 Gang Wu1†
Gang Wu1† Maoteng Li
Maoteng Li- 1School of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China
- 2National Key Lab of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, China
Raphanus sativus is an important Brassicaceae plant and also an edible vegetable with great economic value. However, currently there is not enough transcriptome information of R. sativus tissues, which impedes further functional genomics research on R. sativus. In this study, RNA-seq technology was employed to characterize the transcriptome of leaf tissues. Approximately 70 million clean pair-end reads were obtained and used for de novo assembly by Trinity program, which generated 68,086 unigenes with an average length of 576 bp. All the unigenes were annotated against GO and KEGG databases. In the meanwhile, we merged leaf sequencing data with existing root sequencing data and obtained better de novo assembly of R. sativus using Oases program. Accordingly, potential simple sequence repeats (SSRs), transcription factors (TFs) and enzyme codes were identified in R. sativus. Additionally, we detected a total of 3563 significantly differentially expressed genes (DEGs, P = 0.05) and tissue-specific biological processes between leaf and root tissues. Furthermore, a TFs-based regulation network was constructed using Cytoscape software. Taken together, these results not only provide a comprehensive genomic resource of R. sativus but also shed light on functional genomic and proteomic research on R. sativus in the future.
Introduction
As an important Brassicaceae plant with great economic value, Raphanus sativus was widely cultivated throughout the world. It was also reported that R. sativus has been used as a medicinal plant due to its exceptional medicinal properties (Watt and Breyer-Brandwijk, 1962). For instance, the radish roots contain various peroxidases and could be used for medicinal purposes (Wang et al., 2002). With the development of next-generation sequencing technology, genetic information of R. sativus was gradually revealed in the recent years (Wang et al., 2013a; Xu et al., 2013; Zhang et al., 2013). For example, the transcriptome profiles of leaf tissue of R. sativus were analyzed by RNA-seq technology in our previous study. However, only 22 million clean reads were used and the unigenes generated in this study only represents a small proportion of R. sativus transcriptome because the sequencing coverage was simply not deep enough (Zhang et al., 2013). Therefore, there is an urgent need to analyze more transcriptome data of R. sativus tissues, which will facilitate their potential use as general reference for other related R. sativus studies.
Next-generation sequencing technology has provided a novel method both for gene mapping and transcriptome analysis (Schuster, 2008; Shendure and Ji, 2008; Wang et al., 2009). RNA-seq is not dependent on prior information of the genomic sequence of the target species, which has been widely applied for transcriptome-related studies in many Brassicaceae plant species (Paritosh et al., 2013; Wang et al., 2013b; Kim et al., 2014; Mudalkar et al., 2014). The de novo assembly of sequencing reads is an important step to obtain genome information, such as novel gene discovery, transcription factor (TF) discovery, Simple Sequence Repeat (SSR) mining, and gene expression profile analysis (Powell et al., 1996; Bouché et al., 2002; Heim et al., 2003; Jiao et al., 2003; Knowles and McLysaght, 2009; Zhang et al., 2012b). For example, it has been reported that 30 TF families containing approximately 1500 potential TFs were identified after the completion of the Arabidopsis thaliana genome sequencing project (Riechmann et al., 2000; Mitsuda and Ohme-Takagi, 2009). Also, SSRs are abundantly applied in molecular mapping, genetic diversity research, cultivar fingerprinting due to multiple advantages including co-dominant inheritance nature, high variability, high feasibility of detection, and good transferability (Li et al., 2004). Next-generation sequencing technology for SSR mining has been used in various plant species such as rubber tree (Li et al., 2012), castor bean (Qiu et al., 2010), sesame (Zhang et al., 2012a), and some Brassicaceae plants (Wang et al., 2007; Yamane et al., 2009; Kamei et al., 2010; Ohsako et al., 2010). However, so far, little has been reported about the TF and SSR profiles due to limited genome information in R. sativus.
In this study, we applied the RNA-Seq technology to deeply sequence the transcriptome of leaf tissue in R. sativus. A total of 70 million clean pair-end reads were obtained and used for the de novo assembly of transcriptome of R. sativus. The sequencing coverage was comprehensive enough to identify most unigenes and major metabolic pathways. The de novo assembly obtained a total of 68,086 unigenes with an average length of 576 bp. We further downloaded root sequencing data (SRX256970 and SRX263753) and merged the clean reads of leaf and root sequencing data for de novo assembly using Trinity and Oases programs. The Oases-assembled unigenes were used for functional annotation and downstream analyses. Furthermore, 31,875 potential SSRs with different motifs were detected. Finally, a large number of transcription factors and enzymes were identified by comparing with available databases. These results provide a comprehensive genomic resource for gene expression analysis and functional genomics research on R. sativus in the future.
Materials and Methods
Plant Material and Transcriptome Sequencing
The R. sativus big root radish (No specific permissions were required to use this R. sativus species) was provided by Prof. Jiangsheng Wu of Huazhong Agricultural University, which was cultivated in the experimental field of Huazhong University of Science and Technology in the year of 2012 and 2013. The method for total RNA isolation of leaf tissues was according to the methods that described earlier (Zhang et al., 2013). The isolated RNAs were prepared for the construction of RNA-seq libraries. In short, the mRNAs collected from leaf tissues of R. sativus were first fragmented and further used to synthesize first-strand cDNAs with hexamer and reverse transcriptase (Promega). Subsequently, the second-strand cDNAs were synthesized with DNA polymerase I and RNase H. The obtained cDNA fragments were then purified, end-repaired, A-tailed, and ligated to index adapters (Illumina). The ligation products were amplified by PCR and sequenced using the Illumina GAIIx and a 100 bp pair-end sequencing protocol was employed.
De novo Transcriptome Assembly of R. sativus
Raw reads were initially processed by Illumina Pipeline Software to get sequence information. Then the reads were cleaned by removing adaptor sequences and low quality reads with ambiguous sequences “N.” The Trinity program (Grabherr et al., 2011) was carried out to assemble the clean reads to obtain non-redundant unigenes. Clean reads with a certain overlaps were combined to form longer contiguous sequences (contigs), which were joined into scaffolds that were further assembled by gap filling to acquire unigenes. Transcriptome coverage is highly variable owing to differential gene expression in cells, so there is no single absolutely optimal k-mer length for transcriptome assembly. In this study, 25 was set as the default k-mer size for the de novo assembly of the transcriptome, while other parameters used default values. The length of the assembled unigenes for further study should be no less than 200. For Oases assembly, Velvet was first run using k-mer length of 31 along with other default parameters. The contigs produced by Velvet were post-processed using Oases with k-mer of 31.
