 Xiaochen Chen
Xiaochen Chen Qiushi Li
Qiushi Li Ying Li
Ying Li Jianping Han
Jianping Han- Center for Computational Biology and Bioinformatics, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
The chloroplast genome (cp genome) of Aconitum barbatum var. puberulum was sequenced using the third-generation sequencing platform based on the single-molecule real-time (SMRT) sequencing approach. To our knowledge, this is the first reported complete cp genome of Aconitum, and we anticipate that it will have great value for phylogenetic studies of the Ranunculaceae family. In total, 23,498 CCS reads and 20,685,462 base pairs were generated, the mean read length was 880 bp, and the longest read was 2,261 bp. Genome coverage of 100% was achieved with a mean coverage of 132× and no gaps. The accuracy of the assembled genome is 99.973%; the assembly was validated using Sanger sequencing of six selected genes from the cp genome. The complete cp genome of A. barbatum var. puberulum is 156,749 bp in length, including a large single-copy region of 87,630 bp and a small single-copy region of 16,941 bp separated by two inverted repeats of 26,089 bp. The cp genome contains 130 genes, including 84 protein-coding genes, 34 tRNA genes and eight rRNA genes. Four forward, five inverted and eight tandem repeats were identified. According to the SSR analysis, the longest poly structure is a 20-T repeat. Our results presented in this paper will facilitate the phylogenetic studies and molecular authentication on Aconitum.
Introduction
Aconitum barbatum var. puberulum (Niubian) belongs to the Aconitum subgenus Lycoctonum (Ranunculaceae) and most species of Lycoctonum are low-temperature resistant. However, aconitine is a kind of highly toxic alkaloid, which mainly exists in the plants of Aconitum. Identification and phylogeny studies of Aconitum and the Ranunculaceae family thus are particularly important (Xiao et al., 2005). He et al. (2010) applied the chloroplast genome (cp genome) intergenic region psbA-trnH as a barcode to identify 19 species in Aconitum, and Johansson (Johansson, 1995) used chloroplast DNA restriction site variation among 31 genera of the Ranunculaceae to conduct phylogenetic analyses. However, more in-depth studies of the cp genome are needed.
Chloroplasts possess their own genome and genetic system, which plays an important role in photosynthesis. The first chloroplast genome to be sequenced was that of Nicotiana tabacum, which heralded a new age of chloroplast studies in photobiology, phylogenetic biology, evolutionary biology and even chloroplast genetic engineering (Shinozaki et al., 1986; Hiratsuka et al., 1989; Daniell et al., 1998; Pfannschmidt et al., 1999; Moore et al., 2010; Wu et al., 2012). Some researchers (Chen et al., 2014; Li et al., 2014b) advocated the cp genome as a new DNA barcode to distinguish closely related plants. The typical cp genome structure of higher plants is circular with a length of 120–160 kb, containing approximately 130 genes (Sugiura, 1992). Two inverted repeats (IRs), a large single-copy region (LSC), and a small single-copy region (SSC) constitute the complete cp genome (Zhang et al., 2012). With the development of the next-generation sequencing technology, increasing numbers of species have been sequenced, including duckweed, palm, and others (Jordan et al., 1996; Uthaipaisanwong et al., 2012). Although the interest in the cp genome has increased in the past few decades, with 486 complete cp genome sequences deposited in GenBank (By 2014-7-6), there are still challenges and opportunities to develop a simple and rapid method for sequencing cp genomes. One common strategy is the use of a complete set of universal primers to amplify an entire cp genome and then perform the sequencing (Dong et al., 2013). Another frequently used strategy is “whole-genome sequencing”, which uses the total genome DNA to recover the cp genome through massively parallel sequencing (McPherson et al., 2013). This strategy is quite simple and effective, particularly as the cost of high-throughput sequencing decreases. In the present study, we used purified chloroplast DNA as the template for sequencing with the aim of developing a practical strategy involving the use of multiple samples to sequence the cp genome on the PacBio RS platform.
The third-generation PacBio system is based on the single-molecule real-time (SMRT) sequencing approach (Eid et al., 2009). Second-generation sequencing introduced a novel, rapid method for whole-genome sequencing (Mardis, 2008a,b; Metzker, 2009; Gilles et al., 2011; Kircher et al., 2011). In comparison, the SMRT approach requires no amplification, produces less compositional bias (Schadt et al., 2010), reduces the time required from sample to sequence (Chin et al., 2011; Rasko et al., 2011) and reduces the costs (Rusk, 2009). However, the main advantage of third-generation sequencing is the long read length, which was reported to be as long as 3,000 bp on average, and some reads might be 20,000 bp or longer. The long read length provides an important benefit for de novo assemblies, it allows the discovery of large structural variants, and it provides accurate microsatellite lengths, sensitive SNP detection and haplotype blocks (Metzker, 2009; Roberts et al., 2013; Li et al., 2014a). Because of the unique circular structure of the cp genome, the four junctions between the inverted regions and the single-copy regions have hampered our ability to provide accurate cp genome assemblies. However, the long reads somehow will promote and heighten the accuracy of the assembly (Bashir et al., 2012; Chin et al., 2013). SMRT sequencing combined with circular consensus sequencing (CCS) is thought to be an effective approach. This sequencing method provides multiple reads of individual templates, resulting in a higher per-base sequencing accuracy and a reduced error rate. A PacBio-only assembly could be completed without the need to construct specialized fosmid libraries or other similar assemblies using second-generation sequencing technologies. We sought to investigate whether third-generation sequencing could be used for rapid sequencing of whole cp genomes and eliminate the need to fill in the gaps that exist in the assembled genome.
In the present study, we report the completed cp genome of A. barbatum var. puberulum. To our knowledge, this is the first completed cp genome of Aconitum using the third-generation sequencing platform. Our results demonstrate that the SMRT CCS sequencing strategy is a viable option for rapidly sequencing cp genomes.
