 Lingzhe Huang1†
Lingzhe Huang1† Zefeng Li2†Jianxiang Wu1Yi Xu1
Zefeng Li2†Jianxiang Wu1Yi Xu1 Xiuling Yang1Longjiang Fan2Rongxiang Fang3*
Xiuling Yang1Longjiang Fan2Rongxiang Fang3* Xueping Zhou1*
Xueping Zhou1*- 1State Key Laboratory of Rice Biology, Institute of Biotechnology, Zhejiang University, Hangzhou, People’s Republic of China
- 2Institute of Crop Science and Institute of Bioinformatics, Zhejiang University, Hangzhou, People’s Republic of China
- 3State Key Laboratory of Plant Genomics, Institute of Microbiology, Chinese Academy of Sciences, Beijing, People’s Republic of China
Plant RNA viruses often generate diverse populations in their host plants through error-prone replication and recombination. Recent studies on the genetic diversity of plant RNA viruses in various host plants have provided valuable information about RNA virus evolution and emergence of new diseases caused by RNA viruses. We analyzed and compared the genetic diversity of Rice stripe virus (RSV) populations in Oryza sativa (a natural host of RSV) and compared it with that of the RSV populations generated in an infection of Nicotiana benthamiana, an experimental host of RSV, using the high-throughput sequencing technology. From infected O. sativa and N. benthamiana plants, a total of 341 and 1675 site substitutions were identified in the RSV genome, respectively, and the average substitution ratio in these sites was 1.47 and 7.05 %, respectively, indicating that the RSV populations from infected N. benthamiana plant are more diverse than those from infected O. sativa plant. Our result gives a direct evidence that virus might allow higher genetic diversity for host adaptation.
Introduction
RNA viruses are known for their high evolutionary potential due mainly to their error-prone replication, and fast rate of genome replication (Domingo and Holland, 1997; Roossinck, 1997; Elena and Sanjuán, 2007; Lauring and Andino, 2010; Sanjuan et al., 2010). In addition, template-switching of viral RNA-dependent RNA polymerase (RdRp) during virus replication, known as RNA recombination, also functions to accelerate virus evolution (Nagy and Simon, 1997; Garcia-Arenal et al., 2001). Using these strategies, viruses are capable of bringing beneficial mutations into and removing deleterious mutations from their genomes (Worobey and Holmes, 1999). Mutation in the viral genomic sequence may lead to changes of virus host range, disease symptoms, and emergence of new viruses in nature. Previous reports have indicated that mutant viruses often appeared soon after the RNA virus infected its host plant and to form diverse populations in infected cells, and these studies were analyzed through sequencing random selected clones derived from an infected plant (Domingo and Holland, 1997; Roossinck, 1997; Schneider and Roossinck, 2000; Garcia-Arenal et al., 2001, 2003).
Rice stripe virus (RSV) is one of the most damaging rice pathogens in China and was firstly identified in rice field in China in 1963 (Lin et al., 1990). It is now identified in rice fields in 16 provinces of China and caused significant yield losses (Xiong et al., 2008; Wei et al., 2009). RSV is the type member of the genus Tenuivirus and infects mainly rice and a few other species in the family Poaceae including maize, oat and wheat. In field, RSV is transmitted efficiently by small brown plant hopper (Laodelphax striatellus) in a persistent manner. RSV can propagate within the insect vector and transmitted to its progenies (Koganezawa et al., 1975; Falk and Tsai, 1998). Under the laboratory conditions, RSV can be transmitted to an experimental host, Nicotiana benthamiana, through mechanical inoculation (Kong et al., 2014).
Rice stripe virus is a single-stranded RNA virus containing four genome segments, designated as RNA 1, 2, 3, and 4 (Figure 1; Zhu et al., 1991, 1992; Takahashi et al., 1993; Toriyama et al., 1994). RNA 1 is in negative sense and encodes a putative protein of approximately 337 kDa. This protein was suggested to be part of the RSV RdRp complex and is associated with the RSV filamentous ribonucleoprotein (Toriyama et al., 1994). The other three RNA segments of RSV are all ambisense and each RNA segment contains two ORFs, one is in the viral sense (vRNA) and the other in the viral complementary-sense (vcRNA; Ramirez and Haenni, 1994). The vRNA 2 ORF encodes a membrane-associated protein of 22.8 kDa and the vcRNA 2 ORF encodes a poly-glycoprotein of 94 kDa. Functions of these proteins are still unknown (Takahashi et al., 1993; Falk and Tsai, 1998). The vRNA 3 ORF encodes a 23.9-kDa suppressor of RNA silencing (Xiong et al., 2009) and the vcRNA 3 ORF encodes a nucleocapsid protein of 35 kDa (Kakutani et al., 1991; Zhu et al., 1991). The vRNA 4 ORF encodes a major non-capsid protein of 21.5 kDa that functions in disease symptom development (Kong et al., 2014). The vcRNA 4 ORF encodes a protein of 32.5 kDa and is required for RSV cell-to-cell movement in infected plant (Xiong et al., 2008).