Transcriptome Annotation of R. sativus
All assembled unigenes were compared with the NCBI non-redundant protein (Nr), Swissprot, Pfam and Trembl databases by using blastx with a typical cut-off E-value of 1 e−5 to search for homologs. Based on the results of the Nr database annotation, BLAST2GO (Conesa et al., 2005) was used to obtain Gene Ontology (GO) annotation of assembled unigenes for describing cellular component, molecular function, and biological process. WEGO (Ye et al., 2006) was employed to perform the GO functional classification for understanding the distribution of gene functions at the macro level. The unigenes were also searched against the Cluster of Orthologous Groups (COG) database to classify their functions. The KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway of the assembled unigenes was annotated by mapping the sequences obtained from BLAST2GO to the contents of the KEGG metabolic pathway database (http://david.abcc.ncifcrf.gov/).
Identification of Differentially Expressed Genes in the Leaf and Root Transcriptomes
Reads Per kb per Million reads (RPKM) was used to calculate gene expression level (Mortazavi et al., 2008). The differentially expressed genes (DEGs) was determined with a log-fold expression change (log FC) greater than 2 or less than −2 using a threshold of false discovery rates (FDR < 0.001) and a greater statistically significant value (P < 0.005).
SSR Mining of R. sativus
The SSR mining was performed following the method described previously (Zhang et al., 2012a). In this study, the SSRs were considered to include with five motifs: dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide. For each motif of SSR, the number of contiguous repeat units was more than five. Statistical analysis was performed to summarize the number of SSRs with each type of motif and the length distribution of repeat units.
Identification of Transcription Factors of R. sativus
In order to identify the transcription factors represented in R. sativus transcriptome, all assembled unigenes were searched against the plant transcription factor database (PlnTFDB; http://plntfdb.bio.uni-potsdam.de/v3.0/downloads.php) by using blaxtx with a cut-off E-value of 1 e−5 (Kalra et al., 2013).
Detection of Enzymes of R. sativus
For predicting the potential enzymes in the unigenes, the transcripts were probed against all the enzyme protein sequences at Enzyme nomenclature database (ENZYME; http://enzyme.expasy.org/) by using blaxtx with a cut-off E-value of 1 e−5 (Kalra et al., 2013).
Transposable Element Analysis
We run RepeatMasker (http://www.repeatmasker.org/) with options “quick search” and species “Arabidopsis” to identify repetitive and transposable elements (TE) in R. sativus transcriptome. RepeatMasker version open-3.2.9, rmblastn version (1.2) 2.2.23 and RepBase update 20090604 were used in this study.
Results and Discussion
RNA-Seq and de novo Transcriptome Assembly of R. sativus
Next generation sequencing technology has been widely applied to characterize the transcriptome profiles in multiple non-model plants. Our previous study has analyzed the transcriptome of leaf tissues in R. sativus by RNA sequencing (Zhang et al., 2013). However, due to the low coverage of sequencing reads, this study had been not portrayed as a representative transcriptome assembly project but rather a survey of leaf tissues transcripts. Therefore, to obtain highly reliable de novo transcriptome assembly of leaf tissues in R. sativus, we reconstructed a cDNA library and performed transcriptome sequencing using Illumina sequencing platform. Initially, the base quality of the sequenced reads was evaluated. Approximately, 95,847,818 raw pair-end reads were generated, resulting in a total of 70,879,904 clean reads with a length of 90 bp. We further mapped the clean reads to Brassica rapa genome and found that the clean reads were mainly distributed in the coding sequence region (Figure 1A), which indicated that transcriptome sequencing had good quality. All the clean reads were de novo assembled by Trinity programs (Grabherr et al., 2011), which generated a total of 68,086 unigenes with an average length of 576 bp and an N50 of 773 bp (Figure 1B). Among the unigenes, the shortest and longest unigenes are 201 and 7884 bp, respectively. Moreover, 34,090 unigenes were within the 200–400 bp, and 24,262 unigenes were within the 400–1000 bp. In the meanwhile, we observed that part of unigenes (9734) were over 1000 bp, which is very helpful to further annotation and functional analysis of unigenes. Meanwhile, we made a comparison of de novo assembly between the earlier and current sequencing data from leaf tissues. A better de novo assembly was obtained in current sequencing data and the results are shown in Supplementary Table S1.
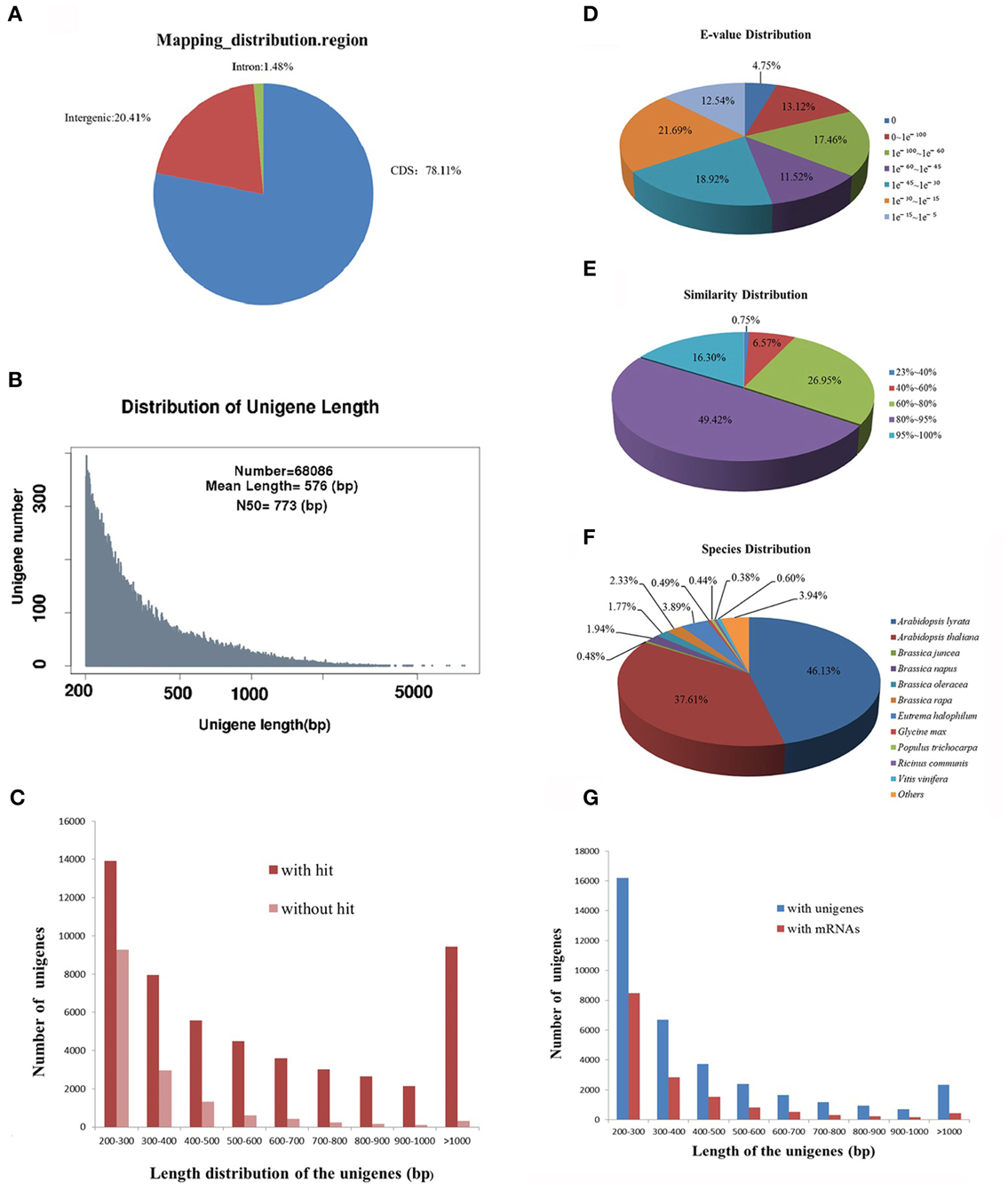
Figure 1. Sequencing, de novo assembly and annotation of leaf transcriptome of R. sativus transcriptome. (A) The clean reads distribution map of leaf tissue on B. rapa genome. (B) The de novo assembly result of leaf transcriptome. (C) Comparative analysis of unigenes length with or without hit against Nr and Swissprot databases. (D) E-value distribution of BLAST hits for each unique sequence against the Nr database. (E) Similarity distribution of the top BLAST hits for each sequence against the Nr database. (F) Species distribution is shown as a percentage of the total homologous sequences against the Nr database. (G) Comparative analysis of the leaf unigenes with the unigene and mRNA databases of R. sativus.
