant-Downton and Rodriguez-Enriquez, 2012). Hence, we proceeded with the examination of putative targets of differentially expressed miRNAs in parental tissues and the progeny. Since the endosperm was the most responsive tissue to stress at the level of gene and smRNA expression (see Figures 1, 4), it conceivably led to the highest number of smRNA/mRNA target pairs identified. Six smRNA/mRNA target pairs were identified in the endosperm of stressed plants (Table 3), with the most intriguing of them being bra-miR168/AGO1 (Max identity = 95%, E-value = 2E-99 for atAGO1 and braAGO1 BLAST search). Curiously, the expression of braAGO1 was also significantly altered in the embryo of stressed plants as compared to controls (Table 4, the Benjamini-Hochberg method). Overall, we observed a bimodal tissue-specific trend in the expression of braAGO1 after HS stress in parental plants. Whereas an insignificant down-regulation was observed in leaves, unfertilized and fertilized ovules, the up-regulation was detected in the inflorescence meristem and pollen (Table 4, the Benjamini-Hochberg method). The absence of negative correlation for significantly overexpressed miR168 and braAGO1, observed in our study in pollen, is in contrast with the previous report showing that the regulation of AGO1 expression by miR168 is active in mature Arabidopsis pollen grains (Grant-Downton et al., 2009a).
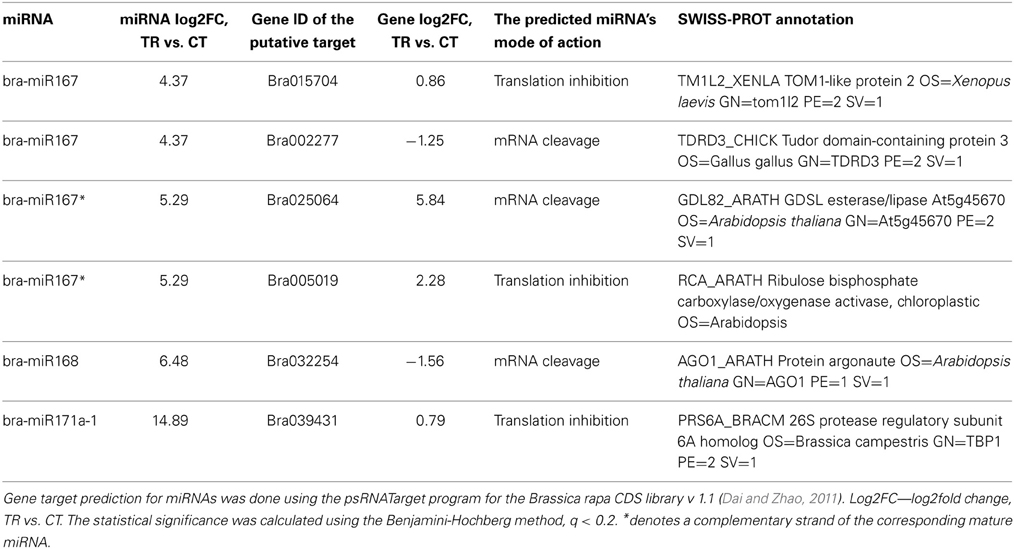
Table 3. Changes in the expression of miRNAs and their putative targets in response to heat stress in the endosperm of B. rapa plants.
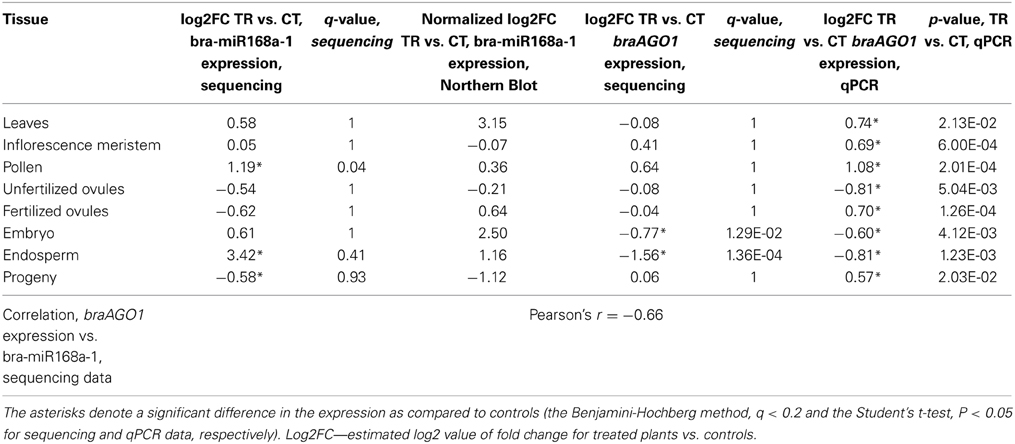
Table 4. The validation of bra-miR168a-1 and braAGO1 expression in tissues of heat-shock-stressed B. rapa parental plants and untreated progeny.
The expression of braAGO1 was similar in the progeny of treated and untreated plants, albeit a significant down-regulation of miR168 was detected (Table 4, the Benjamini-Hochberg method). An inverse correlation for miR168 expression and atAGO1 transcript levels under stress conditions is not always obvious in Arabidopsis, because both atAGO1 and miR168 promoters are activated under abiotic stress conditions, suggesting that an increase in the miR168 level is essential for retaining a stable AGO1 transcript level during stress response (Li et al., 2012). We also observed a moderate negative correlation for bra-miR168a-1 and braAGO1 expression in all tissues of stressed parental plants and untreated progeny of B. rapa (Pearson's r = −0.66, Table 4).
