 Xiaoshuang Li
Xiaoshuang Li Daoyuan Zhang
Daoyuan Zhang Haiyan Li
Haiyan Li Bei Gao
Bei Gao Honglan Yang
Honglan Yang Yuanming Zhang1
Yuanming Zhang1 Andrew J. Wood
Andrew J. Wood- 1Key Laboratory of Biogeography and Bioresource in Arid Land, Xinjiang Institute of Ecology and Geography – Chinese Academy of Sciences, Ürümqi, China
- 2Department of Plant Biology, Southern Illinois University, Carbondale, IL, USA
Syntrichia caninervis is the dominant bryophyte of the biological soil crusts found in the Gurbantunggut desert. The extreme desert environment is characterized by prolonged drought, temperature extremes, high radiation and frequent cycles of hydration and dehydration. S. caninervis is an ideal organism for the identification and characterization of genes related to abiotic stress tolerance. Reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) expression analysis is a powerful analytical technique that requires the use of stable reference genes. Using available S. caninervis transcriptome data, we selected 15 candidate reference genes and analyzed their relative expression stabilities in S. caninervis gametophores exposed to a range of abiotic stresses or a hydration-desiccation-rehydration cycle. The programs geNorm, NormFinder, and RefFinder were used to assess and rank the expression stability of the 15 candidate genes. The stability ranking results of reference genes under each specific experimental condition showed high consistency using different algorithms. For abiotic stress treatments, the combination of two genes (α-TUB2 and CDPK) were sufficient for accurate normalization. For the hydration-desiccation-rehydration process, the combination of two genes (α-TUB1 and CDPK) were sufficient for accurate normalization. 18S was among the least stable genes in all of the experimental sets and was unsuitable as reference gene in S. caninervis. This is the first systematic investigation and comparison of reference gene selection for RT-qPCR work in S. caninervis. This research will facilitate gene expression studies in S. caninervis, related moss species from the Syntrichia complex and other mosses.
Introduction
Syntrichia caninervis is a desert moss and the dominant bryophyte of the biological soil crusts found in the Gurbantunggut desert of Northwestern China (Zhang, 2005). The Gurbantunggut has a mean annual precipitation of ∼79.5 mm and a mean annual evaporation of 2,606.6 mm (Zhang, 2005; Zhang et al., 2011a). S. caninervis has gained particular attention due to its extreme desiccation tolerance (DT; Wood, 2007; Yang et al., 2012) and is closely related to Tortula ruralis. T. ruralis is a model DT moss and many desiccation-related genes have been isolated and analyzed in this species T. ruralis (Chen et al., 2002; Chen and Wood, 2003; Peng et al., 2005). EST data from desiccated and rehydrated T. ruralis gametophytes indicated many novel genes exist in this DT species (Wood et al., 1999; Oliver et al., 2004). S. caninervis, as compared to T. ruralis, is reported to be more tolerant to desiccation stress and have a quicker recovery rate from complete water loss (Oliver et al., 1993; Zhang et al., 2011b). Furthermore, S. caninervis is tolerant to multiple stresses including drought, high/low temperature, high radiation and frequent cycles of hydration and dehydration (Yang et al., 2012). S. caninervis has been systematically studied at the morphological (Stark et al., 2004, 2005, 2009; Stark and McLetchie, 2006; Zhang et al., 2007; Xu et al., 2009a; Zheng et al., 2011; Tao and Zhang, 2012), physiological (Xu et al., 2009b; Li et al., 2010; Zhang et al., 2011b; Wu et al., 2012) and molecular levels (Yang et al., 2012), and is being developed as another model moss for studying the mechanisms of DT and the identification of stress-related genes.
Reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) is one of the most widely used technologies for gene expression studies because of its quantitative accuracy, high sensitivity and high-throughput capabilities (Bustin, 2000; Huggett et al., 2005). The utilization of stable reference genes are a prerequisite for RT-qPCR, although the quality and quantity of the mRNA template and reaction efficiency variations also influence the reliability and accuracy of the technique (Vandesompele et al., 2002; Udvardi et al., 2008; Bustin et al., 2009). RT-qPCR data must be normalized with more than one internal reference gene (Hu et al., 2009) and some reports have suggested that at least three reference genes be combined to normalize the results of RT-qPCR (Hong et al., 2010; Jacob et al., 2013). Reference genes may show different stability patterns even within the same plant, and the results of RT-qPCR cannot be extrapolated to other experimental conditions (Exposito-Rodriguez et al., 2008). Consequently, it is recommended that suitable reference genes should be established for each species and tested for specific experimental conditions to ensure their stability (Li et al., 2012).
Studies on reference gene identification and selection for RT-qPCR have focused on humans and other animal model organisms (Shen et al., 2010; Ponton et al., 2011). In plant science, growing efforts have been made in recent years. Stable reference genes have been identified in variety of plants, such as Arabidopsis (Lilly et al., 2011), grasses (Hong et al., 2008; Lee et al., 2010), fruits (Clancy et al., 2013; Die and Rowland, 2013; Imai et al., 2014), vegetables (Wan et al., 2010; Xu et al., 2012), commercial agricultural crops (Figueiredo et al., 2013; Wang et al., 2013b), and some desert plants (Li et al., 2012; Shi et al., 2012; Zhu et al., 2013). The development of next generation sequencing technology provides new opportunities to explore the genetic resources from an expanding selection of plants (Czechowski et al., 2005; Hong et al., 2010; Narsai et al., 2010; Demidenko et al., 2011; Feng et al., 2013). Furthermore, with the increasing awareness of importance of suitable reference genes, different statistical algorithms like geNorm (Vandesompele et al., 2002), NormFinder (Andersen et al., 2004), BestKeeper (Pfaffl et al., 2004), and RefFinder (Xie et al., 2012) have been developed to determine which reference gene(s) is most suitable for transcript normalization in a given experiment condition for a specific species.
Reference gene identification and selection has been conducted in a variety of plant species, however, studies are limited in non-vascular plants to the moss Physcomitrella patens (Le Bail et al., 2013) and the brown algae Ectocarpus siliculosus (Kianianmomeni and Hallmann, 2013). To date, no study has been performed to evaluation stable reference gene for a desiccation tolerant moss species in response to abiotic stress. The 18S rRNA (18S) has been utilized as a RT-qPCR reference gene in S. caninervis (Yang et al., 2012); however a systematic investigation and stability comparison of reference genes has not been done in S. caninervis.
In this study, 15 reference gene candidates were selected including 12 traditional reference genes (ACT, ARP, α-TUB1, α-TUB2, β-TUB, HIS3, 18S, SPT, UBR1, UBR2, GAPDH1, GAPDH2) and three new reference genes (CDPK, F-BOX, SAND) which demonstrated stable expression in the plants P. patens and Arabidopsis. The expression stabilities of these 15 genes were evaluated in S. caninervis gametophytes subjected to 11 abiotic stress conditions (include one non-stress control) and four samples involved in hydration-desiccation-rehydration process (H-D-R). RT-qPCR data were analyzed using two most widely used algorithms geNorm and NormFinder to determine sets of reference genes suitable for gene expression studies in different experiment conditions. Additionally, a comprehensive reference gene stability analysis tool RefFinder was used to confirm the ranking results obtained from geNorm and NormFinder. This work will facilitate future work on gene expression studies in S. caninervis and also benefit other species of the mosses genus such as T. ruralis.
Materials and Methods
Plant Materials and Treatment
Syntrichia caninervis gametophytes were collected from the Gurbantunggut Desert of Xinjiang Uyghur Autonomous Region of China (Fukang County, 44°32′30′′N, 88°6′42′′E). This sand dune was identified as a permanent research site since 2003 (Wu et al., 2012). The collected moss gametophytes were air-dried and kept in a black bag at room temperature for 1 week.
Syntrichia caninervis gametophytes were exposed to 11 different treatments (i.e., stress condition) and a hydration-desiccation-rehydration process to evaluate the stabilities of the tested reference genes. For the hydration-desiccation-rehydration process, dry gametophores were fully hydrated by placing upon MINIQ-filtered water saturated filter paper (8 mL) in glass petri dishes for 24 h at 25°C (150 μm m-2 s-1; Zhang et al., 2011b), then transfer to clean dishes and dried at room temperature (air dry) for 6 h (∼25°C, RH = 25%; Yang et al., 2012), then rehydrated by transferring to new petri plates and the filter paper was saturated with 8 mL filtered water at 25°C for 2 and 6 h. For abiotic stress treatments, the fully hydrated gametophytes (hydrated for 24 h) were transferred to new petri plates and the filter paper was saturated with 8 mL of one of the following solutions at 25°C: MINIQ-filtered water (control), 20% (w/v) PEG6000 (osmotic stress), 250 mM NaCl (salt stress), 50 mM H2O2 (oxidative stress), 500 μM CuSO4 (metal stress), and 100 μM ABA (exogenous ABA application). For UV exposure, fully hydrated gametophores were exposed to 0.5 w/m2 UV-B irradiation. For wounding, fully hydrated gametophores were cut into small pieces with a razor blade. For cold and heat stresses, gametophores were placed in petri plates on water saturated filter paper and incubated at either 4 or 42°C. For the combination of osmotic stress and elevated temperature stress, fully hydrated gametophores were transferred to petri plates saturated with 8 mL of 10% PEG6000 and incubated at 42°C; all the samples were harvested at 6 h after treatment, and gametophyte shoots (removal of rhizoid, i.e., 100 mg FW) were collected for each sample. Harvested samples were flash frozen in liquid nitrogen and stored at –80°C prior to RNA extraction.
