 Junjie Hu
Junjie Hu Christof Rampitsch
Christof Rampitsch Natalia V. Bykova
Natalia V. Bykova- 1Department of Biology, Memorial University of Newfoundland, St. John’s, NL, Canada
- 2Cereal Proteomics, Cereal Research Centre, Agriculture and Agri-Food Canada, Morden, MB, Canada
Abiotic and biotic stresses constrain plant growth and development negatively impacting crop production. Plants have developed stress-specific adaptations as well as simultaneous responses to a combination of various abiotic stresses with pathogen infection. The efficiency of stress-induced adaptive responses is dependent on activation of molecular signaling pathways and intracellular networks by modulating expression, or abundance, and/or post-translational modification (PTM) of proteins primarily associated with defense mechanisms. In this review, we summarize and evaluate the contribution of proteomic studies to our understanding of stress response mechanisms in different plant organs and tissues. Advanced quantitative proteomic techniques have improved the coverage of total proteomes and sub-proteomes from small amounts of starting material, and characterized PTMs as well as protein–protein interactions at the cellular level, providing detailed information on organ- and tissue-specific regulatory mechanisms responding to a variety of individual stresses or stress combinations during plant life cycle. In particular, we address the tissue-specific signaling networks localized to various organelles that participate in stress-related physiological plasticity and adaptive mechanisms, such as photosynthetic efficiency, symbiotic nitrogen fixation, plant growth, tolerance and common responses to environmental stresses. We also provide an update on the progress of proteomics with major crop species and discuss the current challenges and limitations inherent to proteomics techniques and data interpretation for non-model organisms. Future directions in proteomics research toward crop improvement are further discussed.
Introduction
Understanding all levels that regulate adaptive mechanisms and the resilience of crop plants in the context of climate changes is absolutely essential to reach significant achievements in genomics-driven breeding of major crops for high productivity and stress tolerance. A new pattern of frequently occurring extreme weather events has already been taking a toll on agricultural production systems. In addition to increasing amount of genomic information available for both model and non-model plants, the parallel development of bioinformatics techniques and analytical instrumentation makes proteomics an essential approach to reveal major signaling and biochemical pathways underlying plant life cycle, interaction with the environment, and responses to abiotic and biotic stresses. High-throughput proteomic studies have gone beyond simple identification of individual proteins to quantitative profiling, analysis of dynamic post-translational modifications (PTMs), subcellular localization and compartmentalization, protein complexes, signaling pathways, and protein–protein interactions (for reviews refer to Agrawal et al., 2011, 2013; Matros et al., 2011; Pflieger et al., 2011; Hossain et al., 2012).
Growing in the field or cultivated in the laboratory, plant development and productivity are inevitably controlled by various extreme environmental factors such as drought, heat, salinity, cold, or pathogen infection, which may delay or induce seed germination, reduce seedling growth, and decrease crop yields. Proteomics studies can substantially contribute to revealing virtually every aspect of cellular function in plant stress responses, unraveling possible relationships between protein abundance and/or modification and plant stress tolerance. An increasing number of studies have been discussing the contribution of proteomics to deeper insights into the molecular mechanisms of plant responses to stresses and signaling pathways linking changes in protein expression to cellular metabolic events, such as studies using model plants Arabidopsis (Wienkoop et al., 2010), rice (see for reviews Singh and Jwa, 2013; Kim et al., 2014) and sorghum (Ngara and Ndimba, 2014). Attributed to the improvement in diverse proteomic technology platforms that combined classical two-dimensional electrophoresis (2-DE) gel-based techniques with mass spectrometry (MS)-based quantitative approaches as well as the accessibility of protein databases of various plant species, major monocotyledonous cereals and dicotyledonous legumes (e.g., maize, wheat, barley, soybeans etc.) have been widely used to study quantitative changes in protein abundance related to different abiotic stresses (Li et al., 2013a; Jacoby et al., 2013a; Deshmukh et al., 2014; Wu et al., 2014a; Kamal et al., 2015).
In the agricultural environment crop plants are subject to a complex set of abiotic and biotic stresses. In addition to studying effects of various stresses applied individually under laboratory controlled conditions, recent evidence shows that simultaneous occurrence of multiple stresses affecting crop growth, yield and physiological traits can cause plants to activate intricate metabolic pathways involved in specific programming of gene expression that uniquely respond to different combinations of stresses (Atkinson and Urwin, 2012). Several different signaling pathways involved in multiple stress-responding mechanisms have been revealed in transcriptome, metabolome, and proteome analysis of various crop plants subjected to different stress combinations, suggesting a complex regulatory network orchestrated by hormone signals, transcription factors, antioxidants, kinase cascades, reactive oxygen species (ROS), and osmolyte synthesis (Suzuki et al., 2014).
Fundamentally, crop growth depends on efficient production of energy and nutritional compounds regulated through different organs, which are equipped with various organelles and organ-specific sets of cytosolic proteins, hormones and metabolites (Komatsu and Hossain, 2013). The responses of plant cells to abiotic stresses vary in different organs. Organ-specific proteomics combined with subcellular organelle proteomic studies of developmental mechanisms from leaf to root can provide more detailed information for understanding of cellular mechanisms that regulate stress response and signal transduction in various organelles (Hossain et al., 2012; Komatsu and Hossain, 2013; Table 1). Tissue-targeted seed proteomic studies of different developmental stages under abiotic stresses have contributed to increasing our depth of knowledge about the processes controlling seed development, dormancy and germination by analyzing spatial and functional sub-proteomes (Finnie et al., 2011a). In this article we provide an update on the progress of proteomics with major crop species and discuss the current challenges and limitations inherent to proteomics techniques and data interpretation for non-model organisms.
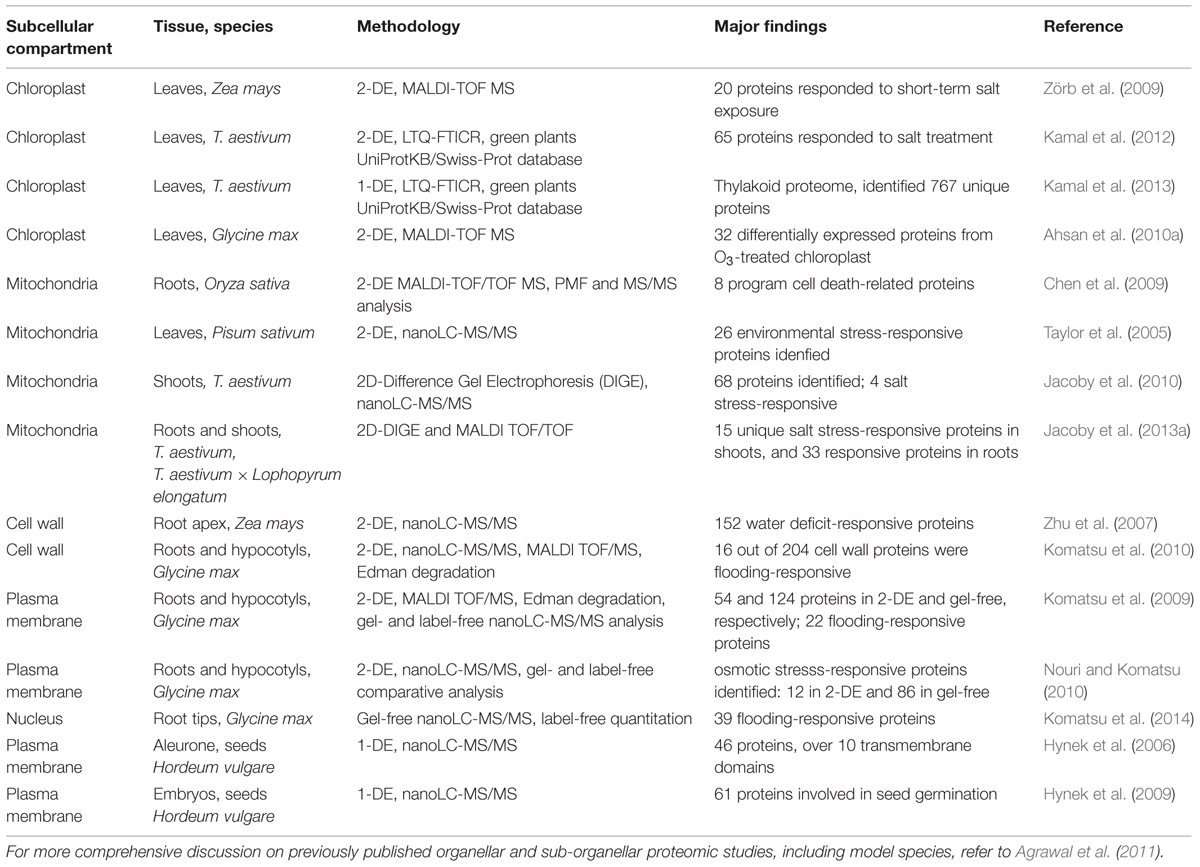
TABLE 1. Overview of approaches used for subcellular proteomic studies in crop plants under abiotic stress.
Approaches and Challenges in Crop Plant Proteomics
With the completion of genome sequences in model species such as dicotyledonous plant Arabidopsis thaliana, monocotyledonous crop plant rice (Oryza sativa ssp. japonica and ssp. indica), model grass Brachypodium distachyon, legume species soybean (Glycine max), Medicago truncatula and Lotus japonicus, cereal crops Zea mays, and Hordeum vulgare, other crop species to follow (NCBI list of sequenced plant genomes), and with recently released chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome (International Wheat Genome Sequencing Consortium [IWGSC], 2014), attention has been focused on linking genomic data and transcriptomic profiles to the spatial and temporal expression, biological function and functional network of proteins. Moreover, dealing with complex and dynamic plant proteomes, it is crucial to choose suitable proteomic approaches targeting the identification of proteins and their modification that may contribute to crop improvement. In the recent years quantitative proteomic studies using high resolution and mass accuracy instruments have been contributing with important information to our understanding of plant growth, development, and interactions with the environment. This capability is especially useful for crops as it may not only contribute to increasing nutritional value and yield, but also to understanding mechanisms of crop adaptation to responses to abiotic stresses.
Recent Innovative Technologies used in Quantitative Plant Proteomics
Driven by innovations in MS-based technologies and rapid development of quantitative methods, proteomics has emerged as a complementary technique to other approaches such as transcriptomics and metabolomics in the post-genomic era (Wienkoop et al., 2010). Proteomic analysis is achieved through (1) separation and identification of proteins based on 2-DE or coupled gel-free shotgun liquid chromatography tandem mass spectrometry (LC–MS/MS) platforms; (2) elucidation of protein functions and protein functional networks in plant metabolic and signaling pathways through the analysis of protein mapping, characterization of PTMs and protein–protein interactions; (3) bioinformatic strategies and the use of databases for both model and non-model plant species (Holman et al., 2013). Recently, the application of gel-free protein separation approaches and ‘second generation’ proteomic techniques such as multidimensional protein identification technology (MudPIT), quantitative proteomic approaches including isotope-coded affinity tags (ICATs), targeted mass tags (TMTs), and isobaric tags for relative and absolute quantitation (iTRAQ) have been widely used in descriptive and comparative proteomic studies of plant development and metabolic strategies in abiotic stress adaptation. Advances in liquid chromatography-based separation and label-free quantitative proteomic analysis of large number of proteins derived from complex plant samples have recently been discussed (Matros et al., 2011). Although modern gel-free quantitative proteomic approaches, label-based and label-free, are considered to be more advanced and can provide more information on comparative changes in protein expression than one and 2-DE gel-based methods, they have limitations. One obvious limitation in terms of global proteome coverage is the fact that they are designed for less hydrophobic, more aqueous buffer-soluble sub-proteomes, whereas the buffers and detergents used in gel-based protein separation techniques can be quite powerful and efficient in solubilization of more hydrophobic protein groups, especially in conjunction with organellar and sub-organellar pre-fractionation. The development and application of label-free quantitative analysis techniques in combination with one and 2-DE gels was the new realization that helped to solve one of the main drawbacks of gel-based separation and quantitative analysis of proteins related to co-focusing/co-localization of several proteins and their modified forms in one spot or band on the gel.