Materials and Methods
Chloroplast DNA Isolation, Sequencing, Assembly, and Validation
Fresh leaves were collected from Donglingshan Mountain, Beijing. Total cpDNA was extracted from approximately 100 g fresh leaves using a sucrose gradient centrifugation method that was described by Li et al. (2012). A total of 700 ng cp genomic DNA was sheared to a target size of 2 kb in an AFA clear mini-tube using a Covaris S2-focused ultrasonicator (Covaris Inc.) to construct the libraries according to the Pacific Biosciences SMRT Sequencing instruction manual. A 0.6X volume of pre-washed AMPure XP magnetic beads was added to the solution of sheared DNA. After concentrating the DNA, an Agilent 2100 and a Qubit fluorometer were used to perform qualitative and quantitative analyses. The samples were incubated at 25°C for 15 min to end-repair the DNA using the PacBio DNA Template Pre Kit 2.0. Then the end-repaired DNA was purified by adding a 0.6X volume of pre-washed AMPure XP magnetic beads. Blunt ligation was performed to obtain the SMRTbellTM Templates, followed by the addition of exonuclease to remove failed ligation products. The SMRTbellTM Templates were then purified in two steps. Before annealing the sequencing primer and binding polymerase to the SMRTbell templates, an Agilent 2100 and a Qubit fluorometer were used to perform the qualitative and quantitative analysis. PacBio DNA/polymerase Binding Kit 2.0 was used to anneal and bind the SMRTbellTM Templates. Two SMRT cells were used with C2 chemistry to sequence the SMRT-bellTM library. Two 45-min windows were captured for sequencing the chloroplast genome. After the CCS reads were derived from the multiple alignments of sub-reads, a quality control step was performed for the downstream assembly: SMRT Portal software (v2.0.0) was used to filter out the sequencing adapters and low-quality sequences (default parameters: sub-read length ≥ 50 bp; polymerase read quality ≥ 0.75; polymerase read length ≥ 50 bp; Li et al., 2014a). The reads were then used to assemble the chloroplast genome according to the strategy described in Qian et al. (2013). First, a workflow was designed to assemble the chloroplast genome: algorithms for greedy assembly, mapping, and consensus calling were used sequentially. Second, BLAST was used to compare the sequences from the greedy workflow, and the results of the alignment were used to construct the raw cp genome. The reads were mapped to the raw cp genome using the BWA tool, and the final cp genome sequence was generated using CAP3-based consensus calling (Altschul et al., 1997; Huang and Madan, 1999; Li and Durbin, 2010). To verify the genome sequence, PCR-based conventional Sanger sequencing was performed on six chloroplast genes (cemA, psbB, psbC, rpoA, rpoC1, and rps4; Cronn et al., 2008). The four junctions between the single-copy regions and the IRs were validated using PCR. The amplified sequences and the SMRT sequencing-based reads were aligned using Mega 5.2.2 (Tamura et al., 2011).
Genome Annotation and Codon Usage
The cp genome was annotated using the program DOGMA (Wyman et al., 2004; default parameters: the percent identity cutoff for protein coding genes=60%, the percent identity cutoff for RNAs = 80%, the E-value = 1e-5 and the number of blast hits to return = 5.), and the position of each gene was determined using a blast method with the complete cp genome sequence of Ranunculus macranthus (GenBank Acc. No. NC_008796) as a reference sequence. Manual corrections for start and stop codons and for intron/exon boundaries were performed by referencing the Chloroplast Genome Database (ChloroplastDB; Cui et al., 2006). The tRNA genes were identified using DOGMA and tRNAscan-SE (Schattner et al., 2005). The circular cp genome map of A. barbatum var. puberulum was drawn using the OrganellarGenome DRAW tool (ORDRAW; Lohse et al., 2007). Codon usage and GC content were analyzed by Mega 5.2.2.
Repeat Analysis
REPuter (Kurtz et al., 2001) was used to assess both direct and IRs according to the following criteria: cutoff n ≥ 30 bp and 90% sequence identities (Hamming distance equal to 3). Tandem Repeats Finder (TRF) v4.04 (Benson, 1999) was used to analyze tandem repeats with the settings reported by Nie et al. (2012). Simple sequence repeats (SSRs) were detected using MISA (http://pgrc.ipk-gatersleben.de/misa/), with thresholds of eight, four and three units for mono-, di-, and trinucleotide SSRs and tetra-, penta-, and hexanucleotide SSRs, respectively.
Results
PacBio RS Output and Genome Validation
Quantitative analysis using an Agilent 2100 showed that the average length of the sheared DNA fragments was approximately 1 kb. In total, 23,498 CCS reads and 20,685,462 base pairs were generated, the mean read length was 880 bp, and the longest read was 2,261 bp. Genome coverage of 100% was achieved with a mean coverage of 132× and no gaps. Detailed information is listed in Table 1. Six conserved genes with poly-structures (cemA, psbB, psbC, rpoA, rpoC1, and rps4) and four junction regions were validated using Sanger sequencing. The validated genes amounted to 7,341 bp, and a comparison of the assembled cp genome sequence with the Sanger sequencing results in these regions showed two mismatches in psbB, giving an error rate of 0.027%.