RNA Extraction and cDNA Synthesis
Total RNA was extracted using TRIzol reagent (Qiagen, USA). Genomic DNA contamination was eliminated using RNase-free DNaseI (Takara, Japan). RNA concentration, purity, and integrity were determined using a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, USA) and visually assessed via gel electrophoresis (1.2% agarose). Only RNA samples with a 260/280 ratio between 1.8 and 2.1 and 260/230 ratio higher than 1.8 were used for subsequent analyses. First strand cDNA was synthesized from 1 μg total RNA, l μl oligo-dT, 1 μl random hexamers, and 4 μl 5 × Primerscript Buffer using PrimeScriptTM RT reagent kit (perfect Real time; Takara, Japan). The reverse transcription was carried out at 37°C for 30 min on a C1000TM Thermal cycler (Bio-Rad, USA) in a final volume of 20 μl, and inactivation of the enzyme was achieved at 85°C for 5 min. All cDNA were stored at –20°C until use.
Reference Genes Selection, PCR Primer Design, and Testing
Based on previous RT-qPCR reports in the model plants Arabidopsis and P. patens, we selected 15 reference genes spanning a range of biological functions as reference gene candidates in S. caninervis. The genes are: 18S ribosomal RNA (18S), actin (ACT), actin-related protein (ARP), alpha tubulin (α-TUB1 and α-TUB2), beta tubulin (β-TUB), calmodulin-like domain protein kinase (CDPK), F-box/kelch-repeat protein (F-BOX), glyceraldehyde-3-phosphate dehydrogenase (GAPDH1 and GAPDH2), histone H3 (HIS3), SAND protein family (SAND), suppressor of Ty (SPT), and ubiquitin protein ligase (UBR1 and UBR2). Most of them are well characterized, plant classic reference genes candidates such as 18S, ACT, and α-TUB genes. Three are newly identified candidates which have demonstrated good stability in plants, including SAND, CDPK, and F-BOX. The 18S gene (KJ398837) was cloned previously by our lab, and the other 14 genes were obtained from our transcriptional data of S. caninervis (Gao et al., 2014). RT-qPCR primers were designed with Primer Premier 5.0 using the following criteria: amplicon length from 100 to 300 bp and a Tm of 58 ± 3°C. The designed primer sets were BLASTed against the local transcriptional data of S. caninervis to verify primer specificity. All RT-qPCR experiments conformed to the MIQE guidelines (Bustin et al., 2009). Amplification efficiency (E) was evaluated using a standard curve generated by RT-qPCR using a 10-fold dilution series (1, 1/10, 1/100, 1/1000, 1/10000, 1/100000) over at least four dilution points that were measured in triplicate. Primer specificity was assessed using melting-curve analysis after RT-qPCR and gel electrophoresis analysis of the amplicons. Primer pairs performed well which showed single product and no product amplified in no-template control (NTC) were further sequenced to exclude the amplification of high identity homologs.
Real-Time Quantitative PCR
cDNA were diluted five times for RT-qPCR. Real-time PCR reactions were carried out in 96-well plates with CFX96 Real-Time PCR Detection System (Bio-Rad, USA) using SYBR Premix Ex TaqTM (Takara, Japan). The reaction mixture consisted of 2 μl 1:5 diluted cDNA samples, 0.4 μl each of the forward and reverse primers (10 μM), 10 μl real-time master mix and 7.2 μl PCR-grade water in a final volume of 20 μl. Two biological replicates for all of the samples and three technical replicates of each biological replicate with a NTC were also used. The RT-qPCR protocol was as follows: 30 s initial denaturation at 95°C, 40 cycles of 94°C for 5 s and 58–61°C for 30 s. To verify the specificity of each primer, a melting-curve analysis was included (65–95°C with fluorescence measured every 0.5°C).
Analysis of Gene Stability
To rank the stability of the tested genes, the two publicly available tools geNorm (Version 3.5) and NormFinder were used. Additionally, a comprehensive web-based tool RefFinder (http:www.leonxie.com/referencegene.php) was used to integrate and confirm the results obtained using geNorm and NormFinder. These three programs can evaluate the expression stability of reference genes from different aspects. GeNorm is a popular algorithm to determine the most stable reference genes and the optimal number of genes needed for accurate normalization. The raw Cq values were imported into Microsoft Excel and transformed into relative quantities using the formula Q = 2(mincq -samplecq), then imported into geNorm to analysis gene expression stability. The sample with the highest expression level (the minimum Cq value) was used as a calibrator and was set to 1, subsequently the expression level of other samples were converted to a relative expression. According to the geNorm manual, the expression stability value (M) and pairwise variation value (V) for each reference gene with all other genes were automatically analyzed and ranked according to their expression stability. Then, the optimal number of reference genes for normalization was determined. As suggested by geNorm, the cutoff of M value was set as 1.5, a lower value of average expression stability (M) indicated more stable gene expression, The pairwise variation (Vn/Vn+1) was analyzed to determine the optimal number of reference genes for accurate normalization. The cutoff value was proposed to be 0.15, below which the inclusion of an additional reference genes is not necessary, while it is suggested that this cutoff should not be too strict (Vandesompele et al., 2002). The NormFinder program identified the genes with optimal normalization among a set of candidate genes according to intra and inter variations. The lowest stability value (the least intra and inter-group variations) indicates the most stable expression within the gene set examined. RefFinder is a comprehensive tool which integrates commonly used reference gene evaluation programs together, including geNorm, NormFinder, Bestkeeper, and the comparative delta Ct methods. RefFinder generated the final overall ranking of tested reference genes based on the geometric mean of the weights of every gene calculating by each program. Raw Cq values (untransformed data) were used directly for data importing of RefFinder program.
Results
Selection of Candidate Reference Genes and Gene Sequence Analysis
The cDNA fragments of the 15 reference genes ranged from 480 bp for SAND to 2390 bp for CDPK. BLASTP demonstrated that all S. caninervis references genes had maximum identity with similar deduced polypeptides from P. patens, (identity ranged from 58 to 100%), (Table S1). For example, ScCDPK share 90% sequence identity with PpCDPK (XP_001776407), 62% with AtCDPK (NP_196107; Table S1), and 99% identity with TrCDPK (GenBank No AAB70706) in desiccation model moss T. ruralis. BLASTN demonstrated that Sc18S had 97% identity with Arabidopsis 18S gene, and shared 90% identity with 18S genes in both P. patens and T. ruralis. All cDNA sequences were deposited to the GenBank database under accession numbers KJ398821 to KJ398834.
Verification of Primer Specificity and Efficiency
The RT-qPCR primer sequences and amplicon characteristics of 15 candidate reference genes are described in Table 1. The sequence length was ranged from 117 to 277 bp. The primer efficiency (E) for each primer pair was greater than 90% and varied from 91.6% for ACT to 103.2% for GAPDH1, and correlation coefficients (R) ranged from 0.992 for SAND to 1.0 for ACT, HIS3, 18S, and CDPK, respectively (Table 1). All primer sets were BLAST searched against the non-redundant (nr) database in NCBI (primer-BLAST program) as well as the local S. caninervis transcriptome data to verify primer specificity. Primer specificity was further assessed by melting-curves and gel electrophoresis analysis. RT-PCR and RT-qPCR products for each primer pair amplified only a single product of the predicted size, and a single peak was obtained by melting-curve analysis (Figure S1). The amplicons were further sequenced to exclude the amplification of homologs with high identities. These analyses demonstrated that each primer pairs used in this study is specific to the candidate genes.

TABLE 1. Primer sequences and amplicon characteristics of 15 reference genes for RT-qPCR analysis.