One of the most socio-economically important agricultural crops is wheat. However, until recently, large-scale proteomic studies with this organism were difficult to realize due to the lack of genomic information available to facilitate protein identifications. The publication of the first draft of completely sequenced wheat genome (International Wheat Genome Sequencing Consortium [IWGSC], 2014) is inspirational to plant proteomics researchers, although the annotation of complete wheat genome assembly will remain a challenging task. Until recently proteomic studies have been using alternative available bioinformatics resources (Table 2) that included wheat EST-based databases (Bykova et al., 2011a,b; Pascovici et al., 2013), or available closely related Brachypodium distachyon model plant genome, or D-genome progenitor Aegilops tauschii, as well as a composite database of available cereals sorghum, maize and rice (Pascovici et al., 2013), a translated database of the low copy number genome assemblies of T. aestivum (Alvarez et al., 2014), and proteins from monocot family Poaceae (Kang et al., 2015). Pascovici et al. (2013) have evaluated the most effective pipeline for large-scale shotgun quantitative experiments using bread wheat (T. aestivum), iTRAQ multirun quantitative approach, and the available resources for bioinformatics data analysis and downstream functional interpretation. The study emphasized many challenges related to the repetitiveness/redundancy/polymorphism of bread wheat genome and therefore extremely large size of the corresponding EST-based database that could not be readily manipulated by the available bioinformatics tools, and the stochastic aspect of protein grouping across multiple runs. The use of smaller databases was demonstrated as alternative pragmatic approaches to reliably identify proteins and proceed with functional annotations (Table 2).
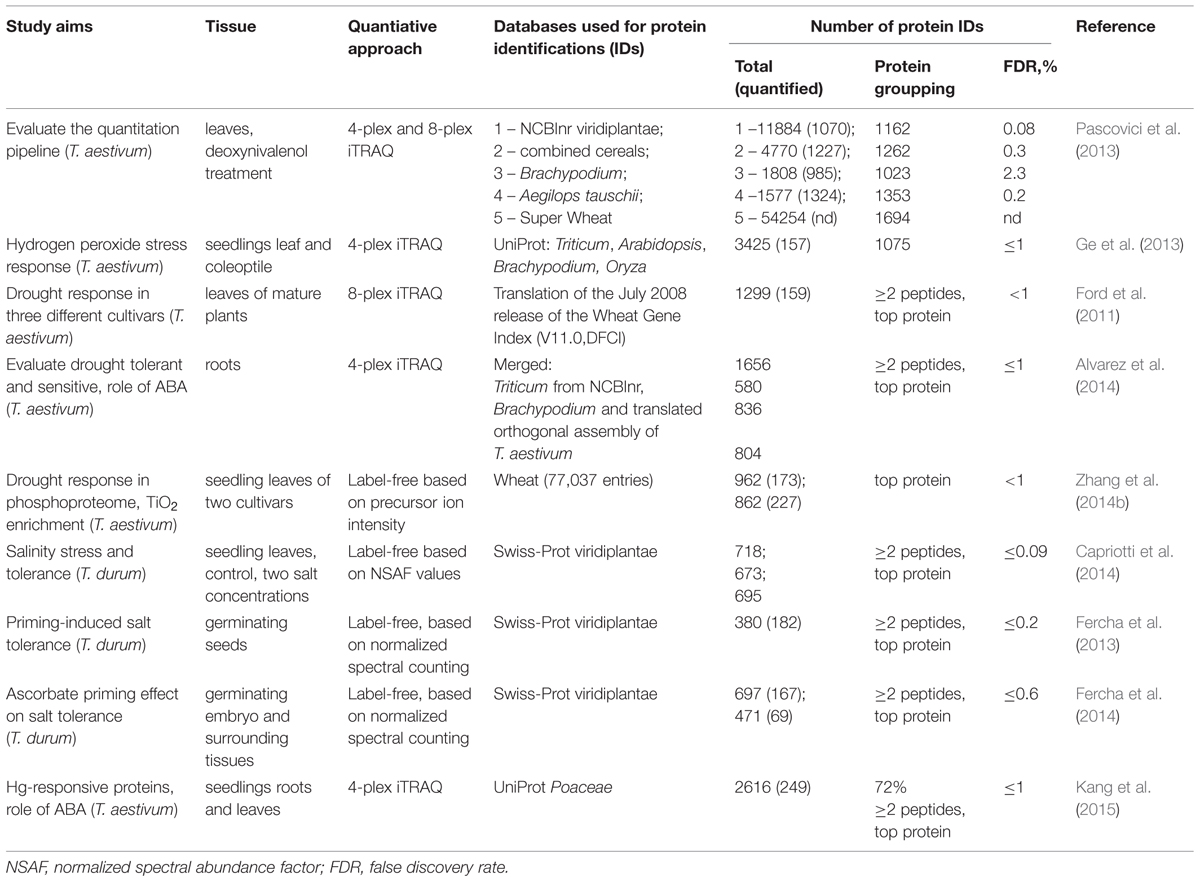
TABLE 2. Recent large-scale gel-free quantitative proteomic studies on wheat.
More recently, targeted MS-based quantitative approaches such as multiplexed selective reaction monitoring (SRM) have proven to be powerful for identification of specific proteins with causative functions in agronomically important traits (Jacoby et al., 2013b). Attributable to outstanding sensitivity of this methodology to selective quantitation of low abundance protein components in complex mixtures, SRM technique is seen by researchers as an alternative to antibody-based immunodetection assays (Picotti et al., 2013). This SRM approach is based on highly specific detection and quantitation of proteotypic couples comprised of target precursor and corresponding fragment ions and until recently it was exclusively based on triple quadrupole MS platforms due to the necessity of two stages of mass filters. The advantages and limitations of this application have been discussed and demonstrated experimentally elsewhere. The new generation of Orbitrap-technology instruments such as Q Exactive hybrid quadrupole-Orbitrap provided an efficient and user-friendly alternative for further application of SRM method in quantitative assay development with high potential for large-scale targeted proteomics experiments (Wu et al., 2014b). The purpose of SRM methodology in plant proteomics is biomarker validation in crops, which follows the discovery phase with more explorative qualitative and quantitative comparative proteomic studies aimed at finding potential candidates important in stress responses. This highly selective and sensitive quantitative approach can be powerful not only for biomarker validation but also for the development of new stress tolerance assessment methods, which will facilitate the identification of genotypes with improved resistance and ultimately discovery of gene targets for marker-assisted breeding.
Another approach has recently been developed for label-free shotgun proteomics based on data independent (MSE) acquisition protocols that has a potential to identify peptides from complex samples in a rapid, consistent, and sensitive way, and offers a higher dynamic range for peptide quantification (Buts et al., 2014). In this approach, ultra performance liquid chromatography is used, which is coupled to an LC–MS/MS run where an alternating energy level allows to obtain accurate precursor masses at low energy, and to take fragmentation spectra of all parent masses at high collision energy in one analytical run. However, this approach is rather at the early stages of development, and has many challenges related to the difficulties in interpretation of very complex composite fragmentation spectra resulting in poor protein identification rate. At present, parallel data-dependent runs are needed in order to acquire all necessary information, build the databases of individual peptide fragmentation spectra, and link them to the MSE (Buts et al., 2014). This approach was successfully used in quantitative analysis of important allergenic proteins in wheat grain extracts, in identification of gliadins and glutenins in wheat grain and quantitation of proteins associated with celiac disease and baker’s asthma (Uvackova et al., 2013a,b).
An alternative strategy that combines high specificity data-independent acquisition method with a novel targeted data extraction approach to mine the resulting fragment ion data sets was recently demonstrated (Gillet et al., 2012). This method, termed SWATH MS, is based on sequential time-and mass-segmented acquisition, which generates fragment ion spectra of all precursors in two user-defined dimensions, retention time and m/z space, resulting in complex fragment ion maps. The interpretation of highly specific multiplexed data sets required the development of fundamentally different data analysis strategy, which uses previously acquired information contained in spectral libraries to mine the fragment ion maps for targeted extraction and quantitation of specific peptides of interest. The accuracy and consistency of SWATH MS was demonstrated to be comparable to SRM approach (Gillet et al., 2012). One of the important advantages of the former, alleviating most constrains of present proteomics methods, is the iterative retrospective re-mining of the acquired data sets for targeted extraction. This approach offers unprecedented possibilities for the qualitative and quantitative profiling not only in proteomics but also in metabolomics and lipidomics. One of the main bottlenecks for proteomics development is the lack of robust bioinformatics tools with novel algorithmic solutions to processing of MS data, which are lagging behind the substantial advances occurring in instrumentation and protocols (Cappadona et al., 2012; Smith et al., 2014). It remains to be seen how this breakthrough technology will evolve into a powerful tool utilized throughout plant sciences.
From Cellular Proteome to Subcellular Protein catalogs
The major advances in organelle-based proteomics have not only provided a deeper insight into protein localization and organelle-specific function in plant biological processes, but also a better understanding of the functions of organelles in metabolic processes involved in plant development and growth (for a recent review refer to Agrawal et al., 2011). The accurate description of an organelle proteome requires the ability to purify organelles from cellular mixture and identify low abundance proteome extracted from these organelles. Several quantitative proteomic studies have focused on spatial subcellular proteomes with particular methods designed for different organelles (references in Table 1).
Proteomic analysis of wheat chloroplasts using a combination of two complementary approaches Tricine SDS–PAGE and 2-DE coupled to a high throughput MS methods (LTQ-FTICR and MALDI-TOF/TOF) has contributed to a better understanding of the responsive proteins in photosynthesis during abiotic stress in plastids (Kamal et al., 2012, 2013). Chloroplast proteomics study of soybean leaves under ozone stress by 2-DE and MALDI-TOF MS approach identified increased expression level of proteins involved in antioxidant defense and carbon metabolism (Ahsan et al., 2010a). Proteomic studies on mitochondrial organelles provided information on both individual proteins and protein complexes that participate in salinity response mechanisms in rice through 2-DE and MALDI-TOF MS (Chen et al., 2009), and in wheat through 2-DE and LC–MS/MS analysis (Jacoby et al., 2010, 2013a). Moreover, the effects of drought, cold and herbicide treatments on mitochondrial proteomics in pea were also analyzed by 2-DE Blue Native PAGE with Q-TOF MS (Taylor et al., 2005). Cell wall proteomic studies of major crops such as rice (using 2-DE and nanoLC–MS/MS), soybean (using 2-DE, MALDI-TOF MS, and nanoLC–MS/MS), and maize (using 2-DE and Q-TOF MS), provided insights into either dehydration- or water stress-responsive proteomes involved in a variety of functions, including carbohydrate metabolism, cellular defense through redox mechanisms, cell wall modification, and cell signaling pathways (Zhu et al., 2007; Komatsu et al., 2010). Plasma membrane, as a primary interface between the cellular cytoplasm and the extracellular environment, plays a vital role in signaling, communication and uptake of nutrients (Alexandersson et al., 2004). Due to their low abundance and low solubility, nanoLC–MS/MS-based proteomic approaches have been used to study osmotic stress-induced proteins in soybean, which are mostly involved in antioxidative defense system (Komatsu et al., 2009; Nouri and Komatsu, 2010). Changes in nuclear proteins under the flooding stress in soybean root tips, studied by gel-free nano-LC MS/MS, revealed differentially regulated responding proteins (Komatsu et al., 2014). Moreover, reversed-phase chromatography, SDS–PAGE and LC–MS/MS have been used to prepare and analyze integral plasma membrane-enriched tissue fractions from barley aleurone layers and germinated embryos, providing more information about aleurone layer as a secretory tissue during seed germination (Hynek et al., 2006, 2009). The accurate description of an organelle proteome requires the ability to identify genuine protein residents (Table 1). Although many challenges remain, quantitative proteomic profiling of organelles has been developed to reliably identify the protein complement of whole organelles, as well as for protein assignment to subcellular location and relative protein quantification, which are improving our understanding of protein functions and dynamics in plant cells.
