 Kiruba S. Arun-Chinnappa
Kiruba S. Arun-Chinnappa David W. McCurdy
David W. McCurdy- Centre for Plant Science, School of Environmental and Life Sciences, The University of Newcastle, Newcastle, NSW, Australia
Vicia faba (L.) is an important cool-season grain legume species used widely in agriculture but also in plant physiology research, particularly as an experimental model to study transfer cell (TC) development. TCs are specialized nutrient transport cells in plants, characterized by invaginated wall ingrowths with amplified plasma membrane surface area enriched with transporter proteins that facilitate nutrient transfer. Many TCs are formed by trans-differentiation from differentiated cells at apoplasmic/symplasmic boundaries in nutrient transport. Adaxial epidermal cells of isolated cotyledons can be induced to form functional TCs, thus providing a valuable experimental system to investigate genetic regulation of TC trans-differentiation. The genome of V. faba is exceedingly large (ca. 13 Gb), however, and limited genomic information is available for this species. To provide a resource for future transcript profiling of epidermal TC differentiation, we have undertaken de novo assembly of a genome-wide transcriptome map for V. faba. Illumina paired-end sequencing of total RNA pooled from different tissues and different stages, including isolated cotyledons induced to form epidermal TCs, generated 69.5 M reads, of which 65.8 M were used for assembly following trimming and quality control. Assembly using a De-Bruijn graph-based approach generated 21,297 contigs, of which 80.6% were successfully annotated against GO terms. The assembly was validated against known V. faba cDNAs held in GenBank, including transcripts previously identified as being specifically expressed in epidermal cells across TC trans-differentiation. This genome-wide transcriptome map therefore provides a valuable tool for future transcript profiling of epidermal TC trans-differentiation, and also enriches the genetic resources available for this important legume crop species.
Introduction
The introduction of RNA sequencing (RNA-Seq) has widened the use of plant species for molecular genetic analyses beyond those for which full genome sequences are available. De novo assembly of transcriptomes from such species has enabled gene discovery and marker identification in a wide variety of non-genome-sequenced agricultural species such as common wheat (Duan et al., 2012), peanut (Zhang et al., 2012), and rubber tree (Mantello et al., 2014), but especially in crop legumes such as pea (Franssen et al., 2011), yellow lupin (Parra-González et al., 2012), lentils (Verma et al., 2013), and chick pea (Garg et al., 2011a,b), which are important to agriculture but genetic studies have been limited due to their often large genomes. In combination with de novo assembly of novel transcriptomes, RNA-Seq provides a platform for transcript profiling in these non-annotated species, thus enabling a deeper understanding at the transcriptional level of diverse but little studied biological processes in plants, such as fiber development in ramie (Boehmeria nivea L.; Chen et al., 2014), cell wall synthesizing enzymes in paper mulberry (Xianjun et al., 2014), biomass production in Panicum hallii (Meyer et al., 2012), and plant sex determination in cucumber (Guo et al., 2010).
Vicia faba (L.) is an important cool-season grain legume species used widely for human nutrition, fodder for livestock and as a rotation crop for nitrogen replenishment of soils (Torres et al., 1993; El-Rodeny et al., 2014). In addition to its importance as a crop species worldwide, V. faba is also widely used in plant physiology research to study such processes as guard cell dynamics in leaves (Fukuda et al., 1998; Fellè et al., 2000), transport processes in phloem (Thorpe et al., 2010; Hafke et al., 2013), and importantly for this study, transfer cell (TC) development in cotyledons (Offler et al., 2003). In the latter case, when isolated cotyledons are placed in culture, adaxial epidermal cells trans-differentiate to become functional epidermal TCs (Farley et al., 2000; Dibley et al., 2009). This cotyledon culture system has been used extensively to investigate the development and composition of the reticulate wall ingrowth network that defines TC identity in these cells (Talbot et al., 2001, 2007a,b; Vaughn et al., 2007), in addition to defining the role of inductive signals such as sugars (Wardini et al., 2007), auxin (Dibley et al., 2009), ethylene (Zhou et al., 2010; Andriunas et al., 2011), reactive oxygen species (Andriunas et al., 2012; Xia et al., 2012), and calcium (Zhang et al., 2015) in polarized wall ingrowth deposition. In an initial study to analyse transcriptional changes occurring across induction and development of adaxial epidermal TCs in cultured cotyledons, Dibley et al. (2009) used cDNA-amplified fragment length polymorphism (AFLP) to detect 5795 transcript-derived fragments (TDFs), of which 264 showed epidermal-specific changes in expression. However, due to the limited capacity of cDNA-AFLP for high-throughput analysis, only 112 TDFs from this cohort of 264 were sequenced to provide a glimpse of the diversity of genes involved in signaling, metabolism, cell division, vesicle trafficking, and cell wall biosynthesis putatively involved in TC trans-differentiation (Dibley et al., 2009). Importantly, however, this study estimated that TC formation in this system may involve differential expression of approximately 650 different genes (Dibley et al., 2009), a total approaching the minimum 815 genes determined from a 12 K microarray analysis to be differentially expressed during nucellar projection and endosperm TC development in barley grains (Thiel et al., 2008).
RNA-Seq provides an attractive technical platform for deep transcript profiling of the trans-differentiation of epidermal TCs in V. faba cotyledons. However, the genome size of V. faba is estimated to be 13 Gb (Kaur et al., 2012), one of the largest described to date for crop legumes, thus making this species an unattractive target for genome sequencing but nonetheless compatible for de novo transcriptome assembly (Kaur et al., 2012). To provide a platform for future RNA-Seq analysis of gene expression during TC differentiation, in this communication we report the de novo assembly of a genome-wide transcriptome map of V. faba using Illumina-based 100 bp paired-end sequencing. Total RNA was isolated from diverse tissues and organs of V. faba, including isolated cotyledons induced to form epidermal TCs. Therefore, this genome-wide transcriptome database would be expected to contain genes involved in the trans-differentiation of epidermal TCs in V. faba cotyledons, and thus provide a reference transcriptome map suitable for transcript profiling of this process by RNA-Seq.