Unexpectedly, in the most of differentially expressed miRNAs, we did not find an obvious negative correlation with their putative gene targets in the endosperm (Table 3). On the contrary, both miRNAs and their target genes demonstrated an increase in the expression following HS. It is possible that these miRNAs function at the level of translational inhibition rather than the level of mRNA cleavage, although the latter mechanism is believed to prevail in plants (Axtell, 2013).
A detailed analysis of mature miRNA sequences belonging to the miR168 gene family revealed a common 20-nt-long core sequence between 3 miRNAs with only a variable 3′-terminal nucleotide (Supplementary Table 8). The validation of miR168 expression was performed using smRNA Northern blot analysis with a probe designed to recognize the consensus sequence of the three mature miRNAs (Supplementary Figure 7).
Discussion
The Embryo and Endosperm Demonstrate the Most Pronounced Oscillations in the Trancriptome Profile after Heat Shock Stress
A tissue-specific perturbation of gene expression in response to stress exposure has been previously shown in a number of plant species including Arabidopsis (Prandl et al., 1995; Nylander et al., 2001; Iyer-Pascuzzi et al., 2011), wine grape Vitis vinifera (Tillett et al., 2011), diploid cotton Gossypium arboretum (Zhang et al., 2013), Nicotiana plumbaginifolia (Castresana et al., 1990) and Brassica napus (Dong et al., 2004). In the current study, we observed tissue-dependent fluctuations of the transcriptome in response to stress in B. rapa plants (Figures 1, 2). Additionally, we demonstrated a comprehensive profile of gene expression following heat shock in somatic and reproductive parental tissues and in the untreated progeny of exposed plants. More importantly, we found that the highest oscillations of gene expression were observed not in parental tissues that were directly exposed to stress (such as leaves) but in the developmentally distant untreated seeds, suggesting the existence of a mitotically and meiotically transmitted signal of plant stress response (Figure 1). A handful of messengers have been implicated in heat stress response (HSR) in plants that include reactive oxygen species (ROS) (Larkindale and Huang, 2004; Larkindale et al., 2005a), Ca2+ cations (Liu et al., 2005), and phytohormones such as abscisic acid (ABA), salicylic acid (SA) and ethylene (Larkindale and Huang, 2004; Larkindale et al., 2005a,b). At present, only ROS was shown to mediate systemic signaling in response to heat stress (Miller et al., 2009), albeit the other mediators are also likely contribute to long-distance signaling in plants (Heil and Ton, 2008; Jung et al., 2009). Regardless of the signal's nature, in our experiments, their action resulted in the priming of an array of genes in somatic and reproductive tissues of stressed plants that enigmatically culminated in a burst of transcription changes in the embryo and endosperm (Figure 1). Alternatively, less pronounced HSR of gene expression observed in the inflorescence meristem and reproductive tissues of stressed B. rapa plants indicates a more stringent regulation of gene expression in these tissues as compared to the embryo and endosperm (Figure 1).
Unfortunately, reports indicating transcriptome changes in reproductive tissues of plants in response to stress are scarce. A single study conducted on mature pollen treated with 0°C for 72 h reported insignificant oscillations in the transcriptome profile of pollen as compared to vegetative leaf tissues (Lee and Lee, 2003). This is consistent with our data demonstrating only three genes to be differentially expressed following HS treatment in pollen (Figure 1). On the other hand, the lack of substantial oscillations in the transcriptome profile in exposed ovules as compared to controls can be simply due to the abortion of severely affected ovules to facilitate shunting of resources from reproductive activities into metabolic reactions that increase stress tolerance (Sun et al., 2004; Young et al., 2004; Hedhly, 2011). The remaining ovules that survived apparently acquired an epigenetic signal that was transmitted to the embryo and endosperm. Curiously, 1240 of differentially expressed genes were common in the stressed embryo and endosperm, (94.58 and 19.48%, respectively), suggesting that the additional maternal genome contributes to substantial fluctuations in the endosperm transcriptome (Figures 2C,D). This finding is in agreement with the previous report demonstrating that the maternal genome in the Arabidopsis endosperm is substantially less methylated than the paternal genome in the CpG context (Ibarra et al., 2012).
Numerous examples of transgenerational inheritance in angiosperms undoubtedly suggest the existence of certain messengers in plants (Boyko et al., 2010; Bilichak et al., 2012; Luna et al., 2012; Rasmann et al., 2012; Slaughter et al., 2012). As such, they presumably act to prime the stress-specific genes for providing faster and more pronounced changes in transcription if akin exposure is encountered by offspring of stressed plants (Kathiria et al., 2010; Luna et al., 2012). Consistent with this notion, we detected a higher enrichment of stress-related genes in the fraction of differentially expressed genes in the progeny of stressed plants as compared to the parental tissues, which argues against a stochastic nature of epigenome variability (54.29% of stress-related genes out of the total number of differentially expressed genes, Supplementary Figure 3C).
