 Katja Witzel
Katja Witzel Susanne Neugart1
Susanne Neugart1 Silke Ruppel
Silke Ruppel Melanie Wiesner
Melanie Wiesner Susanne Baldermann
Susanne Baldermann- 1Leibniz-Institute of Vegetable and Ornamental Crops Großbeeren/Erfurt e.V., Großbeeren, Germany
- 2Institute of Nutritional Science, University of Potsdam, Nuthetal, Germany
Continuing advances in ‘omics methodologies and instrumentation is enhancing the understanding of how plants cope with the dynamic nature of their growing environment. ‘Omics platforms have been only recently extended to cover horticultural crop species. Many of the most widely cultivated vegetable crops belong to the genus Brassica: these include plants grown for their root (turnip, rutabaga/swede), their swollen stem base (kohlrabi), their leaves (cabbage, kale, pak choi) and their inflorescence (cauliflower, broccoli). Characterization at the genome, transcript, protein and metabolite levels has illustrated the complexity of the cellular response to a whole series of environmental stresses, including nutrient deficiency, pathogen attack, heavy metal toxicity, cold acclimation, and excessive and sub-optimal irradiation. This review covers recent applications of ‘omics technologies to the brassicaceous vegetables, and discusses future scenarios in achieving improvements in crop end-use quality.
Introduction
Brassicaceous vegetables, which are cultivated worldwide, belong to the taxa Brassica oleracea (cabbage, broccoli, cauliflower, kale, Brussels sprouts, collard greens, savoy, kohlrabi, and Chinese kale) and B. rapa (turnip, mizuna, napa cabbage, cime di rapa, and turnip rape). Just as for crops generally, maintaining their productivity in the light of incipient climate change and the dynamically changing pest and pathogen community represents a major challenge for the biotechnologist and the plant breeder (Augustine et al., 2014). The importance of this class of vegetables lies not only in their contribution to the vitamin and mineral components of the human diet, but also in their beneficial effect on human health, which reflects the action of the glucosinolates, a group of secondary plant metabolites almost exclusively associated with this plant family (Higdon et al., 2007; Jeffery and Araya, 2009; Wu et al., 2013). The aliphatic glucosinolates (and their break down products) have attracted scientific attention (Reichelt et al., 2002; Halkier and Gershenzon, 2006; Sonderby et al., 2010; Wittstock and Burow, 2010); of particular note in this context is the anti-carcinogen isothiocyanate sulforaphane, the major break down product of glucoraphanin in broccoli (Fahey et al., 1997; Shapiro et al., 2001, 2006).
The use of ‘omics technologies, which current gather information either at the DNA, RNA, protein or metabolite levels, can potentially provide a comprehensive picture of cellular physiology. DNA sequence (although not the epigenome) is largely independent of the growing environment, while the transcriptome, proteome and metabolome are all highly responsive (Figure 1). Tailoring a crop cultivar to a specific environment is the central challenge for the plant breeder, and reflects the reality that genotype on its own will not generally be sufficient to support a biotechnology-driven crop improvement program. Rather, a combination of one or more of the ‘omics platforms is required to deliver reliable information. Such multiple ‘omics data sets tend to be very large, as they represent a series of time point and/or treatment samplings; their analysis can only be addressed computationally (Bieda, 2012; Kohl et al., 2014; Schumacher et al., 2014). The demand for better data processing led to the release of new software packages freely available or provided by vendors (examples are given in Katajamaa and Oresic, 2007), however, the visualization of multi-omic data sets remains an important task for bioinformatics. Practical tools should create clear and meaningful visualizations without being overwhelmed by the complexity of the data sets (recent approaches are discussed by Gehlenborg et al., 2010).
FIGURE 1. The application of ‘omics technologies in brassicaceous vegetables. Arrow size reflect the complexity of the molecular data and the impact on phenotype.
The prior acquisition of a full genome sequence aids materially in the interpretation of such data. The genome sequence of the model plant Arabidopsis thaliana, a member of the Brassicaceae family, has been known for nearly 15 years (Kaul et al., 2000), and the full genome sequences of both B. rapa (Wang et al., 2011) and B. oleracea (Ayele et al., 2005; Liu et al., 2014b) have been published more recently. Substantial amounts of transcript-based data have been acquired for B. oleracea (Gao et al., 2014; Izzah et al., 2014; Kim et al., 2014).
