 Bei Li
Bei Li Junwei Zhang
Junwei Zhang Wei Zhang
Wei Zhang- Key Laboratory of Horticultural Plant Biology, Ministry of Education, College of Horticulture and Forestry Sciences, Huazhong Agricultural University, Wuhan, China
Petunias are important ornamentals with the capacity for cold acclimation. So far, there is limited information concerning gene regulation and signaling pathways associated with the cold stress response in petunias. A custom-designed petunia microarray representing 24816 genes was used to perform transcriptome profiling in petunia seedlings subjected to cold at 2°C for 0.5 h, 2 h, 24 h, and 5 d. A total of 2071 transcripts displayed differential expression patterns under cold stress, of which 1149 were up-regulated and 922 were down-regulated. Gene ontology enrichment analysis demarcated related biological processes, suggesting a possible link between flavonoid metabolism and plant adaptation to low temperatures. Many novel stress-responsive regulators were revealed, suggesting that diverse regulatory pathways may exist in petunias in addition to the well-characterized CBF pathway. The expression changes of selected genes under cold and other abiotic stress conditions were confirmed by real-time RT-PCR. Furthermore, weighted gene co-expression network analysis divided the petunia genes on the array into 65 modules that showed high co-expression and identified stress-specific hub genes with high connectivity. Our identification of these transcriptional responses and groups of differentially expressed regulators will facilitate the functional dissection of the molecular mechanism in petunias responding to environment stresses and extend our ability to improve cold tolerance in plants.
Introduction
The petunia (Petunia hybrida) is an important ornamental that is widely used in landscaping around the world. It is also an excellent model system for studying the evolutionary development of floral organs and the biosynthesis of flavonoids. Nevertheless, because the plant originated from South America, petunias are native to warm habitats. Late-spring freezes can cause substantial damage to petunias as they are less tolerant of cold conditions and lack rapid acclimation response mechanisms as compared with species tolerant of ice in their tissues, such as the spinach and Brassicas (Pennycooke et al., 2003). Thus, frozen conditions and low temperatures are significant limiting factors for its productivity, growing period and geographical location, and there is a demand for novel petunia cultivars with improved cold tolerance.
Cold stress, including chilling (<20°C) and/or freezing (<0°C) temperatures, adversely affects plant growth and development and causes a wide range of physiological and biochemical responses in plants. These responses include transient increases in hormone levels (e.g., ABA) (Chandler and Robertson, 1994), changes in the compositions of membrane lipid (Lynch and Steponkus, 1987), the accumulation of compatible osmolytes, such as soluble sugars, betaine, and praline (Koster and Lynch, 1992; Nomura et al., 1995; Doerffling et al., 1997), and increases in the level of antioxidants (Pennycooke et al., 2005). However, tolerance to freezing can be enhanced when plants are prior exposed to a period of low, but non-freezing temperatures, which is called cold acclimation (CA) (Weiser, 1970). CA activates the expression of a diverse array of plant genes (Thomashow, 2001). In the past decades, numerous cold-responsive genes have been identified and their functions have been characterized using gene transfer, resulting in increased plant stress tolerance (Sanghera et al., 2011). To date, the mechanisms of CA have been extensively investigated in the model plant Arabidopsis thaliana (Gilmour et al., 1998), as well as in other crucial crop species such as barley and maize (Stanca et al., 1996; Fernandes et al., 2008). The most deeply studied mechanism involves a class of ethylene response factors (ERFs), that is, CBFs (C-repeat binding factors, also called DREB1s or dehydration-responsive element-binding protein 1s), which can bind to cis-elements in the promoters of their downstream target genes to execute a highly coordinated transcriptional response to low temperature signals (Chinnusamy et al., 2007). In contrast, less information is available for petunias.
With the advancement of molecular biology technologies and “omics” tools, microarrays have been devised as a powerful strategy for the analysis of transcript abundance in plants. In recent years, transcriptional profiling of diverse plant species under defined stimuli such as hormone or abiotic stresses using microarray analysis has been particularly informative. For example, the expression patterns of genes were identified in rice and Arabidopsis under abscisic acid or cold, drought and high-salinity stresses (Seki et al., 2002; Rabbani et al., 2003). The transcriptomic identification of candidate genes involved in response to salt stress in cotton has also been reported (Rodriguez-Uribe et al., 2011). Additionally, transcriptome profiling of cassava treated with low temperatures highlighted the dynamic responses of tropical plants to cold stress (An et al., 2012). To date, several studies have investigated petunias using genomic tools to develop novel technologies and knowledge. For example, a cDNA library was constructed from petunias for the isolation of genes responsible for benzylbenzoate formation (Boatright et al., 2004). The capacity of a targeted transcriptomics approach to disambiguate regulation of a particular biosynthetic process was illustrated by the identification of the MYB transcription factor ODORANT1, a key regulator of floral scent biosynthesis, using a dedicated petunia microarray (Verdonk et al., 2005). Analysis based on a petunia small RNA library identified unusual tissue-specific expression patterns of conserved miRNAs and a 24-mer RNA (Tedder et al., 2009). Moreover, the petunia transcriptome has recently been employed to gain insight into potential shifts in symbiotic signaling, metabolic activity, and the defense status of plants exposed to high P levels (Breuillin et al., 2010). Nevertheless, genome-wide expression profiling data for petunias under cold stress is still deficient, thereby limiting our capacity to decipher the molecular mechanisms associated with different stress responses. To address this need for a popular ornamental, comprehensive transcriptomic data describing the response of petunias to cold stress treatment were obtained and analyzed using a custom Roche Nimblegen petunia microarray. The results revealed a set of candidate regulators. Our study provides a first glimpse into the understanding of the gene regulation pathways involved in the adaptation of this plant species to cold stress, as well as the potential applications of these data in systems biology and molecular physiology.
Materials and Methods
Plant Materials used for Microarray Hybridization
More than 20 inbred lines were established by successive generations of selfing. To compare the differences of cold resistance among those inbred lines, they were planted in open field in winter under natural low temperatures as well as in artificial climate chambers. One line (coded H) with the best cold tolerance was screened out.
In vitro seedlings of line H were planted in plastic pots at 25°C under a 14 h photoperiod with a light intensity of 2000–2500 lux provided by cool-white fluorescent lights for 1 month in the tissue culture room. Plants at 4–5 pairs of true leaf stage with a uniform growth status were transferred to a chamber for cold treatment at 2°C under weak light (cool-white fluorescent light at approximately 500–1000 lux). Whole plants were harvested after 0.5 h, 2 h, 24 h, and 5 d of treatment, then frozen in liquid nitrogen and held at −80°C prior to transportation to the Bioassay Laboratory of CapitalBio Corporation (Beijing, China) for RNA extraction. Untreated whole plants were harvested as controls (0 h). More than three plants were harvested and pooled for each time point, and the collection was repeated three times as biological replicates.
Microarray Preparation, Hybridization, and Data Extraction
The custom-designed four-plex microarray with 72,000 features was constructed based on the EST sequences generated in Breuillin's et al. (2010) study together with all of the P. hybrida and P. axillaris sequences available from Genbank, TIGR, and the Solanaceae genomics network SGN. The compiled sequence set was ultimately composed of 24,816 non-redundant unique sequences (unigenes). Total RNA was extracted from both the control and cold-treated samples with Trizol Reagent (Invitrogen, http://www.invitrogen.com). The RNA quality was checked on a 1.2% agarose gel using an RNase-free electrophoresis system. RNA labeling and hybridization were conducted by the Bioassay Laboratory of CapitalBio Corporation (Beijing, China) following the manufacturer's instructions. Total RNA was used to generate Cy5/Cy3-labeled DNA using a cRNA amplification labeling kit (CapitalBio). Briefly, the 1st–strand cDNA was reverse transcribed with the T7 Oligo (dT) primer, then the 2nd–strand was synthesized and purified as the template for cRNA transcription with the T7 Enzyme Mix. Two micrograms of cRNA was reverse transcribed to cDNA with random primers, and the cDNA was used as the template for Cy5/Cy3-labeled DNA synthesis with Cy5/Cy3-dCTP. The Cy5/Cy3-labeled DNA in 80 μL hybridization solution (3 × SSC, 0.2% SDS, 5 × Denhart's, and 25% formamide) was hybridized to the 4 × 72K Petunia Array (Roche Nimblegen). After hybridization at 42°C overnight, the samples were washed with 2 × SSC (plus 0.2% SDS) and 0.2 × SSC for 5 min each. The GeneChips were used for scanning with the LuxScan 10 KA (CapitalBio). Three biological replicates were included in this study.
Data Collection and Identification of Differentially Expressed Genes
The probe intensity files resulting from RNA hybridization were first loaded into R. Then, background correction, quantile normalization and summarization were performed using the Robust Multi-array Average (RMA) method included in the Bioconductor Affy package (Bolstad et al., 2003; Irizarry et al., 2003; Gautier et al., 2004; Gentleman et al., 2004) based on the homemade chip description library. The correlation coefficient of paired samples was calculated to test the repeatability between samples. Then, euclidean distances were calculated, and the clustering tree was constructed using the hierarchical cluster method of “complete linkage clustering.”
Differentially expressed genes were calculated using the Bioconductor RankProd package function. RP. Rank product is a non-parametric method returning up- and down-regulated genes, their fold changes (FC), p-values and the percentages of false predictions (PFP). This method was shown to perform better than other methods, including significance analysis of microarrays (SAM), Fisher's Inverse χ2 test and t-based hierarchical modeling (Hong and Breitling, 2008). A gene was considered to be up- or down-regulated if the p-value of the RankProd analysis was <0.05 and the fold change of average expression was >2 (log-fold change >1 for up-regulated genes and log-fold change < −1 for down-regulated genes).