Expression Profiling of the Candidate Reference Genes (RT-qPCR Assay)
The expression levels of 15 candidate reference genes were determined as quantification cycles (Cq value; Figure 1; Table S2). The mean Cq values for reference genes ranged between 12 and 30, with most lying between 20 and 25 across all tested samples. The average Cq value of all the tested samples was 22.8 cycles. UBR1 had the highest median Cq value (29.98), which indicated relatively low expression. In contrast, 18S gene was highly expressed compared to the protein coding genes (Cq = 12.3). ARP, α-TUB1, and GAPDH2 each had low gene expression variation (below three cycles), while 18S and SPT showed higher gene expression variation (above six cycles; Table S2). The variable transcript abundance of the 15 reference genes demonstrated that different reference genes showed varied expression levels under the same experiment set. Even for the same reference gene, gene expression was varied across different S. caninervis samples. This confirmed that no individual gene shows constant expression under all experimental conditions, and stable reference gene selection is necessary for each specific experimental condition in S. caninervis.
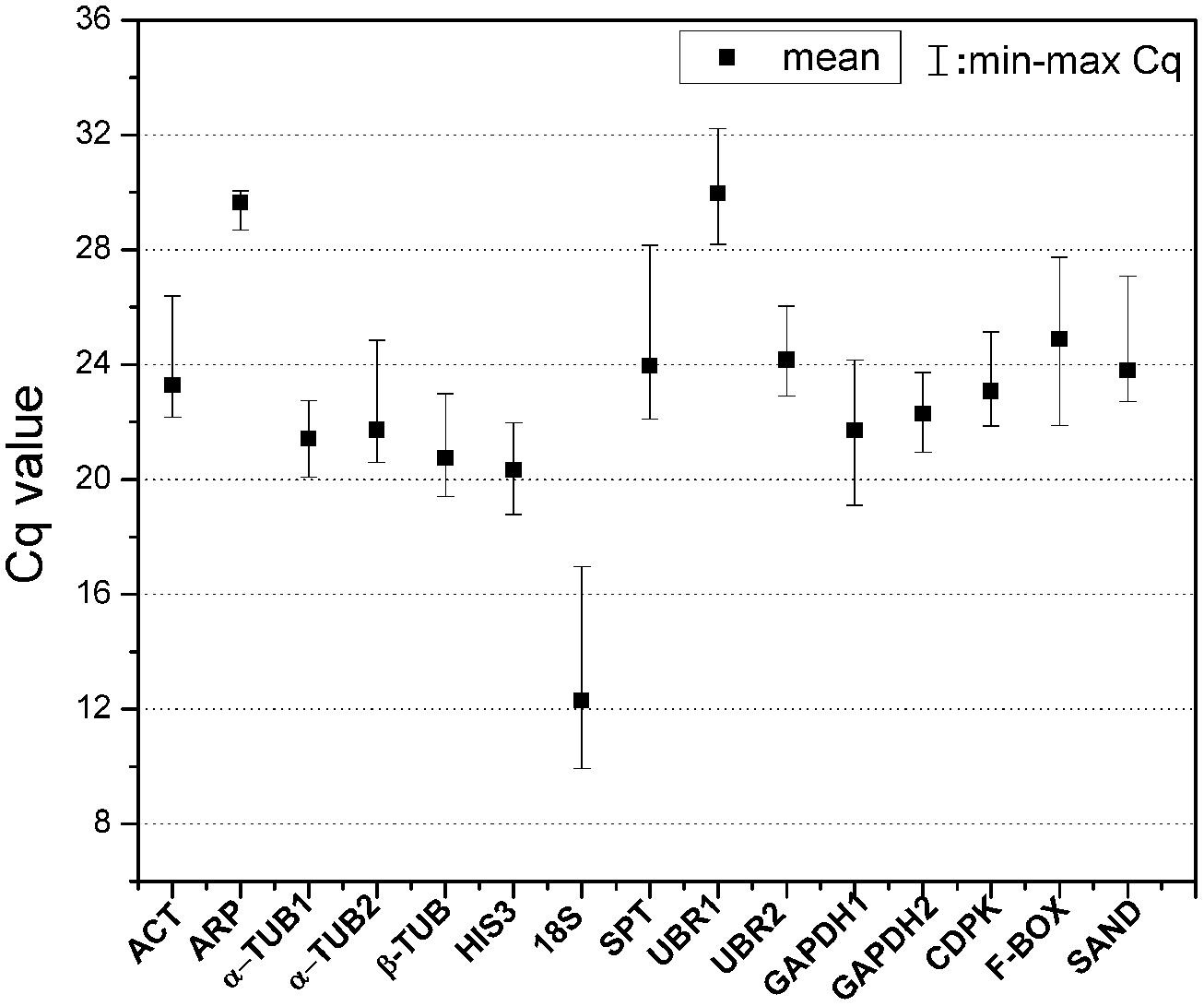
FIGURE 1. Expression levels of 15 candidate reference genes in 15 different samples. The median value and the minimum and maximum Cq of the 15 samples were calculated. Two biological replicates for all of the samples and three technical replicates of each biological replicate were used.
Expression Stability Analysis of the Candidate Reference Genes
The expression levels of tested reference genes were analyzed and ranked using three programs, geNorm, NormFinder, and RefFinder individually.
GeNorm analysis
GeNorm ranked the candidate genes based on the assumption that the expression ratio of two ideal reference genes should be constant throughout different test samples (Vandesompele et al., 2002). The expression stability value (M) and pairwise variation value (V) for each reference gene with all other genes were automatically analyzed and ranked. It is recommend using an M value below the threshold of 1.5 (Vandesompele et al., 2002; the lower the M value, the higher the gene’s expression stability). In our analysis, all 15 candidate reference genes had an M value less than 1.5 (Figure 2) which indicated that all these genes were acceptable stable reference gene candidates. For all the tested samples, α-TUB2 and SAND were the most stable two genes, while 18S was the least stable. The gene stability ranking (from the most stable to the least stable) across all the samples was: α-TUB2/SAND > CDPK > ACT > β-TUB > GAPDH2 > α-TUB1 > UBR2 > HIS3 > F-BOX > GAPDH1 > ARP > UBR1 > SPT > 18S (Figure 2A). Similar results were obtained for abiotic stress treated samples, with slight changes in the order of ranking. The gene stability ranking (from the most stable to the least stable) for stress samples were: α-TUB2/CDPK > SAND > ACT > GAPDH2 > β-TUB > α-TUB1 > UBR2 > HIS3 > GAPDH1 > ARP > F-BOX > SPT > UBR1 > 18S (Figure 2B). For the H-D-R process, different ranking results were obtained as compared to the tested samples and stress-treated samples (from the most stable to the least stable): α-TUB1/ACT > CDPK > GAPDH2 > HIS3 > β-TUB > F-BOX > UBR2 > SAND > α-TUB2 > GAPDH1 > UBR1 > SPT >ARP > 18S (Figure 2C).
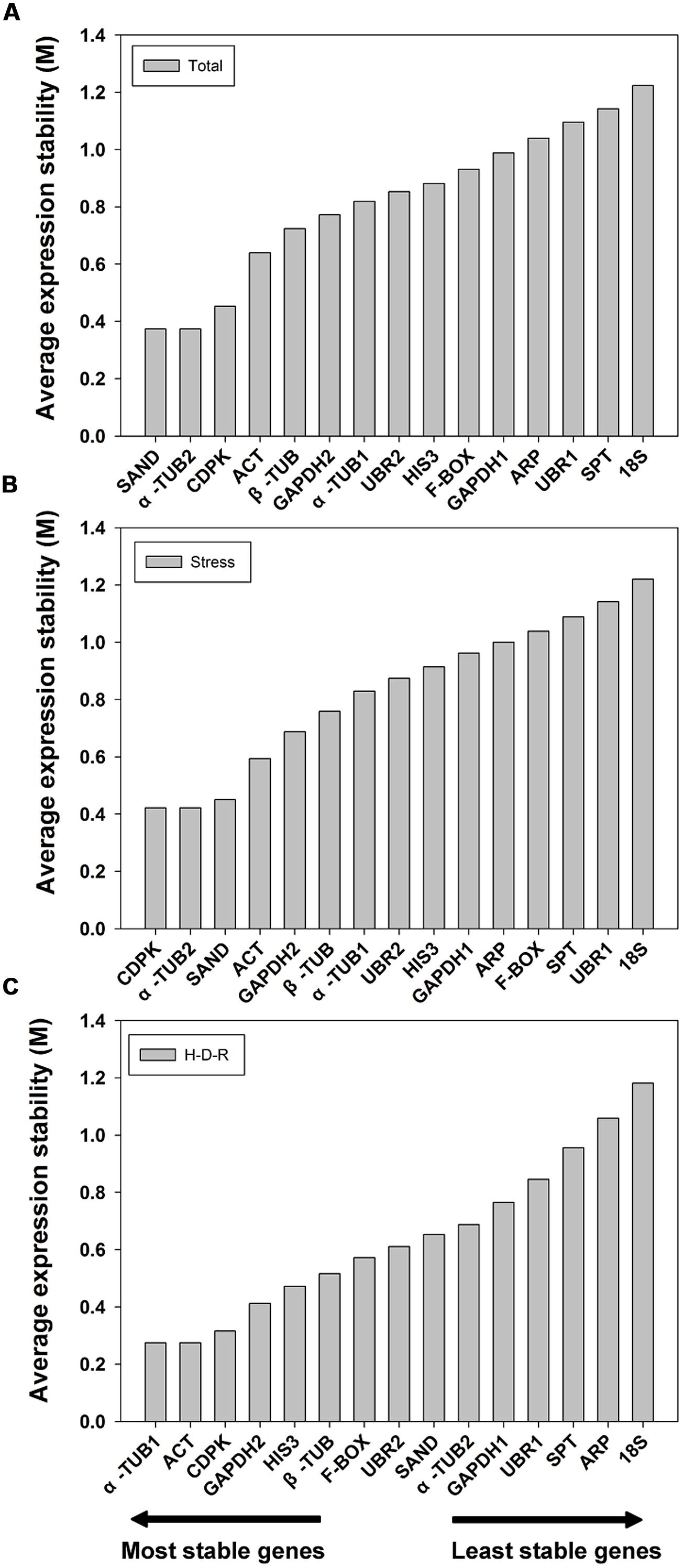
FIGURE 2. Expression stability values (M) and ranking of the candidate reference genes based on geNorm algorithm. The cutoff M value was proposed to be 1.5, a lower M value indicated more stable gene expression. The most stable genes are listed on the left and the least stable on the right. (A) All tested samples, (B) abiotic stresses, (C) hydration-desiccation-rehydration process.