Pollen Exhibits Tissue-Dependent smRNAome Fluctuations in Response to Heat Shock
In numerous flowering plants examined to date, 24-nt-long heterochromatic siRNAs (hc-siRNA) comprise the overwhelming majority of smRNA transcriptome (Nobuta et al., 2008; Wu et al., 2010; Korbes et al., 2012; Axtell, 2013). In our study, a vast amount of smRNAs were in the 24-nt-long fraction followed by the 21-nt-long fraction of reads in all libraries except leaves (Supplementary Figure 4).
Mapping of smRNAs to transposable elements and genic regions has confirmed the previous findings that the 24-nt smRNAs encompass a major fraction of siRNAs for all tissues except pollen (Supplementary Figures 5, 6). Pollen-derived sequencing reads demonstrated a singularity of smRNA length distribution mapped to the gene and transposon regions as compared to other B. rapa tissues. Gene-mapped smRNAs demonstrated the accumulation of sequencing reads with a length less than 19-nt (Supplementary Figure 5C), conceivably the products of mRNA degradation or as-yet-undiscovered pollen-specific regulatory RNAs. In Arabidopsis pollen, the 21-22-nt-long smRNAs are predominantly generated in the vegetative nucleus which is sacrificed by allowing rampant transposon expression concomitant with global DNA demethylation (Slotkin et al., 2009; Calarco et al., 2012). Subsequently, these 21-22-nt siRNAs guide RNA-dependent DNA methylation at non-symmetrical CpHpH sequences in sperm cells in order to reinforce the silencing of transposons. Neither unfertilized nor fertilized ovules responded with significant fluctuations in the smRNA length distribution, albeit the smRNA pathways were shown to be functional in Arabidopsis egg cells (Supplementary Figures 4D,E) (Wuest et al., 2010). Curiously, the fertilization of the embryo sac resulted in slight perturbations in the smRNA profile with a vast majority of repeat-derived sequencing reads (19 out of 27 unique sequencing reads) as compared to controls and unfertilized ovules (Figure 3). Whereas a comparison of unique altered sequencing reads between pollen and fertilized ovules returned only one common siRNA, seven out of 14 genes were found to be a common source of siRNAs in pollen and fertilized ovules. This finding partially confirms a previous report that the vegetative nucleus which does not contribute genetic material to the progeny is the primary source of smRNAs in pollen (Calarco et al., 2012). Also, it may indicate a transcriptionally quiescent response of the embryo sac to stress in plants that can be a prerequisite for the maintenance of genome stability in the harsh environmental conditions.
miR168 is a Putative Messenger of Transgenerational Stress Memory Inheritance in B. rapa Plants
Whereas siRNAs, with a few exceptions (Dunoyer et al., 2010; Mccue et al., 2012), are known to suppress predominantly TE activity in the genome, miRNAs are well-characterized regulatory elements of gene expression in plants and animals (Axtell, 2013).
The Arabidopsis genome encodes 10 AGO proteins, most of which demonstrate a clear bias toward a specific class of smRNAs depending on the size and 5′-terminal nucleotide composition (Vaucheret, 2008). One of the AGO proteins—AGO1—plays a principal role in both the siRNA- and miRNA-guided modulation of gene activity (Bohmert et al., 1998; Morel et al., 2002; Kidner and Martienssen, 2004). As a result of the global importance of AGO1 in plant homeostasis and development, its expression is firmly modulated by negative feedback loops involving miR168 and AGO1-derived siRNAs (Vaucheret et al., 2004, 2006; Mallory and Vaucheret, 2009; Mallory et al., 2009). The former regulation pathway is of a particular interest since miR168 expression is altered by numerous environmental perturbations in a number of plant species (Li et al., 2008, 2012; Ding et al., 2009; Jia et al., 2009, 2010; Zhou et al., 2010; Sunkar et al., 2012). Additionally, the involvement of AGO1 and the microRNA pathway in the adaptation to repeated heat stress has been demonstrated at the physiological and molecular level in Arabidopsis (Stief et al., 2014). In our study, we observed a differential expression of bra-miR168 following HS in the parental tissues that was negatively correlated with braAGO1 transcript levels in the corresponding tissues (Table 4). Although it still remains to be validated whether braAGO1 maintains the same functions as the Arabidopsis homolog, it is an interesting finding that conceivably suggests that miR168 and AGO1 are possible bandmasters of transgenerational stress memory inheritance in plants. Consistent with this notion, we observed an inverse Pearson's correlation for the expression of braAGO1 gene and alterations in the transcriptome profile of the corresponding tissues in the parental exposed plants and untreated progeny (r = −0.89, for the total number of differentially expressed genes and braAGO1 log2fold changes, TR vs. CT in the corresponding tissues). At the same time, we did not observe a Pearson's correlation comparable with the braAGO1 for the other epigenetic-related genes: putative DNA-DIRECTED RNA POLYMERASE E, r = 0.65; putative methyltransferase CMT2, r = 0.63; and putative lysine-specific demethylase JMJ14, r = −0.67.
Consistent with the previous study in Arabidopsis (Zhong et al., 2013) the overwhelming majority of differentially expressed smRNAs were down-regulated in the progeny of stressed plants as compared to controls (338 out of 376, Figure 4), which was concomitant with a slight up-regulation of gene expression (Figure 1). Taking into consideration that smRNAs act strictly by the down-regulation of gene expression, we can speculate that their progressive depletion provides a capacity for an organism to up-regulate rapidly the required gene expression under stress conditions without the ultimate smRNA-mediated transcript degradation. Furthermore, the removal of such post-transcriptional restraints along with locus-specific demethylation can be the cause of transgenerational priming of stress-responsive genes previously described in offspring of stressed plants (Luna et al., 2012).