Organ-Specific Proteome Analysis of Abiotic Stress Responses in Crop Plants
Proteomics of Leaf Photosynthesis and Senescence to Understand Crop Productivity
Leaf photosynthesis is the main source of plant biomass influencing potential crop yield. Highly abundant chlorophyll in leaves plays essential roles in light harvesting and energy transfer during photosynthesis, therefore, chlorophyll metabolism contributes to photosynthetic efficiency during leaf development. Recently, Chu et al. (2015) analyzed changes in protein profiles upon the development of chlorophyll deficiency in Brassica napus leaves and provided new insights into the regulation of chlorophyll biosynthesis and photosynthesis in crops (Chu et al., 2015). Moreover, the levels of chlorophylls were also shown to be associated with the maintenance of photosynthetic rate of CO2 consumption during the grain-filling period, and with the rate of leaf senescence in different rice cultivars (Panda and Sarkar, 2013). Leaf senescence is featured with loss of photosynthetic activities and hydrolysis of macromolecules followed by the degeneration of chloroplasts and remobilization of the hydrolyzed nutrients to young leaves and developing seeds (Guo, 2013). Several studies have focused on the proteomics of leaf senescence, mainly on the investigation of nitrogen mobilization from leaves during leaf senescence (Bazargani et al., 2011; Avice and Etienne, 2014). The results of these studies suggest the importance of proteolysis, chloroplast degradation and nitrogen remobilization during this process (Figure 1). Chloroplast contains up to 75% of leaf nitrogen in the form of Rubisco enzyme components in the stroma and complex of photosystem II in the thylakoid membrane (Roberts et al., 2012). Advances in organelle proteomic studies integrated with large-scale genomic approaches and determination of enzymes with proteolytic activity have addressed the complexity of chloroplastic proteolytic machinery during leaf senescence and investigated different classes of senescence-associated proteases with unique physiological roles according to their expression profiles along the senescence progress (Liu et al., 2008; Roberts et al., 2012). Moreover, the regulation of photosynthetic carbon metabolism has also been investigated during leaf senescence in rice by a comparative proteomic approach, contributing to a deeper insight into the enzymatic regulation involved in the Calvin cycle during senescence featured with down-regulated photorespiration (Zhang et al., 2010). Furthermore, sucrose as the main photosynthetic product is rich with carbon and energy, is a key component in carbon metabolism, and is essential for both plant growth and the synthesis of storage reserves, such as starch and oil (Barsan et al., 2012). Glycolytic enzymes involved in sucrose synthesis are of particular interest with respect to crop yield, and have been identified by subcellular proteomic studies of senescence, photosynthesis, and stress-responding processes in rice leaves (Zhang et al., 2010).
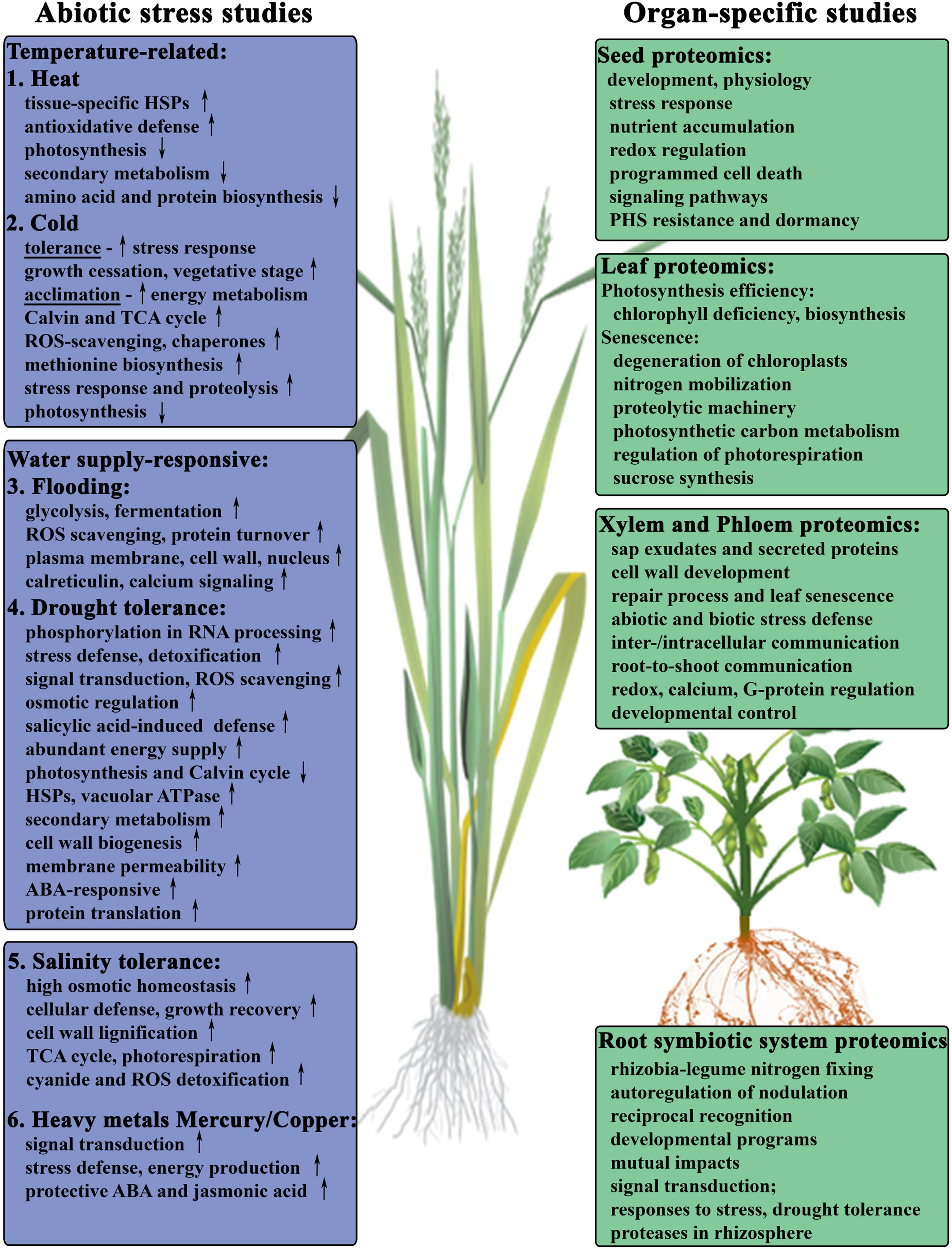
FIGURE 1. Proteomic studies-driven insights into cellular activities during abiotic stress response of major crops. Summary of the abiotic stress studies published in: (1) Heat in soybean (Ahsan et al., 2010b). (2) Cold in winter wheat (Vítámvás et al., 2012; Kosová et al., 2013). (3) Flooding and anoxia in soybean seedlings (Nanjo et al., 2011 review; Komatsu et al., 2009, 2010; Yin et al., 2014). (4) Drought tolerance in wheat roots (Alvarez et al., 2014) and leaves (Ford et al., 2011; Kang et al., 2012; Zhang et al., 2014b). (5) Salinity in wheat (Guo et al., 2012; Jacoby et al., 2013a; Capriotti et al., 2014; Fercha et al., 2014), soybean (Ma et al., 2012), rice (Ruan et al., 2011; Song et al., 2011; Liu et al., 2013b), and barley (Witzel et al., 2009; Gao et al., 2013). (6) Metal ions stress in wheat (Li et al., 2013b; Kang et al., 2015).
Xylem and Phloem Proteomics of Root-to-Leaf Signaling Pathways During Stress
Maximizing crop yields also depends on the leaves receiving an optimal supply of nutrients from the root system via the xylem vessels. The xylem sap is a dynamic fluid circulated between root and leaf, featured with a high content of secreted proteins undergoing changes in its proteome upon abiotic and biotic stresses (Ligat et al., 2011; de Bernonville et al., 2014; Zhang et al., 2014a). Xylem proteomic and secretomic studies have recently become one of the major areas of interest in understanding plant development and responses to environmental perturbations, and illustrated several types of xylem sap-containing proteins that participate in cell wall development and repair process (Kalluri et al., 2009; Ligat et al., 2011; Zhang et al., 2014a), leaf senescence (Wang et al., 2012), abiotic stress responses (Alvarez et al., 2008), biotic stress defense mechanisms (González et al., 2012), and intercellular and intracellular communication (Agrawal et al., 2010). Additional studies of protein and metabolite composition of xylem sap and apoplast in soybean (Glycine max) provide further investigation of expression profiles and signaling roles of corresponding proteomes, and ultimately reveal more root contributions to pathogenic and symbiotic microbe interactions, and root-to-shoot communication (Djordjevic et al., 2007; Subramanian et al., 2009; Krishnan et al., 2011).
The phloem tissue isolated from broccoli (Brassica oleracea) was found to be enriched in proteins associated with biotic and abiotic stress responses and structural proteins (Anstead et al., 2013). Other proteomic studies were predominantly focused on the analysis of phloem sap exudates from agriculturally important plants oilseed rape (Giavalisco et al., 2006), rice (Aki et al., 2008), pumpkin (Lin et al., 2009; Cho et al., 2010; Fröhlich et al., 2012), and melon (Malter and Wolf, 2011), identifying several hundred physiologically relevant proteins and ribonucleoprotein complexes. The phloem sap proteomes showed enhanced presence of proteins involved in redox regulation, defense and stress responses, calcium regulation, RNA metabolism and G-protein signaling (Figure 1). Some of the important insights into the operation of the sieve tube system were revealed through proteomics studies. The findings indicate likely occurrence of protein synthesis and turnover within the angiosperm phloem translocation stream, processes that were thought to be absent in enucleate sieve elements (Lin et al., 2009). There is also indication that phloem may exert some level of control over flowering time (Giavalisco et al., 2006), seed development (Lin et al., 2009), other developmental processes via gibberellin biosynthesis or modification (Cho et al., 2010).
Root Proteomics of Symbiotic Systems to Improve Legume Productivity
In associations between plants and soil microorganisms, only symbiotic interactions have beneficial effects for the host plants while pathogenic interactions lead to severe damages (Schenkluhn et al., 2010). Rhizobia-legume symbiosis is the best characterized beneficial plant–bacterial mutualistic interaction, which represents one of the most productive nitrogen-fixing systems and effectively renders legumes being independent of other nitrogen sources (Mathesius, 2009; Colditz and Braun, 2010). Legume plants maintain control of nodulation to balance the nitrogen gains with their energy needs and developmental costs through a systemic mechanism known as the autoregulation of nodulation that involves peptide hormones, receptor kinases, and small metabolites for long-distance signaling control (Reid et al., 2011, 2012). Legumes are among the most economically important crops due to their high protein content. Model legume plant M. truncatula is closely related to major legume crops such as soybean (Glycine max) and pea (Pisum sativum), therefore its investigation is of high relevance for agriculture. Recently, comparative and quantitative proteomic studies provided in-depth characterizations of M. truncatula proteome focused on symbiosis- or pathogenesis-induced changes of root system (Schenkluhn et al., 2010; Molesini et al., 2013). Moreover, the establishment and maintenance of rhizobium–legume symbiosis require reciprocal recognition with exchanges of signal molecules and complex developmental programs between the organisms, which leads to the formation of nodules on the legume root and the differentiation of rhizobial cells into bacteroids (del Giudice et al., 2011). A number of proteomic studies have attempted to investigate mutual impacts between symbiotic or pathogenic bacteria and the root of host plant in the rhizosphere under a multitude of biotic and abiotic stresses from the soil (for a recent review refer to Knief et al., 2011). Especially under drought stress, symbiotic nitrogen fixation is one of the physiological processes to first show stress responses in nodulated legumes (Figure 1). Differential plant and bacteroid responses to drought stress have been revealed by proteomic analysis of root nodule in M. truncatula (Larrainzar et al., 2007). Large-scale phosphoproteome analyses focused on nitrogen-fixing root nodules in M. truncatula (Wienkoop et al., 2008; Grimsrud et al., 2010) and root hairs in soybean (Nguyen et al., 2012) investigated phosphorylation-mediated signal transduction cascades. Symbiotic signals produced by the rhizobia during the initiation of symbiosis and the development of nodules were revealed, suggesting a complex network of kinase-substrate and phosphatase-substrate interactions in response to rhizobial infections. Furthermore, improved knowledge of root proteases associated with rhizosphere and drought tolerance reinforces the importance of their role in endocytosis of proteins, peptides and microbes, or root exudate-mediated nitrogen uptake mechanisms, which can contribute to a systematic study of root proteases in crop improvement (Adamczyk et al., 2010; Kohli et al., 2012). Additionally, numerous studies have shown that root sensing of stresses from soil drying and salinization can regulate chemical root-to-leaf signaling through altering the complexity of constituents and their interactions in xylem sap, which ultimately reduce leaf transpiration and growth, and therefore, influence crop productivity (Bazargani et al., 2011; Martin-Vertedor and Dodd, 2011).