NormFinder analysis
The 15 candidate reference genes were further evaluated using NormFinder method. NormFinder evaluates the stability of reference genes based on the expression variations of candidate reference genes. The program calculates a stability value for each reference gene and the lower stability value indicated the higher stability (Andersen et al., 2004). NormFinder analysis was performed without grouping. The results are shown in Figure 3. For all tested samples, NormFinder demonstrated that CDPK was the most stable (ranked third by geNorm) followed by α-TUB2, β-TUB, SAND. Similar to geNorm, 18S was the least stable gene. The stability ranking order (from the most stable to the least stable) was: CDPK > α-TUB2 > β-TUB > SAND > GAPDH2 > UBR2 > ACT > α-TUB1 > HIS3 > F-BOX > GAPDH1 > SPT > UBR1 > ARP > 18S (Figure 3A). For abiotic stress, NormFinder suggested that α-TUB2 and CDPK were the most stable genes, and 18S and UBR1 were the least stable. These results are consistent with those obtained by geNorm. The stability ranking order (from the most stable to the least stable) was: α-TUB2 > CDPK > β-TUB > GAPDH2 > SAND > ACT > UBR2 > α-TUB1 > HIS3 > GAPDH1 > SPT > ARP > F-BOX > UBR1 > 18S (Figure 3B). For H-D-R process, β-TUB was the most stable gene. Notably, both by geNorm and NormFinder demonstrated that 18S, ARP, SPT, UBR1, and GAPDH1 were the five least stable genes in response to hydration-desiccation-rehydration. The stability ranking order (from the most stable to the least stable) was: β-TUB > CDPK > α-TUB1 > F-BOX > SAND > ACT > α-TUB2 > UBR2 > HIS3 > GAPDH2> GAPDH1> UBR1>SPT>ARP > 18S (Figure 3C).
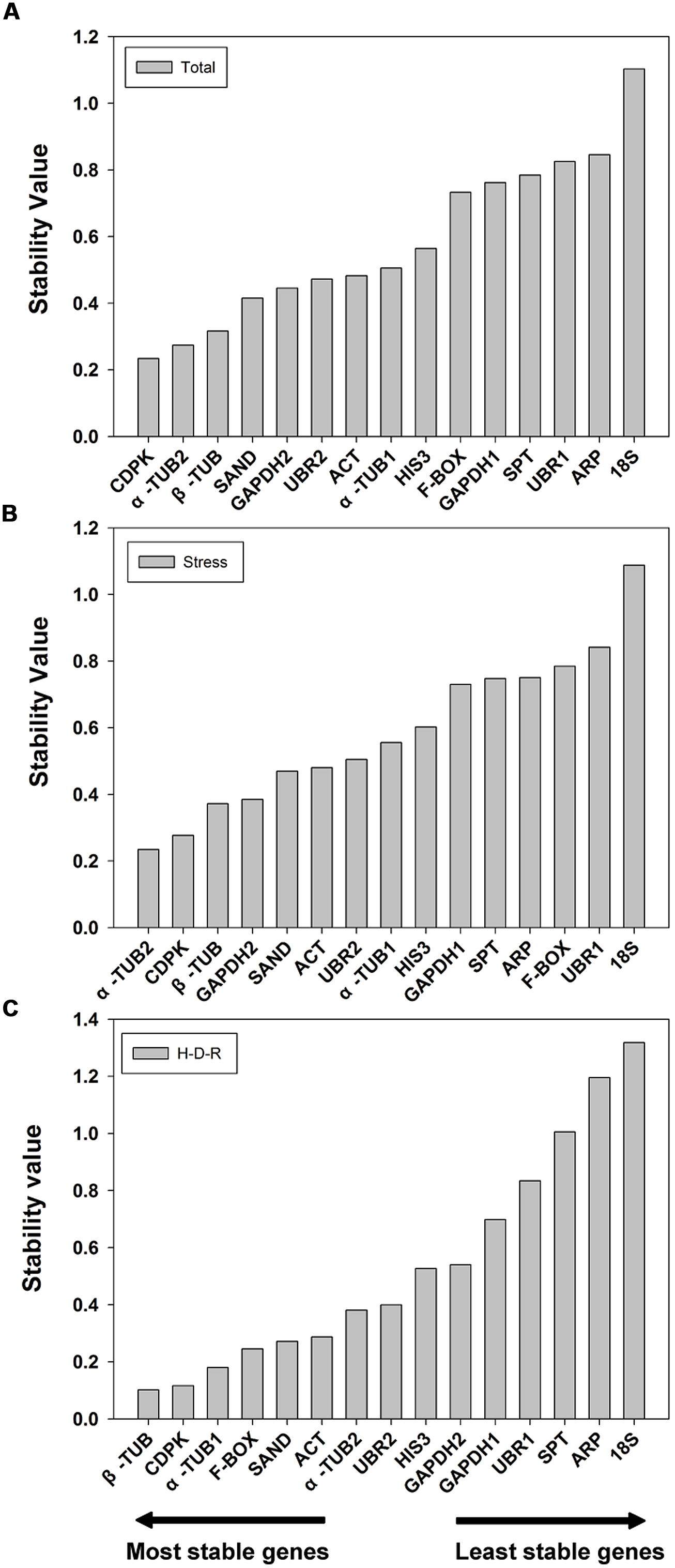
FIGURE 3. Stability value and ranking of the candidate reference genes based on NormFinder algorithm. The lowest stability value indicates the most stable expression within the gene set examined. The most stable genes are listed on the left and the least stable on the right. (A) All tested samples, (B) abiotic stresses, (C) hydration-desiccation-rehydration process.
RefFinder analysis
The results obtained from geNorm and NormFinder were further confirmed using the comprehensive ranking platform RefFinder (Xie et al., 2012). RefFinder is a web-based tool which integrates four current computing programs to compare and re-rank the tested reference genes based on the geometric mean of the weights of every single gene calculating by each program. The final ranking results are shown in Figure 4. For all the tested samples, a similar ranking order was obtained using RefFinder as compared to NormFinder (from the most stable to the least stable): CDPK > α-TUB2 > β-TUB > SAND > GAPDH2 > ACT > UBR2 > ARP > α-TUB1 > HIS3 > F-BOX > GAPDH1 > SPT > UBR1 > 18S (Figure 4A). For stress samples, the ranking result obtained from RefFinder was also more consistent with NormFinder. The ranking order (from the most stable to the least stable) was: α-TUB2 > CDPK > β-TUB > GAPDH2 > SAND > ACT > ARP > α-TUB1 > UBR2 > GAPDH1 > HIS3 > F-BOX > SPT> UBR1> 18S (Figure 4B). For the H-D-R process, the ranking order suggested by RefFinder was more similar to geNorm. The top three stable genes were α-TUB1, ACT, and CDPK (Figure 4C). The ranking order was: α-TUB1 > CDPK > ACT > β-TUB > GAPDH2 > F-BOX > HIS3 > SAND > ARP > UBR2 > UBR1 >α-TUB2 > GAPDH1 > SPT > 18S.