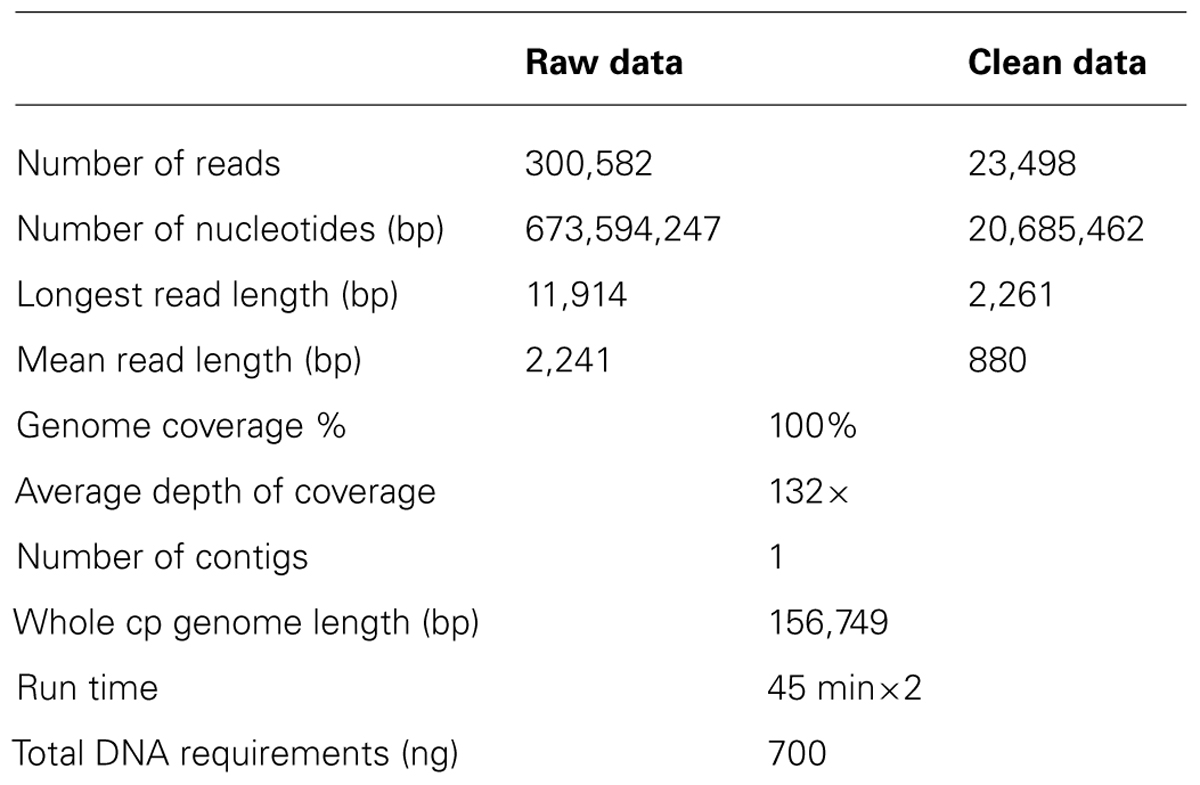
TABLE 1. Summary of the sequencing data for PacBio SMRT.
Genome Features
The complete cp genome of A. barbatum var. puberulum (GenBank acc. No. KC844054) was 156,749 bp in length with the common quadripartite structure found in most land plants (Figure 1), which included a LSC of 87,630 bp and a SSC of 16,941 bp separated by two IRs of 26,089 bp. In accordance with most chloroplast genomes, the nucleotide composition of A. barbatum var. puberulum was biased toward A+T (Sato et al., 1999; Nie et al., 2012; Pan et al., 2012; Yi and Kim, 2012). Overall, the A. barbatum var. puberulum cp genome A+T content was 61.3%, and the LSC and SSC regions (63.9 and 67.3%, respectively) were higher in A+T content than the IR regions (57.0%; Table 2).
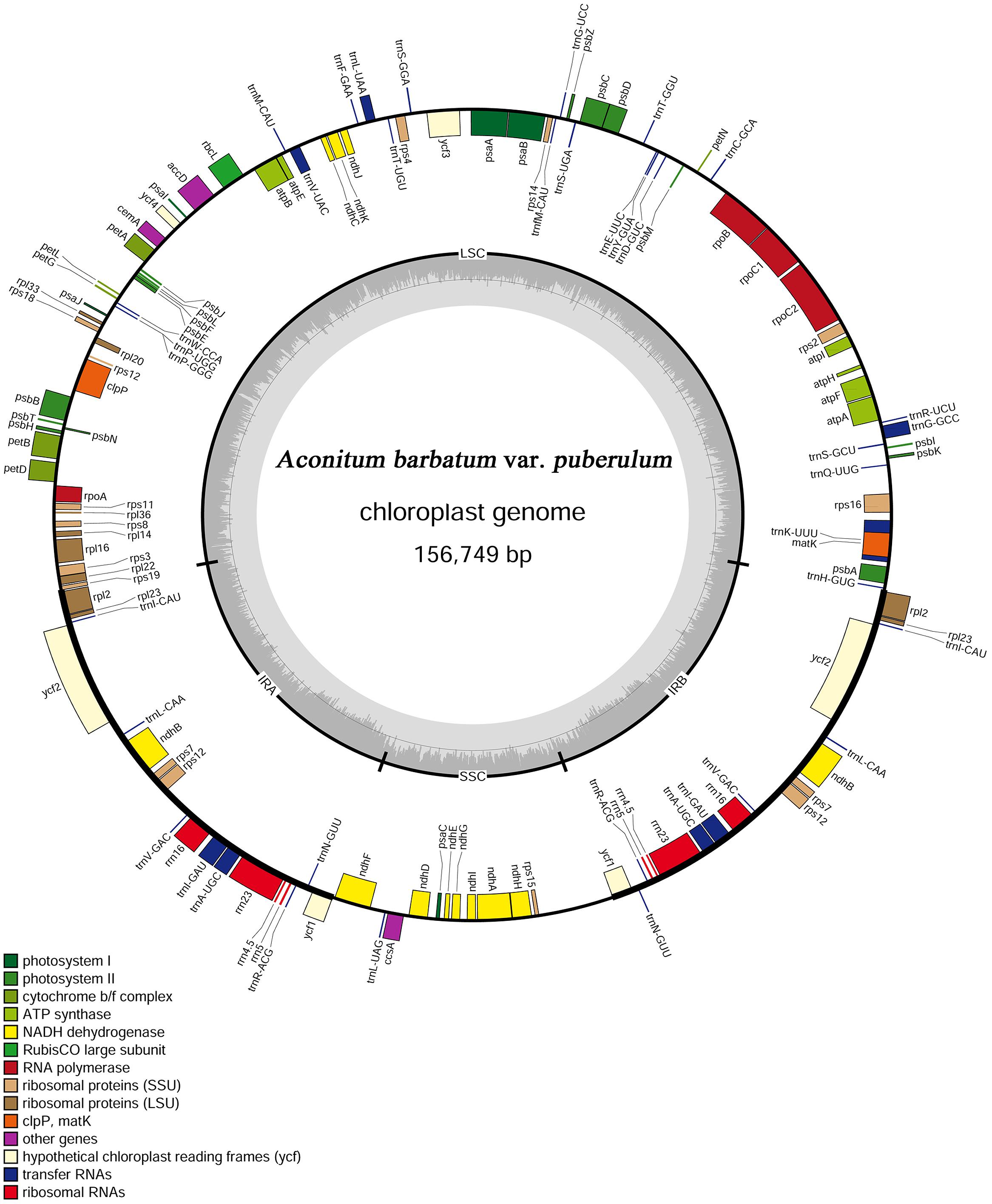
FIGURE 1. Gene map of the Aconitum barbatum var. puberulum chloroplast genome.