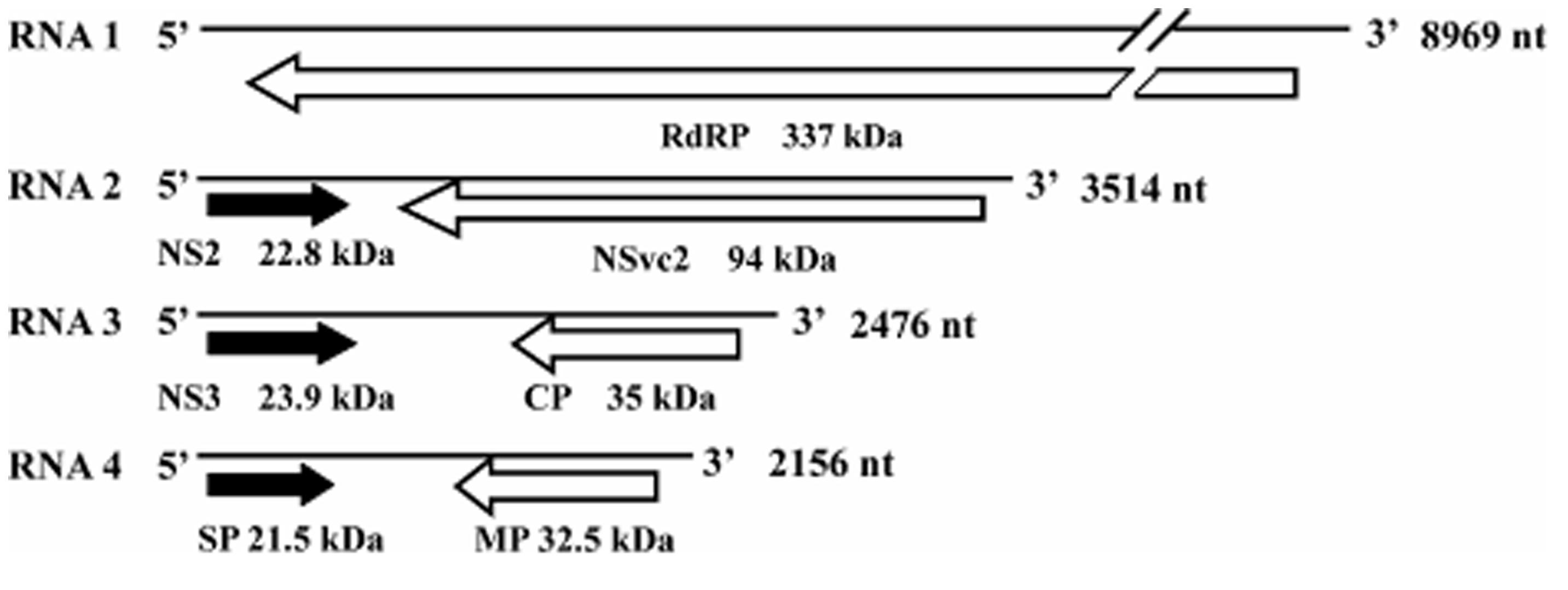
FIGURE 1. Genome organization of Rice stripe virus (RSV). ORFs are indicated as black arrows on the viral-sense RNAs and white arrows on the viral complementary-sense RNAs. Direction of arrowheads indicates the direction of translation. RdRp, RNA-dependent RNA polymerase; CP, coat protein; SP, disease-specific protein; MP, movement protein; NS2 and NS3, non-structural proteins on the viral-sense RNA 2 and 3; NSvc2, non-structural protein on the viral complementary-sense RNA2.
Determination of the complexity of RNA virus population in their host plants requires a rapid, reliable, and cost-effective sequencing method. Because viral RNAs with specific mutations may accumulate to lower levels than the most frequent viral RNAs in infected cells, traditional small-scale sequencing used in many earlier studies might fail to detect these non-conserved or lowly accumulated viral RNAs. Although the evolution of RSV over long periods of time and in areas covering large geographical districts in China has been studied (Wei et al., 2009), the population diversity of RSV within a plant is still unknown. In this study, we used the high-throughput sequencing technology (also known as the second generation sequencing technology) to investigate the diverse population of RSV in an infected Oryza sativa plant (a natural host) and compare it with that observed in an infected N. benthamiana plant (an experimental host) and we found that the RSV populations from the infected N. benthamiana plant are more diverse than those from the infected O. sativa plant. Our results give a direct evidence that virus might allow higher genetic diversity for host adaptation.
Materials and Methods
Virus Source, Host Plants, and Virus Inoculation
Infected O. sativa plants with typical RSV symptoms were collected from rice fields in Jiangsu Province, China. A single plant with only RSV infection was identified through RT-PCR using specific primers and ELISA using specific antibodies as described previously (Wang et al., 2004) and leaves of this plant were then stored at -80°C till use. N. benthamiana seedlings were grown inside an insect-free room with a constant temperature at 25°C and a 16 h light supply. Leaves from the RSV-infected O. sativa plant were ground in 0.1 M phosphate buffer, pH 7.0, and the crude extract was mechanically inoculated to leaves of N. benthamiana seedlings at six- to seven-leaf stages. Young systemic leaves of one N. benthamiana plant showing yellow vein symptoms were harvested at 30 days post inoculation (dpi). The harvested fresh N. benthamiana leaves and the frozen RSV-infected O. sativa leaves were used for the viral population analysis.
Viral cDNA Library Construction and Sequencing
Total RNA was extracted from 1g leaf tissues of the frozen O. sativa and fresh N. benthamiana using TRIzol reagent following manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA), respectively, and cDNA libraries of RSV were then constructed. First strand cDNA was synthesized using the SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA), primers used for first strand cDNA synthesis were designed according to the highly conserved 3′ terminal regions of the four RSV RNAs: 5′-acacaaagtccagaggaaaacaa-3′ for RNA 1, 5′-acacaaagtctgggtataacttctt-3′ for RNA 2, 5′-acacaaagtcctgggtaaaatag-3′ for RNA 3, and 5′-acacaaagtccagggcatttgt-3′ for RNA 4. Second strand cDNAs were synthesized using DNA polymerase I and RNase H. Ends of double strand cDNAs were repaired using T4 DNA polymerase and Klenow DNA polymerase. cDNA pair-end libraries were prepared using standard Illumina protocols and then the libraries were sequenced using Illumina Genome Analyzer II instrument (Illumina, San Diego, CA, USA).
Bioinformatics Analysis
To minimize artificial mistakes from sequencing errors, short reads were pre-processed using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) by removing the low quality reads (below Q30) and adaptors. The reads representing each sample were mapped onto the reference sequences of RSV genome segments (NC_003753.1, NC_003754.1, NC_003755.1, and NC_003776.1) using the MAQ program (Mapping and Assembly with Qualities, Version 0.7.1) with default parameters (Li et al., 2008).
Results
High-throughput Sequencing of RSV Genome from Infected O. sativa and N. benthamiana Plants
The cDNA libraries derived from RSV-infected O. sativa plant and that prepared from RSV-infected N. benthamiana plant were sequenced, using the Illumina/Solexa platform. The sequences have been submitted to Sequence Read Archive (SRA) with accession number SRP051574. A total of 27,965,556 reads were obtained and 6.67% of them were mapped to the RSV genomic sequence. The average sequencing depths of the RSV genome segments in O. sativa were from 953.8 (RNA2) to 2129.1 (RNA4) and those in N. benthamiana were from 3260.2 (RNA1) to 7330.5 (RNA3). The number of mapped bases and the sequencing depth of each RSV RNA segment are shown in Table 1. Our results indicate that the data we obtained are sufficient to represent the RSV populations in a single infected plant.