Materials and Methods
Plant Growth and Harvesting
Vicia faba L. (cv. Fiord) plants were grown in environmentally-controlled glasshouse conditions as previously described (Farley et al., 2000). To survey V. faba transcripts, selected tissues were harvested from flowering plants including expanding and fully expanded leaves, elongating and fully elongated stems, and closed and open flowers. Whole roots including root hairs were isolated from 10-day old seedlings germinated on filter paper. At least three independent biological samples of each tissue type were harvested and snap frozen in liquid nitrogen.
To enrich the transcriptome with genes involved in induction and wall ingrowth deposition in epidermal TCs, isolated cotyledons cultured for 0, 3, 9, and 24 h were also sampled. For this process, V. faba cotyledons (100–120 mg FW) were removed from pods and either fixed immediately (t = 0 h) in ice-cold ethanol:acetic acid (3:1 v/v) for 1 h, or placed adaxial surface down on filter paper soaked in Murashige and Skoog medium (MS; Sigma Australia) in Petri dishes (Dibley et al., 2009). The Petri dishes were sealed with tape and incubated in darkness at 22°C for 3, 9, or 24 h, then the cotyledons were fixed in ice-cold ethanol:acetic acid as described above. Fixation of cotyledons in this manner was used to rapidly inhibit RNA degradation (see Dibley et al., 2009). Fixed cotyledons were rinsed briefly in distilled water, snap frozen in liquid nitrogen and stored at −80°C before isolation of total RNA. A minimum of 3 cotyledons derived from separate pods were sampled for each time point for this analysis.
Total RNA Isolation and cDNA Library Preparation
Total RNA was independently extracted from each batch of isolated organs/tissues (harvested as described above) using an RNeasy Plant RNA isolation kit (Qiagen) incorporating on-column DNA digestion. For transcriptome assembly, up to 1 μg of total RNA from each organ/tissue sample type was pooled to achieve 20 μg in total, of which at least 4 μg was derived from cultured cotyledon tissue. The pooled total RNA was subjected to QA analysis using an Agilent 2100 Bioanalyzer. A single cDNA library for 100-bp paired-end sequencing was constructed from this pooled total RNA using the Illumina TrySeq Library kit and sequencing was performed using the Illumina HiSeq-2000 platform. QA analysis, library construction, and sequencing were performed by the Beijing Genome Institute (Hong Kong).
De novo Assembly of Contigs
Illumina sequencing generated 69,543,694 100-bp paired-end reads from the pooled total RNA used for transcriptome assembly. Sequence filtering was performed by trimming adapter sequences, excluding low quality reads, and ribosomal-derived reads and removing the last 10 bases from the 3′ end of each read to increase sequence confidence. The resulting 65,795,198 90-bp reads were then assembled using a De-Bruijn graph-based de novo assembly program in CLC Genomics Workbench 6.1 run on an Intel® Xeon® workstation with 64 Gb RAM. Word size (k-mer) and bubble size were varied to obtain optimum de novo assembled contigs (Henkel et al., 2012), based on assembly output parameters such as high N50, low total number of contigs, high average contig length, and high percentage of mapped reads (Garg et al., 2011a; Annadurai et al., 2012).
Validation of de novo Assembled Contigs
The optimum assembly (47×300—see Results) derived from comparing k-mer and bubble size was used to create a BLAST database in CLC Genomics Workbench 6.1. Contig validation using 32 full-length V. faba cDNA sequences from GenBank was then performed by executing a BLASTN search for each cDNA against the 47×300 assembly and choosing the top two contigs with highest bit scores (Mizrachi et al., 2010). The outcome of BLASTN alignment parameters for each of the 32 cDNAs with their best hit contig was assessed for validating the contigs derived from the 47×300 assembly.
Functional Annotation
Contigs derived from the 47×300 assembly were functionally annotated by BLASTX analysis against both the Viridiplantae and Arabidopsis databases using default parameters in CLC Genomics Workbench 6.1. The BLASTX output files from both searches were then used to assign the three GO terms of Biological Process (BP), Molecular Function (MF), and Cellular Component (CC) separately using the Blast2GO plugin in CLC Genomics Workbench 6.1 (Conesa et al., 2005). The annotations from both datasets were merged to yield at least one annotation for most of the contigs. The “GO slim” routine in Blast2GO was performed on the merged annotation dataset in order to merge specific GO terms into higher-order generic terms for each contig. This merging eases the process of creating a “combined graph,” which is a directed acrylic graph that summarizes the functional relationship within and between all three GO terms (BP, MF, CC) using Blast2GO (Conesa et al., 2005; Gotz et al., 2008).