Functional Enrichment Analysis
All probe set sequences were annotated by Blast2GO by searching against the nr database using Blastx with parameter E = 10e-3. Then, all probe sets were annotated with three principal GO categories: biological process, molecular function and cellular component. We used the terms in the biological process category for GO analysis. Finally, GO enrichment analysis was performed using a weighted method in combination with Fisher's exact test with a P-value of 0.05 provided by the Bioconductor topGO package (Alexa et al., 2006).
RNA Preparation and Real-Time RT-PCR
To validate the expression patterns of the differentially expressed genes identified from the microarray analysis, real-time RT-PCR was performed using plant materials from in vitro shoot cultures. In vitro seedlings with 4–5 pairs of true leaves were transferred into plastic jars containing 100 mL of MS solution (1 × Murashige and Skoog basal salts and 3% sucrose, pH 6.0) in a chamber at 25°C, 70% relative humidity, and a light intensity of 2000–2500 lux on a 14 h light/10 h dark cycle for 5 days. Then, the solution was replaced with fresh MS medium (pH 6.0) supplemented with 200 mM NaCl, 400 mM mannitol, 100 μM ABA, or 100 μM MeJA. For cold stress treatment, the seedlings were directly transferred into a 2°C chamber. For dehydration stress treatment, the seedlings were transferred and dehydrated onto dried filter paper in covered plastic dishes. All abiotic stress samplings were harvested from whole seedlings and frozen in liquid nitrogen after 3, 6, and 12 h and stored at −80°C for abiotic stress analysis. Plants in the same tissue culture system with no stress treatment were collected and used as controls. All of the stress treatments included three biological and temporal replicates.
The expression of cold-responsive genes was validated by real-time RT-PCR using RNA samples extracted with the EASYspin Plant RNA Mini kit according to the manufacturer's protocol (Aidlab, Beijing, China). The RNA concentration and integrity were estimated with the NanoDrop 2000 UV-Vis Spectrophotometer (Thermo scientific, USA) and denaturing agarose gel electrophoresis. Reverse transcription was performed using the Prime Script® RT reagent kit with gDNA Eraser (Perfect Real Time) (TaKaRa, Dalian, China) according to the manufacturer's protocol. Gene-specific primers (Table S1) were designed with ABI primer express 3.0 software (Applied Biosystems, USA). The product size from each amplicon ranged from 105 to 215 base pairs. The real-time RT-PCR was performed with SYBR® Premix Ex Taq™ (Perfect Real Time) (TaKaRa, Dalian, China) as described in the users' instructions for the ABI 7500 Fast Real-Time PCR System (Applied Biosystems, USA). The PCR reaction mixture contained 1 μl cDNA template, 5 μl SYBR®Premix Ex Taq™ (2x), 0.2 μl forward and reverse primer (10 μmol/μl each), 0.2 μl ROX Reference Dye II (50x), and water up to 10 μl. The amplification conditions were as follows: an initial incubation at 95°C for 30 s, followed by 40 cycles of 95°C for 3 s, 60°C for 30 s, and 72°C for 20 s. A melting curve (60°–95°C with a heating rate of 0.05°C·s−1 and a continuous fluorescence measurement) was performed for each qRT-PCR reaction to determine PCR performances. The glyceraldehyde-3-phosphatedehydrogenase gene (GAPDH) was used as the internal control. All of the samples were measured in triplicate, and the experiments were performed on three biological replicates. The comparative Ct method was adopted to calculate the relative gene expression level across the samples. The relative expression level of each gene in one sample (ΔCt) was calculated as follows: Ct target gene – Ct GAPDH. The relative expression of each gene in two diverse samples (ΔΔCt) was calculated as follows: ΔCt (sample 1) – ΔCt (sample 2).
Co-Expression Network Analysis
To identify co-expression of the petunia genes on the array, log-transformed gene expression values for each condition were used in a meta-analysis to perform Weighted Gene Co-expression Network Analysis (WGCNA) (Langfelder and Horvath, 2008). Briefly, WGCNA can be used for finding clusters (modules) of highly correlated genes, for summarizing such clusters using the module eigengene or an intramodular hub gene, for calculating module membership measures, and for relating modules to one another and to external sample traits (using eigengene network methodology). The blockwiseModules function of the WGCNA package in R was used to generate the modules with a power of six. The correlation coefficient relationship of the module eigengene was calculated, then euclidean distances were calculated and the clustering tree was constructed using the hierarchical cluster method of “complete linkage clustering” included with the R package.
Results
To explore the transcriptomic changes in petunias in response to cold stress, a custom four-plex microarray with 72,000 features generated by Roche NimbleGen was employed in the current study. The detailed protocol for designing the petunia microarray has been described by Breuillin et al. (2010). Our microarray data have been deposited to Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64963) and the accession number is GSE64963. The results of hierarchical clustering indicated the clear separation of the samples from various time points (Figure 1A), which suggested that the whole experiment from sample collection to data extraction was reliable and reproducible.
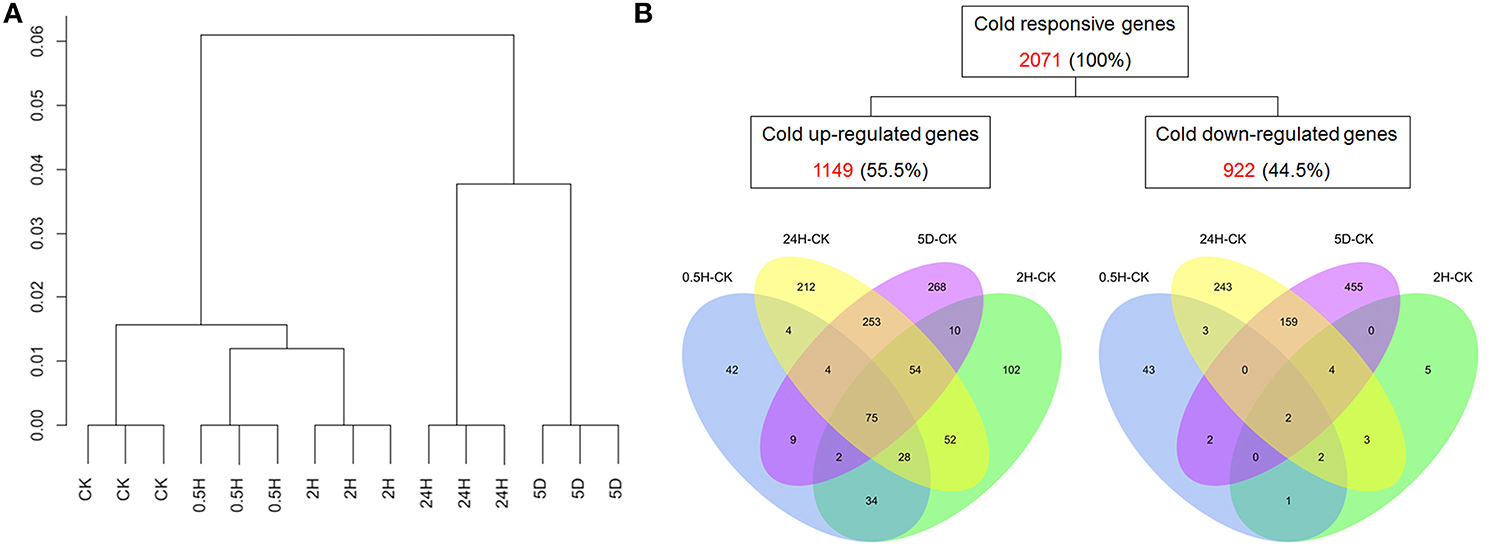
Figure 1. Expression profiling of cold-regulated genes in petunia seedlings. (A) Hierarchical cluster analysis. (B) Venn diagrams showing cold-regulated genes across four comparisons (0.5 h/0 h, 2 h/0 h, 24 h/0 h, and 5 d/0 h). The red numbers are the total numbers of differentially expressed genes (DEGs); the percentages in parentheses were calculated as the ratio of regulated genes to the total number of cold-regulated genes (2071).
Transcriptomic Responses of Petunia Seedlings to Cold Treatment
Two filtering criteria were used in our data analysis to define differentially expressed genes (DEGs): a two-fold or greater change in transcript levels between any two time points and a P-value <0.05. To analyze the differences and similarities among the cold-responsive transcriptomes, a hierarchical clustering approach was utilized to depict the transcripts of all of the differentially expressed probes of the three replicates at 0 h, 0.5 h, 2 h, 24 h, and 5 d. These results demonstrated distinct differences in gene expression profiles between the treatments of 5 d and 0 h or 24 h and 0 h that were in contrast to the relatively highly similar expression profiles between 2 h and 0 h or 0.5 h and 0 h (Figure 1A). Among the DEGs, 198, 357, 682, and 675 genes were considered to be up-regulated and 53, 17, 416, and 622 genes were considered to be down-regulated based on the analysis of 0.5 h/0 h, 2 h/0 h, 24 h/0 h, and 5 d/0 h, respectively (Table S2). Notably, compared with only 251 and 374 genes that showed differential expression at 0.5 h/0 h and 2 h/0 h, 1098, and 1297 genes were found to be differentially expressed at 24 h/0 h and 5 d/0 h, suggesting that prolonged stress treatment triggered the expression of more stress-related genes. To identify both unique and common genes showing differential expression patterns at all time points compared with 0 h, the numbers were calculated and presented using a Venn diagram (Figure 1B). The results showed that 75 DEGs were commonly induced in all of the comparisons for each time point compared with 0 h, which demonstrated a strong linkage among the four stressed comparison points and a progressive biological process. In contrast, 42, 102, 212, and 268 DEGs exhibiting up-regulated expression were unique at 0.5 h/0 h, 2 h/0 h, 24 h/0 h, and 5 d/0 h; thus, these genes may play special roles at those time points. Only 2 suppressed DEGs were found to be common to all of the comparisons for each time point compared with 0 h, whereas approximately 81% (43/53), 29% (5/17), 58% (243/416), and 73% (455/622) of DEGs exhibiting down-regulated expression were unique at 0.5 h/0 h, 2 h/0 h, 24 h/0 h, and 5 d/0 h, respectively.