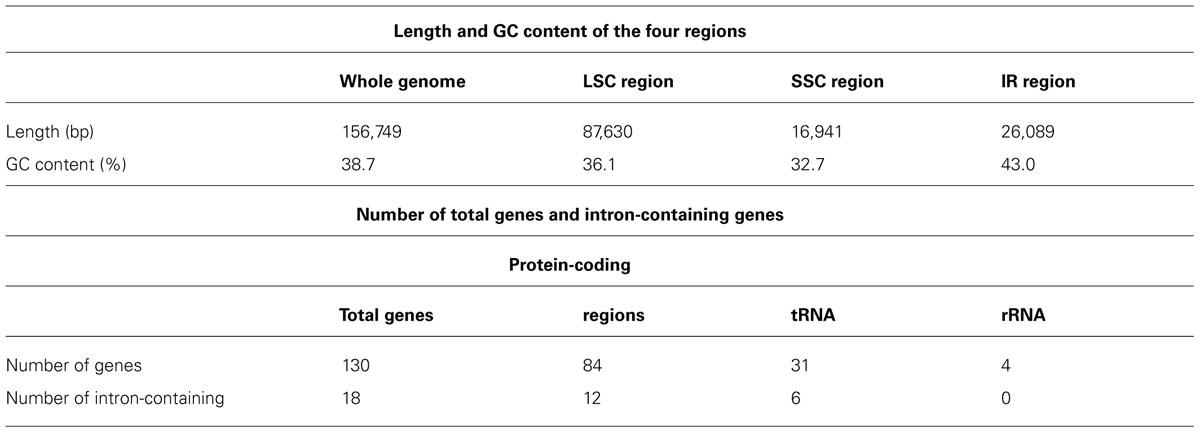
TABLE 2. General information of the Aconitum barbatum var. puberulum chloroplast genome.
The A. barbatum var. puberulum cp genome contained 84 protein-coding regions, including seven genes (rpl2, rpl23, ycf2, ndhB, rps7, rps12, and ycf1) that were duplicated in the IR regions. In total, 31 unique tRNA genes (including seven tRNA genes located in the IR regions, trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG and trnN-GUU) were distributed throughout the cp genome, and four rRNA genes were duplicated in the IR regions. In summary, the cp genome of A. barbatum var. puberulum contained 130 genes, 18 of which were intron-containing genes (Table 3). Three of the intron-containing genes (ycf3, clpP, and rps12) had two introns, and the other 15 had only one intron. The 5′ end of rps12 was located in the LSC region, and the 3′ end was located in the IR region, which caused trans-splicing in rps12. In addition, the sequence of psbD in the cp genome of A. barbatum var. puberulum differed from that in the reference sequence from R. macranthus (GenBank: NC_008796), which was found to be complementary. Moreover, infA was not present in the cp genome of A. barbatum var. puberulum; this gene codes for translation initiation factor 1 and is suspected to be an example of chloroplast-to-nucleus gene transfer (Millen et al., 2001). The codon usage and codon-anticodon recognition pattern of the cp genome are summarized in Table 4. The 31 unique tRNA genes included codons for all 20 amino acids necessary for biosynthesis. Leucine and serine (three of the 31, respectively) were the two most common amino acids represented by the codons of the tRNA in the cp genome.
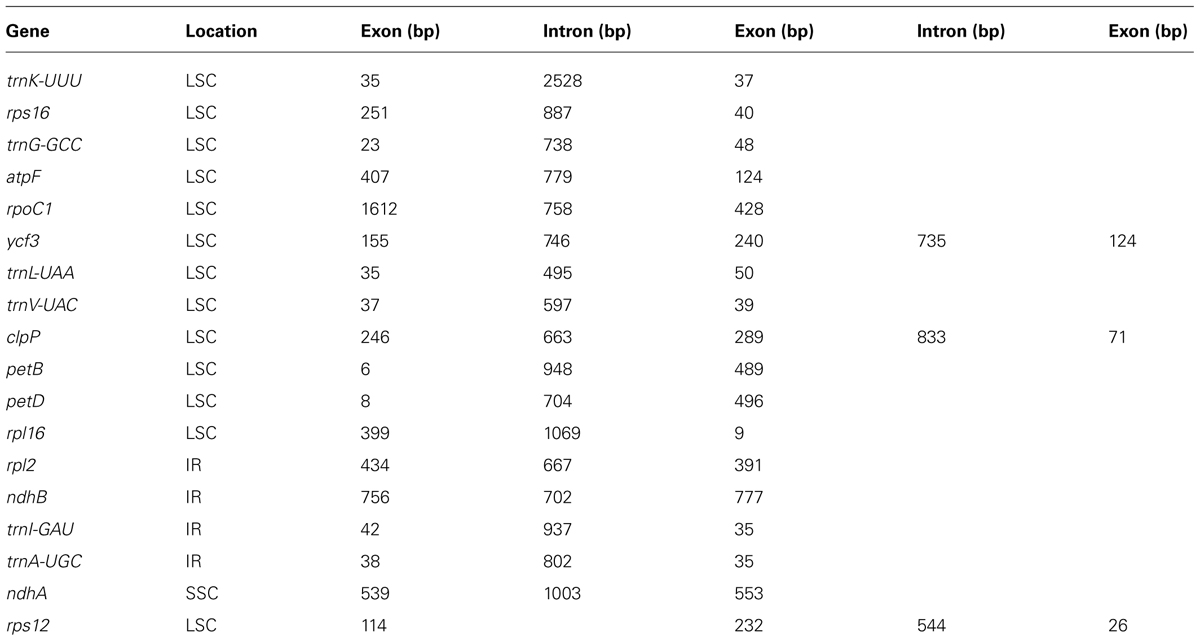
TABLE 3. The genes with introns in the A. barbatum var. puberulum chloroplast genome and the length of the exons and introns.