Proteomic Studies of the Interaction Between Seed Viability, Seedling Growth and Abiotic Stress
Tissue-Specific Proteomic Studies of Seed Developmental Stages During Abiotic Stresses
Seed developmental events are programmed to occur as a result of expression and activation of different proteins in distinct seed compartments (i.e., embryo, endosperm, and caryopsis coat) and even within specific regions (e.g., apical meristem), at distinct developmental stages (Itoh et al., 2005). Tissue- and organelle-specific proteomic studies relevant to seed development focused on characterization of temporal and spatial proteomes together with PTMs in metabolic and molecular events that occur at different seed developmental stages and the transit phases between stages (for review refer to Finnie et al., 2011a). These studies provided an insight into the physiological and biochemical pathways, stress-responsive mechanisms, nutrient accumulation and its regulation, redox regulation in programmed cell death, ferredoxin/thioredoxin-linked metabolic processes and signaling pathways, which are specific to embryo (Huang et al., 2012; Xu et al., 2012; Domínguez and Cejudo, 2014; Wolny et al., 2014), aleurone layer (Hägglund et al., 2010; Finnie et al., 2011b; Barba-Espín et al., 2014), endosperm (Vensel et al., 2005; Balmer et al., 2006), and peripheral layers (Jerkovic et al., 2010; Tasleem-Tahir et al., 2011; Miernyk and Johnston, 2013), and whole kernel solubility-based protein groups (Yang et al., 2011). These proteomic studies elucidated the molecular pathways underlying the control of seed development and physiological transitions, which contribute to the advancement of valuable and potentially agriculturally important strategies for improving yield, quality, and stress tolerance in cereals and legumes.
Pre-harvest sprouting (PHS) causes substantial losses in grain yield and quality, and therefore, it is one of the major factors negatively affecting the quality of crops in the areas with high levels of precipitation during grain maturation. Cereal crops with low levels of seed dormancy are susceptible to PHS when wet and moist conditions occur prior to harvest. A defined level of seed dormancy is under genetic, epigenetic regulation and environmental control, and it is an essential component of seed quality (Graeber et al., 2012). PHS resistance is a complex trait that is determined by genotype together with a number of other factors: stage of maturity, environmental conditions during grain ripening, crop morphology, biotic and abiotic stress (Fofana et al., 2008; Rikiishi and Maekawa, 2014). Proteomics offers the opportunity to examine simultaneous changes and to characterize temporal patterns of protein accumulation occurring during seed dormancy maintenance or release (Rajjou et al., 2011). Given that PHS is closely associated with seed dormancy, it is important to gain a deeper understanding of biomarker proteins and molecular signaling mechanisms involved in dormancy regulation, which will contribute to the breeding of PHS resistant cultivars during crop production. Recent proteomics and transcriptomics studies have indicated that antioxidant defense mechanisms, redox regulation of seed mRNAs and protein thiols, integrated with hormonal signaling play a key role in controlling seed dormancy in wheat (Bykova et al., 2011a,b; Liu et al., 2013a), barley (Bahin et al., 2011; Ishibashi et al., 2012), and rice (Huh et al., 2013; Liu et al., 2014). These findings will significantly contribute to the development of efficient strategies for breeding of PHS tolerant crops.
Proteomic Analysis of Crop Seedlings Subjected to Abiotic Stress Conditions
Seeds break dormancy and restart their metabolism to prepare for germination then proceed with seedling establishment when environmental conditions are suitable for seedling growth and development (Arc et al., 2011). However, crop seedlings in their early growth stage are subjected to various abiotic stresses in the field, which will lead to lower yields and possible crop failure. Quantitative proteomic analysis of soybean seedlings and wheat roots subjected to the flooding or osmotic stresses has revealed the metabolic pathways of flooding-responsive proteome reacting to excess water supply and anoxia, while osmosis-related proteins were responding to the various stresses such as drought, cold, and salinity stresses (Nanjo et al., 2011 and references therein). The initial changes in soybean root tip proteome under flooding stress indicated an important role of calcium signaling in the early responses (Yin et al., 2014), and the exogenous calcium treatment was shown to have a recovery effect on the expression of proteins involved in cell wall, hormone metabolisms, protein degradation and synthesis, and DNA synthesis in soybean roots under flooding stress (Oh et al., 2014). A comparative proteomic study of tissue-specific proteome in soybean seedlings under heat stress indicated common defense and adaptive mechanisms associated with elevated induction of several tissue-specific heat shock proteins and proteins involved in antioxidative defense (Figure 1). The down-regulated proteins were associated with photosynthesis, secondary metabolism, and amino acid and protein biosynthesis in response to heat stress (Ahsan et al., 2010b). Salinity stress-responsive proteins in seedlings have been investigated through a comparative proteomic analysis of salt-tolerant and salt-sensitive varieties of wheat (Guo et al., 2012) and soybean (Ma et al., 2012). A significant number of salt tolerance-related proteins were identified in wheat seedling roots, including signal transduction-associated proteins, carbon, amino acid and nitrogen metabolism proteins, detoxification and defense-related proteins (Guo et al., 2012). In a recent proteomics study of soybean seedling leaves, a salt stress-responsive protein network was proposed, including proteins responsible for redox homeostasis, as well as accelerated proteolysis, which was accompanied by reduced biosynthesis of proteins, impaired photosynthesis and energy supply, and enhanced ethylene biosynthesis (Ma et al., 2012). Recently, a comparative label-free shotgun proteomic analysis on wheat leaves of salt-stress tolerant genotype of durum wheat (T. durum Desf.; Table 2) subjected to increasing salinity levels revealed major changes in proteins involved in primary metabolism, energy production, protein metabolism and cellular defense, as well as tolerance-related high capacity for osmotic homeostasis, and increased cell wall lignification allowing for higher growth recovery potential (Capriotti et al., 2014). In order to understand the role of ascorbate priming in boosting the salt stress tolerance during germination and early seedling growth in durum wheat, a label-free quantitative analysis of whole seeds (Fercha et al., 2013), as well as seed embryos and surrounding tissues was performed to study tissue-specific variation of metabolic proteomes (Fercha et al., 2014). It was demonstrated that ascorbate pretreatment prevents the effect of salinity by specifically changing the abundance of proteins involved in metabolism, protein destination and storage categories, which could be modulated by methionine, auxin and other hormones metabolism, as well as ROS managing and signaling systems (Fercha et al., 2014). These studies provided insights into possible management strategy of cellular activities occurring in salt-responding seedlings of major crops (Figure 1).
Among the cereals, rice (Oryza sativa L.) is a salt-sensitive crop. High salinity causes delayed seed germination, slow seedling growth, and reduced rate of seed maturation, leading to decreased rice yield. In the early period of growth, rice seedling roots, leaf sheath, and leaf blade are highly sensitive to salt stress signal, which is perceived via plasma membrane receptors and can rapidly initiate an intracellular signal to elicit an adaptive response (Ruan et al., 2011). Abbasi and Komatsu (2004) conducted a proteomic analysis of rice leaf sheath, leaf blades, and roots to investigate relative abundance of salt-responsive proteins, which changed according to intracellular ion homeostasis caused by continuous excessive ion uptake. In another study by Yan et al. (2005), it was suggested that energy production was activated in rice roots subjected to salt stress due to the disruption of enzyme activities and basic metabolism. Kim et al. (2005) elucidated differentially expressed proteins in rice seedling leaves during salt stress as being functionally important in major photosynthetic metabolic processes and in oxidative damage processes. Another study supported findings of the relationship between enzymes involved in carbohydrate and energy metabolism and the increased production of antioxidants that mediate maintenance of cellular homeostasis (Liu et al., 2013b). Song et al. (2011) predicted an extracellular salt stress-responsive apoplastic protein network in shoot stems of rice seedlings, suggesting candidate proteins involved in initial perception of salt stress, such as enzymes involved in carbohydrate metabolism, the intracellular equilibrium between ROS production and ROS scavenging, and protein processing and degradation. Moreover, transgenic rice seedlings overexpressing the cyclophilin OsCYP2 showed improved tolerance to salt stress, with an increased antioxidant enzyme activity and a decreased lipid peroxidation, which indicated a role for OsCYP2 in controlling oxidative damages by modulating activities of antioxidant enzymes at translational level (Ruan et al., 2011). Barley (Hordeum vulgare L.), on the other hand, is a salt-tolerant cereal crop and shows variation for tolerance toward salinity stress. MS-based proteomic analysis revealed cultivar-specific and salt stress-responsive protein expression patterns, indicating that proteins involved in the glutathione-based detoxification of ROS were highly expressed in the salt-tolerant genotype, while proteins involved in iron uptake were abundantly expressed in the salt-sensitive genotype. This study emphasized the role of proteins involved in ROS detoxification during salinity stress, and identified potential candidates for increasing salt tolerance in barley (Witzel et al., 2009). Moreover, a comparative transcriptional profiling of barley seedlings under salt stress elucidated a large number of hormone-related kinases and protein phosphatases involved in defense responses against salinity (Gao et al., 2013). These findings provide important information that will be used toward improving salt tolerance of cereals.
More recent proteomics research on the effect of environmental changes and abiotic stress in wheat can be grouped into studies focused on either water supply-responsive proteome reacting to drought (Bazargani et al., 2011; Ford et al., 2011; Ge et al., 2012; Kang et al., 2012; Budak et al., 2013; Alvarez et al., 2014; Zhang et al., 2014b), or temperature-related changes from extreme heat (Yang et al., 2011) to cold (Rinalducci et al., 2011a,b; Kosová et al., 2013) and frost tolerance (Vítámvás et al., 2012; Han et al., 2013), or salinity tolerance and stress (Kamal et al., 2012; Guo et al., 2012; Fercha et al., 2013, 2014; Jacoby et al., 2013a; Capriotti et al., 2014), and heavy metal toxicity stress (Li et al., 2013b; Kang et al., 2015). Availability of moisture during early growing season is critical to wheat production and prolonged drought conditions due to climate change have major impact on the crop yield. Using large-scale isobaric tags-based shotgun quantitative approach, differences in drought stress-responsive proteomes were studied in leaves from mature plants of three wheat varieties, fast and slow responding drought tolerant and intolerant (Ford et al., 2011; Table 2; Figure 1). An increase in proteins involved in ROS scavenging and a down regulation of proteins involved in photosynthesis and the Calvin cycle were observed in cellular response of all three varieties with the tolerant variety showing significant protein changes during early response and increases in proteins involved in cell detoxification. In a recent study Alvarez et al. (2014) used two wheat varieties adapted to different environmental conditions, drought tolerant and sensitive, to quantitatively evaluate inherent differences in protein expression patterns overall and in variety-specific effect of abscisic acid (ABA) on the root proteome (Table 2; Figure 1). It was revealed that the tolerant wheat variety had significantly higher number of ABA-responsive and ABA-induced proteins, most of those in the categories representing response to environmental stress and oxidation-reduction processes, which can play an important role in drought adaptation. Another study showed the effect of salicylic acid treatment on the induction of drought tolerance in wheat seedlings and identified salicylic acid-responsive protein interaction network suggesting more effective defense systems, efficient photosynthesis, active anabolism, and abundant energy supply (Kang et al., 2012). Significant changes in the phosphoproteome under drought stress were demonstrated in seedling leaves of two bread wheat cultivars using gel-free TiO2 phosphopeptide enrichment and label-free relative quantiative approach (Zhang et al., 2014b). Those mainly concerned proteins involved in RNA transcription/processing, stress/detoxification/defense, and signal transduction, drought tolerance and osmotic regulation.