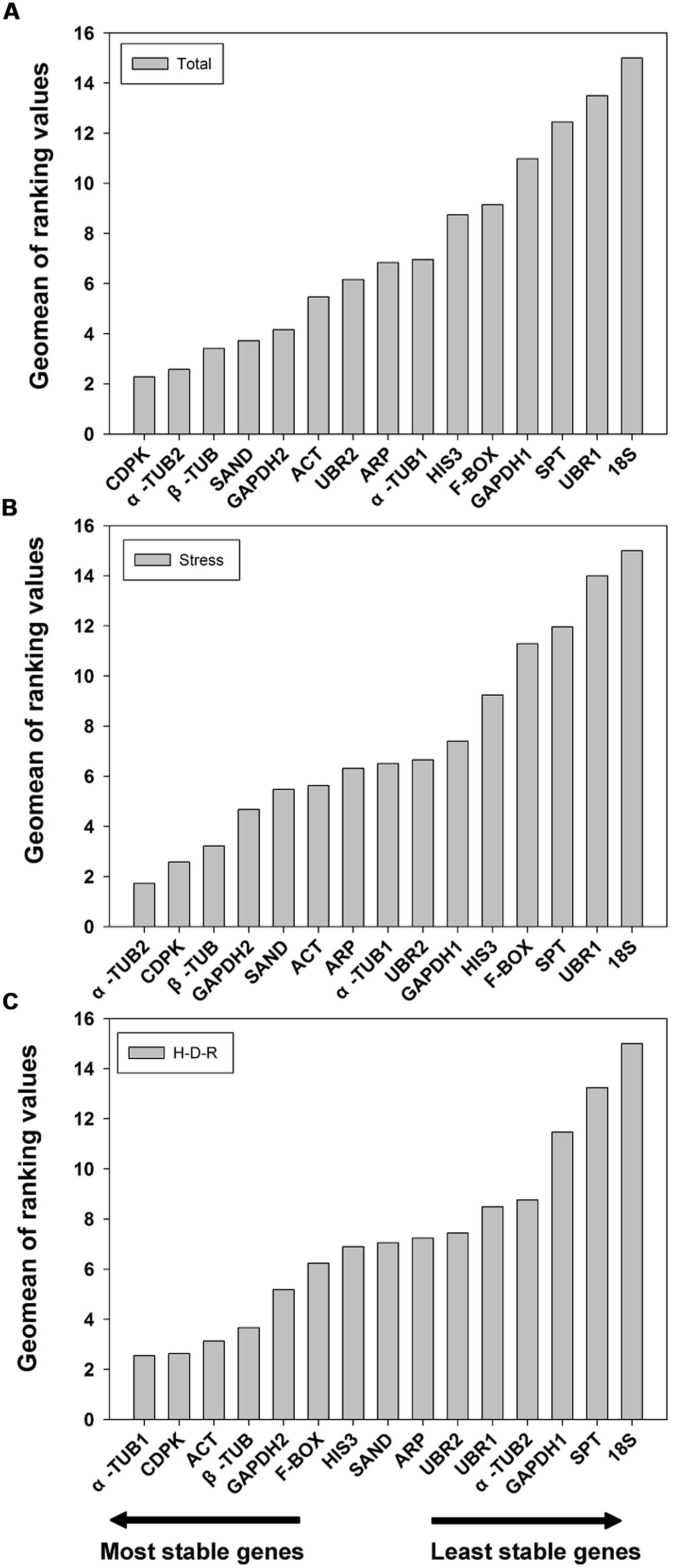
FIGURE 4. Geomean of ranking values of the candidate reference genes based on RefFinder algorithm. RefFinder is a comprehensive tool which generated the final overall ranking of tested reference genes based on the geometric mean of the weights of every gene calculating by each program including geNorm, NormFinder, Bestkeeper, and the comparative delta Ct methods. The most stable genes are listed on the left and the least stable on the right. (A) All tested samples, (B) abiotic stresses, (C) hydration-desiccation-rehydration process.
The stability of reference genes were assessed and ranked using three different algorithms (i.e., geNorm, NormFinder, and RefFinder; Table 2). The rankings are consistent between the algorithms, especially for the top six most stable genes (Italics) and the four least stable genes (underlined). For example, geNorm, NormFinder, and RefFinder all demonstrated that α-TUB2, CDPK, β-TUB were the most stable genes, while 18S, UBR1, SPT were always low ranked and were the least stable genes for various abiotic stress treatment samples.

TABLE 2. Consensus stability ranking of the reference genes evaluated by geNorm, NormFinder, and RefFinder tools.
Optimal Number of Reference Genes for RT-qPCR Normalization
The optimal number of reference genes for accurate normalization of RT-qPCR was also recommended by geNorm through calculating the pairwise variation Vn/Vn+1 value using 0.15 as the proposed cutoff value (Vandesompele et al., 2002). A Vn/Vn+1 value less than 0.15 means the inclusion of the n+1 reference genes is not required, and the top n reference genes are adequate for accurate normalization of RT-qPCR results. When considering all samples, pairwise variation analysis demonstrated that V2/3 exceeded the recommend cutoff value (0.15; Figure 5) and the inclusion of the third a nd fourth stable reference genes (i.e., SAND and ACT) were needed to improve the accuracy of normalization. For stress and hydration-desiccation-rehydration process conditions, the V2/3 value was 0.139 and 0.102, respectively, which indicated that the two most stable reference genes were sufficient for reliable normalization. For the H-D-R process subset, all pairwise variation values (from V2/3 to V14/15) were below the proposed cutoff 0.15. For stress conditions, the combination of CDPK and α-TUB2 are sufficient for normalization while the combination of α-TUB1 and CDPK are sufficient for the H-D-R process.
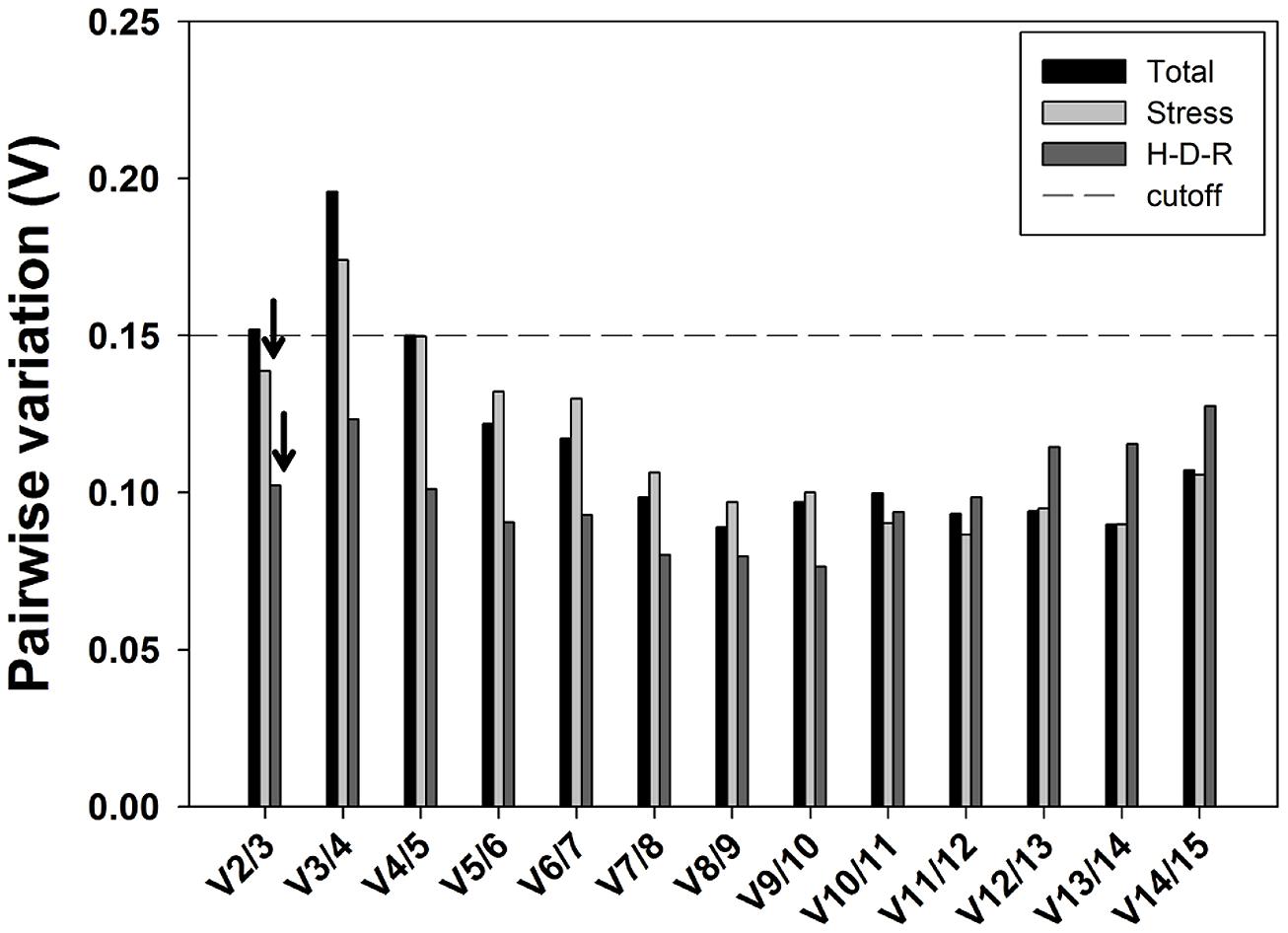
FIGURE 5. Pairwise variation (Vn/Vn+1) analysis of the candidate reference genes. The pairwise variation (Vn/Vn+1) was analyzed based on geNorm algorithm to determine the optimal number of reference genes for accurate normalization. The cutoff value was proposed to be 0.15, below which the inclusion of an additional reference genes is not necessary. Arrows indicated the optimal number of reference genes.