Overall, by using a massive parallel sequencing technology, we provide evidence of transgenerational stress memory inheritance both at the transcriptome and smRNAome levels in plants. More importantly, we also suggest that miR168 is a possible messenger that mediates meiotic epigenetic inheritance in plants. Further experiments on transgenerational stresses involving the Arabidopsis hypomorphic ago1 mutants will shade a new light on its contribution to epigenetic inheritance in plants.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Valentina Titova for proofreading the manuscript. We acknowledge the financial support of Alberta Innovates Biosolutions and National Science and Engineering Research Council of Canada grants to Igor Kovalchuk and Alberta Innovates Technology Features for scholarship to Andriy Bilichak.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2015.00005/abstract
Footnotes
1. ^Oliveros, J. VENNY. An interactive tool for comparing lists with Venn diagrams.
References
Agrawal, A. A. (2001). Transgenerational consequences of plant responses to herbivory: an adaptive maternal effect? Am. Nat. 157, 555–569. doi: 10.1086/319932
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amoah, S., Kurup, S., Rodriguez Lopez, C. M., Welham, S. J., Powers, S. J., Hopkins, C. J., et al. (2012). A hypomethylated population of Brassica rapa for forward and reverse epi-genetics. BMC Plant Biol. 12:193. doi: 10.1186/1471-2229-12-193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Axtell, M. J. (2013). Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 64, 137–159. doi: 10.1146/annurev-arplant-050312-120043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B (Methodological) 57, 289–300.
Bilichak, A., Ilnystkyy, Y., Hollunder, J., and Kovalchuk, I. (2012). The progeny of Arabidopsis thaliana plants exposed to salt exhibit changes in DNA methylation, histone modifications and gene expression. PLoS ONE 7:e30515. doi: 10.1371/journal.pone.0030515
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bohmert, K., Camus, I., Bellini, C., Bouchez, D., Caboche, M., and Benning, C. (1998). AGO1 defines a novel locus of Arabidopsis controlling leaf development. EMBO J. 17, 170–180. doi: 10.1093/emboj/17.1.170
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyko, A., Blevins, T., Yao, Y., Golubov, A., Bilichak, A., Ilnytskyy, Y., et al. (2010). Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 5:e9514. doi: 10.1371/annotation/726f31b5-99c4-44e9-9cd6-b8d66b3f6038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buckley, B. A., Burkhart, K. B., Gu, S. G., Spracklin, G., Kershner, A., Fritz, H., et al. (2012). A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature 489, 447–451. doi: 10.1038/nature11352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burge, S. W., Daub, J., Eberhardt, R., Tate, J., Barquist, L., Nawrocki, E. P., et al. (2013). Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 41, D226–D232. doi: 10.1093/nar/gks1005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calarco, J. P., Borges, F., Donoghue, M. T., Van Ex, F., Jullien, P. E., Lopes, T., et al. (2012). Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151, 194–205. doi: 10.1016/j.cell.2012.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carlsbecker, A., Lee, J. Y., Roberts, C. J., Dettmer, J., Lehesranta, S., Zhou, J., et al. (2010). Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 465, 316–321. doi: 10.1038/nature08977
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castresana, C., De Carvalho, F., Gheysen, G., Habets, M., Inzé, D., and Van Montagu, M. (1990). Tissue-specific and pathogen-induced regulation of a Nicotiana plumbaginifolia beta-1,3-glucanase gene. Plant Cell 2, 1131–1143.
Chen, C. J., Liu, Q., Zhang, Y. C., Qu, L. H., Chen, Y. Q., and Gautheret, D. (2011). Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biol. 8, 538–547. doi: 10.4161/rna.8.3.15199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chi, X., Yang, Q., Chen, X., Wang, J., Pan, L., Chen, M., et al. (2011). Identification and characterization of microRNAs from peanut (Arachis hypogaea L.) by high-throughput sequencing. PLoS ONE 6:e27530. doi: 10.1371/journal.pone.0027530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daugherty, C. J., and Musgrave, M. E. (1994). Characterization of populations of rapid-cycling Brassica-Rapa L selected for differential waterlogging tolerance. J. Exp. Bot. 45, 385–392. doi: 10.1093/jxb/45.3.385
Ding, D., Zhang, L., Wang, H., Liu, Z., Zhang, Z., and Zheng, Y. (2009). Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 103, 29–38. doi: 10.1093/aob/mcn205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dinneny, J. R., Long, T. A., Wang, J. Y., Jung, J. W., Mace, D., Pointer, S., et al. (2008). Cell identity mediates the response of Arabidopsis roots to abiotic stress. Science 320, 942–945. doi: 10.1126/science.1153795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, J. Z., Keller, W. A., Yan, W., and Georges, F. (2004). Gene expression at early stages of Brassica napus seed development as revealed by transcript profiling of seed-abundant cDNAs. Planta 218, 483–491. doi: 10.1007/s00425-003-1124-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dunoyer, P., Brosnan, C. A., Schott, G., Wang, Y., Jay, F., Alioua, A., et al. (2010). An endogenous, systemic RNAi pathway in plants. EMBO J. 29, 1699–1712. doi: 10.1038/emboj.2010.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Emde, A. K., Grunert, M., Weese, D., Reinert, K., and Sperling, S. R. (2010). MicroRazerS: rapid alignment of small RNA reads. Bioinformatics 26, 123–124. doi: 10.1093/bioinformatics/btp601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fahlgren, N., Howell, M. D., Kasschau, K. D., Chapman, E. J., Sullivan, C. M., Cumbie, J. S., et al. (2007). High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE 2:e219. doi: 10.1371/journal.pone.0000219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gehring, M., Bubb, K. L., and Henikoff, S. (2009). Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 324, 1447–1451. doi: 10.1126/science.1171609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grandbastien, M.-A. (1998). Activation of plant retrotransposons under stress conditions. Trends Plant Sci. 3, 181–187. doi: 10.1016/S1360-1385(98)01232-1