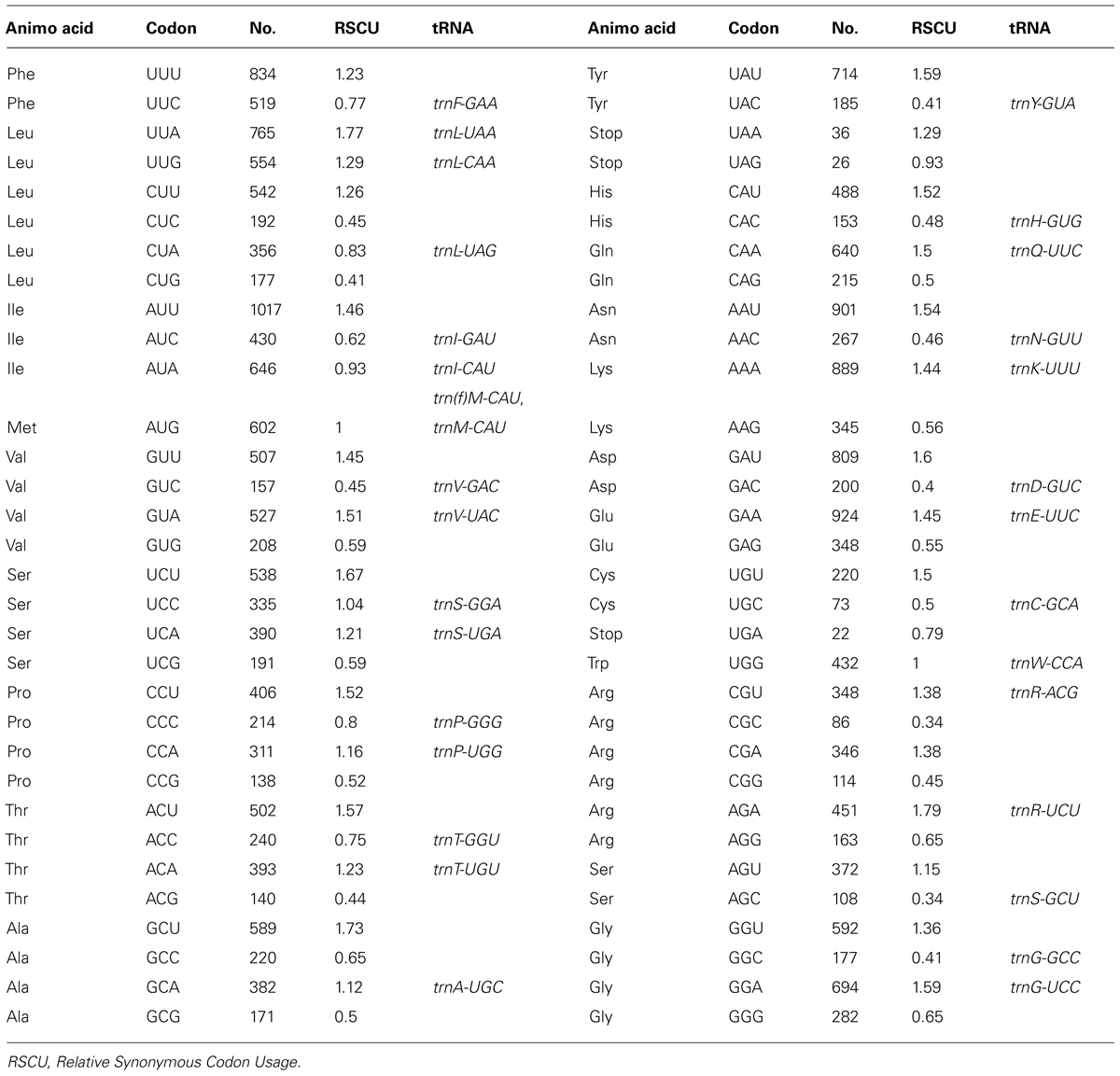
TABLE 4. The codon–anticodon recognition pattern and codon usage for the A. barbatum var. puberulum chloroplast genome.
Repeat Analysis
Four forward, five inverted and eight tandem repeats were identified by REPuter and TRF with a copy size 30 bp or longer (Table 5). Most repeats possessed lengths between 30 and 40 bp, and the longest repeat was 52 bp as a forward repeat located the LSC region (psaA, psaB, CDS). All tandem repeats were found to be repeated twice in the whole cp genome, and six of these were located in intergenic spacer regions, with the left two located within ycf2 (CDS) and rps16 (intron), respectively.