Two wheat cultivars of contrasting temperature growth habits, frost tolerant winter wheat and frost sensitive spring wheat, were used recently to study cold-induced changes in the proteomes of crown tissues (Kosová et al., 2013). Proteins involved in the regulation of stress response, growth cessation and maintenance of vegetative stage were specifically increased in the winter cultivar, whereas proteins involved in restoration of cell division, plant growth and development, and progression to reproductive stage were induced by cold treatment in the spring cultivar. In an earlier study proteomic analysis of the changes induced by a prolonged treatment of vernalization-requiring winter wheat with low temperatures demonstrated down-regulation of several photosynthesis-related proteins and a concomitant increase in abundance of some Calvin and TCA cycle enzymes, and proteolysis (Rinalducci et al., 2011a). Another study on the effect of long-term cold acclimation and dynamics of cold tolerance on crown proteome composition in winter wheat cultivars (Vítámvás et al., 2012) showed that cold acclimation is an active energy-demanding process accompanied by profound changes in carbohydrate metabolism and ROS-scavenging enzymatic system, increased biosynthesis of methionine and S-adenosylmethionine, chaperones, enzymes involved in protein turnover and stress-responsive proteins. Molecular mechanisms helping a cold-sensitive spring wheat cultivar to survive long term exposure to suboptimal temperatures were also investigated from the proteomics point of view (Rinalducci et al., 2011b). Quantitative differences in protein abundance indicated reinforcement in ascorbate recycling, protein processing, and accumulation of the enzyme involved in tetrapyrrole resynthesis, whereas key Krebs cycle enzymes and many photosynthesis related proteins were down-regulated (Figure 1). A short term exposure of spring wheat plants to freezing stress caused extensive changes in major metabolic processes with enhanced accumulation of key proteins involved in ROS detoxification, signal transduction, stress and disease resistance (Han et al., 2013).
Recent plant proteomics studies have also been focused on heavy metal stress responses, biochemical mechanisms involved in cellular detoxification and heavy metal tolerance (Li et al., 2013b; Kang et al., 2015). The study of copper-induced stress responses reflected in proteomes of leaves and roots from young common wheat seedlings identified significant enhancement in the abundance of proteins involved in signal transduction, stress defense, and energy production, whereas carbohydrate, protein and photosynthetic metabolisms were severely reduced (Li et al., 2013b). Under copper stress conditions, exogenous jasmonic acid application had a protective effect, and activated transcription of glutathione-S-transferase. Organ-specific differences in adaptation to high-level mercury (Hg) stress were revealed for majority of proteins with differentially altered abundance in wheat seedling roots and leaves (Kang et al., 2015). The identified Hg-responsive proteins were associated with the main biological processes (Figure 1). Furthermore, application of exogenous ABA facilitated protection against Hg stress identifying potential network of key interacting proteins. These studies provide new insights and will lead to better understanding of heavy-metal stress responses in crop plants.
Concluding Remarks and Future Challenges
Proteomic data that are particularly informative include quantitative protein profiles, profiles of regulatory modifications and protein interaction networks. Gel-based 2-DE proteomic approaches combined with gel-free MS-based quantitative proteomic techniques have been widely used for crop proteome analysis. The complex mixtures of proteins with the dynamic range of protein concentrations in plant cells have been analyzed more in-depth using a combination of separation techniques based on subcellular proteomics in different stress-responding organs and tissues. Improved protein extraction protocols, advanced gel-free quantitative techniques and bioinformatics approaches to the identification and analysis of complex proteomes at both subcellular and whole plant proteome levels in different crops have significantly advanced our understanding of developmental process, abiotic stress sensing mechanisms, and intracellular signal transduction mechanisms. The recent proteomic studies have contributed to elucidation of complex relationship between stress tolerance and crop productivity, which would enable the development of novel breeding strategies resulting in an increase in crop productivity and environmental performance.
Proteomics is rapidly becoming an indispensable tool for global phenotypic characterization at the molecular level that provides invaluable information about novel gene identifications, role of PTMs and protein interactions, linking genotype and its functionality. This type of information is especially useful in breeding programs offering specific identifications of potential biomarkers for isolation of candidate genes that can be integrated through proteomic-based marker-assisted selection and marker-based gene pyramiding (Figure 2). Furthermore, proteomics programs contribute to the analysis of advanced mapping populations such as hybrid doubled haploid lines, near isogenic lines and recombinant inbred lines, which further verify the correlations between responsive quantitative trait loci and stress tolerant phenotypes. One of the challenges is to convert massive data into knowledge that can be readily applied into crop improvement programs. The solution can be found in interdisciplinary approaches, creating sufficient genetic resources, robust bioinformatics tools with novel algorithmic solutions.
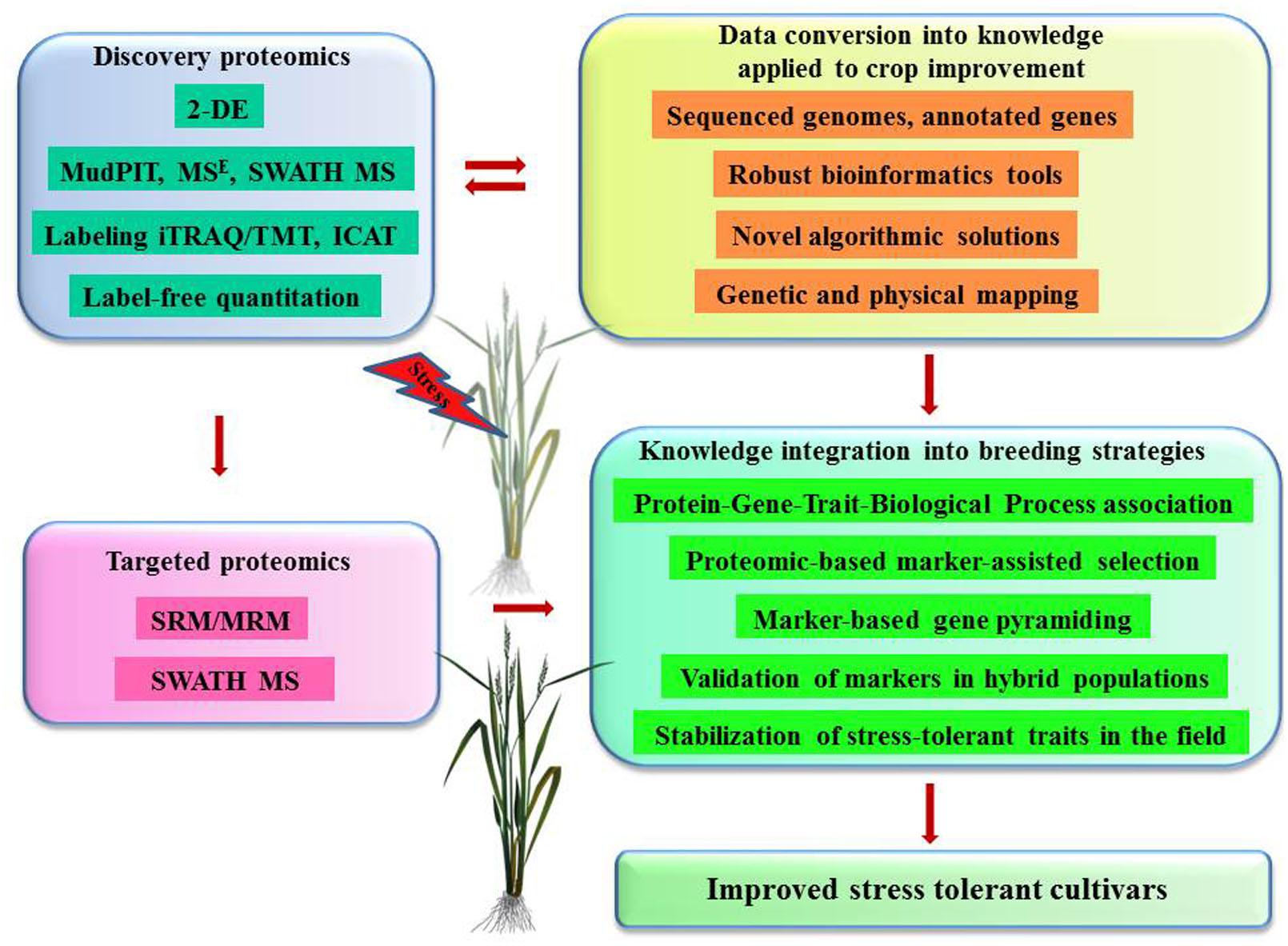
FIGURE 2. Proteomics contribution to biotechnology of crop improvement – future challenges and prospects. Discovery-based proteomics strategies contribute with important information on candidate proteins, biological processes, and mechanisms controlling tolerance to abiotic stresses. This information is further converted into specific knowledge on OTLs, genes, alleles, and their physical chromosomal locations using modern mathematical algorithms and bioinformatics tools. Targeted proteomics strategies facilitate the deployment of knowledge linking genotype and its functionality into breeding programs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors acknowledge Agriculture and Agri-Food Canada for funding support.
References
Abbasi, F. M., and Komatsu, S. (2004). A proteomic approach to analyze salt-responsive proteins in rice leaf sheath. Proteomics 4, 2072–2081. doi: 10.1002/pmic.200300741
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Adamczyk, B., Smolander, A., Kitunen, V., and Godlewski, M. (2010). Proteins as nitrogen source for plants: a short story about exudation of proteases by plant roots. Plant Signal. Behav. 5, 817–819. doi: 10.4161/psb.5.7.11699
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Agrawal, G. K., Bourguignon, J., Rolland, N., Ephritikhine, G., Ferro, M., Jaquinod, M., et al. (2011). Plant organelle proteomics: collaborating for optimal cell function. Mass Spectrom. Rev. 30, 772–853. doi: 10.1002/mas.20301
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Agrawal, G. K., Jwa, N. S., Lebrun, M. H., Job, D., and Rakwal, R. (2010). Plant secretome: unlocking secrets of the secreted proteins. Proteomics 10, 799–827. doi: 10.1002/pmic.200900514
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Agrawal, G. K., Sarkar, A., Righetti, P. G., Pedreschi, R., Carpentier, S., Wang, T., et al. (2013). A decade of plant proteomics and mass spectrometry: translation of technical advancements to food security and safety issues. Mass Spectrom. Rev. 32, 335–365. doi: 10.1002/mas.21365
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ahsan, N., Nanjo, Y., Sawada, H., Kohno, Y., and Komatsu, S. (2010a). Ozone stress-induced proteomic changes in leaf total soluble and chloroplast proteins of soybean reveal that carbon allocation is involved in adaptation in the early developmental stage. Proteomics 10, 2605–2619. doi: 10.1002/pmic.201000180
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ahsan, N., Donnart, T., Nouri, M. Z., and Komatsu, S. (2010b). Tissue-specific defense and thermo-adaptive mechanisms of soybean seedlings under heat stress revealed by proteomic approach. J. Proteome Res. 9, 4189–4204. doi: 10.1021/pr100504j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aki, T., Shigyo, M., Nakano, R., Yoneyama, T., and Yanagisawa, S. (2008). Nano scale proteomics revealed the presence of regulatory proteins including three FT-Like proteins in phloem and xylem saps from rice. Plant Cell Physiol. 49, 767–790. doi: 10.1093/pcp/pcn049
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alexandersson, E., Saalbach, G., Larsson, C., and Kjellbom, P. (2004). Arabidopsis plasma membrane proteomics identifies components of transport, signal transduction and membrane trafficking. Plant Cell Physiol. 45, 1543–1556. doi: 10.1093/pcp/pch209
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alvarez, S., Marsh, E. L., Schroeder, S. G., and Schachtman, D. P. (2008). Metabolomic and proteomic changes in the xylem sap of maize under drought. Plant Cell Environ. 31, 325–340. doi: 10.1111/j.1365-3040.2007.01770.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alvarez, S., Roy Choudhury, S., and Pandey, S. (2014). Comparative quantitative proteomics analysis of the ABA response of roots of drought-sensitive and drought-tolerant wheat varieties identifies proteomic signatures of drought adaptability. J. Proteome Res. 13, 1688–1701. doi: 10.1021/pr401165b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Anstead, J. A., Hartson, S. D., and Thompson, G. A. (2013). The broccoli (Brassica oleracea) phloem tissue proteome. BMC Genomics 14:764. doi: 10.1186/1471-2164-14-764
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arc, E., Galland, M., Cueff, G., Godin, B., Lounifi, I., Job, D., et al. (2011). Reboot the system thanks to protein post-translational modifications and proteome diversity: how quiescent seeds restart their metabolism to prepare seedling establishment. Proteomics 11, 1606–1618. doi: 10.1002/pmic.201000641
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atkinson, N. J., and Urwin, P. E. (2012). The interaction of plant biotic and abiotic stresses: from genes to the field. J. Exp. Bot. 63, 3523–3543. doi: 10.1093/jxb/ers100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Avice, J. C., and Etienne, P. (2014). Leaf senescence and nitrogen remobilization efficiency in oilseed rape (Brassica napus L.). J. Exp. Bot. 65, 3813–3824. doi: 10.1093/jxb/eru177
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bahin, E., Bailly, C., Sotta, B., Kranner, I., Corbineau, F., and Leymarie, J. (2011). Crosstalk between reactive oxygen species and hormonal signaling pathways regulates grain dormancy in barley. Plant Cell Environ. 34, 980–993. doi: 10.1111/j.1365-3040.2011.02298.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Balmer, Y., Vensel, W. H., DuPont, F. M., Buchanan, B. B., and Hurkman, W. J. (2006). Proteome of amyloplasts isolated from developing wheat endosperm presents evidence of broad metabolic capability. J. Exp. Bot. 57, 1591–1602. doi: 10.1093/jxb/erj156
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barba-Espín, G., Dedvisitsakul, P., Hägglund, P., Svensson, B., and Finnie, C. (2014). Gibberellic acid-induced aleurone layers responding to heat shock or tunicamycin provide insight into the n-glycoproteome, protein secretion, and endoplasmic reticulum stress. Plant Physiol. 164, 951–965. doi: 10.1104/pp.113.233163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barsan, C., Zouine, M., Maza, E., Bian, W., Egea, I., Rossignol, M., et al. (2012). Proteomic analysis of chloroplast-to-chromoplast transition in tomato reveals metabolic shifts coupled with disrupted thylakoid biogenesis machinery and elevated energy-production components. Plant Physiol. 160, 708–725. doi: 10.1104/pp.112.203679
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bazargani, M. M., Sarhadi, E., Bushehri, A. A., Matros, A., Mock, H. P., Naghavi, M. R., et al. (2011). A proteomics view on the role of drought-induced senescence and oxidative stress defense in enhanced stem reserves remobilization in wheat. J. Proteomics 74, 1959–1973. doi: 10.1016/j.jprot.2011.05.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Budak, H., Akpinar, B. A., Unver, T., and Turktas, M. (2013). Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanoLC-ESI-MS/MS. Plant Mol. Biol. 83, 89–103. doi: 10.1007/s11103-013-0024-25
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buts, K., Michielssens, S., Hertog, M. L., Hayakawa, E., Cordewener, J., America, A. H., et al. (2014). Improving the identification rate of data independent label-free quantitative proteomics experiments on non-model crops: a case study on apple fruit. J. Proteomics 105, 31–45. doi: 10.1016/j.jprot.2014.02.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bykova, N. V., Hoehn, B., Rampitsch, C., Banks, T., Stebbing, J. A., Fan, T., et al. (2011a). Redox-sensitive proteome and antioxidant strategies in wheat seed dormancy control. Proteomics 11, 865–882. doi: 10.1002/pmic.200900810
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bykova, N. V., Hoehn, B., Rampitsch, C., Hu, J., Stebbing, J. A., and Knox, R. (2011b). Thiol redox-sensitive seed proteome in dormant and non-dormant hybrid genotypes of wheat. Phytochemistry 72, 1162–1172. doi: 10.1016/j.phytochem.2010.12.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cappadona, S., Baker, P. R., Cutillas, P. R., Heck, A. J., and van Breukelen, B. (2012). Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino Acids 43, 1087–1108. doi: 10.1007/s00726-012-1289-1288
Capriotti, A. L., Borrelli, G. M., Colapicchioni, V., Papa, R., Piovesana, S., Samperi, R., et al. (2014). Proteomic study of a tolerant genotype of durum wheat under salt-stress conditions. Anal. Bioanal. Chem. 406, 1423–1435. doi: 10.1007/s00216-013-7549-y.