In order to assess the quality of de novo assembly of R. sativus, the clean reads were mapped to the assembled unigenes. We observed that 45,296,360 (64%) reads were mapped to the assembled unigenes. Among the mapped reads, 29,997,587 (66.23%) reads could uniquely map to the unigenes, while 15,298,773 (33.77%) reads could map to multiple locations on unigenes. In addition, to estimate the level of transcripts coverage of R. sativus, indirect evaluation was performed as described (Zhang et al., 2013). The result showed that the de novo assembly generated in this study was estimated to cover 47.2% of the R. sativus transcriptome. Given that only one tissue (leaf) was used for sequencing and de novo assembly, 47.2% trasncriptome coverage should be appropriate for the annotation and functional research in the future.
For further homology analysis, we compared the assembled leaf unigenes with the CDS sequences of A. thaliana, B. rapa and Thellungiella halophila. Among the 68,086 unigenes, a total of 53,384, 56,695, and 54,003 sequences were mapped to the CDS region in A. thaliana, B. rapa, and T. halophila, respectively. The identity of the significant hits against A. thaliana ranged from 77.65 to 100%, with an average value of 88.61% and an N50-value of 88.67%. The identity of the significant hits against B. rapa varied from 77.97 to 100%, with an average value of 93.02% and an N50-value of 93.6%. While the identity of the hits against T. halophila ranged from 77.54 to 100%, with an average value of 89.71% and an N50-value of 89.94%. In the meanwhile, we also compared the leaf unigenes with the mRNA sequences of A. thaliana, B. rapa, and T. halophila. Almost similar results were obtained as shown in Supplementary Table S2. Collectively, the results indicated that the leaf transcriptome of R. sativus has high similarity with those of other Brassicaceae species.
The assembled leaf unigenes were annotated by using BLASTx against the NCBI non-redundant protein (Nr) database, the Swiss-Prot protein database, Pfam database, Nt database and Trembl database with an E-value cutoff of 1 e−5. The mapping rates of the annotated unigenes were between 52 and 87% (Table 1). Among the 68,086 unigenes, 52,677 (77.37%) had at least one significant match with an E-value less than 1 e−5 against the Nr database. The mapping rates of unigenes against the Swissprot and Trembl protein databases were 52.75 and 77.15%, respectively. The results indicated that most of the unigenes are protein coding genes. More than 90% of the unigenes over 700 bp in length had homology matches, whereas only less than 60% of the unigenes shorter than 300 bp had significant matches (Figure 1C). These short unigenes may lack a known conserved functional domain or represent non-coding RNAs so that they had no significant matches. Alternatively, these unigenes may contain a known protein domain but too short to display sequence matches, resulting in false-negative results. Moreover, the genomic and transcriptomic information is limited for R. sativus, and many R. sativus lineage-specific genes might not be contained in the database. Based on the annotation results of the Nr database, the E-value distribution analysis showed that 65.77% of the matched sequences had strong homology with the E-value < 1 e−30, and 46.85% of the matched sequences showed strong homology with the E-value < 1 e−45, whereas only 34.23% of the matched sequences had high similarity with the E-value from 1 e−30 to 1 e−5 (Figure 1D). The similarity distribution analysis revealed that 65.72% of the mapped sequences had a similarity higher than 80%, whereas 34.28% of mapped sequences had a similarity ranging from 23 to 80% (Figure 1E). The species distribution analysis revealed that leaf tissue in R. sativus has a number of homologous sequences in many plant species (Figure 1F). For instance, Arabidopsis lyrata genes have the highest similarity with R. sativus unigenes among the various plant species (46.13%), followed by A. thaliana (37.61%), Eutrema halophilum (3.89%), B. rapa (2.33%), Brassica napus (1.94%), and Brassica oleracea (1.77%). This result suggested that the genome of A. lyrata or A. thaliana can be used as a reference for the transcriptome analysis of leaf tissue in R. sativus.

Table 1. Blast results of the assembled leaf unigenes of R. sativus.
In order to validate redundancy of the sequences with publicly available data, we compared the leaf unigenes with the R. sativus unigene database on the Genoscope (http://www.ncbi.nlm.nih.gov/unigene/) using blastn with an E-value cutoff of 1 e−10. Among the 68,086 unigenes, 32,255 (47.37%) had significant similarity to R. sativus unigene database. Meanwhile, we found that a total of 35,831 (52.63%) unigenes had no significant match, which indicated that they were probably the non-redundant unigenes (Zhang et al., 2012b). The length of the non-redundant unigenes varied from 201 to 3594 bp, with an average value of 442 bp and an N50-value of 320 bp (Figure 1G). In addition, the leaf unigenes were blasted against the R. sativus mRNA database with an E-value cutoff of 10−10 for identifying novel transcribed sequences. A total of 15,729 (23.11%) unigenes had no match with the R. sativus mRNA database, and were considered potential novel transcribed sequences. The length of the novel transcribed sequences varied from 201 to 3044 bp, with an average length of 368 bp and an N50-value of 283 bp (Figure 1G).