Grant-Downton, R., Hafidh, S., Twell, D., and Dickinson, H. G. (2009a). Small RNA pathways are present and functional in the angiosperm male gametophyte. Mol. Plant 2, 500–512. doi: 10.1093/mp/ssp003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grant-Downton, R., Le Trionnaire, G., Schmid, R., Rodriguez-Enriquez, J., Hafidh, S., Mehdi, S., et al. (2009b). MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. BMC Genomics 10:643. doi: 10.1186/1471-2164-10-643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grant-Downton, R., and Rodriguez-Enriquez, J. (2012). Emerging roles for non-coding RNAs in male reproductive development in flowering plants. Biomolecules 2, 608–621. doi: 10.3390/biom2040608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gurley, W. B. (2000). HSP101: a key component for the acquisition of thermotolerance in plants. Plant Cell 12, 457–460. doi: 10.1105/tpc.12.4.457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hackenberg, M., Huang, P. J., Huang, C. Y., Shi, B. J., Gustafson, P., and Langridge, P. (2013). A comprehensive expression profile of MicroRNAs and other classes of non-coding small RNAs in barley under phosphorous-deficient and -sufficient conditions. DNA Res. 20, 109–125. doi: 10.1093/dnares/dss037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, X. F., Fang, Y. Y., Feng, L., and Guo, H. S. (2008). Characterization of conserved and novel microRNAs and their targets, including a TuMV-induced TIR-NBS-LRR class R gene-derived novel miRNA in Brassica. FEBS Lett. 582, 2445–2452. doi: 10.1016/j.febslet.2008.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hedhly, A. (2011). Sensitivity of flowering plant gametophytes to temperature fluctuations. Environ. Exp. Bot. 74, 9–16. doi: 10.1016/j.envexpbot.2011.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heil, M., and Ton, J. (2008). Long-distance signalling in plant defence. Trends Plant Sci. 13, 264–272. doi: 10.1016/j.tplants.2008.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holeski, L. M., Jander, G., and Agrawal, A. A. (2012). Transgenerational defense induction and epigenetic inheritance in plants. Trends Ecol. Evol. 27, 618–626. doi: 10.1016/j.tree.2012.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsieh, L. C., Lin, S. I., Shih, A. C., Chen, J. W., Lin, W. Y., Tseng, C. Y., et al. (2009a). Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 151, 2120–2132. doi: 10.1104/pp.109.147280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsieh, T. F., Ibarra, C. A., Silva, P., Zemach, A., Eshed-Williams, L., Fischer, R. L., et al. (2009b). Genome-wide demethylation of Arabidopsis endosperm. Science 324, 1451–1454. doi: 10.1126/science.1172417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ibarra, C. A., Feng, X., Schoft, V. K., Hsieh, T. F., Uzawa, R., Rodrigues, J. A., et al. (2012). Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337, 1360–1364. doi: 10.1126/science.1224839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iyer-Pascuzzi, A. S., Jackson, T., Cui, H., Petricka, J. J., Busch, W., Tsukagoshi, H., et al. (2011). Cell identity regulators link development and stress responses in the Arabidopsis root. Dev. Cell 21, 770–782. doi: 10.1016/j.devcel.2011.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jablonka, E., and Raz, G. (2009). Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 84, 131–176. doi: 10.1086/598822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jia, X., Mendu, V., and Tang, G. (2010). An array platform for identification of stress-responsive microRNAs in plants. Methods Mol. Biol. 639, 253–269. doi: 10.1007/978-1-60761-702-0_15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jia, X., Wang, W. X., Ren, L., Chen, Q. J., Mendu, V., Willcut, B., et al. (2009). Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Mol. Biol. 71, 51–59. doi: 10.1007/s11103-009-9508-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johnson-Brousseau, S. A., and McCormick, S. (2004). A compendium of methods useful for characterizing Arabidopsis pollen mutants and gametophytically-expressed genes. Plant J. 39, 761–775. doi: 10.1111/j.1365-313X.2004.02147.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jullien, P. E., Susaki, D., Yelagandula, R., Higashiyama, T., and Berger, F. (2012). DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana. Curr. Biol. 22, 1825–1830. doi: 10.1016/j.cub.2012.07.061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung, H. W., Tschaplinski, T. J., Wang, L., Glazebrook, J., and Greenberg, J. T. (2009). Priming in systemic plant immunity. Science 324, 89–91. doi: 10.1126/science.1170025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kathiria, P., Sidler, C., Golubov, A., Kalischuk, M., Kawchuk, L. M., and Kovalchuk, I. (2010). Tobacco mosaic virus infection results in an increase in recombination frequency and resistance to viral, bacterial, and fungal pathogens in the progeny of infected tobacco plants. Plant Physiol. 