Results
Generation of a de novo-Assembled, Genome-Wide Transcriptome Map of V. faba
To construct a genome-wide transcriptome map of V. faba, total RNA was isolated from all major organs across different stages of development, including isolated cotyledons cultured for 0, 3, 9, and 24 h (see Materials and Methods). Illumina sequencing of this pooled total RNA yielded approximately 69.5 M paired-end reads, and after sequence filtering and quality control, a total of nearly 65.8 M 90-bp paired-end reads were used for de novo assembly using CLC Genomics Workbench 6.1 (see Materials and Methods). De novo assembly was optimized by comparing word size (k-mer) and bubble size on assembly outcomes (Henkel et al., 2012). From 24 different assemblies (see Supplementary Table 1), assembly 47×300 (k-mer 47, bubble size 300) was chosen based on the maximum number of optimum parameters such as high N50 value (1245), low total number of contigs (21,297), high values for both longest contig length (10,528), and percentage of mapped reads (82%) contributing to the assembly (see Table 1; Garg et al., 2011a; Annadurai et al., 2012; Henkel et al., 2012; Dorn et al., 2013). The parameter Reads Mapped Back to Transcripts (RMBT) is an indication of completeness of assembly for de novo assembled transcripts (Moreton et al., 2014), and assembly 47×300 returned a high score of 82% (see Table 1). This assembly was chosen in preference to assembly 20×300 due to the latter's low value for percentage mapped reads (see Supplementary Table 1). Furthermore, a k-mer value of 47 is consistent with previous studies using this value as an optimum word size for de novo transcriptome assembly in other non-sequenced species such as chick pea (Cicer arietinum; Garg et al., 2011a) and insulin plant (Costus pictus; Annadurai et al., 2012). Contig length in assembly 47×300 varied from 432 bp to 10,528 bp, with an average of 1114 bp. Total number of contigs decreased with increasing contig length, with the majority of assembled contigs belonging to the size range 500–800 bp and >1500 bp (see Figure 1), while total reads mapped per contig ranged from 33 to 1,015,710 with an average of 2528 (see Table 2). A majority of the reads were mapped to assembled contigs greater than 1500 bp length, with the least amount of reads mapping to contigs of 400–600 bp (see Table 2). The contigs making up assembly 47×300 have been submitted to the Cool Season Food Legume Genome Database (www.coolseasonfoodlegume.org), while the 69.5 M paired-end raw reads have been submitted to Short Read Archive database (NCBI; accession number SRP055969).

Table 1. Statistics of RNA-Seq and de novo assembly (47×300) of Vicia faba transcriptome map.
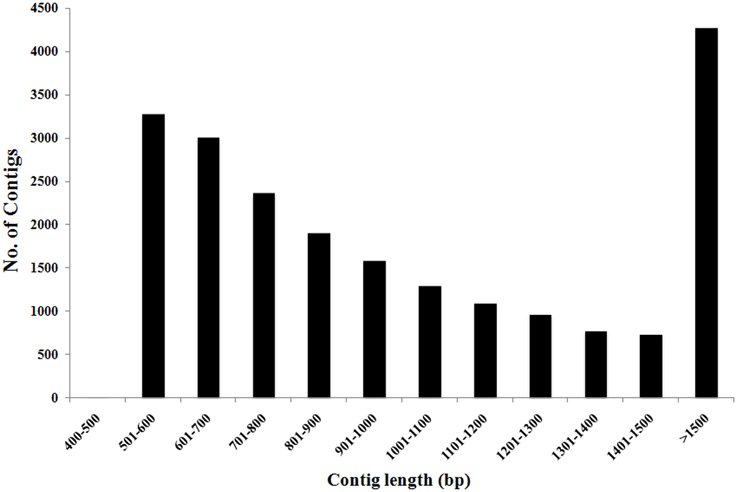
Figure 1. Distribution of total number of V. faba contigs vs. contig length. Most contig lengths were in the range of 500–800 bp (50%) and above 1500 bp (20%). Total number of contigs decreased as contig length increased.
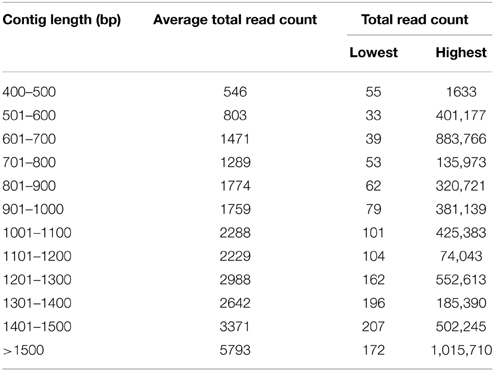
Table 2. Statistics of mapped reads assembled into contigs in assembly 47×300.
Validation of the Genome-Wide Transcriptome Map
To assess the validity of assembly 47×300, 32 full-length V. faba cDNAs available in GenBank were compared by BLASTN against the 21,297 contigs that make up assembly 47×300. Of this cDNA collection, ranging in length from 259 to 3426 bp, 29 cDNAs returned matches of 97–100% identity, while only three returned matches between 90 and 94% (see Table 3). This result indicates that the assembled contig closely matched the relevant cDNA across the length of the transcript-derived contig. Several of the cDNAs that best aligned to contigs in assembly 47×300 corresponded to V. faba cDNAs previously shown to have putative roles in signaling epidermal TC trans-differentiation (see Table 3; Zhou et al., 2010; Andriunas et al., 2012). For example, three ethylene response factor genes (VfERF1-3; Zhou et al., 2010), and two putative respiratory burst oxidase homolog genes (VfrbohA, VfrbohC; Andriunas et al., 2012), each of which shows epidermal-specific change in expression accompanying the trans-differentiation of epidermal TCs, all matched with 98–100% identity across corresponding contig sequence coverage present in assembly 47×300 (see Table 3).