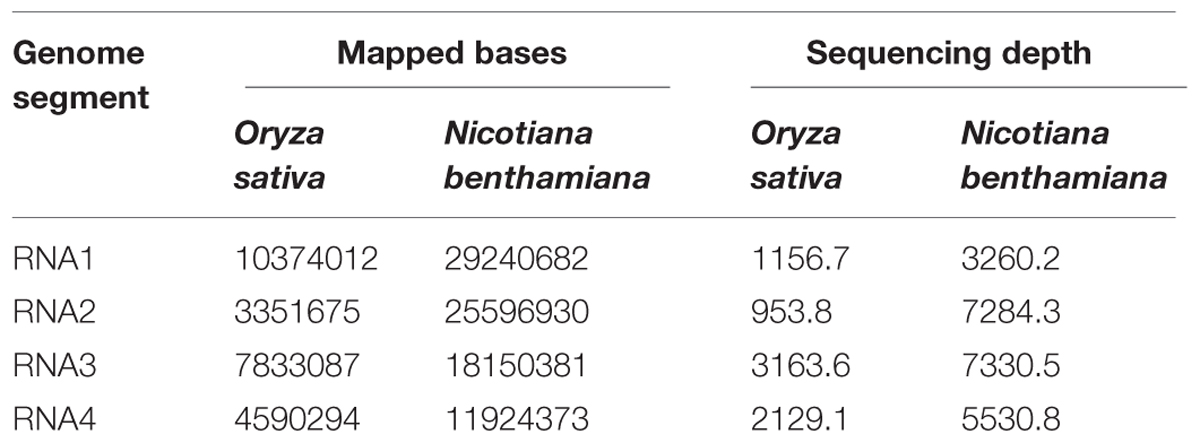
TABLE 1. Number of mapped bases and sequencing depth of RSV genome segments.
Genetic Diversity of RSV Populations in O. sativa
With the high-throughput sequencing results, a “two-dimensional view” of RSV populations is drawn. This includes average nucleotide diversity (number of sites with substitution/number of total sites of corresponding sequence) and substitution ratio within each site (number of substituted nucleotide observed/number of total nucleotide sequenced). Substitutions determined in the RSV populations from the infected O. sativa plant located at 123 different sites in RNA 1 (1.37%), 22 sites in RNA 2 (0.63%), 76 sites in RNA 3 (3.07%), and 60 sites in RNA 4 (2.78%). Among the four RSV RNAs, substitutions occurred more densely in RNA 4. There are 13 sites with substitution ratio above 20% in RSV RNAs and all of them were found in RNA 4, except one at the 3570th site in RNA 1 (Figure 2A). Average nucleotide diversities in the NS3, CP, SP, and MP ORFs were found to be much higher than those in the RdRp, NS2, NSvc2 ORFs and non-coding regions (Figure 3A). Although the average nucleotide diversities in the NS3 and CP ORFs were similar to those in the SP and MP ORFs, the average substitution ratios in these two ORFs were much lower (Figure 3B). Interestingly, the average substitution ratio (5.64%) in non-coding regions was higher than any ratios found in the ORFs analyzed. The average nucleotide diversity in non-coding regions was, however, only 0.89%, lower than all ORFs analyzed except the NS2 (Figure 3).
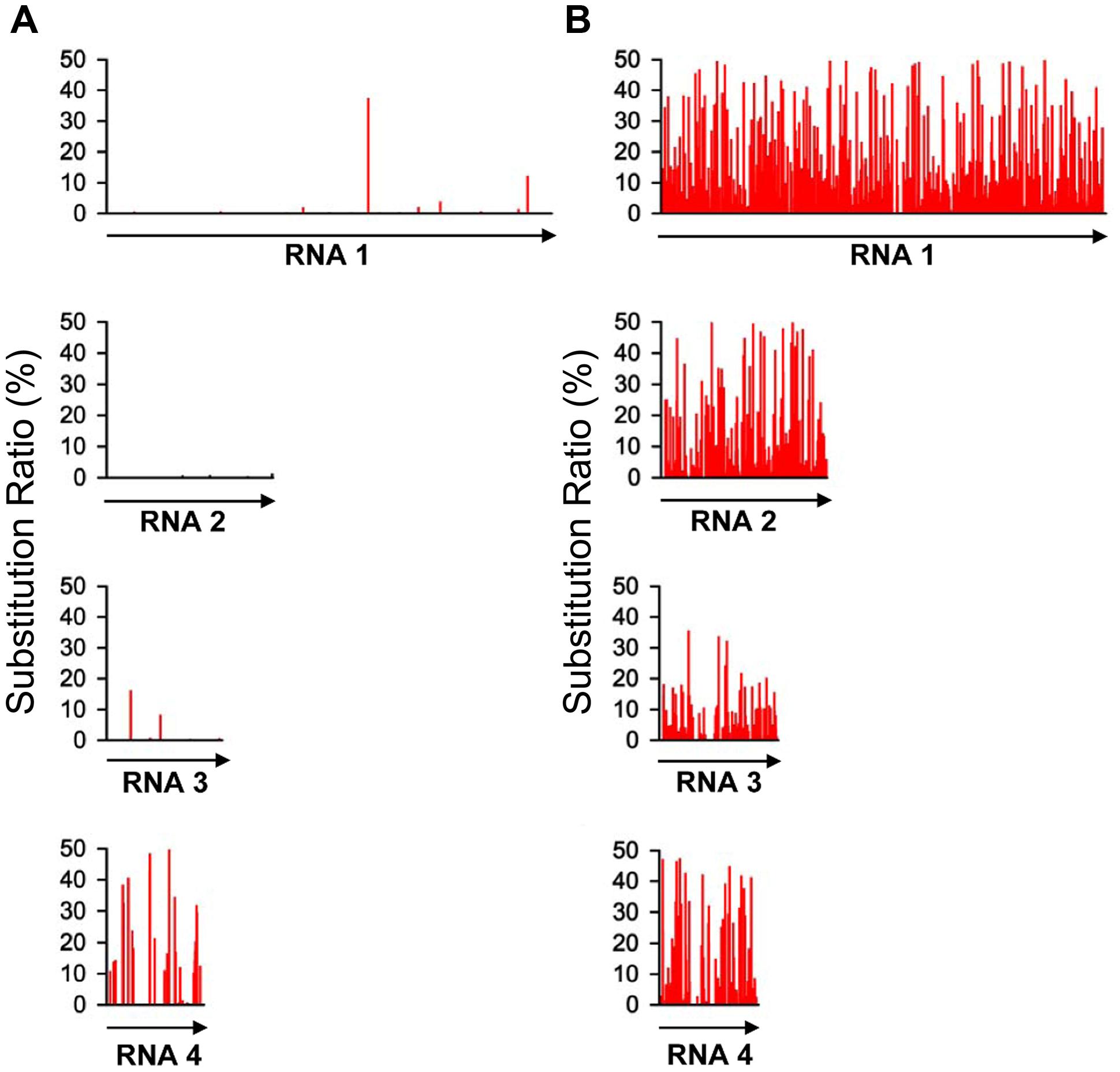
FIGURE 2. Substitution patterns in the RSV genome occurred in Oryza sativa (A) and Nicotiana benthamiana (B). The substitution pattern in each of the four RSV RNA segments is presented. The vertical coordinates represent the substitution ratios derived from the master sequence of each populations at each site (number of substituted nucleotide observed/number of total nucleotide sequenced).