153, 1859–1870. doi: 10.1104/pp.110.157263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khraiwesh, B., Zhu, J. K., and Zhu, J. (2012). Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 1819, 137–148. doi: 10.1016/j.bbagrm.2011.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kidner, C. A., and Martienssen, R. A. (2004). Spatially restricted microRNA directs leaf polarity through ARGONAUTE1. Nature 428, 81–84. doi: 10.1038/nature02366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, S. W., Li, Z., Moore, P. S., Monaghan, A. P., Chang, Y., Nichols, M., et al. (2010). A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic Acids Res. 38, e98. doi: 10.1093/nar/gkp1235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Korbes, A. P., Machado, R. D., Guzman, F., Almerao, M. P., De Oliveira, L. F., Loss-Morais, G., et al. (2012). Identifying conserved and novel microRNAs in developing seeds of Brassica napus using deep sequencing. PLoS ONE 7:e50663. doi: 10.1371/journal.pone.0050663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kotak, S., Larkindale, J., Lee, U., Von Koskull-Doring, P., Vierling, E., and Scharf, K. D. (2007). Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 10, 310–316. doi: 10.1016/j.pbi.2007.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kreps, J. A., Wu, Y., Chang, H. S., Zhu, T., Wang, X., and Harper, J. F. (2002). Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 130, 2129–2141. doi: 10.1104/pp.008532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kruszka, K., Pieczynski, M., Windels, D., Bielewicz, D., Jarmolowski, A., Szweykowska-Kulinska, Z., et al. (2012). Role of microRNAs and other sRNAs of plants in their changing environments. J. Plant Physiol. 169, 1664–1672. doi: 10.1016/j.jplph.2012.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kultz, D. (2005). Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 67, 225–257. doi: 10.1146/annurev.physiol.67.040403.103635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larkindale, J., Hall, J. D., Knight, M. R., and Vierling, E. (2005a). Heat stress phenotypes of arabidopsis mutants implicate multiple signaling pathways in the acquisition of thermotolerance. Plant Physiol. 138, 882–897. doi: 10.1104/pp.105.062257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larkindale, J., and Huang, B. (2004). Thermotolerance and antioxidant systems in Agrostis stolonifera: involvement of salicylic acid, abscisic acid, calcium, hydrogen peroxide, and ethylene. J. Plant Physiol. 161, 405–413. doi: 10.1078/0176-1617-01239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larkindale, J., Mishkind, M., and Vierling, E. (2005b). “Plant responses to high temperature,” in Plant Abiotic Stress, eds M. A. Jenks and P. M. Hasegawa (Oxford, UK: Blackwell Publishing Ltd), 100–144. doi: 10.1002/9780470988503.ch5
Le, B. H., Cheng, C., Bui, A. Q., Wagmaister, J. A., Henry, K. F., Pelletier, J., et al. (2010). Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. U.S.A. 107, 8063–8070. doi: 10.1073/pnas.1003530107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J.-Y., and Lee, D.-H. (2003). Use of serial analysis of gene expression technology to reveal changes in gene expression in Arabidopsis Pollen undergoing cold stress. Plant Physiol. 132, 517–529. doi: 10.1104/pp.103.020511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W., Cui, X., Meng, Z., Huang, X., Xie, Q., Wu, H., et al. (2012). Transcriptional regulation of Arabidopsis MIR168a and argonaute1 homeostasis in abscisic acid and abiotic stress responses. Plant Physiol. 158, 1279–1292. doi: 10.1104/pp.111.188789
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W. X., Oono, Y., Zhu, J., He, X. J., Wu, J. M., Iida, K., et al. (2008). The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20, 2238–2251. doi: 10.1105/tpc.108.059444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, H. T., Sun, D. Y., and Zhou, R. G. (2005). Ca2+ and AtCaM3 are involved in the expression of heat shock protein gene in Arabidopsis. Plant Cell Environ. 28, 1276–1284. doi: 10.1111/j.1365-3040.2005.01365.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Loss-Morais, G., Waterhouse, P. M., and Margis, R. (2013). Description of plant tRNA-derived RNA fragments (tRFs) associated with argonaute and identification of their putative targets. Biol. Direct 8:6. doi: 10.1186/1745-6150-8-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luna, E., Bruce, T. J., Roberts, M. R., Flors, V., and Ton, J. (2012). Next-generation systemic acquired resistance. Plant Physiol. 158, 844–853. doi: 10.1104/pp.111.187468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mallory, A. C., Bartel, D. P., and Bartel, B. (2005). MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 17, 1360–1375. doi: 10.1105/tpc.105.031716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mallory, A. C., Hinze, A., Tucker, M. R., Bouche, N., Gasciolli, V., Elmayan, T., et al. (2009). Redundant and specific roles of the ARGONAUTE proteins AGO1 and ZLL in development and small RNA-directed gene silencing. PLoS Genet. 5:e1000646. doi: 10.1371/journal.pgen.1000646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mallory, A. C., and Vaucheret, H. (2009). ARGONAUTE 1 homeostasis invokes the coordinate action of the microRNA and siRNA pathways. EMBO Rep. 10, 521–526. doi: 10.1038/embor.2009.32