In total, 2071 DEGs, including 1149 cold-inducible and 922 cold-repressed genes, were identified using our array. These genes represent approximately 8.3% (2071/24,816) of the total expressed petunia transcripts on the array. This result indicated that Petunia hybrida is a species with the capacity for cold acclimation (Yelenosky and Guy, 1989) that possess a similarly large amount of cold-responsive genes similar to that reported for other plants (e.g., Arabidopsis). Indeed, the petunia arrays employed here included probes for only a portion of the total genes. Thus, if the probes on the array covered the whole genome, many more genes responsive to cold would have been found. Because differences in the ability of diverse plant species to tolerate cold might not be totally due to the amount of genes responsive to cold stress (Carvallo et al., 2011), it has been suggested that plants require to duly and coordinately mobilize the biological functions and regulatory networks of stress-responsive genes.
Comparison of Significant Gene Ontology Terms of DEG Sets at all Time Points
To gain insight into the putative functions of the 2071 cold-responsive genes identified by the microarray analysis, gene ontology annotation for all of the differentially expressed genes was performed using Blast2gGO against the local nr database. Accordingly, 1388 annotations were obtained based on the gene enrichment analysis that was performed for each comparison group between all time points and 0 h. All three principal GO categories (“Biological Process,” “Molecular Function,” and “Cellular Component”) were collected for each gene, and the biological process was selected for functional enrichment analysis.
We found 198 unique GO terms enriched by DEGs when analyzed gene sets that were up- or down-regulated separately at each time point (Table S3). At 0.5 h/0 h, the most significant GO terms found in the up-regulated gene set were regulation of RNA biosynthetic process (GO:2001141), regulation of RNA metabolic process (GO:0051252) and regulation of transcription, DNA-templated (GO:0006355). The most significant GO terms in the down-regulated sets were transition metal ion transport, and especially zinc ion transmembrane transport (e.g., GO:0000041 and GO:0071577). At 2 h/0 h, only the up-regulated gene sets could be enriched for GO analysis. These DEGs were mainly involved in positive regulation of nucleobase-containing compound metabolic process (GO:0045935), positive regulation of gene expression (GO:0010628), positive regulation of RNA metabolic process (GO:0051254) and positive regulation of transcription, DNA-templated (GO:0045893). The functional enrichment analysis for the down-regulated gene sets was not available, partially due to the lack of annotation for a number of petunia genes. At 24 h/0 h, the terms, “flavone metabolic process” (GO:0051552), “pigment biosynthetic process” (GO:0046148), “flavonol metabolic process” (GO:0051554), “flavone biosynthetic process” (GO:0051553), and “flavonol biosynthetic process” (GO:0051555) were significantly enriched in the up-regulated gene sets (shown in Table 1), whereas responses to various abiotic stresses such as oxygen-containing compound, alcohol and lipid (GO:1901700, GO:0097305 and GO:0033993) and “jasmonic acid biosynthetic process” (GO:0009695) were significantly enriched in the down-regulated gene sets. The top significant GO terms found in the up-regulated gene sets at 5 d/0 h were identical to those at 24 h/0 h (Table S3), whereas the terms significantly enriched in the down-regulated gene sets at 5 d/0 h were mainly represented by response to oxygen-containing compound (GO:1901700).
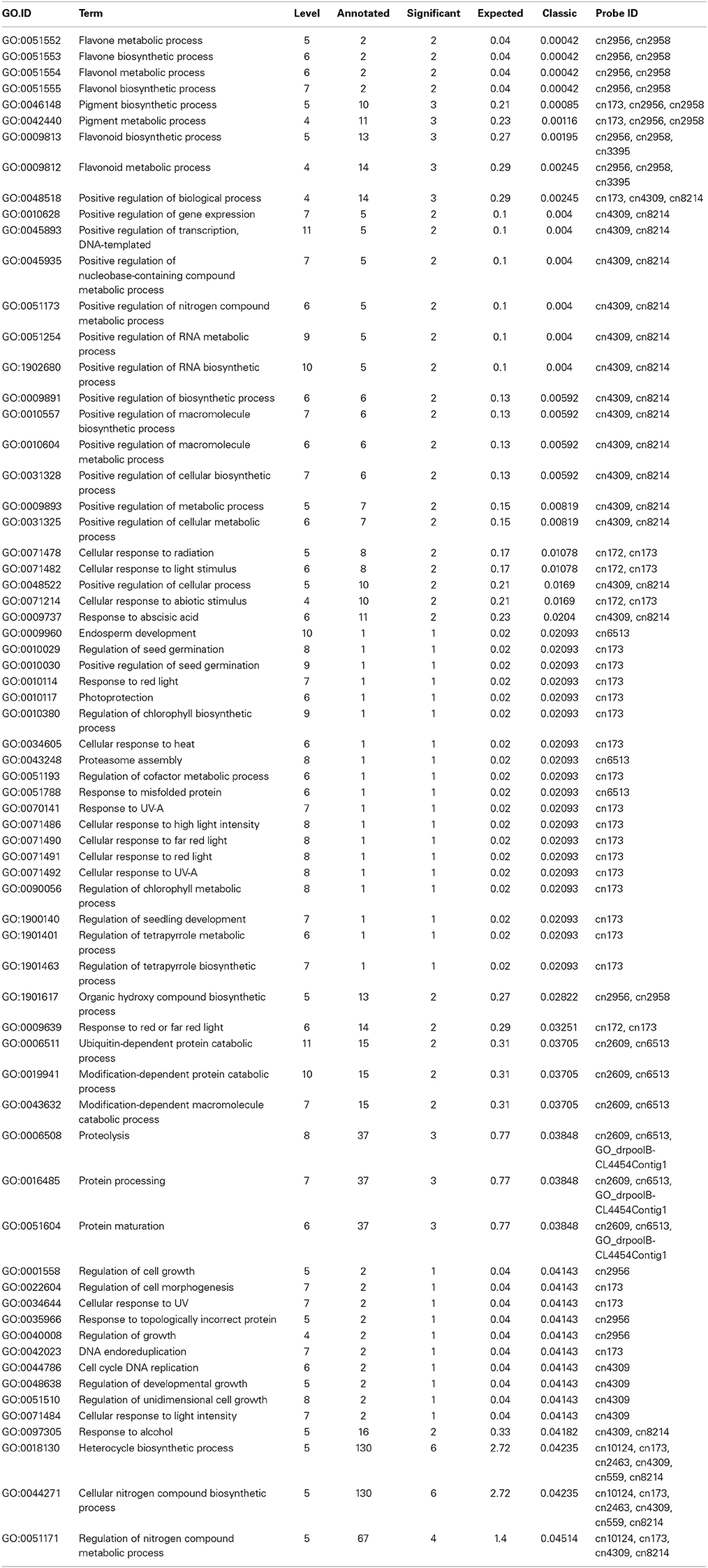
Table 1. Gene ontology (GO) terms enriched by cold induced genes at 24 h/0 h.
Transcription Factor Responses to Cold Stress
We surveyed the putative transcription factors (TFs) that were differentially expressed in petunias at one or more time points under cold treatment based on the annotations (Table S2); a total of 61 DGEs were identified as TFs involved in the cold stress response. The number of up-regulated TFs (48) was prominently greater than the number of down-regulated TFs (13), suggesting that transcriptional activation but not repression was involved. There are more than 2657 predicted TFs in the Arabidopsis genome belonging to 81 gene families that are included in the Plant Transcription Factor Database (PlnTFDB, http://plntfdb.bio.uni-potsdam.de/v3.0/index.php?sp_i-d=ATH) (Perez-Rodriguez et al., 2010). In accordance with this classification, the 61 cold-responsive transcription factors fell into 12 families (Table 2). The six major TF families included AP2-EREBP (APE-TALA2/ET-Responsive Element Binding Protein), zinc finger, MADS-box (MCMI, AG, DEFA, and SRF), MYB (Myeloblastosis)/MYB-related, NAC (NAM, ATAF1/2, CUC2) and GRAS (GAI, RGA, SCR), containing 15, 11, 9, 9, 5, and 5 genes of the 61 total cold-responsive TFs, respectively.