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, X., Wang, Y., Li, J. Y., Jiang, A. L., Cheng, Y. W., and Zhang, W. (2009). Mitochondrial proteome during salt stress-induced programmed cell death in rice. Plant Physiol. Biochem. 47, 407–415. doi: 10.1016/j.plaphy.2008.12.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cho, W. K., Chen, X. Y., Rim, Y., Chu, H., Kim, S., Kim, S. W., et al. (2010). Proteome study of the phloem sap of pumpkin using multidimensional protein identification technology. J. Plant Physiol. 167, 771–778. doi: 10.1016/j.jplph.2010.01.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chu, P., Yan, G. X., Yang, Q., Zhai, L. N., Zhang, C., Zhang, F. Q., et al. (2015). iTRAQ-based quantitative proteomics analysis of Brassica napus leaves reveals pathways associated with chlorophyll deficiency. J. Proteomics 113, 110–126. doi: 10.1016/j.jprot.2014.10.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colditz, F., and Braun, H.-P. (2010). Medicago truncatula proteomics. J. Proteomics 73, 1974–1985. doi: 10.1016/j.jprot.2010.07.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Bernonville, T. D., Albenne, C., Arlat, M., Hoffmann, L., Lauber, E., and Jamet, E. (2014). Xylem sap proteomics. Methods Mol. Biol. 1072, 391–405. doi: 10.1007/978-1-62703-631-3_28
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
del Giudice, J., Cam, Y., Damiani, I., Fung-Chat, F., Meilhoc, E., Bruand, C., et al. (2011). Nitric oxide is required for an optimal establishment of the Medicago truncatula-Sinorhizobium meliloti symbiosis. New Phytol. 191, 405–417. doi: 10.1111/j.1469-8137.2011.03693.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deshmukh, R., Sonah, H., Patil, G., Chen, W., Prince, S., Mutava, R., et al. (2014). Integrating omic approaches for abiotic stress tolerance in soybean. Front. Plant Sci. 5:244. doi: 10.3389/Fpls.2014.00244
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Djordjevic, M. A., Oakes, M., Li, D. X., Hwang, C. H., Hocart, C. H., and Gresshoff, P. M. (2007). The Glycine max xylem sap and apoplast proteome. J. Proteome Res. 6, 3771–3779. doi: 10.1021/pr0606833
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Domínguez, F., and Cejudo, F. J. (2014). Programmed cell death (PCD): an essential process of cereal seed development and germination. Front. Plant Sci. 5:366. doi: 10.3389/fpls.2014.00366
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fercha, A., Capriotti, A. L., Caruso, G., Cavaliere, C., Gherroucha, H., Samperi, R., et al. (2013). Gel-free proteomics reveal potential biomarkers of priming-induced salt tolerance in durum wheat. J. Proteomics 91, 486–499. doi: 10.1016/j.jprot.2013.08.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fercha, A., Capriotti, A. L., Caruso, G., Cavaliere, C., Samperi, R., Stampachiacchiere, S., et al. (2014). Comparative analysis of metabolic proteome variation in ascorbate-primed and unprimed wheat seeds during germination under salt stress. J. Proteomics 108, 238–257. doi: 10.1016/j.jprot.2014.04.040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finnie, C., Sultan, A., and Grasser, K. D. (2011a). From protein catalogues towards targeted proteomics approaches in cereal grains. Phytochemistry 72, 1145–1153. doi: 10.1016/j.phytochem.2010.11.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finnie, C., Andersen, B., Shahpiri, A., and Svensson, B. (2011b). Proteomes of the barley aleurone layer: a model system for plant signaling and protein secretion. Proteomics 11, 1595–1605. doi: 10.1002/pmic.201000656
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fofana, B., Humphreys, G., Rasul, G., Cloutier, S., and Somers, D. (2008). Assessment of molecular diversity at QTLs for preharvest sprouting resistance in wheat using microsatellite markers. Genome 51, 375–386. doi: 10.1139/G08-022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ford, K. L., Cassin, A., and Bacic, A. (2011). Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front. Plant Sci. 2:44. doi: 10.3389/fpls.2011.00044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fröhlich, A., Gaupels, F., Sarioglu, H., Holzmeister, C., Spannagl, M., Durner, J., et al. (2012). Looking deep inside: detection of low-abundance proteins in leaf extracts of Arabidopsis and phloem exudates of pumpkin. Plant Physiol. 159, 902–914. doi: 10.1104/pp.112.198077
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, R., Duan, K., Guo, G., Du, Z., Chen, Z., Li, L., et al. (2013). Comparative transcriptional profiling of two contrasting barley genotypes under salinity stress during the seedling stage. Int. J. Genomics 139, 822–835. doi: 10.1155/2013/972852
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ge, P., Hao, P., Cao, M., Guo, G., Lv, D., Subburaj, S., et al. (2013). iTRAQ-based quantitative proteomic analysis reveals new metabolic pathways of wheat seedling growth under hydrogen peroxide stress. Proteomics 13, 3046–3058. doi: 10.1002/pmic.201300042
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ge, P., Ma, C., Wang, S., Gao, L., Li, X., Guo, G., et al. (2012). Comparative proteomic analysis of grain development in two spring wheat varieties under drought stress. Anal. Bioanal. Chem. 402, 1297–1313. doi: 10.1007/s00216-011-5532-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giavalisco, P., Kapitza, K., Kolasa, A., Buhtz, A., and Kehr, J. (2006). Towards the proteome of Brassica napus phloem sap. Proteomics 6, 896–909. doi: 10.1002/pmic.200500155
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gillet, L. C., Navarro, P., Tate, S., Röst, H., Selevsek, N., Reiter, L., et al. (2012). Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717. doi: 10.1074/mcp.O111.016717
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
González, J. F., Degrassi, G., Devescovi, G., De Vleesschauwer, D., Höfte, M., Myers, M. P., et al. (2012). A proteomic study of Xanthomonas oryzae pv oryzae in rice xylem sap. J. Proteomics 75, 5911–5919. doi: 10.1016/j.jprot.2012.07.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Graeber, K., Nakabayashi, K., Miatton, E., Leubner-Metzger, G., and Soppe, W. J. (2012). Molecular mechanisms of seed dormancy. Plant Cell Environ. 35, 1769–1786. doi: 10.1111/j.1365-3040.2012.02542.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grimsrud, P. A., den Os, D., Wenger, C. D., Swaney, D. L., Schwartz, D., Sussman, M. R., et al. (2010). Large-scale phosphoprotein analysis in Medicago truncatula roots provides insight into in vivo kinase activity in legumes. Plant Physiol. 152, 19–28. doi: 10.1104/pp.109.149625
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, G., Ge, P., Ma, C., Li, X., Lv, D., Wang, S., et al. (2012). Comparative proteomic analysis of salt response proteins in seedling roots of two wheat varieties. J. Proteomics 75, 1867–1885. doi: 10.1016/j.jprot.2011.12.032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, Y. (2013). Towards systems biological understanding of leaf senescence. Plant Mol. Biol. 82, 519–528. doi: 10.1007/s11103-012-9974-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, Q., Kang, G., and Guo, T. (2013). Proteomic analysis of spring freeze-stress responsive proteins in leaves of bread wheat (Triticum aestivum L.). Plant Physiol. Biochem. 63, 236–244. doi: 10.1016/j.plaphy.2012.12.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hägglund, P., Bunkenborg, J., Yang, F., Harder, L. M., Finnie, C., and Svensson, B. (2010). Identification of thioredoxin target disulfides in proteins released from barley aleurone layers. J. Proteomics 73, 1133–1136. doi: 10.1016/j.jprot.2010.01.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holman, J. D., Dasari, S., and Tabb, D. L. (2013). Informatics of protein and posttranslational modification detection via shotgun proteomics. Methods Mol. Biol. 1002, 167–179. doi: 10.1007/978-1-62703-360-2_14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hossain, Z., Nouri, M. Z., and Komatsu, S. (2012). Plant cell organelle proteomics in response to abiotic stress. J. Proteome Res. 11, 37–48. doi: 10.1021/pr200863r
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, H., Møller, I. M., and Song, S. Q. (2012). Proteomics of desiccation tolerance during development and germination of maize embryos. J. Proteomics 75, 1247–1262. doi: 10.1016/j.jprot.2011.10.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huh, S. M., Hwang, Y. S., Shin, Y. S., Nam, M. H., Kim, D. Y., and Yoon, I. S. (2013). Comparative transcriptome profiling of developing caryopses from two rice cultivars with differential dormancy. J. Plant Physiol. 170, 1090–1100. doi: 10.1016/j.jplph.2013.03.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hynek, R., Svensson, B., Jensen, O. N., Barkholt, V., and Finnie, C. (2006). Enrichment and identification of integral membrane proteins from barley aleurone layers by reversed-phase chromatography, SDS-PAGE, and LC-MS/MS. J. Proteome Res. 5, 3105–3113. doi: 10.1021/pr0602850
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hynek, R., Svensson, B., Jensen, O. N., Barkholt, V., and Finnie, C. (2009). The plasma membrane proteome of germinating barley embryos. Proteomics 9, 3787–3794. doi: 10.1002/pmic.200800745
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
International Wheat Genome Sequencing Consortium [IWGSC]. (2014). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788. doi: 10.1126/science.1251788
Ishibashi, Y., Tawaratsumida, T., Kondo, K., Kasa, S., Sakamoto, M., Aoki, N., et al. (2012). Reactive oxygen species are involved in gibberellin/abscisic acid signaling in barley aleurone cells. Plant Physiol. 158, 1705–1714. doi: 10.1104/pp.111.192740
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Itoh, J. I., Nonomura, K. I., Ikeda, K., and Yamaki, S. (2005). Rice plant development: from zygote to spikelet. Plant Cell Physiol. 46, 23–47. doi: 10.1093/pcp/pci501
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jacoby, R. P., Millar, A. H., and Taylor, N. L. (2010). Wheat mitochondrial proteomes provide new links between antioxidant defense and plant salinity tolerance. J. Proteome Res. 9, 6595–6604. doi: 10.1021/pr1007834
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jacoby, R. P., Millar, A. H., and Taylor, N. L. (2013a). Investigating the role of respiration in plant salinity tolerance by analyzing mitochondrial proteomes from wheat and a salinity-tolerant Amphiploid (wheat × Lophopyrum elongatum). J. Proteome Res. 12, 4807–4829. doi: 10.1021/pr400504a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jacoby, R. P., Millar, A. H., and Taylor, N. L. (2013b). Application of selected reaction monitoring mass spectrometry to field-grown crop plants to allow dissection of the molecular mechanisms of abiotic stress tolerance. Front. Plant Sci. 4:20. doi: 10.3389/fpls.2013.00020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jerkovic, A., Kriegel, A. M., Bradner, J. R., Atwell, B. J., Roberts, T. H., and Willows, R. D. (2010). Strategic distribution of protective proteins within bran layers of wheat protects the nutrient-rich endosperm. Plant Physiol. 152, 1459–1470. doi: 10.1104/pp.109.149864
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kalluri, U. C., Hurst, G. B., Lankford, P. K., Ranjan, P., and Pelletier, D. A. (2009). Shotgun proteome profile of Populus developing xylem. Proteomics 9, 4871–4880. doi: 10.1002/pmic.200800854