Given that there are available transcriptome sequencing data of root tissue in R. sativus (Wang et al., 2013a), we thus downloaded root data (SRX256970 and SRX263753) and merged the clean reads of leaf and root sequencing data for de novo assembly using Trinity (Grabherr et al., 2011) and Oases programs (Schulz and Zerbino, 2010). The Trinity assembly generated a total of 103,222 unigenes with an average length of 786 bp and an N50 of 1250 bp (Supplementary Table S3). Among the unigenes, the shortest and longest unigenes are 201 and 15,670 bp, respectively. Statistical analysis showed that 44,479 unigenes were within the 200–400 bp, and 32,168 unigenes were within the 400–1000 bp. Moreover, we observed that 26,575 unigenes were over 1000 bp. Meanwhile, the Oases assembly generated a total of 106,874 unigenes with an average length of 1108 bp and an N50 of 1770 bp. The assembled unigenes generated from Trinity and Oases were deposited in NCBI TSA database with accession number GBZO01 and GBZP01, respectively. Also, the comparison analysis between Trinity and Oases assembly were summarized in Table 2. As the unigenes with longer average length were generated using Oases than Trinity, we thus used Oases-assembled unigenes for further annotation and downstream analyses. The Oases-assembled unigenes were annotated by using BLASTx against the NCBI non-redundant protein (Nr) database, COG database, Swiss-Prot protein database, and Pfam database with an E-value cutoff of 1 e−5. The mapping rates of the annotated unigenes were listed in Table 3.
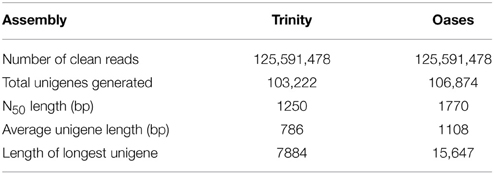
Table 2. De novo assembly analysis using Trinity and Oases.

Table 3. Blast results of Oases-assembled R. sativus unigenes.
Functional Classification of R. sativus Unigenes
Based on sequence homology, 46,586 Oases-assembled R. sativus unigenes were assigned with GO terms. The annotated GO terms were classified into 45 functional groups, which were distributed under the three main categories including molecular function, biological process and cellular component (Figure 2). Within the molecular function category, “catalytic,” “binding” and “transporter” were top three GO terms. Among the biological process, “cellular process,” “metabolic process,” “response to stimulus,” “biological regulation,” “localization,” and “establishment of localization” were major GO terms. In the cellular component, “cell,” “cell part,” “organelle,” and “organelle part” were mainly enriched. Furthermore, only a few unigenes were classified in terms of “synapse,” “synapse part,” “virion,” “virion part,” “protein tag,” “cell killing,” “auxiliary transport protein activity,” and “metallochaperone activity.”
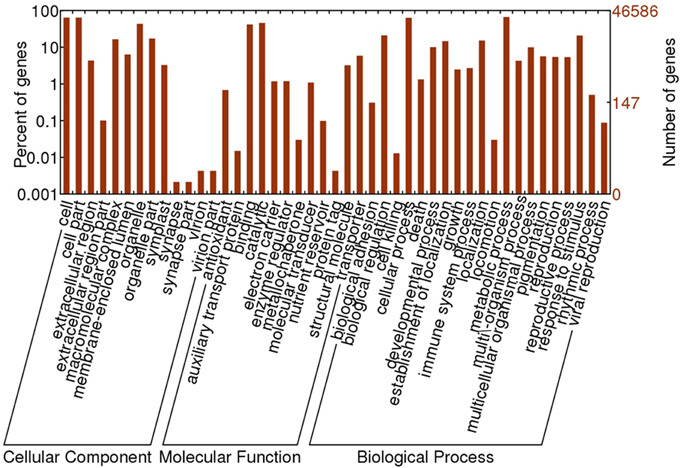
Figure 2. The GO annotation of R. sativus transcriptome.
We further performed a systematic analysis of high-level gene function by assigning R. sativus unigenes to the biochemical pathways in the KEGG database. All Oases-assembled unigenes were compared with the KEGG database to identify the biological pathways. As a result, a total of 42,181 unigenes were annotated in KEGG database, which were associated with 240 KEGG pathways (Supplementary File 1). The top 10 enriched pathways were “Glycolysis/Gluconeogenesis” (660 sequences), “Citrate cycle (TCA cycle)” (331 sequences), “Pentose phosphate pathway” (299 sequences), “Pentose and glucuronate interconversions” (491 sequences), “Fructose and mannose metabolism” (278 sequences), “Galactose metabolism” (314 sequences), “Ascorbate and aldarate metabolism” (346 sequences), “Fatty acid biosynthesis” (122 sequences), “Fatty acid elongation in mitochondria” (11 sequences), “Fatty acid metabolism” (258 sequences). Moreover, KEGG analysis showed that 364 unigenes were involved in the alkaloid biosynthesis. As alkaloids are very necessary for defending against pathogens, the result confirmed that R. sativus is an important source of medicinal compounds (Wang et al., 2002).
Analysis of the Assembled Transcripts in Leaf and Root Tissues
To investigate which unigenes were unique in the leaf tissue, the leaf unigenes (Table 1) were compared to the unigenes of root tissue of R. sativus available in SRA database (Wang et al., 2013a) by using blastx with an E-value cutoff of 1 e−5. As a result, a total of 60,633 (89%) leaf unigenes had perfect match to root unigenes, which indicated that the transcripts of root and leaf tissue of R. sativus are very similar. There were 7453 “leaf-specific” unigenes showing no significant homology to the transcripts of root tissue. In the meanwhile, KEGG analysis showed these unigenes were assigned to 71 pathways (Supplementary File 2). The top five pathways (ranked by p-value) included “Ribosome” (ko03010), “Nitrogen metabolism” (ko00910), “Carbon fixation in photosynthetic organisms” (ko00710), “Photosynthesis” (ko00195) and “Glycolysis/Gluconeogenesis” (ko00010). Ribosome biogenesis is one of the most important cellular processes and makes up the key component to regulate overall protein synthesis and cell growth (Deisenroth and Zhang, 2011). Nitrogen metabolism, Carbon fixation in photosynthetic organisms, and Glycolysis/Gluconeogenesis are all associated with photosynthesis. Photosynthesis is a necessary process used by plants to convert light energy into chemical energy and carbohydrates that can be released to fuel the organisms' activities. Collectively, the results indicated that these leaf-specific unigenes are mainly involved in the photosynthesis pathway. In addition, 15,043 root-specific unigenes were identified. KEGG analysis revealed these root-specific genes were assigned to 96 pathways (Supplementary File 3). The top five pathways (ranked by p-value) included “Phenylalanine metabolism” (ko00360), “Methane metabolism” (ko00680), “Diterpenoid biosynthesis” (ko00904), “Phenylpropanoid biosynthesis” (ko00940), and “Biosynthesis of phenylpropanoids” (ko01061). Among these pathways, phenylalanine metabolism has been proved to play an important role in root growth (Herrig et al., 2002), while methane is metabolized principally by methanotrophs and methanogens in the global carbon cycle, which is helpful for plants to obtain energy for growth (Herrig et al., 2002).