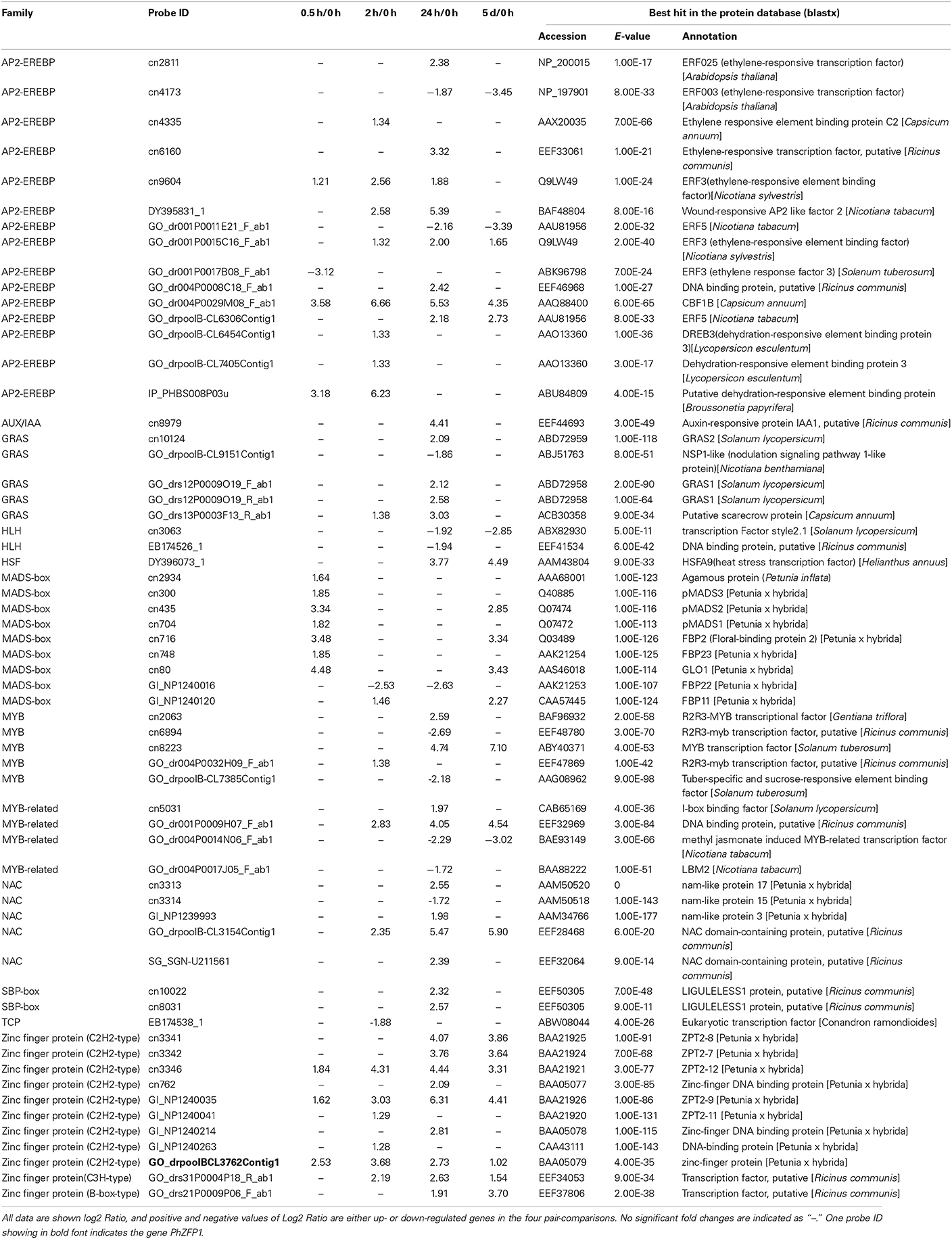
Table 2. Responsive transcription factors during the process of low temperature treatment in petunia seedlings.
Members of the AP2-EREBP family belong to different subfamilies of the ERF/AP2 TF family. For example, AtERF025, AtERF003, CaEREBP-C2, and NsERF3 encode members of the A-4, B-6, B-3, and B-1 subfamilies of the ERF/AP2 TF family. Importantly, we observed that more than half of the members of this family were diversely induced at the early stage (0.5 h or 2 h) during cold treatment, suggesting a relatively fast reaction in triggering transcriptional cascades in the cold response of petunias. We arbitrarily selected two AP2/EREBP TFs (cn9604 and GO_dr001P0015C16_F_ab1) for validation by qRT-PCR. As shown in Table 3, both microarray and qRT-PCR analyses were in agreement concerning the substantial inducibility of the transcripts of these two members following cold treatment. Moreover, the highly induced expression of one CBF homologous gene (GO_dr004P0029M08_F_ab1), named PhCBF1 hereafter, was also verified by real-time RT-PCR (Table 3), implying that the timely response of the DREB/CBF pathway to cold might be similar at least in part between petunias and other plants.
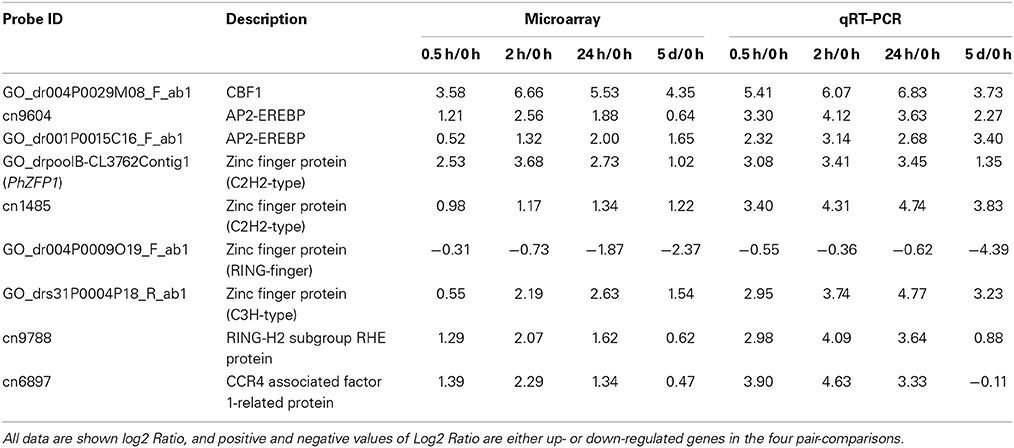
Table 3. Validation of selected array-based gene expression by real-time RT-PCR analysis.
A total of 11 probes fell into three subfamilies of the zinc finger family (C2H2, C3H and B-box). Among these 11 up-regulated zinc finger proteins, we observed that the C2H2 subfamily was particularly enriched in cold-responsive transcripts. Additionally, another C2H2-type zinc finger TF (cn1485) was conspicuously up-regulated following 2 h, 24 h, and 5 d of cold treatment. Because the P-value was higher than 0.05, this TF was excluded from this analysis. However, its cold-induced expression, together with the other four zinc finger proteins (three C2H2 and one B-box), was verified by real-time RT-PCR (Table 3). We also identified one C3H-type zinc finger gene (GO_drs31P0004P18_R_ab1) whose expression level was evidently increased by cold stress and verified by real-time RT-PCR (Table 3).
The majority of MADS-box TFs detected by our array were up-regulated following 0.5 h or 2 h of cold treatment. Only FBP22 was found to be down-regulated after 2 h under cold stress. Interestingly, all of the MADS-box TFs were first identified to be involved in cold responses. Nine MYB or MYB-related family members showed differential expression, with five exhibiting cold-inducible and four cold-repressed expression. Several transcripts (i.e., cn2063 and cn8223) act as transcriptional regulators in phenylpropanoid metabolism. Five transcripts encoding the NAC domain transcription factor were identified, of which only one was down-regulated on the array. Similarly, four GRAS transcription factors detected on our array were up-regulated by cold, whereas one was down-regulated.
In addition to the above-mentioned TFs, we identified two HLHs (Helix-Loop-Helix) that were cold-repressed in our study. In contrast, two probes were found to encode SBP-box proteins, both of which were up-regulated. Additionally, three other types of transcription factor families, including AUX/IAA, HSF, and TCP, were also identified. Previous studies have linked some HSF TFs to cold stress (Carvallo et al., 2011).
Genes Responsive to Cold and Different Abiotic Stresses
To validate the results obtained through microarray analysis, real-time RT-PCR was conducted on 12 selected differentially expressed transcripts, most of which were transcription factors (e.g., AP2-EREBP, C2H2-type, and C3H-type zinc finger protein) and exhibited up-regulated expression (Table 2). The expression of 75% of the cold-responsive genes (9 of 12) based on the microarray data was confirmed using real-time RT-PCR. The log2 ratios from both the results of the microarray and the qRT-PCR analysis for these genes are shown in Table 3. Their expression kinetics from the real-time RT-PCR results were similar to those of the microarray analysis. These results re-confirmed the veracity of our microarray data. However, the fold changes of three cold-responsive genes measured by microarray and by real-time RT-PCR were not consistent. The relative low correspondence may be partially due to the low cut-off value used for the identification of DEGs.
The expression profiles of seven cold-responsive genes were analyzed by real-time RT-PCR using petunia in vitro shoot cultures treated with various stresses, including cold (2°C), dehydration, salinity (200 mM NaCl), osmotic (400 mM mannitol), ABA (100 μM), and JA (100 μM MeJA) for 3 h, 6 h and 12 h (Table 4). From these results, one gene, referred hereafter as PhZFP1 (GO_drpoolB-CL3762Contig1), positively and exclusively responded to cold stress. This gene was predominantly up-regulated by cold and more or less down-regulated by other stresses. Several cold-responsive genes were also strongly induced by the dehydration, salt, osmotic, ABA, or JA stresses. For instance, the transcripts of two genes (GO_dr001P0015C16_F_ab1 and cn9788) were differentially accumulated under dehydration, salinity and osmotic stresses and were remarkably induced by ABA. Two genes encoding zinc finger proteins (cn1485 and GO_drs21P0009P06_F_ab1) were strongly induced by all of the abiotic stresses tested (including both ABA and JA) in this study.