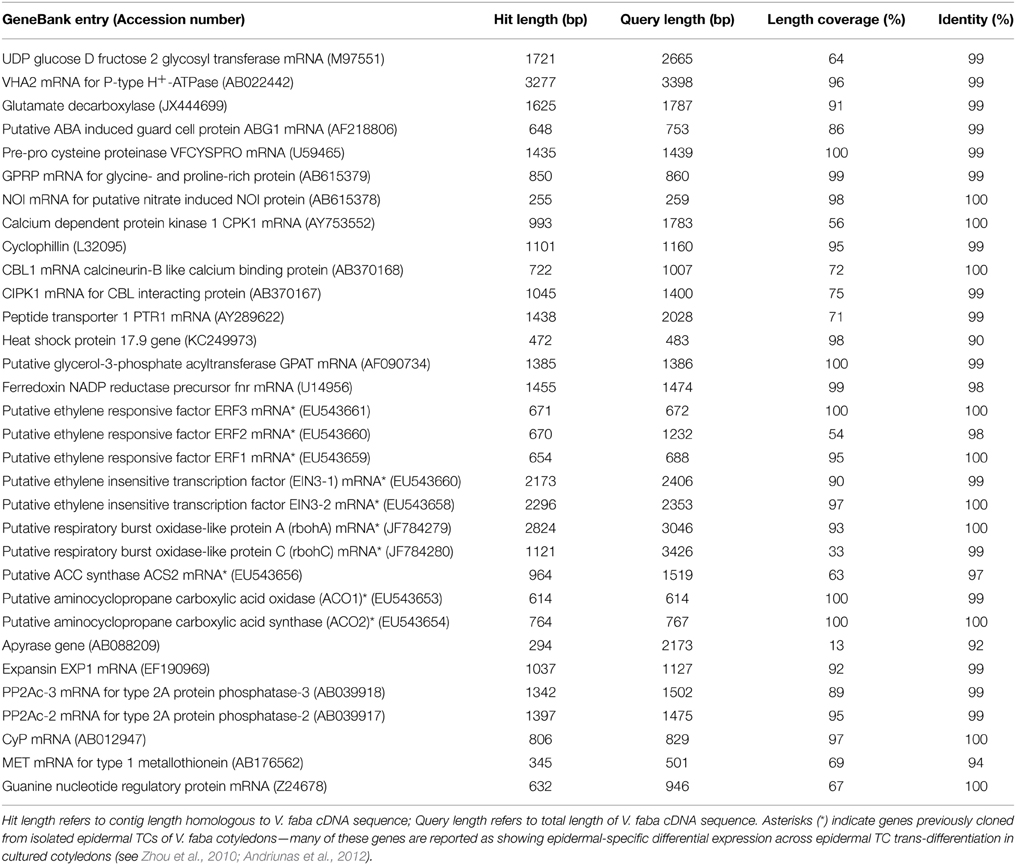
Table 3. BLASTN output for 32 full-length Vicia faba cDNAs assessed against assembly 47×300.
Functional Annotation of Contigs
Functional annotation was undertaken to assign GO terms to the 21,297 contigs comprising assembly 47×300. BLASTX analysis in CLC Genomics Workbench yielded 20,325 and 20,052 annotation hits when compared against the Viridiplantae and Arabidopsis data sets, respectively. The Blast2GO plugin in CLC successfully annotated 12,192 and 15,794 contigs from the Viridiplantae and Arabidopsis datasets, respectively, and thus merging these two annotation datasets (Garg et al., 2011a) yielded at least one annotation term for 17,160 (80.6%) of the 21,297 contigs in 47×300. This total of 80.6% is comparable to annotation statistics for de novo assemblies from other non-sequenced species, or yet to be sequenced at the time of experimentation, such as chick pea (85.5%; Garg et al., 2011a), eucalyptus (Eucalyptus grandis—83.2%; Mizrachi et al., 2010), insulin plant (69.2%; Annadurai et al., 2012), sesame (Sesamum indicum—54.0%; Wei et al., 2011), and sweet potato (Ipomoea batatas—62.0%; Wang et al., 2010). As expected, V. faba transcripts compared against the Viridiplantae database by BLASTX showed highest similarity to closely related legume species for which full genome sequences are now available, with Cicer arietinum (chick pea) and Medicago truncatula genes accounting for 47% and 38% of the annotated hits, respectively (see Figure 2).
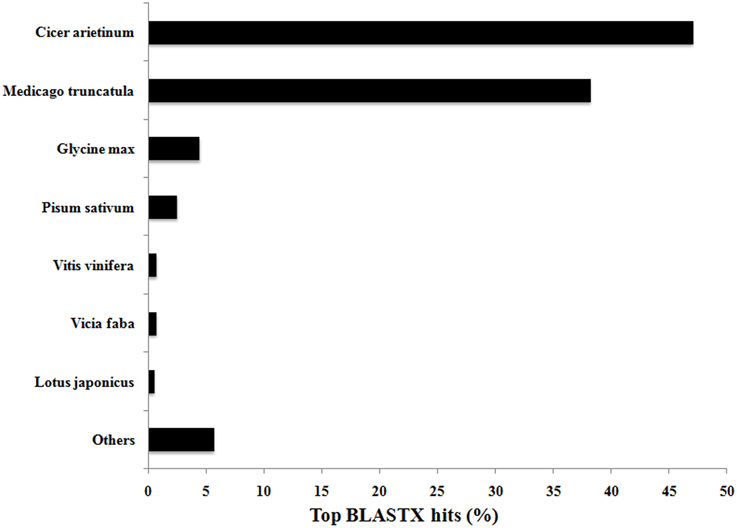
Figure 2. Top BLASTX hits of V. faba contigs against the Viridiplantae database. Contig sequences of V. faba are highly conserved with closely related and sequenced legume species, with 85% of the contigs showing similarity with Cicer arietinum (chick pea) and Medicago truncatula.