The expression levels of root unigenes were defined as control to identify the differentially expressed genes (DEG) between leaf and root tissues in R. sativus, the gene expression levels were calculated using the RPKM method (Reads per Kb per Million mapped reads). A total of 3563 significantly differentially expressed genes (P = 0.05) were identified, including 2335 up-regulated and 1228 down-regulated genes. To achieve a functional annotation of differentially expressed genes, GO classifications were assigned to the differentially expressed genes by using Cytoscape Enrichment Map (http://www.cytoscape.org/). As shown in Figure 3, 2613 DEGs were assigned with Biological Process terms. Within the biological process category, the common GO terms between up-regulated genes and down-regulated genes were “Metabolic process,” “Response to stimulus,” “Cellular process,” “Multi-organism process,” and “Biological regulation.” In the meanwhile, we observed that some DEGs were clustered in different Go terms. For example, 95 genes were up-regulated in leaf tissue and significantly enriched in “Cellular component organization.” Nevertheless, we observed that 172 genes were up-regulated in root tissue and significantly enriched in “Developmental process,” 63 genes were up-regulated in root tissue and significantly enriched in “Reproduction” and 96 genes were up-regulated in root tissue and significantly enriched in “Localization.” We further examined gene functions in these different GO terms by KEGG analysis. The result showed that many genes in “Cellular component organization,” such as AT5G66570 (Oxygen-evolving enhancer protein), AT2G47910 (Chlororespiratory reduction 6) and etc., are involved in photosynthesis pathway in leaf tissue (Supplementary Figure 1), which supports the feasibility of GO classifications in Figure 3. In the meanwhile, we also examined the functions of genes in “Developmental process,” “Reproduction,” and “Localization” by KEGG analysis. The results showed that many genes up-regulated in root tissue are involved in spliceosome pathway, tryptophan metabolism pathway and protein export pathway. Taken together, these KEGG results explain the basis of GO classifications of such DEGs between leaf and root tissues and are helpful for functional genomics study of these unigenes in R. sativus.
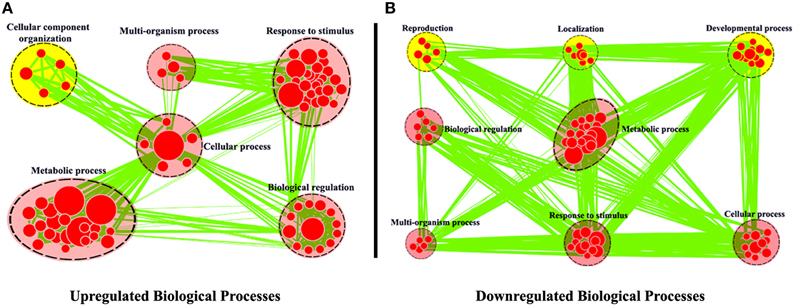
Figure 3. Biological Process analysis of differentially expressed genes between leaf and root tissues. GO modules enriched with up-regulated DEGs (A) and down-regulated DEGs (B) were visualized by the Enrichment Map in Cytoscape. The red and yellow circles indicate the common and different biological processes between up-regulated and down-regulated DEGs, respectively.
SSR Analysis of the Transcripts in R. sativus
Simple sequence repeats (SSRs) have been the most popular molecular-marker for various applications, such as quantitative trait loci (QTL) exploration, genome mapping, and genetic analysis (Li et al., 2002, 2004; Sharopova et al., 2002; Xu et al., 2011; Yu et al., 2011). In this study, we predicted potential SSRs in the assembled unigenes of R. sativus. A total of 31,875 potential SSRs were identified in 13,469 unigenes, of which 10,143 unigenes contained more than one SSR. A comprehensive analysis was performed to describe the type, frequency and distribution of all the potential SSRs. On average, one SSR could be found every 1.87 kb in the unigenes. The SSRs included 15,745 (49.39%) dinucleotide motifs, 15,555 (48.80%) trinucleotide motifs, 552 (1.73%) tetranucleotide motifs, 13 (0.04%) pentanucleotide motifs, and 10 (0.03%) hexanucleotide motifs (Figure 4). The number distribution of repeat units in all SSRs was also summarized (Table 4). The result showed that the repeat number of most SSRs was no more than 10, and no SSRs with more than 20 repeat sequences were observed. In addition, the repeat number and the total repeat length (= type number * type length * type average repeat number) of each SSR type were analyzed (Supplementary File 4). For most dinucleotide type, the repeat number was distributed between 5 and 11, with an average value of 6.31. The average repeat number of trinucleotide and tetranucleotide type was 5.49 and 5.06, respectively. While all the repeat number of pentanucleotide and hexanucleotide type was only five. Collectively, a total of 31,875 potential SSRs were identified from transcriptomic data of R. sativus, which is helpful for different aspects of agronomic research. However, transcriptome-derived SSR identification is mainly based on CDS information, which led to the omission of a fairly large number of SSRs in the intronic and non-coding regions in comparison with genome sequence-derived SSR identification.
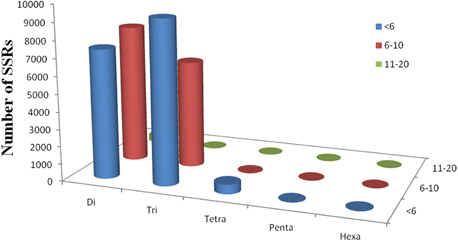
Figure 4. The SSR mining results in R. sativus. The profiles of different SSR types in R. sativus.
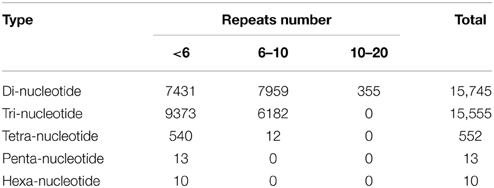
Table 4. SSRs distribution in the unigenes of R. sativus.
Transcription Factors Identification and Network Analysis of the Transcripts
Transcription factors (TFs) play important roles in multiple biological processes. Here, we identified a total of 2440 potential TF unigenes by comparing R. sativus unigenes with the plant transcription factor database. The length of these TF unigenes varied from 200 to 7043 bp, with an average value of 737 bp and an N50-value of 415 bp. The 200–300 bp class was the most enriched in total sequence number (33.37%), followed by >1000 bp (24.76%), 300–400 bp (15.33%), 400–500 bp (7.83%), 500–600 bp (5.41%), 600–700 bp (4.10%), 700–800 bp (3.32%), 800–900 bp class (3.16%), and 900–1000 bp (2.75%) (Figure 5A). The potential TFs were distributed in 78 families, such as HB, MYB, bHLH, C3H, bZIP, NAC, WRKY, and so on (Figure 5B). Among these TF gene families, HB, MYB and bHLH were the most abundant TF families (Supplementary File 5). It was reported that HBs may be involved in cell differentiation and control of cell growth, as well as patterning of diverse organisms. (Kappen, 2000). MYB proteins constitute a diverse class of DNA-binding proteins, which associate with regulation of secondary metabolism, control of cellular morphogenesis and regulation of meristem formation and the cell cycle (Jin and Martin, 1999). For bHLH (basic helix-loop-helix) family, its members were reported to participate in controlling cell proliferation and development of specific cell lineages (Heim et al., 2003).