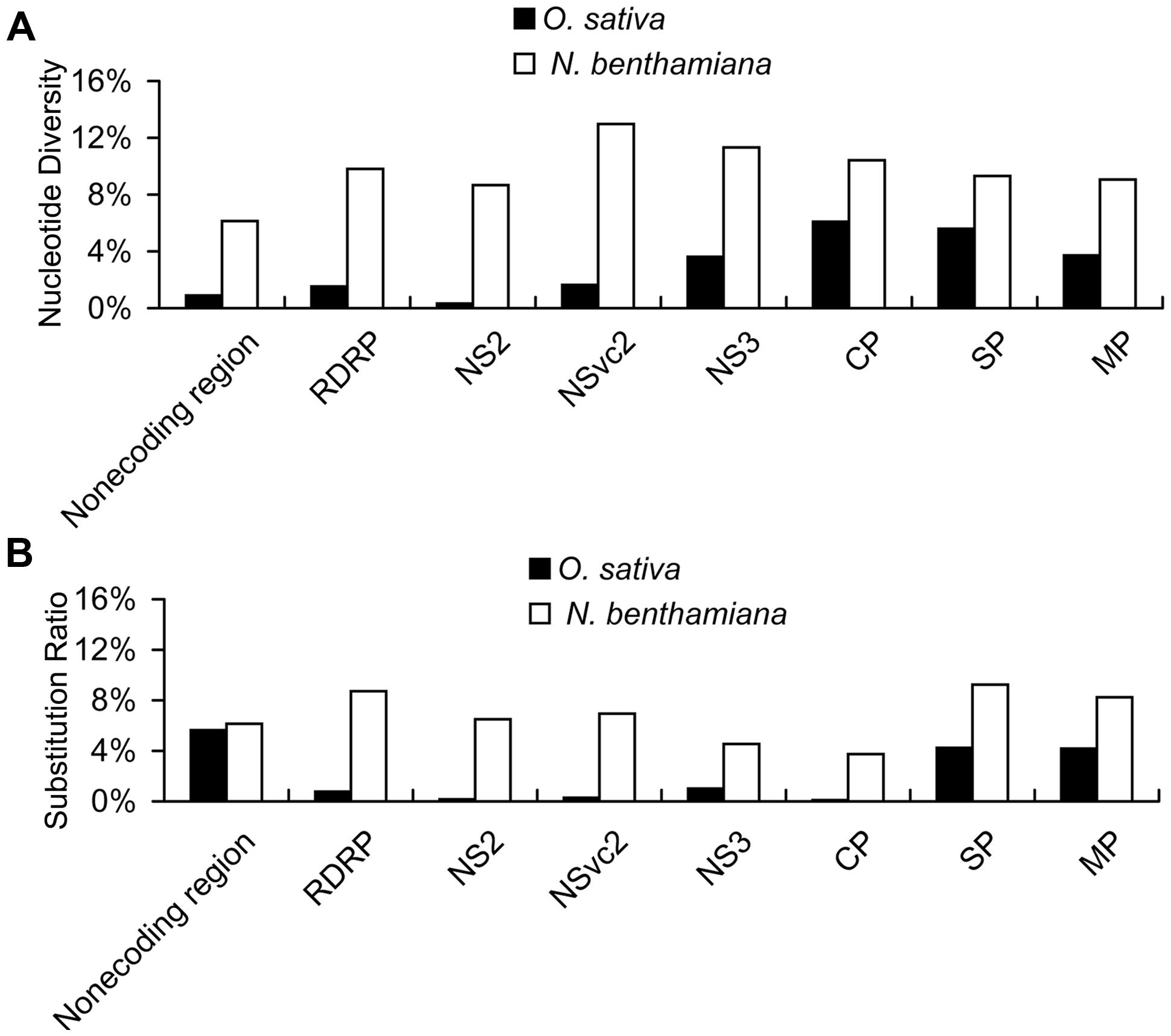
FIGURE 3. Average nucleotide diversity (A) and average substitution ratio (B) of each ORF and non-coding regions in RSV quasispecies from O. sativa and N. benthamiana. Average nucleotide diversity represents number of sites with substitution/number of total sites of corresponding sequence and substitution ratio represents number of substituted nucleotide observed/number of total nucleotide sequenced.
Transitions (i.e., A↔G and C↔T) were detected at 238 sites and transversions (i.e., A ↔ C, A ↔ T, G ↔ C, and G ↔ T) were found at 103 sites. The ratio of transition to transversion was about 2.3 (Table 2). A previous report also indicated that transitions occurred on an average of 2.5 times more frequently than transversions (Lemey et al., 2009). Our result shows that there is no significant difference for substitution tendency among the four transitions (Table 2). At the transition or transversion sites, the average ratio of each substitution varied without clear preferences, e.g., the average ratio of A → T transversion (2.79%) was much higher than A → G transition (0.80%). The average ratio of C → A transversion (2.12%) was similar to C → T transition (2.19%; Table 2).