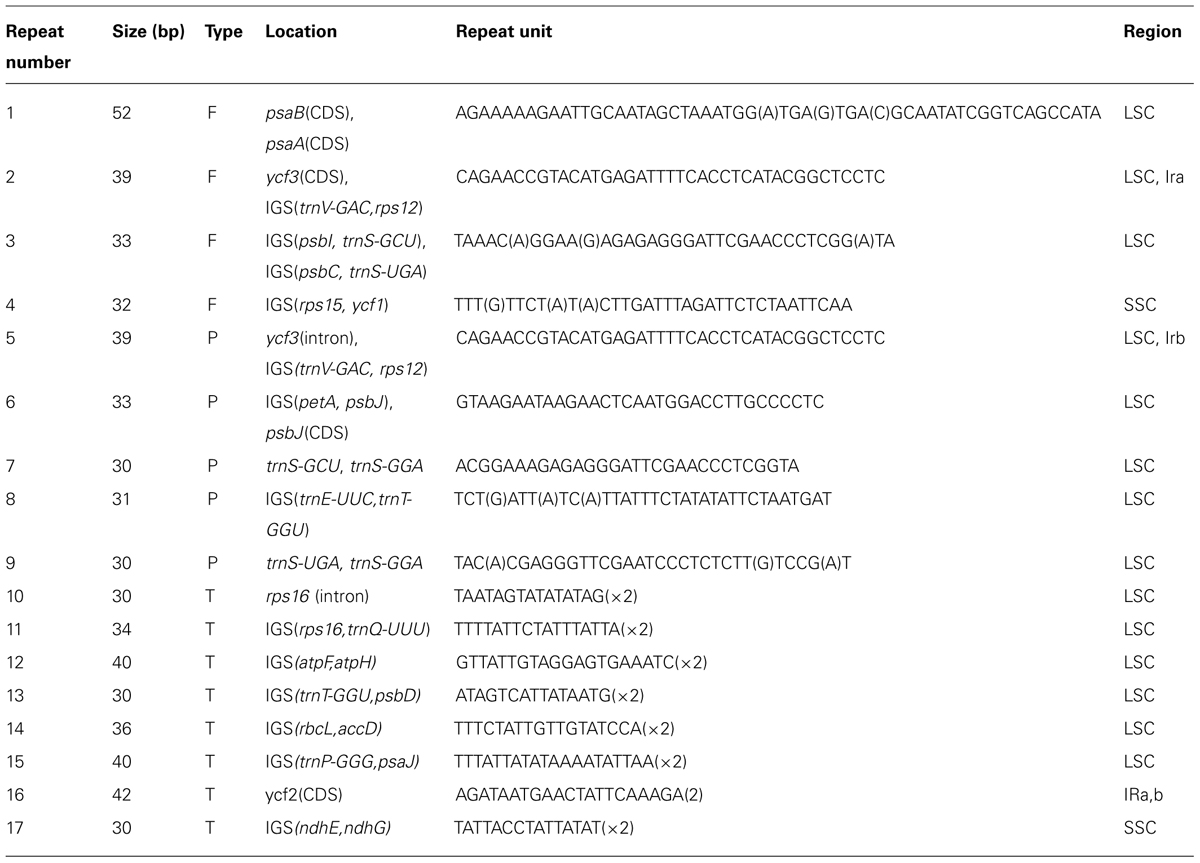
TABLE 5. Repeated sequences in the A. barbatum var. puberulum chloroplast genome.
SSR Analysis
Microsatellites in the chloroplast genome are highly informative about genetic diversity and represent a useful tool for population genetics and evolutionary and ecological studies (Powell et al., 1996; Huang and Sun, 2000; Provan et al., 2001). Thus, the SSRs in the cp genome of A. barbatum var. puberulum were identified for use in future studies. The total number of the mononucleotides (not shorter than 8 bp) was 131, and T represented the highest portion (53.4%) followed by A, C, and G (44.3%, 1.5%, and 0.8%, respectively). The longest poly structure was a 20 T-repeat. In total, 56 dinucleotides were detected throughout the cp genome, and most of them were present as four repetitions (78.6%), e.g., ATATATAT. The combination of AT/TA was the most prevalent dinucleotide (42.9%). Four types of trinucleotide (ATA/ATT/TAT/TTA) were present as multiple A/T nucleotides. Seven tetranucleotides were detected, but no penta- or hexanucleotides (repeated at least three times) were found in the cp genome of A. barbatum var. puberulum. It can be inferred that the SSR loci contribute to the A+T richness of the cp genome. The longest poly-T and poly-A structures (20-nucleotide repeats and 14-nucleotide repeats, respectively) were located in IGS (petA-pabJ), ycf3, and IGS (psaJ-rpl33).
Discussion
Aconitum is Highly Toxic, and Chloroplast Genome is Informative and Referable for Molecular Identification
In recent years, there have been many reports on the improper use of toxic, aconitine-containing plants, which has led to deaths (Poon et al., 2006; Chen et al., 2012). Therefore, Aconitum identification is important. With reductions in sequencing costs, cp genomes could be used as super-barcodes in the near future. After sequencing and analyzing the cp genomes of 37 different Pinus species, Parks et al. (2009) concluded that cp genomes could be used to improve phylogenetic resolution at lower taxonomic levels and could be thought of as species-level DNA barcodes. Li et al. (2014b) also suggest that complete cp genomes have tremendous potential for the identification of closely related species. Aconitum consists of approximately 300 species and its taxonomy has been complex due to the close relationships among different species (Xiao et al., 2005; Jabbour and Renner, 2012). Cp genome regions such as psbA-trnH have been applied, but it cannot be used to identify all the species of Aconitum (He et al., 2010). Hence, whole cp genomes are thought to have the potential in Aconitum identification studies. In our study, the successful use of a third-generation sequencing platform provides a new, rapid way to sequence the Aconitum cp genomes, which could help to lay the foundation for the molecular identification of Aconitum based on its cp genomes.
CCS Reads Proved to Be Reliable via Sanger Sequencing Validation
In this study, we demonstrated the feasibility of sequencing a cp genome using the PacBio SMRT third-generation sequencing platform; use of this platform has been shown to be a rapid approach for sequencing small genomes, such as microbial and plasmid genomes (Chin et al., 2013). We evaluated the error-rate of the PacBio RS data by comparing its results with those obtained by Sanger sequencing. The CCS reads generated in our study had an error rate of approximately 0.027%, which was lower than the rate reported by Cronn et al. (2008) for Illumina sequencing-by-synthesis technology (0.056%). However, some questions remain regarding the error rate of the PacBio system. The observed raw error rate was 12.86%, which was much higher than that of other platforms, such as Illumina MiSeq and Ion Torrent PGM (Quail et al., 2012). To improve this situation, CCS is thought to be an effective approach. CCS is one of the PacBio RS sequencing protocols that performs multiple passes on each molecule that is sequenced. After the application of the necessary QC filters, the result is an error-corrected consensus read with a higher intra-molecular accuracy. This approach results in higher per-base quality and reduced concerns about suspicious results. By generating multiple reads from the same molecule and eliminating errors resulting from single reads, the PacBio system’s inherent error rate can be bypassed. For that reason, in this study, we used the CCS protocol to sequence the A. barbatum var. puberulum cp genome and obtain high-quality reads. The data presented here show that SMRT sequencing using the CCS strategy is a powerful tool for sequencing cp genomes. In addition, in some extreme situations, we suggest completing genome assembly by combining CCS reads with regular long reads. We believe that this strategy would be an effective way to solve the problems associated with assembling large genomes or genomes that contain special structures.