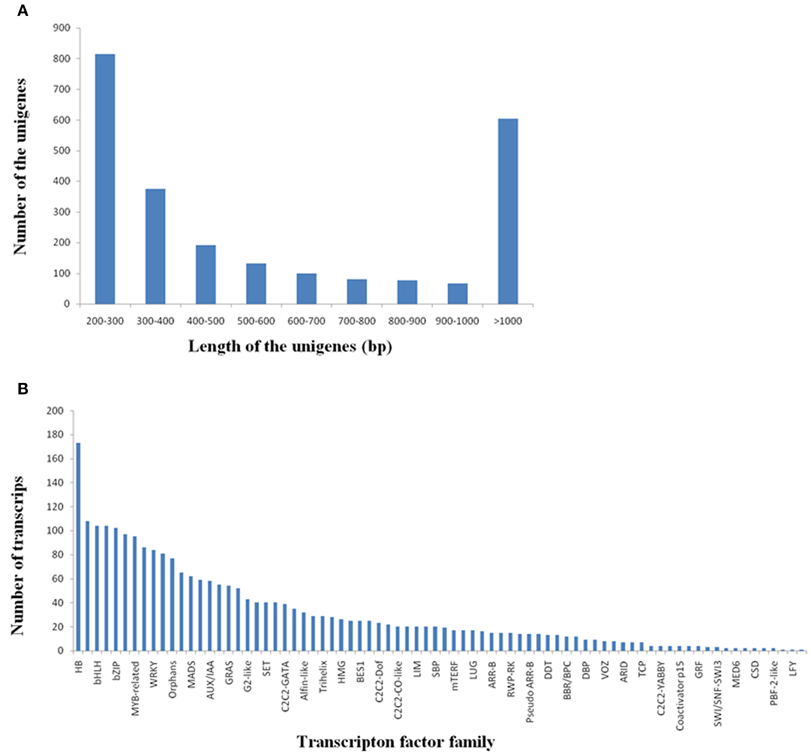
Figure 5. Discovery of the transcripts encoding transcription factors in R. sativus. (A) Length distribution of potential transcription factor genes. (B) Transcripts distribution in various transcription factor families.
Transcription factors play an important role in regulating almost each aspect of the organism's life metabolism. Nevertheless, understanding how TF-based regulation network eventually affect the phenotypes remains still elusive. In order to analyze the interaction between TFs and their gene targets, we used the Cytoscape software to construct a TF-based regulation network with 142 nodes and 460 edges. As shown in Figure 6, a total of 21TFs, such as SPA1, EIN3, WRKY33, and etc., were involved in the complicated regulation network. Network analysis showed that SPA1 interacts with three targets including ATMYC2, CAB1, and COP1. For example, it was reported that ATMYC2 and SPA1 act redundantly to suppress photomorphogenic growth in A. thaliana (Gangappa et al., 2010). We also observed that EIN3 interacts with EIL1, EIN2, EBF1, EBF2, AP2, ARR3, ARR5, and ARR15. Among these target genes, EIL1, EIN2, EBF1, EBF2, and AP2 were all linked with ethylene mediated signaling pathway. Accordingly, ARR3, ARR5, and ARR15 were all involved in the regulation process of circadian rhythm, red light signaling pathway and etc.
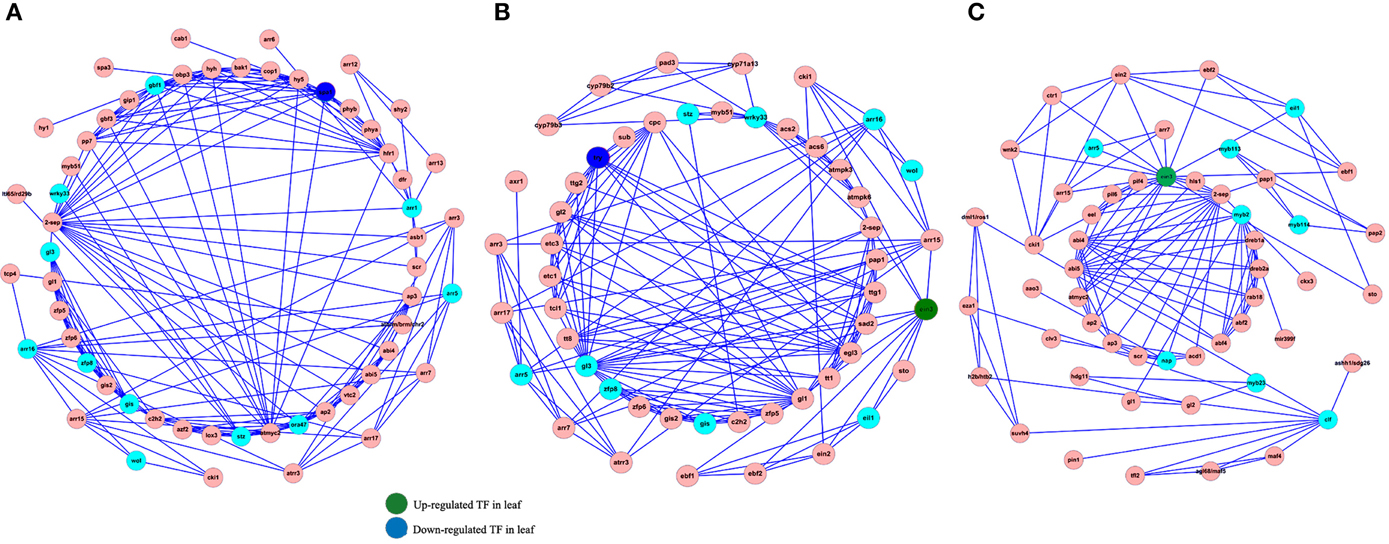
Figure 6. Construction of TFs-based regulation network by Cytoscape software. Sky blue and red circles represent identified TFs and genes involved in the network (A–C), respectively. The blue and green circles represent up-regulated and down-rugulated TFs in leaf.
Enzyme Detection and Characterization of R. sativus
By searching against the available database, a total of 12,238 R. sativus unigenes were annotated with 13,168 enzyme codes. The annotated enzyme codes included six classes: Oxidoreductases (1845 sequences), Transferases (6077 sequences), Hydrolases (3728 sequences), Lyases (537 sequences), Isomerases (494 sequences), and Ligases (487 sequences) (Figure 7). Furthermore, 967 enzyme types were detected. The most abundant enzyme types were non-specific serine/threonine protein kinase (2258 sequences), phosphoprotein phosphatase (491 sequences) and RNA helicase (305 sequences), respectively (Supplementary File 6). These enzymes were associated with various biological processes. For instance, through the phosphorylation of the serine or threonine residues in their substrate, serine/threonine protein kinases play an important role in many cellular processes such as post-translational modification and are potential drug targets in human cancers, inflammation, and metabolic diseases (Mumby and Walter, 1993). In contrast, phosphoprotein phosphatases usually remove phosphate groups from the substrates and are involved in many cellular processes including metabolism, gene transcription and translation (Shi, 2009). Collectively, we identified a bunch of enzyme codes in the transcripts of R. sativus by searching their unigenes against the databases. As enzymes are a type of large biological molecules responsible for thousands of metabolic processes that sustain life, the identification of important enzyme codes might provide key clues to reveal some important functional pathways and metabolic activities between leaf tissue and root tissue in R. sativus. In the meanwhile, we ran RepeatMasker to identify repetitive elements in leaf and root transcriptome of R. sativus. The generated results were listed in Supplementary Table S4.