Discussion
Reference Gene Identification and Selection in Mosses
Reverse transcription quantitative real-time polymerase chain reaction is a powerful technology for gene expression studies, and the utilization of suitable reference genes is a prerequisite to ensure reliable and accurate data. Numerous studies have documented the selection of reference genes in various plants including Arabidopsis (Czechowski et al., 2005; Remans et al., 2008; Hong et al., 2010; Lilly et al., 2011) and crops such as soybean (Libault et al., 2008; Hu et al., 2009; Kulcheski et al., 2010). In this regard, the model moss P. patens has been extensively studied and the vast majority of RT-qPCR gene expression studies employ either ACT (Yamawaki et al., 2011; Cui et al., 2012; Liu et al., 2013) or UBQ (Harries et al., 2005; Dittrich and Devarenne, 2012) as a single reference gene. Recently, reference gene selection in P. patens has focused on hormone treatment and the comparison of developmental stages (Le Bail et al., 2013). Gene expression studies in the moss T. ruralis have employed 18S as an internal control (Zeng and Wood, 2000; Chen et al., 2002; Zeng et al., 2002). Our initial RT-qPCR experiments in S. caninervis employed 18S (Yang et al., 2012) and no other reference genes were tested or compared. However, a growing number of studies have demonstrated that 18S, ACT, and UBQ genes were not stable in many species under non-standard experimental condition (Jian et al., 2008; Lovdal and Lillo, 2009; Yang et al., 2010; Lilly et al., 2011). In P. patens, Wang et al. (2012) reported that ACT and GAPDH genes are up-regulated by abiotic stress. Overall, reference gene research in mosses lags behind other major plant groups. To date, few studies have undertaken a systematic comparison and selection of reference genes in mosses or within moss gametophores exposed to abiotic stress. Very few moss genes deposited in the database is the main reason for the limitation of reference gene study in mosses, more work including reference gene isolation and selection need to increase efforts for mosses gene study especially for extreme stress tolerant species.
Reference Genes Stability in S. caninervis
Recent studies demonstrated that the expression of some classic reference genes, such ACT (Lovdal and Lillo, 2009; Lilly et al., 2011), 18S (Die et al., 2010; Yang et al., 2010), and GAPDH (Li et al., 2012; Wang et al., 2012) can vary greatly and are unsuitable for gene normalization (particularly in response to abiotic stress). In this study, ACT and GAPDH2 genes showed good stability ranking (within the top six stable genes), while 18S was always the least stable gene under three tested experimental conditions. Some studies support the idea that new reference genes can be more stably expressed under specific conditions as compared with classic ones (Czechowski et al., 2005; Libault et al., 2008). In the present study, three new reference genes CDPK, F-BOX, and SAND were tested. Similar to soybean (Libault et al., 2008), our results showed that the CDPK gene was one of the top two stable genes under all experimental conditions as determined by each of the algorithms. In contrast, the F-BOX gene which is reported to be a stable reference gene (Czechowski et al., 2005; Libault et al., 2008) was always amongst the least stable genes in our analysis. The SAND gene is also reported to be a stable reference gene (Czechowski et al., 2005; Demidenko et al., 2011; Lilly et al., 2011; Zhu et al., 2013) and in our study was suitable for analyzing abiotic stress samples.
Comprehensive Assessment of Reference Gene Stability Using Multiple Evaluation Tools
Increasing awareness of the importance of suitable reference genes has lead to the development of different statistical algorithms to determine which reference gene(s) is best suited for transcript normalization under a given experiment condition for a specific species. GeNorm, NormFinder, Bestkeeper are three widely applied algorithms used for the assessment of reference gene stability. Since different algorithms may obtain differing stability rankings, one challenge is to identify the most stable (and therefore most suitable) reference gene. RefFinder, was developed to provide a final comprehensive ranking of reference genes based on the geometric mean of the weights of every gene calculated by each program (Xie et al., 2012). Jacob et al. (2013) recommend more than two algorithms should be used for reference gene stability evaluation. Bestkeeper cannot analyze more than 10 reference genes and was therefore excluded from this study (Pfaffl et al., 2004). The results from geNorm, NormFinder, and RefFinder were consistent (i.e., ranking of the six most stable and four least stable reference genes were identical), although the specific rankings of each reference gene varied. This indicated that the results obtained from these three software application were sufficient for accurate validation in this study.
Transcriptome-Based Systematic Selection is an Important Strategy for Reference Gene Study
The number of reference genes evaluated in plants (especially for non-model plants) is limited and usually does not exceed eight sequences (Yang et al., 2010; Xu et al., 2012; Wang et al., 2013a) due in part to the limited availability of species-specific gene sequences. With access to large-scale datasets, the evaluation and selection of reference genes have proliferated in model plants such as Arabidopsis (Czechowski et al., 2005) and rice (Narsai et al., 2010). As an important model moss for DT research and valuable anti-stress genes discovery, the first large-scale transcriptome data for S. caninervis, consisting of 92,240 unigenes (Gao et al., 2014), has been recently characterized and has served as the source of the reported gene selection. In this study, we perform a systematic comparison and selection of reference genes suitable for a wide range of experimental conditions from the desert plant S. caninervis. Our results demonstrate that transcriptome sequencing data is a useful source for candidate reference genes screening and represents an important strategy for large-scale reference gene selection for non-model plants.
We performed the first systematic selection of reference genes in desert moss S. caninervis. Our results reinforce the idea that reference gene stability should test in specific plant species, under particular experimental conditions, in similar tissues and evaluated with multiple programs. The stably expressed reference genes identified in this study will facilitate future work on gene expression studies in mosses and Syntrichia ssp. in particular.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the National Basic Research Program of China (2014CB954203), the National Natural Science Foundation (U1170304), and the Chinese Scholarship Council (award to Xiaoshuang Li).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2015.00038/abstract
References
Andersen, C. L., Jensen, J. L., and Orntoft, T. F. (2004). Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250. doi: 10.1158/0008-5472.CAN-04-0496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bustin, S. A. (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25, 169–193. doi: 10.1677/jme.0.0250169
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M.,et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, X. B., Kanokporn, T., Zeng, Q., Wilkins, T. A., and Wood, A. J. (2002). Characterization of the V-type H (+)-ATPase in the resurrection plant Tortula ruralis: accumulation and polysomal recruitment of the proteolipid c subunit in response to salt-stress. J. Exp. Bot. 53, 225–232. doi: 10.1093/jexbot/53.367.225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, X. B., and Wood, A. J. (2003). The 26S proteasome of the resurrection plant Tortula ruralis: cloning and characterization of the TrRPT2 subunit. Biol. Plant. 46, 363–368. doi: 10.1023/A:1024369917715
Clancy, M. A., Rosli, H. G., Chamala, S., Barbazuk, W. B., Civello, P. M., and Folta, K. M. (2013). Validation of reference transcripts in strawberry (Fragaria spp.). Mol. Genet. Genomics 288, 671–681. doi: 10.1007/s00438-013-0780-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cui, S., Hu, J., Guo, S., Wang, J., Cheng, Y., Dang, X.,et al. (2012). Proteome analysis of Physcomitrella patens exposed to progressive dehydration and rehydration. J. Exp. Bot. 63, 711–726. doi: 10.1093/jxb/err296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Czechowski, T., Stitt, M., Altmann, T., Udvardi, M. K., and Scheible, W. R. (2005). Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 139, 5–17. doi: 10.1104/pp.105.063743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demidenko, N. V., Logacheva, M. D., and Penin, A. A. (2011). Selection and validation of reference genes for quantitative real-time PCR in buckwheat (Fagopyrum esculentum) based on transcriptome sequence data. PLoS ONE 6:e19434. doi: 10.1371/journal.pone.0019434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Die, J. V., Roman, B., Nadal, S., and Gonzalez-Verdejo, C. I. (2010). Evaluation of candidate reference genes for expression studies in Pisum sativum under different experimental conditions. Planta 232, 145–153. doi: 10.1007/s00425-010-1158-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Die, J. V., and Rowland, L. J. (2013). Superior cross-species reference genes: a blueberry case study. PLoS ONE 8:e73354. doi: 10.1371/journal.pone.0073354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dittrich, A. C., and Devarenne, T. P. (2012). Characterization of a PDK1 homologue from the moss Physcomitrella patens. Plant Physiol. 158, 1018–1033. doi: 10.1104/pp.111.184572