The 17,160 annotated contigs (herein referred to as unigenes) were assigned GO terms in three categories, namely Biological Process, Molecular Function, and Cellular Component (see Figure 3). Within Biological Process, the category “Response to different stress and stimulus,” which incorporates biotic, abiotic, endogenous, and extracellular, was highly represented (25%) followed by “Metabolic process” (13%). Within Molecular Function, processes like “Nucleotide binding” (34%), “Transcriptional regulation and DNA binding” (14%), and “Kinase activity” (14%) were dominant in this category. Similarly, Cellular Component was highly represented by “Plasma membrane” (21%) followed by “Plastid” (20%) subgroups.
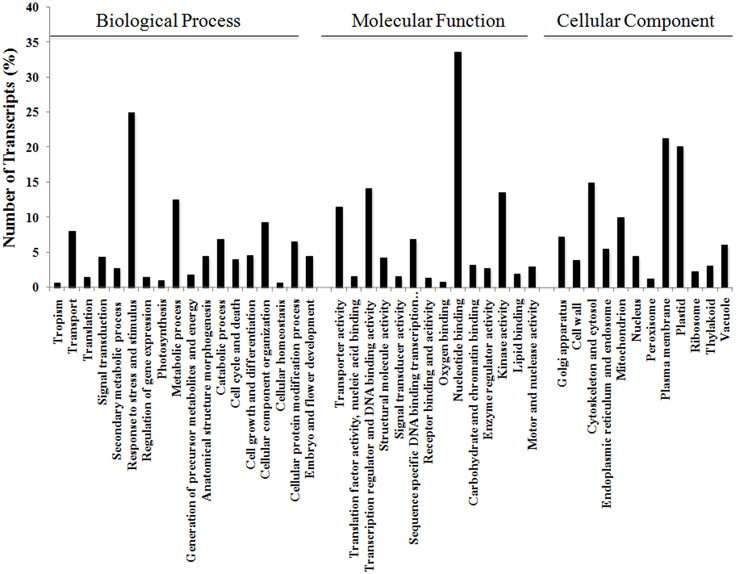
Figure 3. Functional annotation of V. faba contigs. Blast2GO characterization of GO terms in the three categories of Biological Process, Molecular Function, and Cellular Component.
Analysis of the annotated data set showed that 726 unigenes (4.2% of 17,160 in total) were identified as coding for putative transcription factors based on the GO term 0003700 (Transcription factor activity) appearing in the output for Molecular Function category. Of this total, 376 could be assigned to 31 of the 58 families of known plant transcription factors identified in PlnTFDB based on presence of family name in the annotation (see Figure 4; Perez-Rodriguez et al., 2009). A similar representation of plant transcription factor families has been observed in de novo assembled transcriptomes from chick pea (Garg et al., 2011b), soybean (Schmutz et al., 2010), and other legumes (Libault et al., 2009). Of those unigenes assigned to the 31 families, 19% were classified as homeobox transcription factors, with WRKY and AP2-EREBP genes making up the next most prominent families at 8% each (Figure 4).
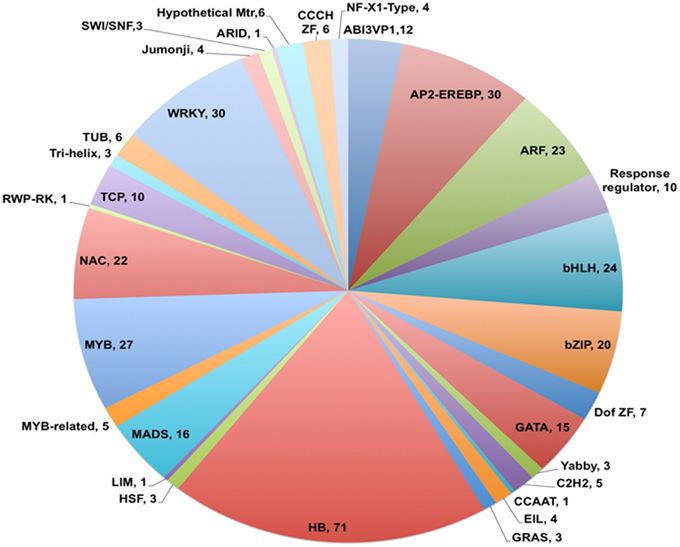
Figure 4. Distribution of V. faba transcripts into different transcription factor families. Pie chart showing families of transcription factors and total number of members identified for each family. Homeobox family members (HB) contributed 19% of the total transcription factors identified and classified, followed by WRKY (8%) and AP2-EREBP (8%) sequences. The number listed after each family name represents the total number of transcription factors assigned to that family.
Discussion
We have generated a genome-wide transcriptome map of V. faba to provide a reference for future RNA-Seq-based transcript profiling of TC development. Despite the large genome size of this important crop legume (ca. 13 Gb; Kaur et al., 2012), the paired-end strategy using Illumina sequencing was successful in generating 21,297 contigs, of which 80.6% (17,160) could be annotated against GO terminology. The total number of gene loci in V. faba is not known, but the 17,160 unigenes identified in this study may represent at least half of the gene space in faba bean given the number of genes identified in flowering plants (~27,000–39,000; TAIR10—www.arabidopsis.org; Rice Genome Annotation Project—http://rice.plantbiology.msu.edu/). This argument is based in part on a similar study by Lehnert and Walbot (2014) who reported that de novo assembly of 20,881 contigs from Dahlia may represent about half the gene loci in this species, which has a genome of approximately 9.4 Gb.