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marin, E., Jouannet, V., Herz, A., Lokerse, A. S., Weijers, D., Vaucheret, H., et al. (2010). miR390, Arabidopsis TAS3 tasiRNAs, and their AUXIN RESPONSE FACTOR targets define an autoregulatory network quantitatively regulating lateral root growth. Plant Cell 22, 1104–1117. doi: 10.1105/tpc.109.072553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mccue, A. D., Nuthikattu, S., Reeder, S. H., and Slotkin, R. K. (2012). Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 8:e1002474. doi: 10.1371/journal.pgen.1002474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, G., Schlauch, K., Tam, R., Cortes, D., Torres, M. A., Shulaev, V., et al. (2009). The plant NADPH oxidase RBOHD mediates rapid systemic signaling in response to diverse stimuli. Sci. Signal. 2, ra45. doi: 10.1126/scisignal.2000448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montgomery, T. A., Howell, M. D., Cuperus, J. T., Li, D., Hansen, J. E., Alexander, A. L., et al. (2008). Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 133, 128–141. doi: 10.1016/j.cell.2008.02.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morel, J. B., Godon, C., Mourrain, P., Beclin, C., Boutet, S., Feuerbach, F., et al. (2002). Fertile hypomorphic ARGONAUTE (ago1) mutants impaired in post-transcriptional gene silencing and virus resistance. Plant Cell 14, 629–639. doi: 10.1105/tpc.010358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moxon, S., Jing, R., Szittya, G., Schwach, F., Rusholme Pilcher, R. L., Moulton, V., et al. (2008). Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 18, 1602–1609. doi: 10.1101/gr.080127.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nobuta, K., Lu, C., Shrivastava, R., Pillay, M., De Paoli, E., Accerbi, M., et al. (2008). Distinct size distribution of endogeneous siRNAs in maize: evidence from deep sequencing in the mop1-1 mutant. Proc. Natl. Acad. Sci. U.S.A. 105, 14958–14963. doi: 10.1073/pnas.0808066105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nylander, M., Svensson, J., Palva, E. T., and Welin, B. V. (2001). Stress-induced accumulation and tissue-specific localization of dehydrins in Arabidopsis thaliana. Plant Mol. Biol. 45, 263–279. doi: 10.1023/A:1006469128280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prandl, R., Kloske, E., and Schoffl, F. (1995). Developmental regulation and tissue-specific differences of heat shock gene expression in transgenic tobacco and Arabidopsis plants. Plant Mol. Biol. 28, 73–82. doi: 10.1007/BF00042039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qi, J., Yu, S., Zhang, F., Shen, X., Zhao, X., Yu, Y., et al. (2010). Reference gene selection for real-time quantitative polymerase chain reaction of mrna transcript levels in chinese cabbage (Brassica rapa L. ssp. pekinensis). Plant Mol. Biol. Rep. 28, 597–604. doi: 10.1007/s11105-010-0185-1
Rajagopalan, R., Vaucheret, H., Trejo, J., and Bartel, D. P. (2006). A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 20, 3407–3425. doi: 10.1101/gad.1476406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rasmann, S., De Vos, M., Casteel, C. L., Tian, D., Halitschke, R., Sun, J. Y., et al. (2012). Herbivory in the previous generation primes plants for enhanced insect resistance. Plant Physiol. 158, 854–863. doi: 10.1104/pp.111.187831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seong, K. H., Li, D., Shimizu, H., Nakamura, R., and Ishii, S. (2011). Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145, 1049–1061. doi: 10.1016/j.cell.2011.05.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Slaughter, A., Daniel, X., Flors, V., Luna, E., Hohn, B., and Mauch-Mani, B. (2012). Descendants of primed Arabidopsis plants exhibit resistance to biotic stress. Plant Physiol. 158, 835–843. doi: 10.1104/pp.111.191593
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Slotkin, R. K., Vaughn, M., Borges, F., Tanurdzic, M., Becker, J. D., Feijo, J. A., et al. (2009). Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136, 461–472. doi: 10.1016/j.cell.2008.12.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smyth, D. R., Bowman, J. L., and Meyerowitz, E. M. (1990). Early flower development in Arabidopsis. Plant Cell 2, 755–767. doi: 10.1105/tpc.2.8.755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Song, C., Wang, C., Zhang, C., Korir, N. K., Yu, H., Ma, Z., et al. (2010). Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 11:431. doi: 10.1186/1471-2164-11-431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stief, A., Altmann, S., Hoffmann, K., Pant, B. D., Scheible, W. R., and Baurle, I. (2014). Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell 26, 1792–1807. doi: 10.1105/tpc.114.123851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stocks, M. B., Moxon, S., Mapleson, D., Woolfenden, H. C., Mohorianu, I., Folkes, L., et al. (2012). The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28, 2059–2061. doi: 10.1093/bioinformatics/bts311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, K., Hunt, K., and Hauser, B. A. (2004). Ovule abortion in Arabidopsis triggered by stress. Plant Physiol. 135, 2358–2367. doi: 10.1104/pp.104.043091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sunkar, R., Li, Y. F., and Jagadeeswaran, G. (2012). Functions of microRNAs in plant stress responses. Trends Plant Sci. 17, 196–203. doi: 10.1016/j.tplants.2012.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szittya, G., Moxon, S., Santos, D. M., Jing, R., Fevereiro, M. P., Moulton, V., et al. (2008). High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics 9:593. doi: 10.1186/1471-2164-9-593