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamal, A. H. M., Cho, K., Choi, J. S., Bae, K. H., Komatsu, S., Uozumi, N., et al. (2013). The wheat chloroplastic proteome. J. Proteomics 93, 326–342. doi: 10.1016/j.jprot.2013.03.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamal, A. H., Cho, K., Kim, D. E., Uozumi, N., Chung, K. Y., Lee, S. Y., et al. (2012). Changes in physiology and protein abundance in salt-stressed wheat chloroplasts. Mol. Biol. Rep. 39, 9059–9074. doi: 10.1007/s11033-012-1777-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamal, A. H. M., Rashid, H., Sakata, K., and Komatsu, S. (2015). Gel-free quantitative proteomic approach to identify cotyledon proteins in soybean under flooding stress. J. Proteomics 112, 1–13. doi: 10.1016/j.jprot.2014.08.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, G., Li, G., Wang, L., Wei, L., Yang, Y., Wang, P., et al. (2015). Hg-responsive proteins identified in wheat seedlings using iTRAQ analysis and the role of ABA in Hg stress. J. Proteome Res. 14, 249–67. doi: 10.1021/pr5006873
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, G., Li, G., Xu, W., Peng, X., Han, Q., Zhu, Y., et al. (2012). Proteomics reveals the effects of salicylic acid on growth and tolerance to subsequent drought stress in wheat. J. Proteome Res. 11, 6066–6079. doi: 10.1021/pr300728y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, D. W., Rakwal, R., Agrawal, G. K., Jung, Y. H., Shibato, J., Jwa, N. S., et al. (2005). A hydroponic rice seedling culture model system for investigating proteome of salt stress in rice leaf. Electrophoresis 26, 4521–4539. doi: 10.1002/elps.200500334
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, S. T., Kim, S. G., Agrawal, G. K., Kikuchi, S., and Rakwal, R. (2014). Rice proteomics: a model system for crop improvement and food security. Proteomics 14, 593–610. doi: 10.1002/pmic.201300388
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Knief, C., Delmotte, N., and Vorholt, J. A. (2011). Bacterial adaptation to life in association with plants - A proteomic perspective from culture to in situ conditions. Proteomics 11, 3086–3105. doi: 10.1002/pmic.201000818
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kohli, A., Narciso, J. O., Miro, B., and Raorane, M. (2012). Root proteases: reinforced links between nitrogen uptake and mobilization and drought tolerance. Physiol. Plant. 145, 165–179. doi: 10.1111/j.1399-3054.2012.01573.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Komatsu, S., Hiraga, S., and Nouri, M. Z. (2014). Analysis of flooding-responsive proteins localized in the nucleus of soybean root tips. Mol. Biol. Rep. 41, 1127–1139. doi: 10.1007/s11033-013-2959-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Komatsu, S., and Hossain, Z. (2013). Organ-specific proteome analysis for identification of abiotic stress response mechanism in crop. Front. Plant Sci. 4:71. doi: 10.3389/Fpls.2013.00071
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Komatsu, S., Kobayashi, Y., Nishizawa, K., Nanjo, Y., and Furukawa, K. (2010). Comparative proteomics analysis of differentially expressed proteins in soybean cell wall during flooding stress. Amino Acids 39, 1435–1449. doi: 10.1007/s00726-010-0608-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Komatsu, S., Wada, T., Abalea, Y., Nouri, M. Z., Nanjo, Y., Nakayama, N., et al. (2009). Analysis of plasma membrane proteome in soybean and application to flooding stress response. J. Proteome Res. 8, 4487–4499. doi: 10.1021/pr9002883
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kosová, K., Vítámvás, P., Planchon, S., Renaut, J., Vanková, R., and Prášil, I. T. (2013). Proteome analysis of cold response in spring and winter wheat (Triticum aestivum) crowns reveals similarities in stress adaptation and differences in regulatory processes between the growth habits. J. Proteome Res. 12, 4830–4845. doi: 10.1021/pr400600g
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krishnan, H. B., Natarajan, S. S., Bennett, J. O., and Sicher, R. C. (2011). Protein and metabolite composition of xylem sap from field-grown soybeans (Glycine max). Planta 233, 921–931. doi: 10.1007/s00425-011-1352-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Larrainzar, E., Wienkoop, S., Weckwerth, W., Ladrera, R., Arrese-Igor, C., and González, E. M. (2007). Medicago truncatula root nodule proteome analysis reveals differential plant and bacteroid responses to drought stress. Plant Physiol. 144, 1495–1507. doi: 10.1104/pp.107.101618
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, J., Dickerson, T. J., and Hoffmann-Benning, S. (2013a). Contribution of proteomics in the identification of novel proteins associated with plant growth. J. Proteome Res. 12, 4882–4891. doi: 10.1021/pr400608d
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, G., Peng, X., Xuan, H., Wei, L., Yang, Y., Guo, T., et al. (2013b). Proteomic analysis of leaves and roots of common wheat (Triticum aestivum L.) under copper-stress conditions. J. Proteome Res. 2, 4846–4861. doi: 10.1021/pr4008283
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ligat, L., Lauber, E., Albenne, C., San Clemente, H., Valot, B., Zivy, M., et al. (2011). Analysis of the xylem sap proteome of Brassica oleracea reveals a high content in secreted proteins. Proteomics 11, 1798–1813. doi: 10.1002/pmic.201000781
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, M.-K., Lee, Y.-J., Lough, T. J., Phinney, B. S., and Lucas, W. J. (2009). Analysis of the pumpkin phloem proteome provides insights into angiosperm sieve tube function. Mol. Cell. Proteomics 8, 343–356. doi: 10.1074/mcp.M800420-MCP200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, A., Gao, F., Kanno, Y., Jordan, M. C., Kamiya, Y., Seo, M., et al. (2013a). Regulation of wheat seed dormancy by after-ripening is mediated by specific transcriptional switches that induce changes in seed hormone metabolism and signaling. PLoS ONE 8:e56570. doi: 10.1371/journal.pone.0056570
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, D. W., Ford, K. L., Roessner, U., Natera, S., Cassin, A. M., Patterson, J. H., et al. (2013b). Rice suspension cultured cells are evaluated as a model system to study salt responsive networks in plants using a combined proteomic and metabolomic profiling approach. Proteomics 13, 2046–2062. doi: 10.1002/pmic.201200425
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, J., Wu, Y. H., Yang, J. J., Liu, Y. D., and Shen, F. F. (2008). Protein degradation and nitrogen remobilization during leaf senescence. J. Plant Biol. 51, 11–19. doi: 10.1007/BF03030735
Liu, Y., Fang, J., Xu, F., Chu, J., Yan, C., Schläppi, M. R., et al. (2014). Expression patterns of ABA and GA metabolism genes and hormone levels during rice seed development and imbibition: a comparison of dormant and non-dormant rice cultivars. J. Genet. Genomics 41, 327–338. doi: 10.1016/j.jgg.2014.04.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, H., Song, L., Shu, Y., Wang, S., Niu, J., Wang, Z., et al. (2012). Comparative proteomic analysis of seedling leaves of different salt tolerant soybean genotypes. J. Proteomics 75, 1529–1546. doi: 10.1016/j.jprot.2011.11.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Malter, D., and Wolf, S. (2011). Melon phloem-sap proteome: developmental control and response to viral infection. Protoplasma 248, 217–224. doi: 10.1007/s00709-010-0215-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martin-Vertedor, A. I., and Dodd, I. C. (2011). Root-to-shoot signaling when soil moisture is heterogeneous: increasing the proportion of root biomass in drying soil inhibits leaf growth and increases leaf abscisic acid concentration. Plant Cell Environ. 34, 1164–1175. doi: 10.1111/j.1365-3040.2011.02315.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mathesius, U. (2009). Comparative proteomic studies of root-microbe interactions. J. Proteomics 72, 353–366. doi: 10.1016/j.jprot.2008.12.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matros, A., Kaspar, S., Witzel, K., and Mock, H. P. (2011). Recent progress in liquid chromatography-based separation and label-free quantitative plant proteomics. Phytochemistry 72, 963–974. doi: 10.1016/j.phytochem.2010.11.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miernyk, J. A., and Johnston, M. L. (2013). Proteomic analysis of the testa from developing soybean seeds. J. Proteomics 89, 265–272. doi: 10.1016/j.jprot.2013.05.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Molesini, B., Cecconi, D., Pii, Y., and Pandolfini, T. (2013). Local and systemic proteomic changes in Medicago truncatula at an early phase of Sinorhizobium meliloti infection. J. Proteome Res. 13, 408–421. doi: 10.1021/pr4009942
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nanjo, Y., Nouri, M. Z., and Komatsu, S. (2011). Quantitative proteomic analyses of crop seedlings subjected to stress conditions; a commentary. Phytochemistry 72, 1263–1272. doi: 10.1016/j.phytochem.2010.10.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ngara, R., and Ndimba, B. (2014). Model plant systems in salinity and drought stress proteomics studies: a perspective on Arabidopsis and Sorghum. Plant Biol. 16, 1029–1032. doi: 10.1111/plb.12247
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nguyen, T. H., Brechenmacher, L., Aldrich, J. T., Clauss, T. R., Gritsenko, M. A., Hixson, K. K., et al. (2012). Quantitative phosphoproteomic analysis of soybean root hairs inoculated with Bradyrhizobium japonicum. Mol. Cell. Proteomics 11, 1140–1155. doi: 10.1074/mcp.M112.018028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nouri, M. Z., and Komatsu, S. (2010). Comparative analysis of soybean plasma membrane proteins under osmotic stress using gel-based and LC MS/MS-based proteomics approaches. Proteomics 10, 1930–1945. doi: 10.1002/pmic.200900632
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oh, M., Nanjo, Y., and Komatsu, S. (2014). Gel-free proteomic analysis of soybean root proteins affected by calcium under flooding stress. Front. Plant Sci. 5:559. doi: 10.3389/fpls.2014.00559
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Panda, D., and Sarkar, R. K. (2013). Natural leaf senescence: probed by chlorophyll fluorescence, CO2 photosynthetic rate and antioxidant enzyme activities during grain filling in different rice cultivars. Physiol. Mol. Biol. Plants 19, 43–51. doi: 10.1007/s12298-012-0142-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pascovici, D., Gardiner, D. M., Song, X., Breen, E., Solomon, P. S., Keighley, T., et al. (2013). Coverage and consistency: bioinformatics aspects of the analysis of multirun iTRAQ experiments with wheat leaves. J. Proteome Res. 12, 4870–4881. doi: 10.1021/pr400531y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pflieger, D., Bigeard, J., and Hirt, H. (2011). Isolation and characterization of plant protein complexes by mass spectrometry. Proteomics 11, 1824–1833. doi: 10.1002/pmic.201000635
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Picotti, P., Bodenmiller, B., and Aebersold, R. (2013). Proteomics meets the scientific method. Nat. Methods 10, 24–27. doi: 10.1038/nmeth.2291