Validation of the 47×300 assembly against full-length V. faba cDNAs in GenBank established that the CLC assembly routine using a De-Bruijn graph-based approach gave 97–100% identity for 29 out of the 32 cDNAs tested across an average “Hit length” of 1157 bp, thus providing high confidence that the 47×300 assembly accurately reflects the component of the V. faba transcriptome captured by the sequencing strategy. Furthermore, the value of the transcriptome map as a resource for future transcript profiling of TC trans-differentiation is indicated by the presence of several transcripts, such as three ethylene response factor genes (VfERF1-3), two respiratory burst oxidase homologs (VfrbohA, VfrbohC), two ethylene insensitive transcription factors (VfEIN3-1, VfEIN3-2), two putative ACC oxidase genes (VfACO1, VfACO2, and two putative ACC synthase genes (VfACS1, VfACS1). Each of these genes has been cloned from isolated epidermal tissue, with many displaying epidermal-specific differential expression during the trans-differentiation of epidermal TCs (Zhou et al., 2010; Andriunas et al., 2012).
A major aim of our future investigations is to identify transcription factors regulating the trans-differentiation of TCs. Annotation of the 47×300 assembly revealed that 4.2% (726) of the 17,160 unigenes encoded putative transcription factors, of which representatives from 31 of the 58 known families of transcription factors in plants were identified. Within this cohort, 19% were identified as homeobox genes, while WRKY and AP2-EREBP members were the next most prominently represented families at 8% each. Homeobox genes are commonly involved in regulating morphogenesis and cell identity, typically via switching on cascades of gene expression (Williams, 1998). WRKY transcription factors are involved in biotic and abiotic stress defense pathways in plants (Rushton et al., 2010), and members of the AP2-EREBP family have roles in stress signaling pathways induced by hormones like ethylene and methyl jasmonate (Dietz et al., 2010). Given that the trans-differentiation of TCs involves differential expression of many hundreds of genes (Thiel et al., 2008; Dibley et al., 2009) in response to biotic (Cabrera et al., 2014) and abiotic stresses that can involve ethylene (Zhou et al., 2010) and methyl jasmonate (Amiard et al., 2007), the diverse representation of transcription factors in this transcriptome map augers well as a reference tool to identify key transcriptional regulators of TC trans-differentiation.
Overall, these considerations support the conclusion that assembly 47×300 represents a high-quality, genome-wide transcriptome map of V. faba and thus provides a validated platform for subsequent transcript profiling of TC trans-differentiation by RNA-Seq.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by a University of Newcastle Faculty of Science and Information Technology Strategic Research Initiative Grant. KA-C was supported by UNIPRS and UNRSC Scholarships. We thank Joe Enright for valuable assistance with plant growth.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2015.00217/abstract
References
Amiard, V., Demmig-Adams, B., Mueh, K. E., Turgeon, R., Combs, A. F., and Adams, W. W. (2007). Role of light and jasmonic acid signaling in regulating foliar phloem cell wall ingrowth development. New Phytol. 173, 722–731. doi: 10.1111/j.1469-8137.2006.01954.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Andriunas, F. A., Zhang, H. M., Weber, H., McCurdy, D. W., Offler, C. E., and Patrick, J. W. (2011). Glucose and ethylene signalling pathways converge to regulate trans-differentiation of epidermal cells in Vicia narbonensis cotyledons. Plant J. 68, 987–998. doi: 10.1111/j.1365-313X.2011.04749.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Andriunas, F. A., Zhang, H. M., Xia, X., Offler, C. E., McCurdy, D. W., and Patrick, J. W. (2012). Reactive oxygen species form part of a regulatory pathway initiating trans-differentiation of epidermal transfer cells in Vicia faba cotyledons. J. Exp. Bot. 63, 3617–3629. doi: 10.1093/jxb/ers029
Annadurai, R. S., Jayakumar, V., Mugasimangalam, R. C., Katta, M. A., Anand, S., Gopinathan, S., et al. (2012). Next generation sequencing and de novo transcriptome analysis of Costus pictus D. Don, a non-model plant with potent anti-diabetic properties. BMC Genomics 13:663. doi: 10.1186/1471-2164-13-663
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cabrera, J., Barcala, M., Fenoll, C., and Escobar, C. (2014). Transcriptomic signatures of transfer cells in early developing nematode feeding cells of Arabidopsis focused on auxin and ethylene signaling. Front. Plant Sci. 5:107. doi: 10.3389/fpls.2014.00107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, J., Pei, Z., Dai, L., Wang, B., Liu, L., An, X., et al. (2014). Transcriptome profiling using pyrosequencing shows genes associated with bast fiber development in ramie (Boehmeria nivea L.). BMC Genomics 15:919. doi: 10.1186/1471-2164-15-919
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Conesa, A., Gotz, S., Garca-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 18, 3674–3676. doi: 10.1093/bioinformatics/bti610
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dibley, S. J., Zhou, Y., Andriunas, F. A., Talbot, M. J., Offler, C. E., Patrick, J. W., et al. (2009). Early gene expression programs accompanying trans-differentiation of epidermal cells of Vicia faba cotyledons into transfer cells. New Phytol. 182, 863–877. doi: 10.1111/j.1469-8137.2009.02822.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dietz, K. J., Vogel, M. O., and Viehhauser, A. (2010). AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 245, 3–14. doi: 10.1007/s00709-010-0142-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dorn, K. M., Fankhauser, J. D., and Marks, M. D. (2013). De novo assembly of the pennycress (Thalaspi arvense) transcriptome provides tools for the development of a winter cover crop and biodiesel feedstock. Plant J. 75, 1028–1038. doi: 10.1111/tpj.12267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duan, J., Xia, C., Zhao, G., Jia, J., and Kong, X. (2012). Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genomics 13:392. doi: 10.1186/1471-2164-13-392