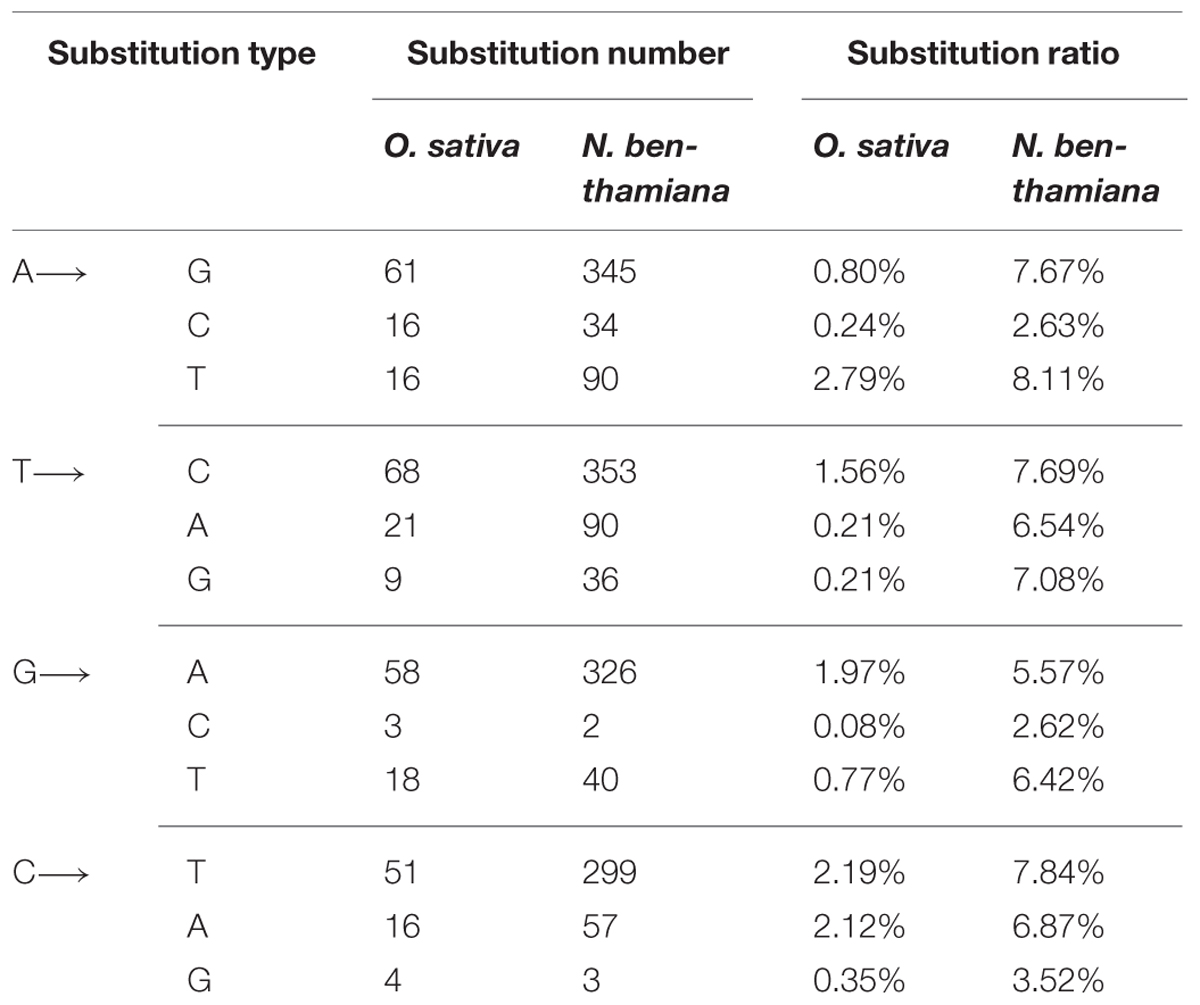
TABLE 2. Substitution tendency among RSV quasispecies in different hosts.
Non-synonymous substitutions were also found in 71 codons located within 6 of the 7 ORFs analyzed. These include 25 codons in RdRp, 7 in NSvc2, 9 in NS3, 12 in CP, 8 in SP, and 10 in MP. No non-synonymous substitution was detected in NS2 ORF. Only 4 out of the 71 codons had substitution ratio above 10%. These include the 150th codon in the NS3 ORF (16.19%, Pro → Leu), the 8th codon in the SP ORF (10.92%, Val → Ile), and the 12th and the 200th codons in the MP ORF (12.62 and 34.45%, Ser → Asn and Ile → Val, respectively; Figure 4A). The sites of synonymous substitutions were 3.5 times higher than those of non-synonymous substitutions. The average substitution ratio of synonymous substitution was 1.64 times higher than that of non-synonymous substitutions.
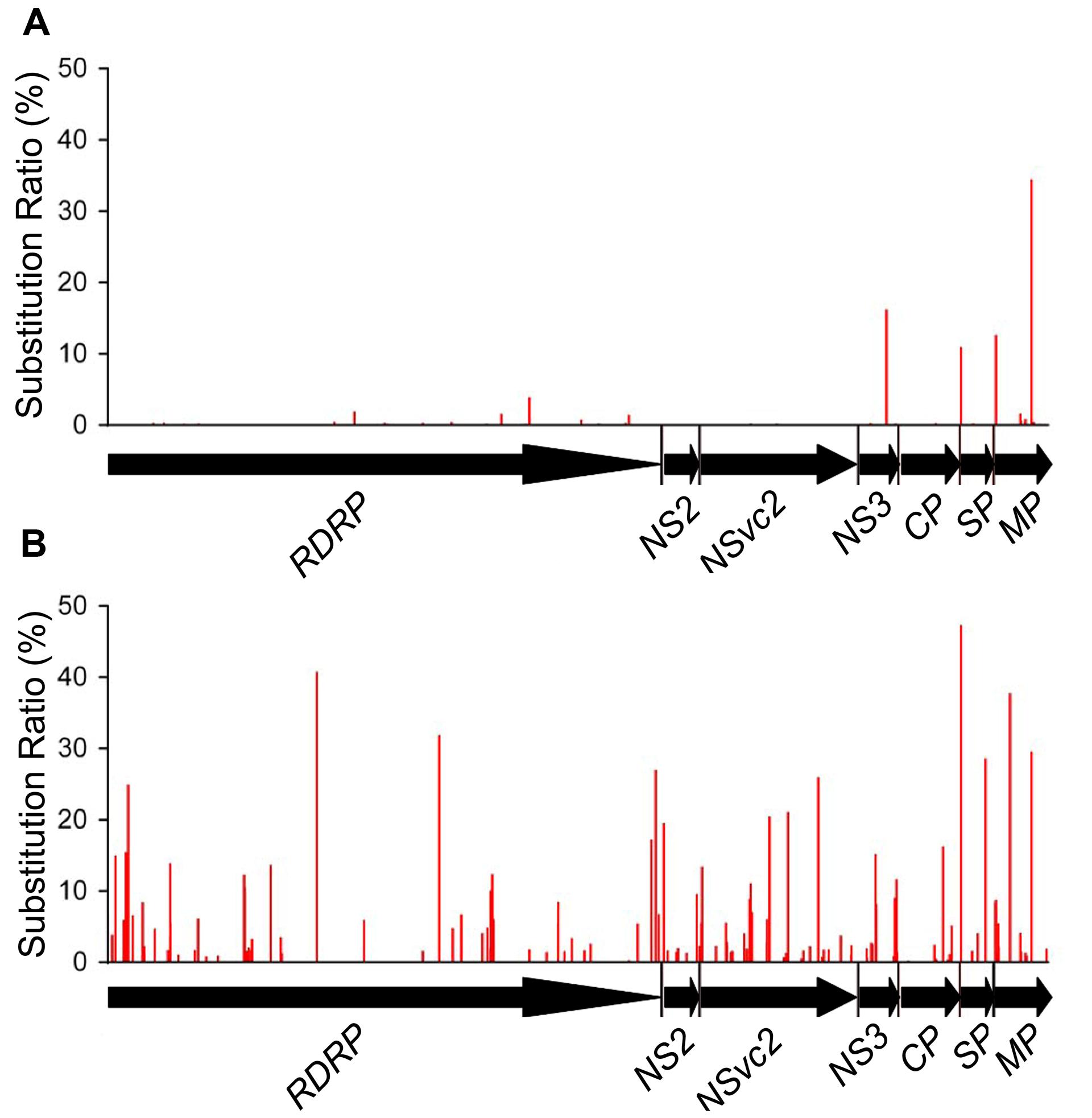
FIGURE 4. Non-synonymous substitution patterns in RSV ORFs from infected O. sativa (A) and N. benthamiana (B) plants. The seven RSV ORFs are shown by horizontal arrows, and the vertical coordinates represent the ratios of substitutions derived from the master sequence of each populations at each site (number of substituted nucleotide observed/number of total nucleotide sequenced).