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Exposito-Rodriguez, M., Borges, A. A., Borges-Perez, A., and Perez, J. A. (2008). Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol. 8:131. doi: 10.1186/1471-2229-8-131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, L., Yu, Q., Li, X., Ning, X., Wang, J., Zou, J.,et al. (2013). Identification of reference genes for qRT-PCR analysis in Yesso Scallop Patinopecten yessoensis. PLoS ONE 8:e75609. doi: 10.1371/journal.pone.0075609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Figueiredo, A., Loureiro, A., Batista, D., Monteiro, F., Varzea, V., Pais, M. S.,et al. (2013). Validation of reference genes for normalization of qPCR gene expression data from Coffea spp. hypocotyls inoculated with Colletotrichum kahawae. BMC Res. 6:388. doi: 10.1186/1756-0500-6-388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, B., Zhang, D. Y., Li, X. S., Yang, H. L., and Wood, J. A. (2014). De novo assembly and characterization of the transcriptome in the desiccation-tolerant moss Syntrichia caninervis. BMC Res. 7:490. doi: 10.1186/1756-0500-7-490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harries, P. A., Pan, A., and Quatrano, R. S. (2005). Actin-related protein2/3 complex component ARPC1 is required for proper cell morphogenesis and polarized cell growth in Physcomitrella patens. Plant Cell 17, 2327–2339. doi: 10.1105/tpc.105.033266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, S. M., Bahn, S. C., Lyu, A., Jung, H. S., and Ahn, J. H. (2010). Identification and testing of superior reference genes for a starting pool of transcript normalization in Arabidopsis. Plant Cell Physiol. 51, 1694–1706. doi: 10.1093/pcp/pcq128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, S. Y., Seo, P. J., Yang, M. S., Xiang, F., and Park, C. M. (2008). Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol. 8:112. doi: 10.1186/1471-2229-8-112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, R., Fan, C., Li, H., Zhang, Q., and Fu, Y. F. (2009). Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol. Biol. 10:93. doi: 10.1186/1471-2199-10-93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huggett, J., Dheda, K., Bustin, S., and Zumla, A. (2005). Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 6, 279–284. doi: 10.1038/sj.gene.6364190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imai, T., Ubi, B. E., Saito, T., and Moriguchi, T. (2014). Evaluation of reference genes for accurate normalization of gene expression for real time-quantitative PCR in Pyrus pyrifolia using different tissue samples and seasonal conditions. PLoS ONE 9:e86492. doi: 10.1371/journal.pone.0086492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacob, F., Guertler, R., Naim, S., Nixdorf, S., Fedier, A., Hacker, N. F.,et al. (2013). Careful selection of reference genes is required for reliable performance of RT-qPCR in human normal and cancer cell lines. PLoS ONE 8:e59180. doi: 10.1371/journal.pone.0059180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jian, B., Liu, B., Bi, Y., Hou, W., Wu, C., and Han, T. (2008). Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol. Biol. 9:59. doi: 10.1186/1471-2199-9-59
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kianianmomeni, A., and Hallmann, A. (2013). Validation of reference genes for quantitative gene expression studies in Volvox carteri using real-time RT-PCR. Mol. Biol. Rep. 40, 6691–6699. doi: 10.1007/s11033-013-2784-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kulcheski, F. R., Marcelino, F. C., Nepomuceno, A. L., Abdelnoor, R. V., and Margis, R. (2010). The use of microRNAs as reference genes for quantitative PCR in soybean. Anal. Biochem. 406, 185–192. doi: 10.1016/j.ab.2010.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Le Bail, A., Scholz, S., and Kost, B. (2013). Evaluation of reference genes for RT qPCR analyses of structure-specific and hormone regulated gene expression in Physcomitrella patens gametophytes. PLoS ONE 8:e70998. doi: 10.1371/journal.pone.0070998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J. M., Roche, J. R., Donaghy, D. J., Thrush, A., and Sathish, P. (2010). Validation of reference genes for quantitative RT-PCR studies of gene expression in perennial ryegrass (Lolium perenne L.). BMC Mol. Biol. 11:8. doi: 10.1186/1471-2199-11-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, X. S., Yang, H. L., Zhang, D. Y., Zhang, Y. M., and Wood, A. J. (2012). Reference gene selection in the desert plant Eremosparton songoricum. Int. J. Mol. Sci. 13, 6944–6963. doi: 10.3390/ijms13066944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y., Wang, Z., Xu, T., Tu, W., Liu, C., Zhang, Y.,et al. (2010). Reorganization of photosystem II is involved in the rapid photosynthetic recovery of desert moss Syntrichia caninervis upon rehydration. J. Plant Physiol. 167, 1390–1397. doi: 10.1016/j.jplph.2010.05.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Libault, M., Bilgin, S., Radwan, D., Benitez, O., Clough, M., and Stacey, S. (2008). Identification of four soybean reference genes for gene expression normalization. Plant Genome 1:44. doi: 10.3835/plantgenome2008.02.0091
Lilly, S. T., Drummond, R. S., Pearson, M. N., and Macdiarmid, R. M. (2011). Identification and validation of reference genes for normalization of transcripts from virus-infected Arabidopsis thaliana. Mol. Plant Microbe Interact. 24, 294–304. doi: 10.1094/MPMI-10-10-0236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y. J., Han, X. M., Ren, L. L., Yang, H. L., and Zeng, Q. Y. (2013). Functional divergence of the glutathione S-transferase supergene family in Physcomitrella patens reveals complex patterns of large gene family evolution in land plants. Plant Physiol. 161, 773–786. doi: 10.1104/pp.112.205815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lovdal, T., and Lillo, C. (2009). Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold, and light stress. Anal. Biochem. 387, 238–242. doi: 10.1016/j.ab.2009.01.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narsai, R., Ivanova, A., Ng, S., and Whelan, J. (2010). Defining reference genes in Oryza sativa using organ, development, biotic, and abiotic transcriptome datasets. BMC Plant Biol. 10:56. doi: 10.1186/1471-2229-10-56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oliver, M. J., Dowd, S. E., Zaragoza, J., Mauget, S. A., and Payton, P. R. (2004). The rehydration transcriptome of the desiccation-tolerant bryophyte Tortula ruralis: transcript classification and analysis. BMC Genomics 5:89. doi: 10.1186/1471-2164-5-89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oliver, M. J., Mishler, B. D., and Quisenberry, J. E. (1993). Comparative measures of desiccation-tolerance in the Tortula ruralis complex. 1. Variation in damage control and repair. Am. J. Bot. 80, 127–136. doi: 10.2307/2445030
Peng, C. A., Oliver, M. J., and Wood, A. J. (2005). Is the rehydrin TrDr3 from Tortula ruralis associated with tolerance to cold, salinity, and reduced pH? Physiological evaluation of the TrDr3-orthologue, HdeD from Escherichia coli in response to abiotic stress. Plant Biol. 7, 315–320. doi: 10.1055/s-2005-865653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfaffl, M. W., Tichopad, A., Prgomet, C., and Neuvians, T. P. (2004). Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515. doi: 10.1023/B:BILE.0000019559.84305.47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ponton, F., Chapuis, M. P., Pernice, M., Sword, G. A., and Simpson, S. J. (2011). Evaluation of potential reference genes for reverse transcription-qPCR studies of physiological responses in Drosophila melanogaster. J. Insect Physiol. 57, 840–850. doi: 10.1016/j.jinsphys.2011.03.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Remans, T., Smeets, K., Opdenakker, K., Mathijsen, D., Vangronsveld, J., and Cuypers, A. (2008). Normalisation of real-time RT-PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta 227, 1343–1349. doi: 10.1007/s00425-008-0706-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shen, Y., Li, Y., Ye, F., Wang, F., Lu, W., and Xie, X. (2010). Identification of suitable reference genes for measurement of gene expression in human cervical tissues. Anal. Biochem. 405, 224–229. doi: 10.1016/j.ab.2010.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, J., Liu, M., Shi, J., Zheng, G., Wang, Y., Wang, J.,et al. (2012). Reference gene selection for qPCR in Ammopiptanthus mongolicus under abiotic stresses and expression analysis of seven ROS-scavenging enzyme genes. Plant Cell Rep. 31, 1245–1254. doi: 10.1007/s00299-012-1245-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stark, L. R., and McLetchie, D. N. (2006). Gender-specific heat-shock tolerance of hydrated leaves in the desert moss Syntrichia caninervis. Physiol. Plant. 126, 187–195. doi: 10.1111/j.1399-3054.2006.00590.x