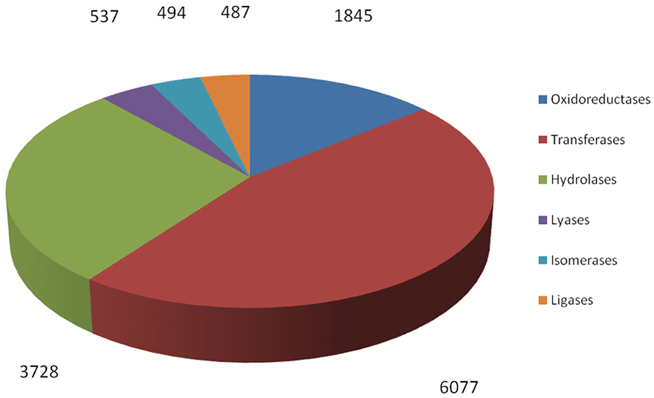
Figure 7. Functional characterization and classification of potential enzyme genes in the transcripts of R. sativus.
Conclusions
The transcriptome of leaf tissue in R. sativus was sequenced by Illumina Solexa technology. Approximately 70 million clean pair-end reads were obtained and used for the de novo assembly by Trinity program, which generated 68,086 unigenes with an average length of 576 bp. In the meanwhile, we merged leaf sequencing data with existing root sequencing data and obtained better de novo assembly of R. sativus using Oases program. Accordingly, potential simple sequence repeats (SSRs), transcription factors (TFs) and enzyme codes were identified in R. sativus. Additionally, we detected a total of 3563 DEGs (P = 0.05) and tissue-specific biological processes between leaf and root tissues. Furthermore, a TFs-based regulation network was constructed using Cytoscape software. Taken together, this study not only provides a critical genomic resource of R. sativus, but also paves a way for gene expression analysis, functional genomics and even proteomics research on R. sativus in the future.
Database Linking
The sequencing data of leaf and root tissues supporting the results of this study were deposited in NCBI Sequence Read Archive (SRA) Sequence Database with accession number SRP038118 and SRX256970.
Author Contributions
LM and ZY conceived and designed the experiments and contributed the reagents. WG and ZL analyzed the data and wrote the manuscript. YY performed the RNA isolation experiment. WJ provided the R. sativus materials and participated in the design of the study. YL helped to analyze the data and draft the manuscript. LM coordinated the study and revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31171582, 31000570), National Key Technology R&D Program (2010BAD01B03), the Natural Science Funds for Distinguished Young Scholars of Hubei Province of China (2010CDA097) and the New Century Talents Support Program by the Ministry of Education of China (NCET110172).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2015.00198
References
Bouché, N., Scharlat, A., Snedden, W., Bouchez, D., and Fromm, H. (2002). A novel family of calmodulin-binding transcription activators in multicellular organisms. J. Biol. Chem. 277, 21851–21861. doi: 10.1074/jbc.M200268200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis of functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deisenroth, C., and Zhang, Y. (2011). The ribosomal protein-Mdm2-p53 pathway and energy metabolism: bridging the gap between feast and famine. Genes Cancer 2, 392–403. doi: 10.1177/1947601911409737
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gangappa, S. N., Prasad, V. B., and Chattopadhyay, S. (2010). Functional interconnection of MYC2 and SPA1 in the photomorphogenic seedling development of Arabidopsis. Plant Physiol. 154, 1210–1219. doi: 10.1104/pp.110.163717
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heim, M. A., Jakoby, M., Werber, M., Martin, C., Weisshaar, B., and Bailey, P. C. (2003). The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 20, 735–747. doi: 10.1093/molbev/msg088
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herrig, V., Ferrarese-Mde, L., Suzuki, L. S., Rodrigues, J. D., and Ferrarese-Filho, O. (2002). Peroxidase and phenylalanine ammonia-lyase activities, phenolic acid contents, and allelochemicals-inhibited root growth of soybean. Biol. Res. 35, 59–66. doi: 10.4067/S0716-97602002000100009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jiao, Y., Yang, H., Ma, L., and Sun, N. (2003). A genome-wide analysis of blue-light regulation of Arabidopsis transcription factor gene expression during seedling development. Plant Physiol. 133, 1480–1493. doi: 10.1104/pp.103.029439
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jin, H., and Martin, C. (1999). Multifunctionality and diversity within the plant MYB-gene family. Plant Mol. Biol. 41, 577–585.
Kalra, S., Puniya, B. L., Kulshreshtha, D., Kumar, S., Kaur, J., Ramachandran, S., et al. (2013). De novo transcriptome sequencing reveals important molecular networks and metabolic pathways of the plant, Chlorophytum borivilianum. PloS ONE 8:e83336. doi: 10.1371/journal.pone.0083336
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamei, A., Tsuro, M., Kubo, N., Hayashi, T., Wang, N., Fujimura, T., et al. (2010). QTL mapping of clubroot resistance in radish (Raphanus sativus L.). Theor. Appl. Genet. 120, 1021–1027. doi: 10.1007/s00122-009-1230-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kappen, C. (2000). The homeodomain: an ancient evolutionary motif in animals and plants. Comput. Chem. 24, 95–103. doi: 10.1016/S0097-8485(99)00049-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, H. A., Lim, C. J., Kim, S., Choe, J. K., Jo, S. H., Baek, N., et al. (2014). High-throughput sequencing and de novo assembly of Brassica oleracea var. capitata L. for transcriptome analysis. PLoS ONE 9:e92087. doi: 10.1371/journal.pone.0092087
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Knowles, D. G., and McLysaght, A. (2009). Recent de novo origin of human protein coding genes. Genome Res. 19, 1752–1759. doi: 10.1101/gr.095026.109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, D. J., Deng, Z., Qin, B., Liu, X. H., and Men, Z. H. (2012). De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 13:192 doi: 10.1186/1471-2164-13-192
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Y. C., Korol, A. B., Fahima, T., Beiles, A., and Nevo, E. (2002). Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol. Ecol. 11, 2453–2465. doi: 10.1046/j.1365-294X.2002.01643.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Y. C., Korol, A. B., Fahima, T., and Nevo, E. (2004). Microsatellites within genes: structure, function, and evolution. Mol. Biol. Evol. 21, 991–1007. doi: 10.1093/molbev/msh073
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mitsuda, N., and Ohme-Takagi, M. (2009). Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiol. 50, 1232–1248. doi: 10.1093/pcp/pcp075
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mudalkar, S., Golla, R., Ghatty, S., and Reddy, A. R. (2014). De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol. Biol. 84, 159–171. doi: 10.1007/s11103-013-0125-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mumby, M. C., and Walter, G. (1993). Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73, 673–699.