Genetic Diversity of RSV Populations in N. benthamiana
To investigate the role of host in RSV evolution, the virus was mechanically inoculated in N. benthamiana, an experimental host of RSV. In infected N. benthamiana plant, substitutions were found at 1675 sites in the RSV populations, approximately five times more than those found in the RSV populations from infected O. sativa plant. The average nucleotide diversity and substitution ratio were also very different between these two groups of RSV populations. For RSV populations from N. benthamiana, substitutions were detected in 874 sites in RNA 1 (9.74%), 368 sites in RNA 2 (10.47%), 204 sites in RNA 3 (8.24%), and 149 sites in RNA 4 (6.91%). The substitutions occurred more densely in RNA 1 and 2. There were 206 substitution sites with the substitution ratios above 20%, and more than half of them were found in RNA 1 (Figure 2B).
Our results also indicate that the average nucleotide diversity in each ORF and non-coding regions is higher for RSV populations from N. benthamiana, especially in the RdRp, NS2, and NSvc2 ORFs and the non-coding region. Consequently the differences of average nucleotide diversities among the ORFs and non-coding regions are decreased. The highest average nucleotide diversity is in the NSvc2 ORF (12.97%) and the lowest average nucleotide diversity is in non-coding regions (6.14%; Figure 3A). The average substitution ratios are also increased in all ORFs and non-coding regions analyzed. The highest average substitution ratio is found in the SP ORF (9.25%) and the lowest in the CP ORF (3.75%). Interestingly, the average substitution ratio is not increased in non-coding regions (Figure 3B).
A total of 1323 transition sites and 352 transversion sites were detected in the sequences of RSV populations from N. benthamiana. The transitions occurred on an average of 3.76 times more frequently than the transversions, but no significant difference in frequency was observed for the four transition groups (Table 2). The average substitution ratios of the four transition groups varied without a clear preference. The highest average substitution ratio was for transversion A → T (8.11%) found in RSV populations from O. sativa (Table 2).
Non-synonymous substitutions were found in 147 codons distributed in the seven ORFs. These include 60 non-synonymous substituted codons in RdRp, 6 in NS2, 35 in NSvc2, 16 in NS3, 11 in CP, 6 in SP, and 13 in MP ORF. Twenty-four of the 147 codons had substitution ratios above 10%, and two of them were detected at the eighth codon in the SP ORF and the 200th codon in the MP ORF, respectively. It is worth to mention that the RSV populations from O. sativa also had high substitution ratios at these two codons. There are 24 unique non-synonymous substitutions in the RSV populations from N. benthamiana, likely to be the mutations caused by the interaction between RSV and N. benthamiana (Figure 4B). The average substitution ratios of the synonymous and non-synonymous substitutions were about five times of those found in the RSV populations from O. sativa.
Population Structures of RSV in Different Host Plants
The master sequences of RSV populations from the infected O. sativa and N. benthamiana plants were identified after mapping the reads from the high-throughput sequencing data on the reference sequences of RSV genome segments. There are 156 different (0.9% of genome nucleotides) substitutions between the two master sequences. The master sequence from infected O. sativa plant is composed of 39.09% GC nucleotides while that from the infected N. benthamiana plant is 38.92%. When the substitution tendency bias was analyzed, more transitions than transversions (approximately fourfold) were observed, and there were remarkably more G/C to A/T substitutions than the reverse substitutions (Table 3). After RSV was transmitted from O. sativa to N. benthamiana, the G/C ↔ A/T substitutions varied among the regions in the RSV genome (Table 4). For example the G/C ↔ A/T substitutions were similar in the non-coding regions of the two master sequences. In the coding regions, the G/C → A/T substitutions seemed to occur more often than the reverse substitutions (Table 4). As reported previously, the rice genome is GC rich and rice coding sequences contain 45–75% G and C, whereas the tobacco genome is GC poor and its coding sequences contain 40–60% G and C (Salinas et al., 1988; Matassi et al., 1989; Carels and Bernardi, 2000). The decreased GC level after the virus was transmitted from O. sativa to N. benthamiana suggests that RSV may adjust its codon usage to ensure its efficient protein translation in a new host.
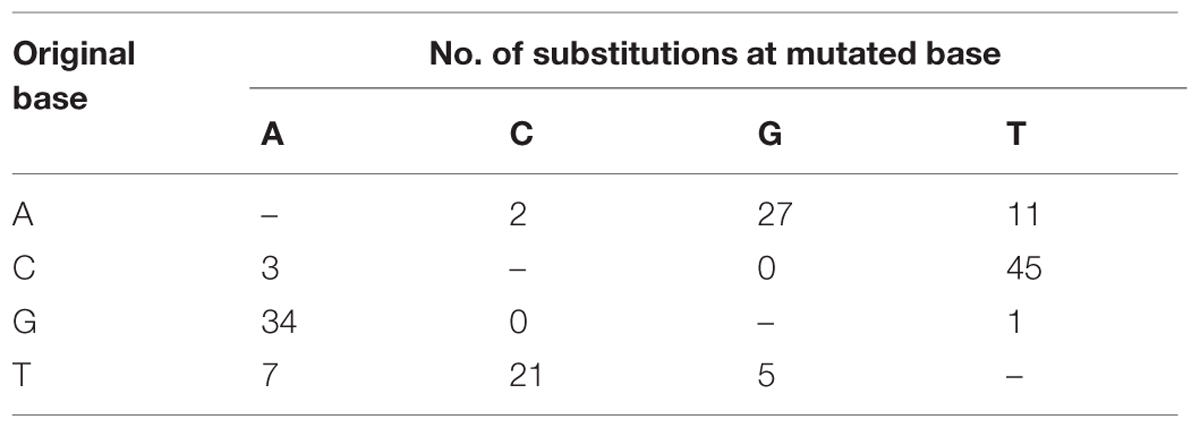
TABLE 3. Substitution tendency of the master sequence after RSV was transmitted from O. sativa to N. benthamiana.