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rajjou, L., Belghazi, M., Catusse, J., Ogé, L., Arc, E., Godin, B., et al. (2011). Proteomics and posttranslational proteomics of seed dormancy and germination. Methods Mol. Biol. 773, 215–236. doi: 10.1007/978-1-61779-231-1_14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reid, D. E., Ferguson, B. J., Hayashi, S., Lin, Y. H., and Gresshoff, P. M. (2011). Molecular mechanisms controlling legume autoregulation of nodulation. Ann. Bot. 108, 789–795. doi: 10.1093/aob/mcr205
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reid, D. E., Hayashi, S., Lorenc, M., Stiller, J., Edwards, D., Gresshoff, P. M., et al. (2012). Identification of systemic responses in soybean nodulation by xylem sap feeding and complete transcriptome sequencing reveal a novel component of the autoregulation pathway. Plant Biotechnol. J. 10, 680–689. doi: 10.1111/j.1467-7652.2012.00706.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rikiishi, K., and Maekawa, M. (2014). Seed maturation regulators are related to the control of seed dormancy in wheat (Triticum aestivum L.). PLoS ONE 9:e107618. doi: 10.1371/journal.pone.0107618
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rinalducci, S., Egidi, M. G., Mahfoozi, S., Godehkahriz, S. J., and Zolla, L. (2011a). The influence of temperature on plant development in a vernalization-requiring winter wheat: a 2-DE based proteomic investigation. J. Proteomics 74, 643–659. doi: 10.1016/j.jprot.2011.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rinalducci, S., Egidi, M. G., Karimzadeh, G., Jazii, F. R., and Zolla, L. (2011b). Proteomic analysis of a spring wheat cultivar in response to prolonged cold stress. Electrophoresis 32, 1807–1818. doi: 10.1002/elps.201000663
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roberts, I. N., Caputo, C., Criado, M. V., and Funk, C. (2012). Senescence-associated proteases in plants. Physiol. Plant. 145, 130–139. doi: 10.1111/j.1399-3054.2012.01574.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ruan, S. L., Ma, H. S., Wang, S. H., Fu, Y. P., Xin, Y., Liu, W. Z., et al. (2011). Proteomic identification of OsCYP2, a rice cyclophilin that confers salt tolerance in rice (Oryza sativa L.) seedlings when overexpressed. BMC Plant Biol. 11:34. doi: 10.1186/1471-2229-11-34
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schenkluhn, L., Hohnjec, N., Niehaus, K., Schmitz, U., and Colditz, F. (2010). Differential gel electrophoresis (DIGE) to quantitatively monitor early symbiosis- and pathogenesis-induced changes of the Medicago truncatula root proteome. J. Proteomics 73, 753–768. doi: 10.1016/j.jprot.2009.10.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Singh, R., and Jwa, N. S. (2013). Understanding the responses of rice to environmental stress using proteomics. J. Proteome Res. 12, 4652–4669. doi: 10.1021/pr400689j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, R., Mathis, A. D., Ventura, D., and Prince, J. T. (2014). Proteomics, lipidomics, metabolomics: a mass spectrometry tutorial from a computer scientist’s point of view. BMC Bioinformatics 15(Suppl. 7):S9. doi: 10.1186/1471-2105-15-S7-S9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Song, Y., Zhang, C., Ge, W., Zhang, Y., Burlingame, A. L., and Guo, Y. (2011). Identification of NaCl stress-responsive apoplastic proteins in rice shoot stems by 2D-DIGE. J. Proteomics 74, 1045–1067. doi: 10.1016/j.jprot.2011.03.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Subramanian, S., Cho, U.-H., Keyes, C., and Yu, O. (2009). Distinct changes in soybean xylem sap proteome in response to pathogenic and symbiotic microbe interactions. BMC Plant Biol. 9:119. doi: 10.1186/1471-2229-9-119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suzuki, N., Rivero, R. M., Shulaev, V., Blumwald, E., and Mittler, R. (2014). Abiotic and biotic stress combinations. New Phytol. 203, 32–43. doi: 10.1111/nph.12797
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tasleem-Tahir, A., Nadaud, I., Girousse, C., Martre, P., Marion, D., and Branlard, G. (2011). Proteomic analysis of peripheral layers during wheat (Triticum aestivum L.) grain development. Proteomics 11, 371–379. doi: 10.1002/pmic.201000333
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taylor, N. L., Heazlewood, J. L., Day, D. A., and Millar, A. H. (2005). Differential impact of environmental stresses on the pea mitochondrial proteome. Mol. Cell. Proteomics 4, 1122–1133. doi: 10.1074/mcp.M400210-MCP200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uvackova, L., Skultety, L., Bekesova, S., McClain, S., and Hajduch, M. (2013a). MS(E) based multiplex protein analysis quantified important allergenic proteins and detected relevant peptides carrying known epitopes in wheat grain extracts. J. Proteome Res. 12, 4862–4869. doi: 10.1021/pr400336f
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uvackova, L., Skultety, L., Bekesova, S., McClain, S., and Hajduch, M. (2013b). The MS(E)-proteomic analysis of gliadins and glutenins in wheat grain identifies and quantifies proteins associated with celiac disease and baker’s asthma. J. Proteomics 93, 65–73. doi: 10.1016/j.jprot.2012.12.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vensel, W. H., Tanaka, C. K., Cai, N., Wong, J. H., Buchanan, B. B., and Hurkman, W. J. (2005). Developmental changes in the metabolic protein profiles of wheat endosperm. Proteomics 5, 1594–1611. doi: 10.1002/pmic.200401034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vítámvás, P., Prášil, I. T., Kosová, K., Planchon, S., and Renaut, J. (2012). Analysis of proteome and frost tolerance in chromosome 5A and 5B reciprocal substitution lines between two winter wheats during long-term cold acclimation. Proteomics 12, 68–85. doi: 10.1002/pmic.201000779
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Li, B., Du, M., Eneji, A. E., Wang, B., Duan, L., et al. (2012). Mechanism of phytohormone involvement in feedback regulation of cotton leaf senescence induced by potassium deficiency. J. Exp. Bot. 63, 5887–5901. doi: 10.1093/jxb/ers238
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wienkoop, S., Baginsky, S., and Weckwerth, W. (2010). Arabidopsis thaliana as a model organism for plant proteome research. J. Proteomics 73, 2239–2248. doi: 10.1016/j.jprot.2010.07.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wienkoop, S., Larrainzar, E., Glinski, M., González, E. M., Arrese-Igor, C., and Weckwerth, W. (2008). Absolute quantification of Medicago truncatula sucrose synthase isoforms and N-metabolism enzymes in symbiotic root nodules and the detection of novel nodule phosphoproteins by mass spectrometry. J. Exp. Bot. 59, 3307–3315. doi: 10.1093/jxb/ern182
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Witzel, K., Weidner, A., Surabhi, G. K., Börner, A., and Mock, H. P. (2009). Salt stress-induced alterations in the root proteome of barley genotypes with contrasting response towards salinity. J. Exp. Bot. 60, 3545–3557. doi: 10.1093/jxb/erp198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wolny, E., Braszewska-Zalewska, A., and Hasterok, R. (2014). Spatial distribution of epigenetic modifications in Brachypodium distachyon embryos during seed maturation and germination. PLoS ONE 9:e101246. doi: 10.1371/journal.pone.0101246
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, D. Z., Shen, Q. F., Qiu, L., Han, Y., Ye, L. Z., Jabeen, Z., et al. (2014a). Identification of proteins associated with ion homeostasis and salt tolerance in barley. Proteomics 14, 1381–1392. doi: 10.1002/pmic.201300221
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, C., Shi, T., Brown, J. N., He, J., Gao, Y., Fillmore, T. L., et al. (2014b). Expediting SRM assay development for large-scale targeted proteomics experiments. J. Proteome Res. 13, 4479–4487. doi: 10.1021/pr500500d
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, H., Zhang, W., Gao, Y., Zhao, Y., Guo, L., and Wang, J. (2012). Proteomic analysis of embryo development in rice (Oryza sativa). Planta 235, 687–701. doi: 10.1007/s00425-011-1535-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yan, S., Tang, Z., Su, W., and Sun, W. (2005). Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics 5, 235–244. doi: 10.1002/pmic.200400853
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, F., Jørgensen, A. D., Li, H., Søndergaard, I., Finnie, C., Svensson, B., et al. (2011). Implications of high-temperature events and water deficits on protein profiles in wheat (Triticum aestivum L. cv. Vinjett) grain. Proteomics 11, 1684–1695. doi: 10.1002/pmic.201000654
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yin, X., Sakata, K., Nanjo, Y., and Komatsu, S. (2014). Analysis of initial changes in the proteins of soybean root tip under flooding stress using gel-free and gel-based proteomic techniques. J. Proteomics 106, 1–16. doi: 10.1016/j.jprot.2014.04.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, A., Lu, Q., Yin, Y., Ding, S., Wen, X., and Lu, C. (2010). Comparative proteomic analysis provides new insights into the regulation of carbon metabolism during leaf senescence of rice grown under field conditions. J. Plant Physiol. 167, 1380–1389. doi: 10.1016/j.jplph.2010.05.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, Z., Xin, W., Wang, S., Zhang, X., Dai, H., Sun, R., et al. (2014a). Xylem sap in cotton contains proteins that contribute to environmental stress response and cell wall development. Funct. Integr. Genomics 15, 17–26. doi: 10.1007/s10142-014-0395-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, M., Lv, D., Ge, P., Bian, Y., Chen, G., Zhu, G., et al. (2014b). Phosphoproteome analysis reveals new drought response and defense mechanisms of seedling leaves in bread wheat (Triticum aestivum L.). J Proteomics 109, 290–308. doi: 10.1016/j.jprot.2014.07.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, J., Alvarez, S., Marsh, E. L., Lenoble, M. E., Cho, I. J., Sivaguru, M., et al. (2007). Cell wall proteome in the maize primary root elongation zone. II. Region-specific changes in water soluble and lightly ionically bound proteins under water deficit. Plant Physiol. 145, 1533–1548. doi: 10.1104/pp.107.107250
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zörb, C., Herbst, R., Forreiter, C., and Schubert, S. (2009). Short-term effects of salt exposure on the maize chloroplast protein pattern. Proteomics 9, 4209–4220. doi: 10.1002/pmic.200800791
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: combinatorial stresses, quantitative techniques, crop productivity, tissue-specific proteomics, subcellular localization
Citation: Hu J, Rampitsch C and Bykova NV (2015) Advances in plant proteomics toward improvement of crop productivity and stress resistance. Front. Plant Sci. 6:209. doi: 10.3389/fpls.2015.00209
Received: 31 December 2014; Accepted: 16 March 2015;
Published online: 14 April 2015
Edited by:
Sabine Lüthje, University of Hamburg, GermanyReviewed by:
Christian Lindermayr, Helmholtz Zentrum München, GermanyJesus V. Jorrin Novo, University of Cordoba, Spain
Copyright © 2015 Hu, Rampitsch and Bykova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalia V. Bykova, Cereal Proteomics, Cereal Research Centre, Agriculture and Agri-Food Canada, 101 Route 100, Morden, MB R6M 1Y5, CanadabmF0YWxpYS5ieWtvdmFAYWdyLmdjLmNh