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
El-Rodeny, W., Kimura, M., Hirakawa, H., Sabah, A., Shirasawa, K., Sato, S., et al. (2014). Development of EST-SSR markers and construction of a linkage map in faba bean (Vicia faba). Breeding Sci. 64, 252–263. doi: 10.1270/jsbbs.64.252
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farley, S. J., Patrick, J. W., and Offler, C. E. (2000). Functional transfer cells differentiate in cultured cotyledons of Vicia faba L. seeds. Protoplasma 214, 102–117. doi: 10.1007/BF02524267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fellè, H. H., Hanstein, S., Steinmeyer, R., and Hedrich, R. (2000). Dynamics of ionic activities in the apoplast of the sub-stomatal cavity of intact Vicia faba leaves during stomatal closure evoked by ABA and darkness. Plant J. 24, 297–304. doi: 10.1046/j.1365-313x.2000.00878.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Franssen, S. U., Shrestha, R. P., Brautigam, A., Bornberg-Bauer, E., and Weber, A. P. M. (2011). Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics 12:227. doi: 10.1186/1471-2164-12-227
Fukuda, M., Hasezawa, S., Asai, N., Nakajima, N., and Kondo, N. (1998). Dynamic organization of microtubules in guard cells of Vicia faba L. with diurnal cycle. Plant Cell Physiol. 39, 80–86. doi: 10.1093/oxfordjournals.pcp.a029293
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garg, R., Patel, R. K., Jhanwar, S., Priya, P., Bhattacharjee, A., Yadav, G., et al. (2011b). Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol. 156, 1661–1678. doi: 10.1104/pp.111.178616
Garg, R., Patel, R. K., Tyagi, A. K., and Jain, M. (2011a). De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 18, 53–63. doi: 10.1093/dnares/dsq028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gotz, S., Garcia-Gomez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guo, S. G., Zheng, Y., Joung, J. G., Liu, S. Q., Zhang, Z. H., Crasta, O. R., et al. (2010). Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types. BMC Genomics 11:384. doi: 10.1186/1471-2164-11-384
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hafke, J. B., Höll, S., Kühn, C., and van Bel, A. (2013). Electrophysiological approach to determine kinetic parameters of sucrose uptake by single sieve elements or phloem parenchyma cells in intact Vicia faba plants. Front. Plant Sci. 4:274. doi: 10.3389/fpls.2013.00274
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Henkel, C. V., Dirks, R. P., and De Wijze, D. L. (2012). First draft genome sequence of the Japanese eel, Anguilla japonica. Gene 511, 195–201. doi: 10.1016/j.gene.2012.09.064
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaur, S., Pembleton, L. W., Cogan, N. O., Savin, K. W., Leonforte, T., and Paull, J. (2012). Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics 13:104. doi: 10.1186/1471-2164-13-104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lehnert, E. M., and Walbot, V. (2014). Sequencing and de novo assembly of a Dahlia hybrid cultivar transcriptome. Front. Plant Sci. 5:340. doi: 10.3389/fpls.2014.00340
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Libault, M., Joshi, T., Benedito, V. A., Xu, D., Udvardi, M. K., and Stacy, G. (2009). Legume transcription factor genes: what makes legumes so special. Plant Physiol. 151, 991–1001. doi: 10.1104/pp.109.144105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mantello, C. C., Cardoso-Silva, C. B., Da Silva, C. C., De Souza, L. M., Scaloppi Junior, E. J., Goncalves, P. S., et al. (2014). De novo assembly and transcriptome analysis of the rubber tree (Hevea brasiliensis) and SNP markers development for rubber biosynthesis pathways. PLoS ONE 9:e102665. doi: 10.1371/journal.pone.0102665
Meyer, E., Logan, T. L., and Juenger, T. E. (2012). Transcriptome analysis and gene expression atlas for Panicum hallii var. filipes, a diploid model for biofuel research. Plant J. 70, 879–890. doi: 10.1111/j.1365-313X.2012.04938.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mizrachi, E., Hefer, C. A., Ranik, M., Joubert, F., and Myburg, A. A. (2010). De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics 11:681. doi: 10.1186/1471-2164-11-681
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moreton, J., Dunham, S. P., and Emes, R. D. (2014). A consensus approach to vertebrate de novo transcriptome assembly from RNA-Seq data: assembly of the duck (Anasplatyrhynchos) transcriptome. Front. Genet. 5:190. doi: 10.3389/fgene.2014.00190
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Offler, C. E., McCurdy, D. W., Patrick, J. W., and Talbot, M. J. (2003). Transfer cells: cells specialized for a special purpose. Annu. Rev. Plant Biol. 54, 431–454. doi: 10.1146/annurev.arplant.54.031902.134812
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parra-González, L., Aravena-Abarzúa, G., Navarro-Navarro, C., Udall, J., Maughan, J., Peterson, L. M., et al. (2012). Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies. BMC Genomics 13:425. doi: 10.1186/1471-2164-13-425
Perez-Rodriguez, P., Riano-Pachon, D. M., Correa, L. G. G., Stefan, A., Rensing, S. A., Kersten, B., et al. (2009). PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, 822. doi: 10.1093/nar/gkp805