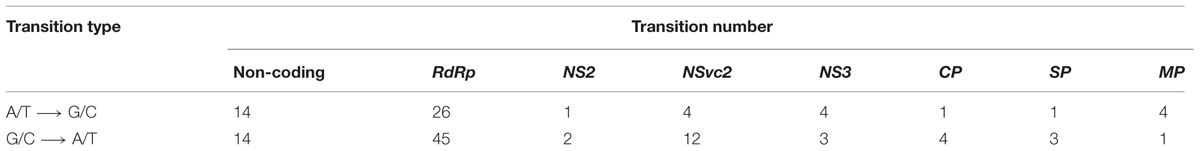
TABLE 4. Number of A/T ↔ G/C substitutions in the master sequence after RSV was transmitted from O. sativa to N. benthamiana.
Discussion
Since the first introduction of quasispecies hypothesis by Eigen and Schuster (1977), this population-based framework has been used extensively to study RNA virus evolution. Based on the complete genomic sequences or partial genomic sequences of RSV isolates, RSV isolates in China were mainly divided into two subtypes: subtype II from Yunnan province and subtype I from rest parts of China, RSV isolates in subtype II were more genetically diverse than those in subtype I, and the mean genetic distance of RSV genes between two subtypes, within subtype I and subtype II ranged from 0.0529 to 0.0865, 0.0123 to 0.0256, and 0.0183 to 0.0387, respectively (Wei et al., 2009; Huang et al., 2013). In this study, we used the high-throughput sequencing technology to analyze and profile RSV populations from infected O. sativa or N. benthamiana plants. Two parameters, average nucleotide diversity and average substitution ratio at single sites, were obtained through high-throughput sequencing and used to evaluate genetic diversities in RSV populations from an infected O. sativa or N. benthamiana plant. For the whole RSV genome, the average nucleotide diversity and average substitution ratio were 1.99% and 1.47% from O. sativa, and 9.79% and 7.05% from N. benthamiana, respectively. RSV isolates in China were reported to have low genetic diversity because of its narrow host range (Wei et al., 2009). Our study find the diversity of RSV populations increased after RSV was transmitted from O. sativa to N. benthamiana. N. benthamiana might provide quite different select pressure from O. sativa, and this observation also gave a direct evidence that virus might allow higher genetic diversity for host adaptation (Fargette et al., 2006).
The two parameters are independent of each other regardless of the origin of the two virus populations. For example, the average nucleotide diversity in the CP ORF was the highest in the RSV populations from the infected O. sativa plant, whereas the average substitution ratio in this ORF was much lower than some of the other ORFs (Figure 3). Also transitions were much higher than transversions in the RSV populations from both infected O. sativa and N. benthamiana plants (Table 4), but the average substitution ratio did not have difference (Figure 2). These results indicate that these two parameters are likely driven by different factors.
The genetic diversity in RNA virus populations was considered to be controlled by the interactions between host and viral factors (Schneider and Roossinck, 2001). So there were at least two steps during RNA virus evolution: substitutions emerged via replication errors and the selection of the original and substituted bases by host. During the second step, the accumulation of the less adaptive one might be repressed by host–virus interaction. So if host–virus interaction is in favor of the substituted base, it will accumulate more than the original base and eventually replace the original one. Otherwise, the substituted base will accumulate at lower level or even be eliminated. So substitution ratio reveals the destiny of each substitutions under host selection. Thus the sites with high substitution ratios may be essential in virus evolution in corresponding host. In this study, sites with high substitution ratio in O. sativa and N. benthamiana were documented. Further work is necessary to confirm their function in virus evolution and host–virus interaction.
The observation that the average substitution ratio of non-coding regions was not changed after RSV was transmitted form O. sativa to N. benthamiana indicates that the RSV non-coding sequences may not play an important role in RSV–host plant interactions. On the other hand both synonymous and non-synonymous substitution ratios were remarkably increased after RSV was transmitted form O. sativa to N. benthamiana. This observation suggests that both synonymous and non-synonymous substitutions are also essential in RSV–host plant interactions. Possible scenarios that may be used to explain our hypothesis are that the non-synonymous substitutions may alter features of RSV protein(s) to better interact with specific host factors during its infection, e.g., replication and movement in plant, while synonymous substitutions alter the codon composition to accord with host codon usage bias.
Conclusion
We find the RSV populations from infected N. benthamiana plant is more diverse than that obtained from rice and the high-throughput sequencing technology is a powerful technology to investigate population genetic diversity of plant RNA viruses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the National High Technology Research and Development Program of China (2012AA101505), the earmarked fund for Modern Agro-industry Technology Research System (CARS-01-19) and the National Natural Science of China (31272015).