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tel-Zur, N., and Goldman, I. L. (2007). Analysis of sub-populations of rapid-cycling Brassica rapa following recurrent bi-directional selection for cotyledon size. Plant Breed. 126, 62–66. doi: 10.1111/j.1439-0523.2007.01317.x
Tillett, R. L., Ergul, A., Albion, R. L., Schlauch, K. A., Cramer, G. R., and Cushman, J. C. (2011). Identification of tissue-specific, abiotic stress-responsive gene expression patterns in wine grape (Vitis vinifera L.) based on curation and mining of large-scale EST data sets. BMC Plant Biol. 11:86. doi: 10.1186/1471-2229-11-86
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tiwari, S. B., Hagen, G., and Guilfoyle, T. (2003). The roles of auxin response factor domains in auxin-responsive transcription. Plant Cell 15, 533–543. doi: 10.1105/tpc.008417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Loon, L. C. (1997). Induced resistance in plants and the role of pathogenesis-related proteins. Eur. J. Plant Pathol. 103, 753–765. doi: 10.1023/A:1008638109140
Vaucheret, H. (2008). Plant ARGONAUTES. Trends Plant Sci. 13, 350–358. doi: 10.1016/j.tplants.2008.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vaucheret, H., Mallory, A. C., and Bartel, D. P. (2006). AGO1 homeostasis entails coexpression of MIR168 and AGO1 and preferential stabilization of miR168 by AGO1. Mol. Cell 22, 129–136. doi: 10.1016/j.molcel.2006.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vaucheret, H., Vazquez, F., Crete, P., and Bartel, D. P. (2004). The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 18, 1187–1197. doi: 10.1101/gad.1201404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walley, J. W., and Dehesh, K. (2010). Molecular mechanisms regulating rapid stress signaling networks in Arabidopsis. J. Integr. Plant Biol. 52, 354–359. doi: 10.1111/j.1744-7909.2010.00940.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, F., Li, L., Liu, L., Li, H., Zhang, Y., Yao, Y., et al. (2012). High-throughput sequencing discovery of conserved and novel microRNAs in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol. Genet. Genomics 287, 555–563. doi: 10.1007/s00438-012-0699-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Williams, P. H., and Hill, C. B. (1986). Rapid-cycling populations of Brassica. Science 232, 1385–1389. doi: 10.1126/science.232.4756.1385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, L., Zhou, H., Zhang, Q., Zhang, J., Ni, F., Liu, C., et al. (2010). DNA methylation mediated by a microRNA pathway. Mol. Cell 38, 465–475. doi: 10.1016/j.molcel.2010.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wuest, S. E., Vijverberg, K., Schmidt, A., Weiss, M., Gheyselinck, J., Lohr, M., et al. (2010). Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr. Biol. 20, 506–512. doi: 10.1016/j.cub.2010.01.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, X., and Li, L. (2011). miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27, 2614–2615. doi: 10.1093/bioinformatics/btr430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Young, L. W., Wilen, R. W., and Bonham-Smith, P. C. (2004). High temperature stress of Brassica napus during flowering reduces micro- and megagametophyte fertility, induces fruit abortion, and disrupts seed production. J. Exp. Bot. 55, 485–495. doi: 10.1093/jxb/erh038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, X., Wang, H., Lu, Y., De Ruiter, M., Cariaso, M., Prins, M., et al. (2012). Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J. Exp. Bot. 63, 1025–1038. doi: 10.1093/jxb/err337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Yao, D., Wang, Q., Xu, W., Wei, Q., Wang, C., et al. (2013). mRNA-seq analysis of the gossypium arboreum transcriptome reveals tissue selective signaling in response to water stress during seedling stage. PLoS ONE 8:e54762. doi: 10.1371/journal.pone.0054762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhong, S. H., Liu, J. Z., Jin, H., Lin, L., Li, Q., Chen, Y., et al. (2013). Warm temperatures induce transgenerational epigenetic release of RNA silencing by inhibiting siRNA biogenesis in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 110, 9171–9176. doi: 10.1073/pnas.1219655110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, L., Liu, Y., Liu, Z., Kong, D., Duan, M., and Luo, L. (2010). Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 61, 4157–4168. doi: 10.1093/jxb/erq237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zinn, K. E., Tunc-Ozdemir, M., and Harper, J. F. (2010). Temperature stress and plant sexual reproduction: uncovering the weakest links. J. Exp. Bot. 61, 1959–1968. doi: 10.1093/jxb/erq053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: epigenetics, small RNAs, transcriptome, B. rapa, transgenerational inheritance
Citation: Bilichak A, Ilnytskyy Y, Wóycicki R, Kepeshchuk N, Fogen D and Kovalchuk I (2015) The elucidation of stress memory inheritance in Brassica rapa plants. Front. Plant Sci. 6:5. doi: 10.3389/fpls.2015.00005
Received: 18 September 2014; Accepted: 05 January 2015;
Published online: 21 January 2015.
Edited by:
Shawn Kaeppler, University of Wisconsin-Madison, USAReviewed by:
Rajandeep Sekhon, Clemson University, USAThelma Farai Madzima, Florida State University, USA
Copyright © 2015 Bilichak, Ilnytskyy, Wóycicki, Kepeshchuk, Fogen and Kovalchuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Igor Kovalchuk, Department of Biological Sciences, University of Lethbridge, University Drive 4401, Lethbridge, AB, T1K 3M4, Canada e-mail:aWdvci5rb3ZhbGNodWtAdWxldGguY2E=