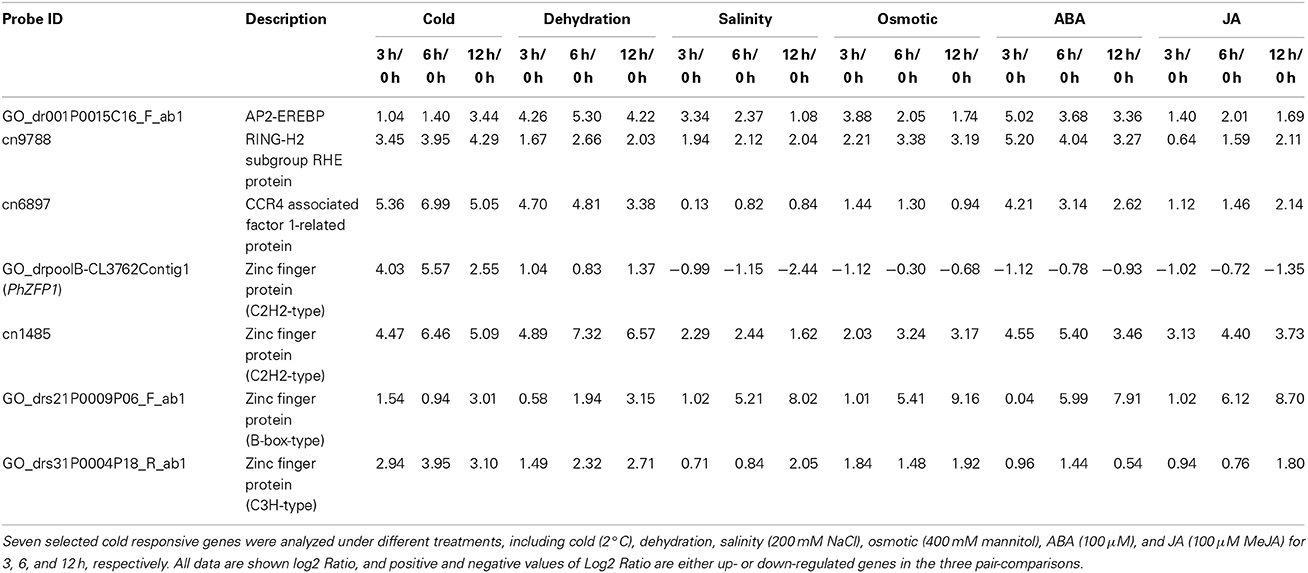
Table 4. Real-time RT-PCR verification of cold-responsive genes under various abiotic stresses.
Network Classification and Identification
To identify sets of functionally related genes, we constructed a petunia gene co-expression network using our microarray samples as input. Prior to the analysis, the gene numbers of each module were transformed to the log2 base scale. Then, a clustering approach of the weighted correlation network was undertaken, resulting in a total of 65 modules of highly correlated genes. In Figure 2, the modules are denoted by different colors. Obviously, the numbers of genes (probe sets) per module varied, and more than half of the modules contained less than 500 genes (probe sets) (Table S4). To explore the co-expression relationships between modules, a module's representative expression pattern was summarized using the first principal component of all of the module's gene members. Using the complete linkage method, all of the module eigengenes were clustered, thereby allowing the characterization of similar structures between the modules (Figure 3).
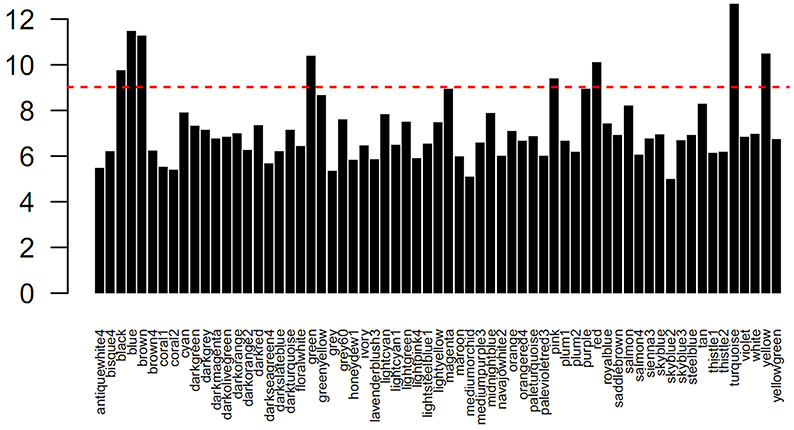
Figure 2. Distribution of genes (probes) in each module. The red line indicates the number of 500 genes.
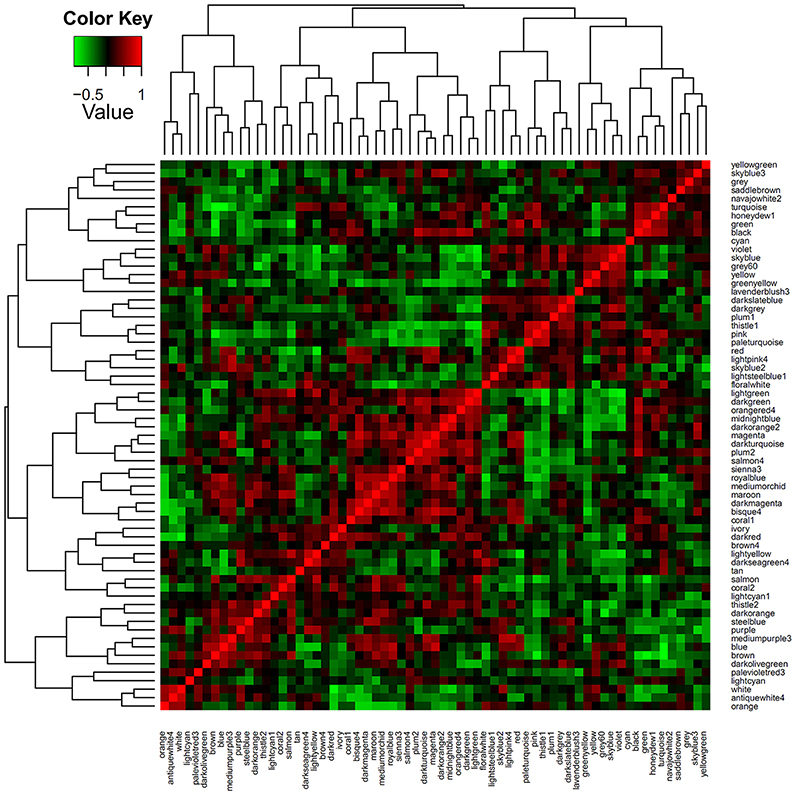
Figure 3. Module eigenvector clustering. The co-expression network with 65 modules and the eigenvectors of each module calculated and clustered using the WGCNA software. The color scale represents the strength of the correlation between the modules (from −1 to 1).
Among the 65 modules, the purple module (size 491) strongly attracted our attention. This module included several transcription factors such as PhZFP1 (GO_drpoolB-CL3762Contig1) and other zinc finger TFs (cn1485 and GO_drs31P0004P18_R_ab1) displaying substantial responsiveness to cold stress in our analysis. PhZFP1 was regarded as a particularly promising candidate gene worthy of further investigation based on its unique stress-induced expression profile compared with other cold-responsive genes tested in our study (Table 4). To quantify the association between PhZFP1 and other petunia genes in the purple module, the top 50 genes positively correlated with PhZFP1 were chosen and are shown in Table S5. Strikingly, PhCBF1 (GO_dr004P0029M08_F_ab1) was also found to be contained in the purple module and included in the top 50 genes positively correlated with PhZFP1. Because PhZFP1 and PhCBF1 were included in the same module and positively correlated with each other, they most likely have a certain relatedness in functionality.
Discussion
Relationship Between Flavonoid Metabolism and Cold Stress Response in Petunias
The results of the biological process for functional enrichment analysis showed that the categories associated with flavonoid metabolism, including “pigment biosynthetic process” and “flavonol metabolic process,” were the most remarkable GO terms. This result suggests that the activation of flavonoid metabolism at the relatively late stage may be an integral part of the plant adaptation to low temperatures in petunias. Previous work has mainly indicated that flavonoids are induced in response to UV irradiation. It was considered that these UV-absorbing compounds could provide protection against cell death stemming from UV-B damage by protecting DNA from dimerization and breakage (Dixon and Paiva, 1995). Other stresses such as cold that induce phenylpropanoids have been less well studied. Increased anthocyanin levels were detected following cold stress in blood oranges (Crifo et al., 2011), but the reasons for this increase are still unclear. In this study, several genes encoding key enzymes implicated in flavonoid metabolism were found among the cold-induced genes at 5 d/0 h (Table S2), such as CHS (chalcone synthase, GI_TC432), CHI (chalcone isomerase, cn302) and F3H (flavanone 3-hydroxylase, cn2956) (Table S2). These genes are held in common in the biosynthetic pathway of anthocyanins, flavonols, and flavan-3-ols. Our results in petunias are to a great extent consistent with previous observations concerning the increased accumulation of two flavonoid pathway proteins (CHS and F3H) in strawberries in response to cold stress (Koehler et al., 2012). Thus, we speculate that the well-defined induction of these genes in petunias subjected to cold stress would probably lead to a rise in flavonoid levels; in turn, the rising flavonoid levels might contribute to the control of cell osmotic potential, thereby effectively coping with the imposed adverse conditions (Chalker-Scott, 1999). However, it is worthwhile to note that in the present study the noticeable major category predicted to be involved in the cold stress response in petunias was associated with flavonoid metabolism, whereas other significant cold-related GO terms were rare. This may be because a large portion of DEGs share homology with genes encoding putative proteins of unknown function or share no significant homology with any database accession. Future studies will be focused on detailing the quantitative and qualitative transcriptomes of petunias under cold stress. These efforts will be greatly facilitated by the completion of petunia genome sequencing and the rapidly developing technologies for RNA sequencing.