References
Domingo, E., and Holland, J. J. (1997). RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151–178. doi: 10.1146/annurev.micro.51.1.151
Eigen, M., and Schuster, P. (1977). The hypercycle. a principle of natural self-organization. Part A: emergence of the hypercycle. Naturwissenschaften 64, 541–565. doi: 10.1007/BF00450633
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Elena, S. F., and Sanjuán, R. (2007). Virus evolution: Insights from an experimental approach. Annu. Rev. Ecol. Evol. Syst. 38, 27–52. doi: 10.1146/annurev.ecolsys.38.091206.095637
Falk, B. W., and Tsai, J. H. (1998). Biology and molecular biology of viruses in the genus Tenuivirus. Annu. Rev. Phytopathol. 36, 139–163. doi: 10.1146/annurev.phyto.36.1.139
Fargette, D., Konate, G., Fauquet, C., Muller, E., Peterschmitt, M., and Thresh, J. M. (2006). Molecular ecology and emergence of tropical plant viruses. Annu. Rev. Phytopathol. 44, 235–260. doi: 10.1146/annurev.phyto.44.120705.104644
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garcia-Arenal, F., Fraile, A., and Malpica, J. M. (2001). Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 39, 157–186. doi: 10.1146/annurev.phyto.39.1.157
Garcia-Arenal, F., Fraile, A., and Malpica, J. M. (2003). Variation and evolution of plant virus populations. Int. Microbiol. 6, 225–232. doi: 10.1007/s10123-003-0142-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, L. Z., Rao, L. X., Zhou, X. P., and Wu, J. X. (2013). Genetic variability and evolution of rice stripe virus. J. Zhejiang Univ. Sci. B 14, 875–885. doi: 10.1631/jzus.B1300014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kakutani, T., Hayano, Y., Hayashi, T., and Minobe, Y. (1991). Ambisense segment 3 of rice stripe virus: the first instance of a virus containing two ambisense segments. J. Gen. Virol. 72, 465–468. doi: 10.1099/0022-1317-72-2-465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koganezawa, H., Doi, Y., and Yora, K. (1975). Purification of Rice stripe virus. Ann. Phytopathol. Soc. Jpn. 41, 148–154. doi: 10.3186/jjphytopath.41.148
Kong, L., Wu, J., Lu, L., Xu, Y., and Zhou, X. (2014). Interaction between Rice stripe virus disease-specific protein and host PsbP enhances virus symptoms. Mol. Plant 7, 691–708. doi: 10.1093/mp/sst158
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lauring, A. S., and Andino, R. (2010). Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 6:e1001005. doi: 10.1371/journal.ppat.1001005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lemey, P., Salemi, M., and Vandamme, A. M. (2009). The Phylogenetic Handbook: a Practical Approach to Phylogenetic Analysis and Hypothesis Testing. Cambridge: Cambridge University Press. doi: 10.1017/CBO9780511819049
Li, H., Ruan, J., and Durbin, R. (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858. doi: 10.1101/gr.078212.108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, Q. Y., Xie, L. H., Zhou, Z. J., Xie, L. Y., and Wu, Z. J. (1990). Studies on rice stripe: i. distribution of and losses caused by the disease. J. Fujian Agric. Coll. 19, 421–425.
Matassi, G., Montero, L. M., Salinas, J., and Bernardi, G. (1989). The isochore organization and the compositional distribution of homologous coding sequences in the nuclear genome of plants. Nucleic Acids Res. 17, 5273–5290. doi: 10.1093/nar/17.13.5273
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nagy, P. D., and Simon, A. E. (1997). New insights into the mechanisms of RNA recombination. Virology 235, 1–9. doi: 10.1006/viro.1997.8681
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramirez, B. C., and Haenni, A. L. (1994). Molecular biology of Tenuiviruses, a remarkable group of plant viruses. J. Gen. Virol. 75, 467–475. doi: 10.1099/0022-1317-75-3-467
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roossinck, M. J. (1997). Mechanisms of plant virus evolution. Annu. Rev. Phytopathol. 35, 191–209. doi: 10.1146/annurev.phyto.35.1.191
Salinas, J., Matassi, G., Montero, L. M., and Bernardi, G. (1988). Compositional compartmentalization and compositional patterns in the nuclear genomes of plants. Nucleic Acids Res. 16, 4269–4285. doi: 10.1093/nar/16.10.4269
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanjuan, R., Nebot, M. R., Chirico, N., Mansky, L. M., and Belshaw, R. (2010). Viral mutation rates. J. Virol. 84, 9733–9748. doi: 10.1128/JVI.00694-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schneider, W. L., and Roossinck, M. J. (2000). Evolutionarily related Sindbis-like plant viruses maintain different levels of population diversity in a common host. J. Virol. 74, 3130–3134. doi: 10.1128/JVI.74.7.3130-3134.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schneider, W. L., and Roossinck, M. J. (2001). Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J. Virol. 75, 6566–6571. doi: 10.1128/JVI.75.14.6566-6571.2001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takahashi, M., Toriyama, S., Hamamatsu, C., and Ishihama, A. (1993). Nucleotide sequence and possible ambisense coding strategy of rice stripe virus RNA segment 2. J. Gen. Virol. 74, 769–773. doi: 10.1099/0022-1317-74-4-769
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Toriyama, S., Takahashi, M., Sano, Y., Shimizu, T., and Ishihama, A. (1994). Nucleotide sequence of RNA 1, the largest genomic segment of rice stripe virus, the prototype of the Tenuiviruses. J. Gen. Virol. 75, 3569–3579. doi: 10.1099/0022-1317-75-12-3569
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, G., Zhou, Y., Chen, Z., and Zhou, X. (2004). Production of monoclonal antibodies to Rice stripe virus and application in virus detection. Acta Phytopathol. Sin. 34, 302–306.
Wei, T. Y., Yang, J. G., Liao, F. R., Gao, F. L., Lu, L. M., Zhang, X. T.,et al. (2009). Genetic diversity and population structure of rice stripe virus in China. J. Gen. Virol. 90, 1025–1034. doi: 10.1099/vir.0.006858-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Worobey, M., and Holmes, E. C. (1999). Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 80, 2535–2543.
Xiong, R., Wu, J., Zhou, Y., and Zhou, X. (2008). Identification of a movement protein of the Tenuivirus rice stripe virus. J. Virol. 82, 12304–12311. doi: 10.1128/JVI.01696-08
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xiong, R., Wu, J., Zhou, Y., and Zhou, X. (2009). Characterization and subcellular localization of an RNA silencing suppressor encoded by rice stripe Tenuivirus. Virology 387, 29–40. doi: 10.1016/j.virol.2009.01.045
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, Y., Hayakawa, T., and Toriyama, S. (1992). Complete nucleotide sequence of RNA 4 of rice stripe virus isolate T, and comparison with another isolate and with maize stripe virus. J. Gen. Virol. 73, 1309–1312. doi: 10.1099/0022-1317-73-5-1309
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Rice strip virus, population, diversity, deep sequencing
Citation: Huang L, Li Z,Wu J, Xu Y, Yang X, Fan L, Fang R and Zhou X (2015) Analysis of genetic variation and diversity of Rice stripe virus populations through high-throughput sequencing. Front. Plant Sci. 6:176. doi: 10.3389/fpls.2015.00176
Received: 31 October 2014; Accepted: 05 March 2015;
Published online: 24 March 2015.
Edited by:
Joanna Marie-France Cross, Inonu, TurkeyReviewed by:
Abdelaleim Ismail ElSayed, Zagazig University, EgyptAngelantonio Minafra, CNR Institute for Sustainable Plant Protection, Italy
Shahin S. Ali, United States Department of Agriculture, USA
Copyright © 2015 Huang, Li, Wu, Xu, Yang, Fan, Fang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueping Zhou, State Key Laboratory of Rice Biology, Institute of Biotechnology, Zhejiang University, Hangzhou 310058, People’s Republic of Chinaenpob3VAemp1LmVkdS5jbg==; Rongxiang Fang, State Key Laboratory of Plant Genomics, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, People’s Republic of ChinaZmFuZ3J4QGltLmFjLmNu
†These authors have contributed equally to this work.