The Long Reads Derived From PacBio Improve Genome Assembly
The long read lengths undoubtedly provide a number of benefits in genome sequencing and assembly. The most obvious benefit is for de novo assemblies. Previous studies have shown that, compared with Illumina data, chloroplast genome assembly using the PacBio RS sequencer generated longer contigs and fewer unresolved gaps (Ferrarini et al., 2013). In this study, we constructed the draft sequence in a step-by-step manner by extending two seed reads on both the 5′ and 3′ ends until they overlapped at the two IR regions. For all CCS sub-reads, the top BLASTn hit for the seed sequence was selected and used to extend the read. The longer reads (an average of 880 bp) made our assembly and analysis more effective. We encountered no problems mapping the seed sequence reads to the repeat regions of the A. barbatum var. puberulum cp genome, which are listed in Table 5. Even without any other biological or phytological information about the target species, it took less than half an hour to finish the genome assembly step. This strategy is clearly a highly effective and accurate method for obtaining plant cp genomes. In addition, one of the features of the cp genome, the two long IR (regions), is also a valuable target for evaluating the PacBio system. As mentioned above, the comparatively longer CCS reads provided more conveniences on dealing with those special structures.
Eliminating the PCR Amplification Step Saves Time
The SMRT method does not require PCR amplification, which reduces the time required for sequencing. In our study, the sequencing reaction time was 90 min, which streamlined the sequencing process by reducing the overall time in the lab. In addition, eliminating the PCR amplification step alleviated the sequencing bias. In some extreme situations, e.g., AT-rich, GC-rich, and repeat-rich regions, the results are unsatisfactory due to the loss of DNA during amplification (Bashir et al., 2012). The sequencing of unamplified molecules will improve genome assembly and allow the detection unique and informative structures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (No. 81473303) and the Major Scientific and Technological Special Project for “Significant New Drugs Creation” (No. 2014ZX09304307001). We thank our colleagues who helped with sample collection, identification, laboratory work and manuscript preparation, including Hui Yao, Yingjie Zhu, Lili Wang, and Baosheng Liao.
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W.,et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bashir, A., Klammer, A. A., Robins, W. P., Chin, C.-S., Webster, D., Paxinos, E.,et al. (2012). A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 30, 701–707. doi: 10.1038/nbt.2288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, S. P., Ng, S. W., Poon, W. T., Lai, C. K., Ngan, T. M., Tse, M. L.,et al. (2012). Aconite poisoning over 5 years: a case series in Hong Kong and lessons towards herbal safety. Drug Saf. 35, 575–587. doi: 10.2165/11597470-000000000-00000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, S., Pang, X., Song, J., Shi, L., Yao, H., Han, J.,et al. (2014). A renaissance in herbal medicine identification: from morphology to DNA. Biotechnol. Adv. 32, 1237–1244. doi: 10.1016/j.biotechadv.2014.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C.,et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chin, C.-S., Sorenson, J., Harris, J. B., Robins, W. P., Charles, R. C., Jean-Charles, R. R.,et al. (2011). The origin of the Haitian cholera outbreak strain. N. Engl. J. Med. 364, 33–42. doi: 10.1056/NEJMoa1012928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cronn, R., Liston, A., Parks, M., Gernandt, D. S., Shen, R., and Mockler, T. (2008). Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 36, e122–e122. doi: 10.1093/nar/gkn502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cui, L., Veeraraghavan, N., Richter, A., Wall, K., Jansen, R. K., Leebens-Mack, J.,et al. (2006). ChloroplastDB: the chloroplast genome database. Nucleic Acids Res. 34, D692–D696. doi: 10.1093/nar/gkj055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daniell, H., Datta, R., Varma, S., Gray, S., and Lee, S.-B. (1998). Containment of herbicide resistance through genetic engineering of the chloroplast genome. Nat. Biotechnol. 16, 345–348. doi: 10.1038/nbt0498-345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, W., Xu, C., Cheng, T., Lin, K., and Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G.,et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferrarini, M., Moretto, M., Ward, J. A., Surbanovski, N., Stevanovic, V., Giongo, L.,et al. (2013). An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genomics 14:670. doi: 10.1186/1471-2164-14-670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilles, A., Meglécz, E., Pech, N., Ferreira, S., Malausa, T., and Martin, J.-F. (2011). Accuracy and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genomics 12:245. doi: 10.1186/1471-2164-12-245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, J., Wong, K.-L., Shaw, P.-C., Wang, H., and Li, D.-Z. (2010). Identification of the medicinal plants in Aconitum L. by DNA barcoding technique. Planta Med. 76, 1622–1628. doi: 10.1055/s-0029-1240967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hiratsuka, J., Shimada, H., Whittier, R., Ishibashi, T., Sakamoto, M., Mori, M.,et al. (1989). The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 217, 185–194. doi: 10.1007/BF02464880
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, J., and Sun, M. (2000). Genetic diversity and relationships of sweet potato and its wild relatives in Ipomoea series batatas (Convolvulaceae) as revealed by inter-simple sequence repeat (ISSR) and restriction analysis of chloroplast DNA. Theor. Appl. Genet. 100, 1050–1060. doi: 10.1007/s001220051386
Huang, X., and Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jabbour, F., and Renner, S. S. (2012). A phylogeny of Delphinieae (Ranunculaceae) shows that Aconitum is nested within Delphinium and that Late Miocene transitions to long life cycles in the Himalayas and Southwest China coincide with bursts in diversification. Mol. Phylogenet. Evol. 62, 928–942. doi: 10.1016/j.ympev.2011.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johansson, J. T. (1995). A revised chloroplast DNA phylogeny of the Ranunculaceae, in systematics and evolution of the Ranunculiflorae. Springer 9, 253–261.