Established and Novel Players Involved in Cold Stress Regulation in Petunias
Transcription factors play important roles in plant development and stress tolerance (Lee et al., 2005), and genes related to “Transcription factor activity” may be central regulators involved in upstream cold signal transduction which are capable of triggering a cascade of downstream gene expression. In this study, we found 61 transcription factor genes that were differentially regulated across different time points. The six most highly represented TF families were AP2-EREBP, zinc finger, MADS-box, MYB/MYB-related, NAC, and GRAS. These cold-responsive TFs (shown in Table 2) represent potentially important regulators of the cold-stress response in petunias.
The AP2-EREBP family plays a predominant role in the early stages of the cold response, which has been evidenced by some of the best characterized CBFs (CRT/DRE binding factors) in the cold regulatory pathway. CBF transcription factors, attached to the A-1 subfamily of the ERF/AP2 TF family, are major regulators that exert their effects via activation of cold-regulated effectors in Arabidopsis and other plants (Gilmour et al., 1998). Here, one CBF homologous gene (GO_dr004P0029M08_F_ab1) PhCBF1, was highly induced, suggesting that the response of the DREB/CBF pathway to cold in petunias is at least partially similar to that observed in other plants, although it is probably not the only pathway involved. Certain member of the AP2-EREBP family has been reported to show responses to cold stress in Capsicum annuum, such as CaEREBP-C2 (http://www.ncbi.nlm.nih.gov/protein/AAX20035). Other members were first identified as being involved in the cold response, such as NtERF5, which was previously characterized as a pathogen-regulated factor (Fischer and Droge-Laser, 2004). Additionally, NsERF3, which is homologous to two AP2/EREBP family members (cn9604 and GO_dr001P0015C16_F_ab1), was shown to be involved in the regulation of gene expression by stress factors as well as components of stress signal transduction pathways. This gene most likely acts as a transcriptional repressor and may regulate other AtERFs (based on similarity), whereas the regulation mechanism remains obscure (Kitajima et al., 2000), it might be of interest to further study these two AP2/EREBP TFs.
Zinc finger family members in Arabidopsis have been demonstrated to be involved in the cold response (Lee et al., 2005). Recently, the genes encoding zinc finger proteins in blueberries were also detected to be cold acclimation-responsive based on transcriptome database mining (Die and Rowland, 2014). In our study, we identified 11 up-regulated zinc finger proteins, among which members of the C2H2 subfamily were especially abundant. Previous studies revealed that many C2H2 zinc finger proteins regulate responses to multiple abiotic stresses, such as ZAT10 and ZAT12 in Arabidopsis (Davletova et al., 2005; Mittler et al., 2006). Thus, C2H2 zinc finger proteins are crucial factors that link various signal transduction pathways in response to different stresses and participate in several cellular processes, whereas only a few C2H2 proteins seem to be associated with responses to one specific stress (Kielbowicz-Matuk, 2012). Consistent with this finding, several petunia cold-responsive C2H2-type zinc finger genes found in our array were induced not only by cold stress but also by other abiotic stresses, including dehydration and salinity. The exception was one C2H2-type zinc finger gene called PhZFP1 (GO_drpoolB-CL3762Contig1). PhZFP1 was exclusively and positively responsive to cold. Interestingly, our findings that PhZFP1 and PhCBF1 were contained in the same module and tightly correlated with each other suggested that this gene may be a critical regulator of the cold stress response that functions in an ABA-independent manner similar to CBFs. Abundant evidence has illustrated that the transcription network of CBF plays a significant role in cold acclimation of evolutionarily different plant species. Nevertheless, gene expression analysis revealed several transcription factors besides CBFs that are induced during cold acclimation. Transgenic analysis of cold-inducible TFs helped to validate their functions in cold tolerance and suggested that other classes of transcription factors also play a significant role in cold acclimation (Chinnusamy et al., 2010). For example, constitutive overexpression of the soybean C2H2-type zinc finger SCOF1 (soybean cold-inducible factor-1) in Arabidopsis transgenic plants elevated the expression of COR (cold-regulated) genes and conferred constitutive freezing tolerance. SCOF1 interacts with soybean G-box binding bZIP transcription factor SGBF1, which is induced by both cold and ABA. It was suggested that SCOF-1 might function as a positive regulator of COR gene expression mediated by the ABA responsive element via protein-protein interactions, which in turn improves cold tolerance of plants (Kim et al., 2001). The large proportion of C2H2-type zinc finger family members with cold-inducible transcripts identified in our study makes this class of TFs an attractive target for further functional characterization. For instance, the question of whether PhZFP1 is involved in the CBF-dependent or CBF-independent transcriptional pathway and the mechanism by which the PhZFP1 transcriptional networks operate during cold acclimation and cold stress tolerance in petunias are unknown. Compared with the relatively large C2H2 subfamily, knowledge about the biological role of the smaller C3H and B-box subfamilies is very limited. C3H-type zinc fingers are a type of plant cold shock domain protein (CSDP). Although their biological function and modes of action are not totally revealed, evidence from Arabidopsis suggests that CSDPs may confer cold tolerance by functioning as RNA chaperones (Kim et al., 2007, 2009). All of these results suggest an important role for zinc finger genes in petunia responses to cold stress.
The notable MADS-box family members are known to be key regulators of several plant development processes. The best studied plant MADS-box TFs are those involved in floral organ identity determination (Parenicova et al., 2003). However, a recent study of the Arabidopsis MADS-box gene SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) revealed that this representative floral activator could also function as a negative regulator of the cold response pathway through the direct repression of CBFs (Seo et al., 2009). This result hints that the responsiveness of the MADS-box transcription factors to cold stress observed in this study might not be a coincidence, and offers the possibility that some MADS-box TFs, for instance, FBP22 (GI_NP1240016) which displays high homology to SOC1 and other SOC1-like proteins are the key regulators of crosstalk between the cold response and flowering time regulation. MYB proteins are crucial factors controlling development, metabolism and responses to biotic and abiotic stresses in regulatory networks (Dubos et al., 2010). A comprehensive analysis of MYB transcription factor gene expression demonstrated that almost all MYB TFs are responsive to hormones or stresses (Chen et al., 2006). In this study, five up-regulated and four down-regulated MYB or MYB-related family members showed diverse expression patterns. One MYB-related transcription factor (GO_dr004P0017J05_F_ab1) was reported to be a regulator of the tobacco defense-related genes (Sugimoto et al., 2000). Several transcripts, such as cn2063 and cn8223, were identified to be involved in the regulation of anthocyanin biosynthesis (Nakatsuka et al., 2008) (http://www.ncbi.nlm.nih.gov/protein/ABY40371). The differential expression of MYB TFs implies that overlapping environmental pressures or certain metabolic pathways may be integrated into petunia responses to cold stress mediated by the regulation of MYBs.
The appearance of five transcripts encoding the NAC domain TF on the list of transcription factors regulated in response to cold stress is in line with the existing knowledge concerning plant-specific NAC proteins, which are renowned for their roles in biotic and abiotic stress responses (Puranik et al., 2012). In contrast, the currently available knowledge concerning the function of the GRAS gene family is not abundant. Most characterized members of this family encode transcriptional regulators which have functions in plant growth and development, such as axillary meristem formation, root radial patterning and gibberellin signal transduction (Tian et al., 2004). It is worth noting that there have been some results supporting a function for GRAS TFs in plant responses to biotic and abiotic stresses (Kim et al., 2001).
An additional five families (HLH, SBP-box, AUX/IAA, HSF, and TCP) accounted for approximately 11.5% of the total number of cold-responsive TFs detected in our array. Although these families play various roles in plant developmental processes and environmental responses, for instance, the SBP-box gene family has been revealed to control flower and fruit development as well as other significant physiological processes (Chen et al., 2010), most have not been linked to cold stress resistance in plants. Nevertheless, it is important to note the knowledge concerning the function of some members in other aspects has started to gradually increase. For example, several grape SBP-box genes were identified and presumed to be potentially involved in the defense against different stresses, including cold treatment (Hou et al., 2013).
A great number of genes (approximately 37.8% of the total 2071 DEGs) encode proteins of unknown function. The study of these genes may reveal new mechanisms that are fundamental to petunia plants coping with low temperatures. Indeed, the microarray that was used to monitor gene expression was based on ESTs from Petunia hybrida and Petunia axillaries, which was not sufficiently reflective of the overall genome. Thus, the present analysis was significantly limited in scope. Nevertheless, our results support the fact that the large-scale dataset strategy can be useful in obtaining meaningful biological information. In this sense, it is possible that our results have identified for the first time a set of potential regulators which may play a role in the regulation of cold stress tolerance in petunias. Further investigations will be focused on the functional validation of candidate genes for improving petunia tolerance through genetic engineering.
Crosstalk Among Different Signaling Systems in Regulating the Cold Stress Response in Petunias
In plants, the existence of complex signaling networks and multiple defense strategies results in their enhanced defense capacity. Because the cold stress-signaling pathway may interact with other signaling systems (i.e., drought, salt, and ABA) (Seki et al., 2002), we conducted qRT-PCR to investigate the expression patterns of seven cold-responsive genes under different abiotic stresses. Most were induced to varying degrees by at least one of the five common abiotic stress treatments, including exposure to dehydration, salinity, osmotic, ABA, and JA in addition to cold, suggesting that they may participate in the regulation of a wide spectrum of responses to diverse abiotic stresses. These results indicate that dynamic crosstalk exists among signaling pathways associated with cold, dehydration, and salt stress in petunias, which may induce common alterations, such as cellular dehydration (Rabbani et al., 2003).