Ohsako, T., Hirai, M., and Yamabuki, M. (2010). Spatial structure of microsatellite variability within and among populations of wild radish Raphanus sativus L. var. hortensis Backer f. raphanistroides Makino (Brassicaceae) in Japan. Breed. Sci. 60, 195–202. doi: 10.1270/jsbbs.60.195
Paritosh, K., Yadava, S. K., Gupta, V., Panjabi-Massand, P., Sodhi, Y. S., Pradhan, A. K., et al. (2013). RNA-seq based SNPs in some agronomically important oleiferous lines of Brassica rapa and their use for genome-wide linkage mapping and specific-region fine mapping. BMC Genomics 14:463. doi: 10.1186/1471-2164-14-463
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Powell, W., Morgante, M., Andre, C., Hanafey, M., Vogel, J., Tingey, S., et al. (1996). The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 2, 225–238. doi: 10.1007/BF00564200
Qiu, L., Yang, C., Tian, B., Yang, J. B., and Liu, A. (2010). Exploiting EST databases for the development and characterization of EST-SSR markers in castor bean (Ricinus communis L.). BMC Plant Biol. 10:278. doi: 10.1186/1471-2229-10-278
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Riechmann, J. L., Heard, J., Martin, G., Reuber, L., Jiang, C., Keddie, J., et al. (2000). Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290, 2105–2110. doi: 10.1126/science.290.5499.2105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schulz, M., and Zerbino, D. (2010). Oases – De Novo Transcriptome Assembler for Very Short Reads. Technical Report, MPI for Molecular Genomics, European Bioinformatics Institute (EMBL-EBI), UC Santa Cruz.
Schuster, S. C. (2008). Next-generation sequencing transforms today's biology. Nat. Methods 5, 16–18. doi: 10.1038/nmeth1156
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharopova, N., McMullen, M. D., Schultz, L., Schroeder, S., Sanchez-Villeda, H., Gardiner, J., et al. (2002). Development and mapping of SSR markers for maize. Plant Mol. Biol. 48, 463–481. doi: 10.1023/A:1014868625533
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shendure, J., and Ji, H. (2008). Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145. doi: 10.1038/nbt1486
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shi, Y. (2009). Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484. doi: 10.1016/j.cell.2009.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, L., Wei, L., Wang, L., and Xu, C. (2002). Effects of peroxidase on hyperlipidemia in mice. J. Agric. Food. Chem. 50, 868–870. doi: 10.1021/jf011130+
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, N., Hu, J., Ohsawa, R., Ohta, M., and Fujimura, T. (2007). Identification and characterization of microsatellite markers derived from expressed sequence tags (ESTs) of radish (Raphanus sativus L.). Mol. Ecol. Notes 7, 503–506. doi: 10.1111/j.1471-8286.2006.01635.x
Wang, Y., Pan, Y., Liu, Z., Zhu, X., Zhai, L., Xu, L., et al. (2013b). De novo transcriptome sequencing of radish (Raphanus sativus L.) and analysis of major genes involved in glucosinolate metabolism. BMC Genomics 14:836. doi: 10.1186/1471-2164-14-836
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Xu, L., Chen, Y., Shen, H., Gong, Y., Limera, C., et al. (2013a). Transcriptome profiling of radish (Raphanus sativus L.) root and identification of genes involved in response to lead (Pb) stress with next generation sequencing. PLoS ONE 8:e66539. doi: 10.1371/journal.pone.0066539
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Watt, J., and Breyer-Brandwijk, M. (1962). The Medicinal and Poisonous Plants of Southern and Eastern Africa, 2nd Edn. Edinburgh; London: E. & S. Livingstone.
Xu, L., Wang, Y., Xu, Y., Wang, L., Zhai, L., Zhu, X., et al. (2013). Identification and characterization of novel and conserved microRNAs in radish (Raphanus sativus L.) using high-throughput sequencing. Plant Sci. 201, 108–114. doi: 10.1016/j.plantsci.2012.11.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, P., Wu, X., Wang, B., Liu, Y., Ehlers, J. D., Close, T. J., et al. (2011). A SNP and SSR based genetic map of asparagus bean (Vigna. unguiculata ssp. sesquipedialis) and comparison with the broader species. PLoS ONE 6:15952. doi: 10.1371/journal.pone.0015952
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamane, K., Lü, N., and Ohnishi, O. (2009). Multiple origins and high genetic diversity of cultivated radish inferred from polymorphism in chloroplast simple sequence repeats. Breed. Sci. 59, 55–65. doi: 10.1270/jsbbs.59.55
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic. Acids Res. 34, 293–297. doi: 10.1093/nar/gkl031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, Y., Yuan, D., Liang, S., Li, X., Wang, X., Lin, Z., et al. (2011). Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between gossypium hirsutum and G. barbadense. BMC Genomics 12:15. doi: 10.1186/1471-2164-12-15
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, H. Y., Wei, L. B., Miao, H. M., Zhang, T. D., and Wang, C. Y. (2012a). Development and validation of genic-SSR markers in sesame by RNA-seq. BMC Genomics 13:316. doi: 10.1186/1471-2164-13-316
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, J., Liang, S., Duan, J., Wang, J., Chen, S., Cheng, Z., et al. (2012b). De novo assembly and characterisation of the transcriptome during seed development and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 13:90. doi: 10.1186/1471-2164-13-90
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, L., Jia, H., Yin, Y., Wu, G., Xia, H., Wang, X., et al. (2013). Transcriptome analysis of leaf tissue of Raphanus sativus by RNA sequencing. PLoS ONE 8:e80350. doi: 10.1371/journal.pone.0080350
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Raphanus sativus, transcriptome, RNA sequencing, transcription factor, simple sequence repeats
Citation: Wu G, Zhang L, Yin Y, Wu J, Yu L, Zhou Y and Li M (2015) Sequencing, de novo assembly and comparative analysis of Raphanus sativus transcriptome. Front. Plant Sci. 6:198. doi: 10.3389/fpls.2015.00198
Received: 23 October 2014; Accepted: 12 March 2015;
Published: 01 April 2015.
Edited by:
Tiegang Lu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Mukesh Jain, National Institute of Plant Genome Research, IndiaPrakash P. Kumar, National University of Singapore, Singapore
Copyright © 2015 Wu, Zhang, Yin, Wu, Yu, Zhou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanhong Zhou and Maoteng Li, School of Life Science and Technology, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan Hubei 430074, ChinaeWh6aG91QG1haWwuaHVzdC5lZHUuY24=;bGltYW90ZW5nNDI2QG1haWwuaHVzdC5lZHUuY24=
†These authors have contributed equally to this work.