Stark, L. R., Mcletchie, D. N., and Roberts, S. P. (2009). Gender differences and a new adult eukaryotic record for upper thermal tolerance in the desert moss Syntrichia caninervis. J. Therm. Biol. 34, 131–137. doi: 10.1016/j.jtherbio.2008.12.001
Stark, L. R., Nichols, L., Mcletchie, D. N., and Bonine, M. L. (2005). Do the sexes of the desert moss Syntrichia caninervis differ in desiccation tolerance? A leaf regeneration assay. Int. J. Plant Sci. 166, 21–29. doi: 10.1086/425671
Stark, L. R., Nichols, L., Mcletchie, D. N., Smith, S. D., and Zundel, C. (2004). Age and sex-specific rates of leaf regeneration in the Mojave Desert moss Syntrichia caninervis. Am. J. Bot. 91, 1–9. doi: 10.3732/ajb.91.1.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tao, Y., and Zhang, Y. M. (2012). Effects of leaf hair points of a desert moss on water retention and dew formation: implications for desiccation tolerance. J. Plant Res. 125, 351–360. doi: 10.1007/s10265-011-0449-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Udvardi, M. K., Czechowski, T., and Scheible, W. R. (2008). Eleven golden rules of quantitative RT-PCR. Plant Cell 20, 1736–1737. doi: 10.1105/tpc.108.061143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A.,et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034 doi: 10.1186/gb-2002-3-7-research0034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wan, H., Zhao, Z., Qian, C., Sui, Y., Malik, A. A., and Chen, J. (2010). Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal. Biochem. 399, 257–261. doi: 10.1016/j.ab.2009.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, L. J., Wang, Y. C., and Zhou, P. (2013a). Validation of reference genes for quantitative real-time PCR during Chinese wolfberry fruit development. Plant Physiol. Biochem. 70, 304–310. doi: 10.1016/j.plaphy.2013.05.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, M., Wang, Q., and Zhang, B. (2013b). Evaluation and selection of reliable reference genes for gene expression under abiotic stress in cotton (Gossypium hirsutum L.). Gene 530, 44–50. doi: 10.1016/j.gene.2013.07.084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Liu, Y., and Yang, P. (2012). Proteomic studies of the abiotic stresses response in model moss - Physcomitrella patens. Front. Plant Sci. 3:258. doi: 10.3389/fpls.2012.00258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wood, A. J. (2007). Invited essay: new frontiers in bryology and lichenology-The nature and distribution of vegetative desiccation-tolerance in hornworts, liverworts and mosses. Bryologist 110, 163–177. doi: 10.1639/0007-2745(2007)110[163:IENFIB]2.0.CO;2
Wood, A. J., Duff, R. J., and Oliver, M. J. (1999). Expressed sequence tags (ESTs) from desiccated Tortula ruralis identify a large number of novel plant genes. Plant Cell Physiol. 40, 361–368. doi: 10.1093/oxfordjournals.pcp.a029551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, N., Zhang, Y. M., Downing, A., Zhang, J., and Yang, C. H. (2012). Membrane stability of the desert moss Syntrichia caninervis Mitt. during desiccation and rehydration. J. Bryol. 34, 1–8. doi: 10.1179/1743282011Y.0000000043
Xie, F. L., Xiao, P., Chen, D. L., Xu, L., and Zhang, B. H. (2012). miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 80, 75–84. doi: 10.1007/s11103-012-9885-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, S. J., Jiang, P. A., Wang, Z. W., and Wang, Y. (2009a). Crystal structures and chemical composition of leaf surface wax depositions on the desert moss Syntrichia caninervis. Biochem. Syst. Ecol. 37, 723–730. doi: 10.1016/j.bse.2009.12.012
Xu, S. J., Liu, C. J., Jiang, P. A., Cai, W. M., and Wang, Y. (2009b). The effects of drying following heat shock exposure of the desert moss Syntrichia caninervis. Sci. Total Environ. 407, 2411–2419. doi: 10.1016/j.scitotenv.2008.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y. Y., Zhu, X. W., Gong, Y. Q., Xu, L., Wang, Y., and Liu, L. W. (2012). Evaluation of reference genes for gene expression studies in radish (Raphanus sativus L.) using quantitative real-time PCR. Biochem. Biophys. Res. Commun. 424, 398–403. doi: 10.1016/j.bbrc.2012.06.119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamawaki, S., Yamashino, T., Nakanishi, H., and Mizuno, T. (2011). Functional characterization of HY5 homolog genes involved in early light-signaling in Physcomitrella patens. Biosci. Biotechnol. Biochem. 75, 1533–1539. doi: 10.1271/bbb.110219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, H. L., Zhang, D. Y., Wang, J. C., Wood, A. J., and Zhang, Y. M. (2012). Molecular cloning of a stress-responsive aldehyde dehydrogenase gene ScALDH21 from the desiccation-tolerant moss Syntrichia caninervis and its responses to different stresses. Mol. Biol. Rep. 39, 2645–2652. doi: 10.1007/s11033-011-1017-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Y., Hou, S., Cui, G., Chen, S., Wei, J., and Huang, L. (2010). Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol. Biol. Rep. 37, 507–513. doi: 10.1007/s11033-009-9703-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeng, Q., Chen, X. B., and Wood, A. J. (2002). Two early light-inducible protein (ELIP) cDNAs from the resurrection plant Tortula ruralis are differentially expressed in response to desiccation, rehydration, salinity, and high light. J. Exp. Bot. 53, 1197–1205. doi: 10.1093/jexbot/53.371.1197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeng, Q., and Wood, A. J. (2000). A cDNA encoding ribosomal protein RPL15 from the desiccation-tolerant bryophyte Tortula ruralis: mRNA transcripts are stably maintained in desiccated and rehydrated gametophytes. Biosci. Biotech. Biochem. 64, 2221–2224. doi: 10.1271/bbb.64.2221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, D. Y., Liu, H. L., Shi, X., Wang, J. C., and Zhang, Y. K. (2011a). Limitations on the recruitment of the rare sand shrubby legume Eremosparton songoricum (Fabaceae) in Gurbantunggut Desert, China. J. Arid Land 3, 75–84. doi: 10.3724/SP.J.1227.2011.00075
Zhang, J., Zhang, Y. M., Downing, A., Wu, N., and Zhang, B. C. (2011b). Photosynthetic and cytological recovery on remoistening Syntrichia caninervis Mitt., a desiccation-tolerant moss from Northwestern China. Photosynthetica 49, 13–20. doi: 10.1007/s11099-011-0002-6
Zhang, Y. M. (2005). The microstructure and formation of biological soil crusts in their early developmental stage. Chinese Sci. Bull. 50, 117–121.
Zhang, Y. M., Chen, J., Wang, L., Wang, X. Q., and Gu, Z. H. (2007). The spatial distribution patterns of biological soil crusts in the Gurbantunggut Desert, Northern Xinjiang, China. J. Arid Environ. 68, 599–610. doi: 10.1016/j.jaridenv.2006.06.012
Zheng, Y. P., Xu, M., Zhao, J. C., Zhang, B. C., Bei, S. Q., and Hao, L. H. (2011). Morphological adaptations to drought and reproductive strategy of the moss Syntrichia caninervis in the Gurbantunggut Desert, China. Arid Land Res. Manag. 25, 116–127. doi: 10.1080/15324982.2011.554956
Zhu, J., Zhang, L., Li, W., Han, S., Yang, W., and Qi, L. (2013). Reference gene selection for quantitative real-time PCR normalization in Caragana intermedia under different abiotic stress conditions. PLoS ONE 8:e53196. doi: 10.1371/journal.pone.0053196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Syntrichia caninervis, quantitative real-time PCR, reference gene, geNorm, NormFinder, RefFinder
Citation: Li X, Zhang D, Li H, Gao B, Yang H, Zhang Y and Wood AJ (2015) Characterization of reference genes for RT-qPCR in the desert moss Syntrichia caninervis in response to abiotic stress and desiccation/rehydration. Front. Plant Sci. 6:38. doi: 10.3389/fpls.2015.00038
Received: 20 October 2014; Accepted: 15 January 2015;
Published online: 05 February 2015.
Edited by:
Sagadevan G. Mundree, Queensland University of Technology, AustraliaReviewed by:
Gong-yin Ye, Zhejiang University, ChinaManoj K. Sharma, Jawaharlal Nehru University, India
Copyright © 2015 Li, Zhang, Li, Gao, Yang, Zhang and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daoyuan Zhang, Key Laboratory of Biogeography and Bioresource in Arid Land, Xinjiang Institute of Ecology and Geography – Chinese Academy of Sciences, Ürümqi 830011, China e-mail:emhhbmdkeUBtcy54amIuYWMuY24=