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rushton, P. J., Somssich, I. E., Ringler, P., and Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15, 247–258. doi: 10.1016/j.tplants.2010.02.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmutz, J., Cannon, S. B., and Schlueter, J. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Talbot, M. J., Franceschi, V. R., McCurdy, D. W., and Offler, C. E. (2001). Wall ingrowth architecture in epidermal transfer cells of Vicia faba cotyledons. Protoplasma 215, 191–203. doi: 10.1007/BF01280314
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Talbot, M. J., Wasteneys, G., McCurdy, D. W., and Offler, C. E. (2007a). Deposition patterns of cellulose microfibrils in flange wall ingrowths of transfer cells indicate clear parallels with those of secondary wall thickenings. Funct. Plant Biol. 34, 307–313. doi: 10.1071/FP06273
Talbot, M. J., Wasteneys, G., Offler, C. E., and McCurdy, D. W. (2007b). Cellulose synthesis is required for deposition of reticulate wall ingrowths in transfer cells. Plant Cell Physiol. 48, 147–158. doi: 10.1093/pcp/pcl046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thiel, J., Weier, D., Sreenivasulu, N., Strickert, M., Weichert, N., Melzer, M., et al. (2008). Different hormonal regulation of cellular differentiation and function in nucellar projection and endosperm transfer cells: a microdissection-based transcriptome study of young barley grains. Plant Physiol. 148, 1436–1452. doi: 10.1104/pp.108.127001
Thorpe, M. R., Furch, A. C. U., Minchin, P. E. H., Föller, J., van Bel, A. J. E., and Hafke, J. B. (2010). Rapid cooling triggers forisome dispersion just before phloem transport stops. Plant Cell Environ. 33, 259–271. doi: 10.1111/j.1365-3040.2009.02079.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Torres, A. M., Weeden, N. F., and Martin, A. (1993). Linkage among isozyme, RFLP and RAPD markers in Vicia faba. Theor. Appl. Genet. 85, 937–945. doi: 10.1007/BF00215032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vaughn, K. C., Talbot, M. J., Offler, C. E., and McCurdy, D. W. (2007). Wall ingrowths in epidermal transfer cells of Vicia faba cotyledons are modified primary walls marked by localized accumulations of arabinogalactan proteins. Plant Cell Physiol. 48, 159–168. doi: 10.1093/pcp/pcl047
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verma, P., Shah, N., and Bhatia, S. (2013). Development of an expressed gene catalogue and molecular markers from the de novo assembly of short sequence reads of the lentil (Lens culinaris Medik.) transcriptome. Plant Biotech. J. 11, 894–905. doi: 10.1111/pbi.12082
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Z., Fang, B., Chen, J., Zhang, X., and Luo, Z. (2010). De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genomics 11:726. doi: 10.1186/1471-2164-11-726
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wardini, T., Talbot, M. J., Offler, C. E., and Patrick, J. W. (2007). Role of sugars in regulating transfer cell development in cotyledons of developing Vicia faba seeds. Protoplasma 230, 75–88. doi: 10.1007/s00709-006-0194-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wei, W., Qi, X., Wang, L., Zhang, Y., Hua, W., Li, D., et al. (2011). Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12:451 doi: 10.1186/1471-2164-12-451
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Williams, R. (1998). Plant homeobox genes: many functions stem from a common motif. Bioessays 20, 280–282.
Xianjun, P., Linhong, T., Xiaoman, W., Yucheng, W., and Shihua, S. (2014). De novo assembly of expressed transcripts and global transcriptomic analysis from seedlings of the paper mulberry (Broussonetia kazinoki x Broussonetia papyifera). PLoS ONE 9:e97487. doi: 10.1371/journal.pone.0097487
Xia, X., Zhang, H.-M., Andriunas, F. A., Offler, C. E., and Patrick, J. W. (2012). Extracellular hydrogen peroxide, produced through a respiratory burst oxidase/superoxidedismutase pathway, directs ingrowth wall formation in epidermal transfer cells of Vicia faba cotyledons. Plant Signal. Behav. 7, 1–4. doi: 10.4161/psb.21320
Zhang, H.-M., Imtiaz, M. S., Laver, D. R., McCurdy, D. W., Offler, C. E., van Helden, D. F., et al. (2015). Polarized and persistent Ca2+ plumes define loci for formation of wall ingrowth papillae in transfer cells. J. Exp. Bot. 66, 1179–1190. doi: 10.1093/jxb/eru460
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, J. A., Liang, S., Duan, J. L., Wang, J., Chen, S. L., Cheng, Z. S., et al. (2012). De novo assembly and characterisation of the transcriptome during seed development, and generation of genetic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 13:90. doi: 10.1186/1471-2164-13-90
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, Y. C., Andriunas, F., Offler, C. E., McCurdy, D. W., and Patrick, J. W. (2010). An epidermal-specific ethylene signal cascade regulates trans-differentiation of transfer cells in Vicia faba cotyledons. New Phytol. 185, 931–943. doi: 10.1111/j.1469-8137.2009.03136.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Vicia faba, transfer cell, wall ingrowths, de novo transcriptome assembly, transcription factors, RNA-Seq
Citation: Arun-Chinnappa KS and McCurdy DW (2015) De novo assembly of a genome-wide transcriptome map of Vicia faba (L.) for transfer cell research. Front. Plant Sci. 6:217. doi: 10.3389/fpls.2015.00217
Received: 02 February 2015; Accepted: 17 March 2015;
Published: 09 April 2015.
Edited by:
Georgina Hernandez, Universidad Nacional Autonoma de Mexico, MexicoReviewed by:
Marc Libault, University of Oklahoma, USAGregorio Hueros, Universidad de Alcalá, Spain
Johannes Thiel, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Germany
Copyright © 2015 Arun-Chinnappa and McCurdy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David W. McCurdy, Centre for Plant Science, School of Environmental and Life Sciences, The University of Newcastle, University Drive, Callaghan, NSW 2308, AustraliaZGF2aWQubWNjdXJkeUBuZXdjYXN0bGUuZWR1LmF1