Induced defense responses are controlled by a network of interconnected signal transduction pathways in which hormonal signals, such as ABA or JA, could coordinately activate the transcription of various defense-related genes (Glazebrook, 2005). Plant hormones offer a vital function in signaling networks implicated in plant responses to a variety of biotic and abiotic stresses. ABA plays a critical role in the abiotic stress response by inducing stomatal closure which results in the reduction of transpiration (Pantin et al., 2013) and by regulating root growth, ion channels and gene expression (Duan et al., 2013), whereas JA plays an important role in the defense response, especially against biotic stresses, such as necrotrophic pathogens and wounding (Thaler et al., 2012). In this study, we determined that the expressions of the majority of the cold-responsive genes analyzed (5/7) were significantly modified by ABA treatment (Table 4). Thus, our result is in agreement with the known effect of ABA on cold stress signaling, which indicates their possible involvement in ABA-dependent pathways. However, recent findings have increasingly suggested that JA also plays a role in the regulation of plant abiotic stress tolerance, including stresses, such as salinity and cold (Kang et al., 2005; Khan et al., 2012). These findings may explain our observation of the substantial elevated expression level of two cold-responsive zinc finger genes (cn1485 and GO_drs21P0009P06_F_ab1) following MeJA treatment (Table 4). For example, it was demonstrated that jasmonate acts as an upstream signal of the ICE-CBF/DREB1 transcriptional pathway to positively regulate freezing stress responses in Arabidopsis (Hu et al., 2013). Because the two zinc finger protein genes mentioned above were also responsive to all of the other abiotic treatments, the result suggests that their involvement in the response to cold stress may be dependent on multiple signaling pathways including MeJA molecular signals. Taken together, our analyses indicate that crosstalk among different signaling systems plays a significant role in regulating the cold stress response in petunias.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported financially by the National Natural Science Foundation of China (31101576), the Research Fund for the Doctoral Program of Higher Education of China (20110146120040) and the Fundamental Research Funds for the Central Universities (2013PY090). The authors wish to acknowledge Dr. Hao Du in the National Key Laboratory of Crop Genetic Improvement in Wuhan, Huazhong Agricultural University for comments and advice on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2015.00118/abstract
Table S1. Primers used for real-time RT-PCR verification.
Table S2. Differentially expressed genes (DEGs) in the four paired comparisons from low temperature-treated samples at 0 h, 0.5 h, 24 h and 5 d.
Table S3. Gene ontology (GO) terms enriched by cold responsive genes at all time points.
Table S4. Gene modules detected by WGCNA in the petunia gene co-expression network.
Table S5. The top 50 genes positively correlated with PhZFP1.
References
Alexa, A., Rahnenführer, J., and Lengauer, T. (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. doi: 10.1093/bioinformatics/btl140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
An, D., Yang, J., and Zhang, P. (2012). Transcriptome profiling of low temperature-treated cassava apical shoots showed dynamic responses of tropical plant to cold stress. BMC Genomics 13:64. doi: 10.1186/1471-2164-13-64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boatright, J., Negre, F., Chen, X. L., Kish, C. M., Wood, B., Peel, G., et al. (2004). Understanding in vivo benzenoid metabolism in petunia petal tissue. Plant Physiol. 135, 1993–2011. doi: 10.1104/pp.104.045468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bolstad, B. M., Irizarry, R. A., Strand, M., and Speed, T. P. (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193. doi: 10.1093/bioinformatics/19.2.185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Breuillin, F., Schramm, J., Hajirezaei, M., Ahkami, A., Favre, P., Druege, U., et al. (2010). Phosphate systemically inhibits development of arbuscular mycorrhiza in Petunia hybrida and represses genes involved in mycorrhizal functioning. Plant J. 64, 1002–1017. doi: 10.1111/j.1365-313X.2010.04385.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carvallo, M. A., Pino, M. T., Jeknic, Z., Zou, C., Doherty, C. J., Shiu, S. H., et al. (2011). A comparison of the low temperature transcriptomes and CBF regulons of three plant species that differ in freezing tolerance: Solanum commersonii, Solanum tuberosum, and Arabidopsis thaliana. J. Exp. Bot. 62, 3807–3819. doi: 10.1093/jxb/err066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chalker-Scott, L. (1999). Environmental significance of anthocyanins in plant stress responses. Photochem. Photobiol. 70, 1–9. doi: 10.1111/j.1751-1097.1999.tb01944.x
Chandler, P. M., and Robertson, M. (1994). Gene expression regulated by abscisic acid and its relation to stress tolerance. Annu. Rev. Plant Physiol. Plant Mol. Biol. 45, 113–141. doi: 10.1146/annurev.pp.45.060194.000553
Chen, X., Zhang, Z., Liu, D., Zhang, K., Li, A., and Mao, L. (2010). SQUAMOSA promoter−binding protein−like transcription factors: star players for plant growth and development. J. Integr. Plant Biol. 52, 946–951. doi: 10.1111/j.1744-7909.2010.00987.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y. H., Yang, X. Y., He, K., Liu, M. H., Li, J. G., Gao, Z. F., et al. (2006). The MYB transcription factor superfamily of arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 60, 107–124. doi: 10.1007/s11103-005-2910-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chinnusamy, V., Zhu, J.-K., and Sunkar, R. (2010). “Gene regulation during cold stress acclimation in plants,” in Plant Stress Tolerance: Methods and Protocols, ed R. Sunkar (Humana Press), 39–55.
Chinnusamy, V., Zhu, J., and Zhu, J. K. (2007). Cold stress regulation of gene expression in plants. Trends Plant Sci. 12, 444–451. doi: 10.1016/j.tplants.2007.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crifo, T., Puglisi, I., Petrone, G., Recupero, G. R., and Lo Piero, A. R. (2011). Expression analysis in response to low temperature stress in blood oranges: implication of the flavonoid biosynthetic pathway. Gene 476, 1–9. doi: 10.1016/j.gene.2011.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davletova, S., Schlauch, K., Coutu, J., and Mittler, R. (2005). The zinc-finger protein Zat12 plays a central role in reactive oxygen and abiotic stress signaling in Arabidopsis. Plant Physiol. 139, 847–856. doi: 10.1104/pp.105.068254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Die, J. V., and Rowland, L. J. (2014). Elucidating cold acclimation pathway in blueberry by transcriptome profiling. Environ. Exp. Bot. 106, 87–98. doi: 10.1016/j.envexpbot.2013.12.017
Dixon, R. A., and Paiva, N. L. (1995). Stress-induced phenylpropanoid metabolism. Plant Cell 7, 1085–1097. doi: 10.1105/tpc.7.7.1085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doerffling, K., Doerffling, H., Lesselich, G., Luck, E., Zimmermann, C., Melz, G., et al. (1997). Heritable improvement of frost tolerance in winter wheat by in vitro-selection of hydroxyproline-resistant proline overproducing mutants. Euphytica 93, 1–10. doi: 10.1023/A:1002946622376
Duan, L. N., Dietrich, D., Ng, C. H., Chan, P. M. Y., Bhalerao, R., Bennett, M. J., et al. (2013). Endodermal ABA signaling promotes lateral root quiescence during salt stress in Arabidopsis seedlings. Plant Cell 25, 324–341. doi: 10.1105/tpc.112.107227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dubos, C., Stracke, R., Grotewold, E., Weisshaar, B., Martin, C., and Lepiniec, L. (2010). MYB transcription factors in Arabidopsis. Trends Plant Sci. 15, 573–581. doi: 10.1016/j.tplants.2010.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandes, J., Morrow, D. J., Casati, P., and Walbot, V. (2008). Distinctive transcriptome responses to adverse environmental conditions in Zea mays L. Plant Biotechnol. J. 6, 782–798. doi: 10.1111/j.1467-7652.2008.00360.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischer, U., and Droge-Laser, W. (2004). Overexpression of NtERF5, a new member of the tobacco ethylene response transcription factor family enhances resistance to Tobacco mosaic virus. Mol. Plant Microbe Interact. 17, 1162–1171. doi: 10.1094/MPMI.2004.17.10.1162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gautier, L., Cope, L., Bolstad, B. M., and Irizarry, R. A. (2004). affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315. doi: 10.1093/bioinformatics/btg405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. doi: 10.1186/gb-2004-5-10-r80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilmour, S. J., Zarka, D. G., Stockinger, E. J., Salazar, M. P., Houghton, J. M., and Thomashow, M. F. (1998). Low temperature regulation of the Arabidopsis CBF family of AP2 transcriptional activators as an early step in cold-induced COR gene expression. Plant J. 16, 433–442. doi: 10.1046/j.1365-313x.1998.00310.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glazebrook, J. (2005). Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227. doi: 10.1146/annurev.phyto.43.040204.135923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hong, F., and Breitling, R. (2008). A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics 24, 374–382. doi: 10.1093/bioinformatics/btm620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hou, H., Li, J., Gao, M., Singer, S. D., Wang, H., Mao, L., et al. (2013). Genomic organization, phylogenetic comparison and differential expression of the SBP-Box family genes in grape. PLoS ONE 8:e59358. doi: 10.1371/journal.pone.0059358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, Y., Jiang, L., Wang, F., and Yu, D. (2013). Jasmonate regulates the inducer of Cbf expression-c-repeat binding factor/dre binding factor1 cascade and freezing tolerance in Arabidopsis. Plant Cell 25, 2907–2924. doi: 10.1105/tpc.113.112631
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Irizarry, R. A., Hobbs, B., Collin, F., Beazer−Barclay, Y. D., Antonellis, K. J., Scherf, U., et al. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. doi: 10.1093/biostatistics/4.2.249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, D. J., Seo, Y. J., Lee, J. D., Ishii, R., Kim, K. U., Shin, D. H., et al. (2005). Jasmonic acid differentially affects growth, ion uptake and abscisic acid concentration in salt-tolerant and salt-sensitive rice cultivars. J. Agron. Crop Sci. 191, 273–282. doi: 10.1111/j.1439-037X.2005.00153.x
Khan, M. I., Syeed, S., Nazar, R., and Anjum, N. (2012). “An insight into the role of salicylic acid and jasmonic acid in salt stress tolerance,” in Phytohormones and Abiotic Stress Tolerance in Plants, eds N. A. Khan, R. Nazar, N. Iqbal and N. A. Anjum (Berlin Heidelberg: Springer), 277–300.