Jordan, W. C., Courtney, M. W., and Neigel, J. E. (1996). Low levels of intraspecific genetic variation at a rapidly evolving chloroplast DNA locus in North American duckweeds (Lemnaceae). Am. J. Bot. 83, 430–439. doi: 10.2307/2446212
Kircher, M., Heyn, P., and Kelso, J. (2011). Addressing challenges in the production and analysis of illumina sequencing data. BMC Genomics 12:382. doi: 10.1186/1471-2164-12-382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Q., Li, Y., Song, J., Xu, H., Xu, J., Zhu, Y.,et al. (2014a). High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 204, 1041–1049. doi: 10.1111/nph.12966
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, X., Hu, Z., Lin, X., Li, Q., Gao, H., Luo, G.,et al. (2012). High-throughput pyrosequencing of the complete chloroplast genome of Magnolia officinalis and its application in species identification. Acta Pharm. Sin. 47, 124–130.
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., and Chen, S. (2014b). Plant DNA barcoding: from gene to genome. Biol. Rev. Camb. Philos. Soc. doi: 10.1111/brv.12104 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mardis, E. R. (2008a). The impact of next-generation sequencing technology on genetics. Trends Genet. 24, 133–141. doi: 10.1016/j.tig.2007.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mardis, E. R. (2008b). Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402. doi: 10.1146/annurev.genom.9.081307.164359
McPherson, H., Van Der Merwe, M., Delaney, S. K., Edwards, M. A., Henry, R. J., Mcintosh, E.,et al. (2013). Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 13:8. doi: 10.1186/1472-6785-13-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Metzker, M. L. (2009). Sequencing technologies—the next generation. Nat. Rev. Genet. 11, 31–46. doi: 10.1038/nrg2626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L.,et al. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.1105/tpc.13.3.645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nie, X., Lv, S., Zhang, Y., Du, X., Wang, L., Biradar, S. S.,et al. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 7:e36869. doi: 10.1371/journal.pone.0036869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pan, I.-C., Liao, D.-C., Wu, F.-H., Daniell, H., Singh, N. D., Chang, C.,et al. (2012). Complete chloroplast genome sequence of an orchid model plant candidate: Erycina pusilla apply in tropical Oncidium breeding. PLoS ONE 7:e34738. doi: 10.1371/journal.pone.0034738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfannschmidt, T., Nilsson, A., and Allen, J. F. (1999). Photosynthetic control of chloroplast gene expression. Nature 397, 625–628. doi: 10.1038/17624
Poon, W. T., Lai, C. K., Ching, C. K., Tse, K. Y., So, Y. C., Chan, Y. C.,et al. (2006). Aconite poisoning in camouflage. Hong Kong Med. J. 12, 456–459.
Powell, W., Machray, G. C., and Provan, J. (1996). Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1, 215–222. doi: 10.1016/1360-1385(96)86898-1
Provan, J., Powell, W., and Hollingsworth, P. M. (2001). Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol. Evol. (Amst.) 16, 142–147. doi: 10.1016/S0169-5347(00)02097-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X.,et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8:e57607. doi: 10.1371/journal.pone.0057607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R.,et al. (2012). A tale of three next generation sequencing platforms: comparison of ion torrent, pacific biosciences and illumina miSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13–341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rasko, D. A., Webster, D. R., Sahl, J. W., Bashir, A., Boisen, N., Scheutz, F.,et al. (2011). Origins of the E. coli strain causing an outbreak of hemolytic–uremic syndrome in Germany. N. Engl. J. Med. 365, 709–717. doi: 10.1056/NEJMoa1106920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roberts, R. J., Carneiro, M. O., and Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biol. 14, 405. doi: 10.1186/gb-2013-14-6-405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rusk, N. (2009). Cheap third-generation sequencing. Nat. Methods 6, 244–244. doi: 10.1038/nmeth0409-244a
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., and Tabata, S. (1999). Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 6, 283–290. doi: 10.1093/dnares/6.5.283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schadt, E. E., Turner, S., and Kasarskis, A. (2010). A window into third-generation sequencing. Hum. Mol. Genet. 19, R227–R240. doi: 10.1093/hmg/ddq416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T.,et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uthaipaisanwong, P., Chanprasert, J., Shearman, J., Sangsrakru, D., Yoocha, T., Jomchai, N.,et al. (2012). Characterization of the chloroplast genome sequence of oil palm (Elaeis guineensis Jacq.). Gene 500, 172–180. doi: 10.1016/j.gene.2012.03.061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, J., Liu, B., Cheng, F., Ramchiary, N., Choi, S. R., Lim, Y. P.,et al. (2012). Sequencing of chloroplast genome using whole cellular DNA and Solexa sequencing technology. Front. Plant Sci. 3:243. doi: 10.3389/fpls.2012.00243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiao, P., Wang, F., Gao, F., Yan, L., Chen, D., and Liu, Y. (2005). A pharmacophylogenetic study of Aconitum L.(Ranunculaceae) from China. Acta Phytotaxon. Sin. 44, 1–46. doi: 10.1360/aps050046
Yi, D.-K., and Kim, K.-J. (2012). Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 7:e35872. doi: 10.1371/journal.pone.0035872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, T., Fang, Y., Wang, X., Deng, X., Zhang, X., Hu, S.,et al. (2012). The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica: insights into the evolution of plant organellar genomes. PLoS ONE 7:e30531. doi: 10.1371/journal.pone.0030531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: chloroplast genome, Aconitum, circular consensus sequencing, PacBio RS, the third generation sequencing
Citation: Chen X, Li Q, Li Y, Qian J and Han J (2015) Chloroplast genome of Aconitum barbatum var. puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Front. Plant Sci. 6:42. doi: 10.3389/fpls.2015.00042
Received: 22 October 2014; Accepted: 15 January 2015;
Published online: 06 February 2015.
Edited by:
Jan Dvorak, University of California, Davis, USAReviewed by:
Xiaowu Wang, Institute of Vegetables and Flowers – Chinese Academy of Agricultural Sciences, ChinaJan Dvorak, University of California, Davis, USA
Copyright © 2015 Chen, Li, Li, Qian and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianping Han, Center for Computational Biology and Bioinformatics, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 151 Malianwa North Road, Haidian, Beijing 100193, China e-mail:anBoYW5AaW1wbGFkLmFjLmNu