Kielbowicz-Matuk, A. (2012). Involvement of plant C2H2-type zinc finger transcription factors in stress responses. Plant Sci. 185, 78–85. doi: 10.1016/j.plantsci.2011.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J. C., Lee, S. H., Cheong, Y. H., Yoo, C. M., Lee, S. I., Chun, H. J., et al. (2001). A novel cold-inducible zinc finger protein from soybean, SCOF-1, enhances cold tolerance in transgenic plants. Plant J. 25, 247–259. doi: 10.1046/j.1365-313x.2001.00947.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J. Y., Park, S. J., Jang, B., Jung, C. H., Ahn, S. J., Goh, C. H., et al. (2007). Functional characterization of a glycine-rich RNA-binding protein 2 in Arabidopsis thaliana under abiotic stress conditions. Plant J. 50, 439–451. doi: 10.1111/j.1365-313X.2007.03057.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, M. H., Sasaki, K., and Imai, R. (2009). Cold shock domain protein 3 regulates freezing tolerance in Arabidopsis thaliana. J. Biol. Chem. 284, 23454–23460. doi: 10.1074/jbc.M109.025791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kitajima, S., Koyama, T., Ohme-Takagi, M., Shinshi, H., and Sato, F. (2000). Characterization of gene expression of NsERFs, transcription factors of basic PR genes from Nicotiana sylvestris. Plant Cell Physiol. 41, 817–824. doi: 10.1093/pcp/41.6.817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koehler, G., Wilson, R. C., Goodpaster, J. V., Sonsteby, A., Lai, X., Witzmann, F. A., et al. (2012). Proteomic study of low-temperature responses in strawberry cultivars (Fragaria x ananassa) that differ in cold tolerance. Plant Physiol. 159, 1787–1805. doi: 10.1104/pp.112.198267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koster, K. L., and Lynch, D. V. (1992). Solute accumulation and compartmentation during the cold acclimation of Puma rye. Plant Physiol. 98, 108–113. doi: 10.1104/pp.98.1.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, B. H., Henderson, D. A., and Zhu, J. K. (2005). The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 17, 3155–3175. doi: 10.1105/tpc.105.035568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lynch, D. V., and Steponkus, P. L. (1987). Plasma membrane lipid alterations associated with cold acclimation of winter rye seedlings (Secale cereale L. cv Puma). Plant Physiol. 83, 761–767. doi: 10.1104/pp.83.4.761
Mittler, R., Kim, Y., Song, L., Coutu, J., Coutu, A., Ciftci-Yilmaz, S., et al. (2006). Gain- and loss-of-function mutations in Zat10 enhance the tolerance of plants to abiotic stress. FEBS Lett. 580, 6537–6542. doi: 10.1016/j.febslet.2006.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakatsuka, T., Haruta, K. S., Pitaksutheepong, C., Abe, Y., Kakizaki, Y., Yamamoto, K., et al. (2008). Identification and characterization of R2R3-MYB and bHLH transcription factors regulating anthocyanin biosynthesis in gentian flowers. Plant Cell Physiol. 49, 1818–1829. doi: 10.1093/pcp/pcn163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nomura, M., Muramoto, Y., Yasuda, S., Takabe, T., and Kishitani, S. (1995). The accumulation of glycinebetaine during cold-acclimation in early and late cultivars of barley. Euphytica 83, 247–250. doi: 10.1007/bf01678137
Pantin, F., Monnet, F., Jannaud, D., Costa, J. M., Renaud, J., Muller, B., et al. (2013). The dual effect of abscisic acid on stomata. New Phytol. 197, 65–72. doi: 10.1111/nph.12013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parenicova, L., De Folter, S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15, 1538–1551. doi: 10.1105/tpc.011544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pennycooke, J. C., Cox, S., and Stushnoff, C. (2005). Relationship of cold acclimation, total phenolic content and antioxidant capacity with chilling tolerance in petunia (Petunia x hybrida). Environ. Exp. Bot. 53, 225–232. doi: 10.1016/j.envexpbot.2004.04.002
Pennycooke, J. C., Jones, M. L., and Stushnoff, C. (2003). Down-regulating alpha-galactosidase enhances freezing tolerance in transgenic petunia. Plant Physiol. 133, 901–909. doi: 10.1104/pp.103.024554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perez-Rodriguez, P., Riano-Pachon, D. M., Correa, L. G., Rensing, S. A., Kersten, B., and Mueller-Roeber, B. (2010). PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, 822–827. doi: 10.1093/nar/gkp805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Puranik, S., Sahu, P. P., Srivastava, P. S., and Prasad, M. (2012). NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 17, 369–381. doi: 10.1016/j.tplants.2012.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rabbani, M. A., Maruyama, K., Abe, H., Khan, M. A., Katsura, K., Ito, Y., et al. (2003). Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA get-blot analyses. Plant Physiol. 133, 1755–1767. doi: 10.1104/pp.103.025742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodriguez-Uribe, L., Higbie, S. M., Stewart, J. M., Wilkins, T., Lindemann, W., Sengupta-Gopalan, C., et al. (2011). Identification of salt responsive genes using comparative microarray analysis in Upland cotton (Gossypium hirsutum L.). Plant Sci. 180, 461–469. doi: 10.1016/j.plantsci.2010.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanghera, G. S., Wani, S. H., Hussain, W., and Singh, N. B. (2011). Engineering cold stress tolerance in crop plants. Curr. Genomics 12, 30–43. doi: 10.2174/138920211794520178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seki, M., Narusaka, M., Ishida, J., Nanjo, T., Fujita, M., Oono, Y., et al. (2002). Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 31, 279–292. doi: 10.1046/j.1365-313X.2002.01359.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seo, E., Lee, H., Jeon, J., Park, H., Kim, J., Noh, Y. S., et al. (2009). Crosstalk between cold response and flowering in Arabidopsis is mediated through the flowering-time gene SOC1 and its upstream negative regulator FLC. Plant Cell 21, 3185–3197. doi: 10.1105/tpc.108.063883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stanca, A., Crosatti, C., Grossi, M., Lacerenza, N., Rizza, F., and Cattivelli, L. (1996). Molecular adaptation of barley to cold and drought conditions. Euphytica 92, 215–219. doi: 10.1007/BF00022847
Sugimoto, K., Takeda, S., and Hirochika, H. (2000). MYB-related transcription factor NtMYB2 induced by wounding and elicitors is a regulator of the tobacco retrotransposon Tto1 and defense-related genes. Plant Cell 12, 2511–2527. doi: 10.1105/tpc.12.12.2511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tedder, P., Zubko, E., Westhead, D. R., and Meyer, P. (2009). Small RNA analysis in Petunia hybrida identifies unusual tissue-specific expression patterns of conserved miRNAs and of a 24mer RNA. RNA 15, 1012–1020. doi: 10.1261/rna.1517209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thaler, J. S., Humphrey, P. T., and Whiteman, N. K. (2012). Evolution of jasmonate and salicylate signal crosstalk. Trends Plant Sci. 17, 260–270. doi: 10.1016/j.tplants.2012.02.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomashow, M. F. (2001). So what's new in the field of plant cold acclimation? Lots! Plant Physiol. 125, 89–93. doi: 10.1104/pp.125.1.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tian, C. G., Wan, P., Sun, S. H., Li, J. Y., and Chen, M. S. (2004). Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol. Biol. 54, 519–532. doi: 10.1023/B:PLAN.0000038256.89809.57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verdonk, J. C., Haring, M. A., Van Tunen, A. J., and Schuurink, R. C. (2005). ODORANT1 regulates fragrance biosynthesis in petunia flowers. Plant Cell 17, 1612–1624. doi: 10.1105/tpc.104.028837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weiser, C. (1970). Cold resistance and injury in woody plants knowledge of hardy plant adaptations to freezing stress may help us to reduce winter damage. Science 169, 1269–1278. doi: 10.1126/science.169.3952.1269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yelenosky, G., and Guy, C. L. (1989). Freezing tolerance of citrus, spinach, and petunia leaf tissue: osmotic adjustment and sensitivity to freeze induced cellular dehydration. Plant Physiol. 89, 444–451. doi: 10.1104/pp.89.2.444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: abiotic stress, microarray, transcriptional profiling, gene expression, cold-responsive regulator, Petunia hybrida
Citation: Li B, Ning L, Zhang J, Bao M and Zhang W (2015) Transcriptional profiling of Petunia seedlings reveals candidate regulators of the cold stress response. Front. Plant Sci. 6:118. doi: 10.3389/fpls.2015.00118
Received: 04 December 2014; Accepted: 12 February 2015;
Published online: 02 March 2015.
Edited by:
Diego Rubiales, Consejo Superior de Investigaciones Cientificas, SpainReviewed by:
Chenglin Chai, University of Missouri-Columbia, USACraita Bita, Radboud University Nijmegen, Netherlands
Copyright © 2015 Li, Ning, Zhang, Bao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Zhang, Key Laboratory of Horticultural Plant Biology, Ministry of Education, College of Horticulture and Forestry Sciences, Huazhong Agricultural University, No.1, Shizishan Street, Hongshan District, Wuhan 430070 Hubei, China e-mail:emhhbmd3QG1haWwuaHphdS5lZHUuY24=