Genomics
Genomic research has a great capability in speeding up breeding processes and several applications for crop improvement, through, e.g., marker-assisted selection and gene pyramiding. In case of brassicaceous vegetables, several populations have been generated to establish linkage maps using simple sequence repeat (SSR), amplified fragment length polymorphism (AFLP), nucleotide binding site or expressed sequence tag (EST) markers with the aim to genetically localize favorable traits by quantitative trait locus (QTL) analysis (summarized in Table 1). A survey of available online tools, covering mapping populations, linkage maps, gene sequences, and QTL, is presented by Li et al. (2013). Linkage maps were generated by crossing different genotypes of B. oleracea (Camargo et al., 1997; Iniguez-Luy et al., 2008) and B. oleracea var. botrytis (Gu et al., 2008). In the previous examples, a segregating offspring population was used to establish marker order and spacing. The same was shown for doubled haploid lines where offspring are homozygous (see Pink et al., 2008 for review). A doubled haploid population derived from crosses of two broccoli cultivars was genotyped by SSR and AFLP markers and QTL analysis identified loci for horticulturally important characteristics (Walley et al., 2012). Plant genetic resources have been characterized using AFLP markers to assess the huge genetic diversity present in gene banks, including Dutch and Italian B. oleracea (van Hintum et al., 2007; Maggioni et al., 2014) and Czech B. oleracea var. capitata accessions (Faltusova et al., 2011). A B. rapa collection consisting of 239 accessions was genotyped using SSR markers and subsequent association mapping identified two markers associated with flowering time (Zhao et al., 2010). Genetic basis of flowering time was also investigated in B. rapa by association mapping using natural variation and recombinant inbred lines (Lou et al., 2011). Phylogenetic relationships were established in B. rapa subspecies by AFLP markers (Takuno et al., 2007). The development of genetic maps from Brassica vegetables paved the way for QTL analysis and, together with accurate phenotyping, morphological, qualitative and yield traits were evaluated in different species. QTL mapping was conducted for eleven yield- and heading-related traits in doubled haploid Chinese cabbage lines and identified 46 main QTL (Liu et al., 2013b). In addition, 27 QTL were found for leaf and heading-related traits in a segregating population of Chinese cabbage (Ge et al., 2011). A segregating population of a cross between a Chinese cabbage with a turnip was used to identify loci related to morphological characteristics of the tap root (Lu et al., 2008). To investigate the genetic background of premature bolting under cold stress, a Chinese cabbage population was constructed based on crossing early and late bolting genotypes and QTL analysis identified 26 QTL (Wang et al., 2014c). Seven morphological traits were screened in a population derived from crossing a Chinese cabbage with a vegetable turnip and resulted in the detection of eight QTL (Kubo et al., 2010). A doubled haploid population of B. oleracea var. capitata served for detecting 13 QTL for heading-related traits (Lv et al., 2014). QTL analysis of a B. oleracea var. italica population, derived from a cross of a heat-sensitive and a heat-tolerant cultivar, identified AFLP markers correlated to floral development under heat stress (Lin et al., 2013). A high-density linkage map was established for a B. oleracea population segregating for carotenoid concentration in florets and three carotenoid QTL were found (Brown et al., 2014). Authors applied the B. napus SNP array and presented a 96% coverage of the B. oleracea genome. Localizing underlying factors affecting another major pigment, chlorophyll, in a Chinese cabbage population revealed five QTL for chlorophyll a and five QTL for chlorophyll b content (Ge et al., 2012). Vegetables with high nutrient use efficiency are also developed in order to reduce fertilizer application. Association mapping of B. oleracea accessions identified some QTL related to potassium concentration in shoot and those were tested using substitution lines (White et al., 2010). The gain of knowledge on the genetic localization of favorable traits is transferred to breeding new lines through marker-assisted selection. Accumulating three major loci for clubroot (Plasmodiophora brassicae) resistance genes resulted in the development of Chinese cabbage lines with strengthened resistance (Matsumoto et al., 2012).
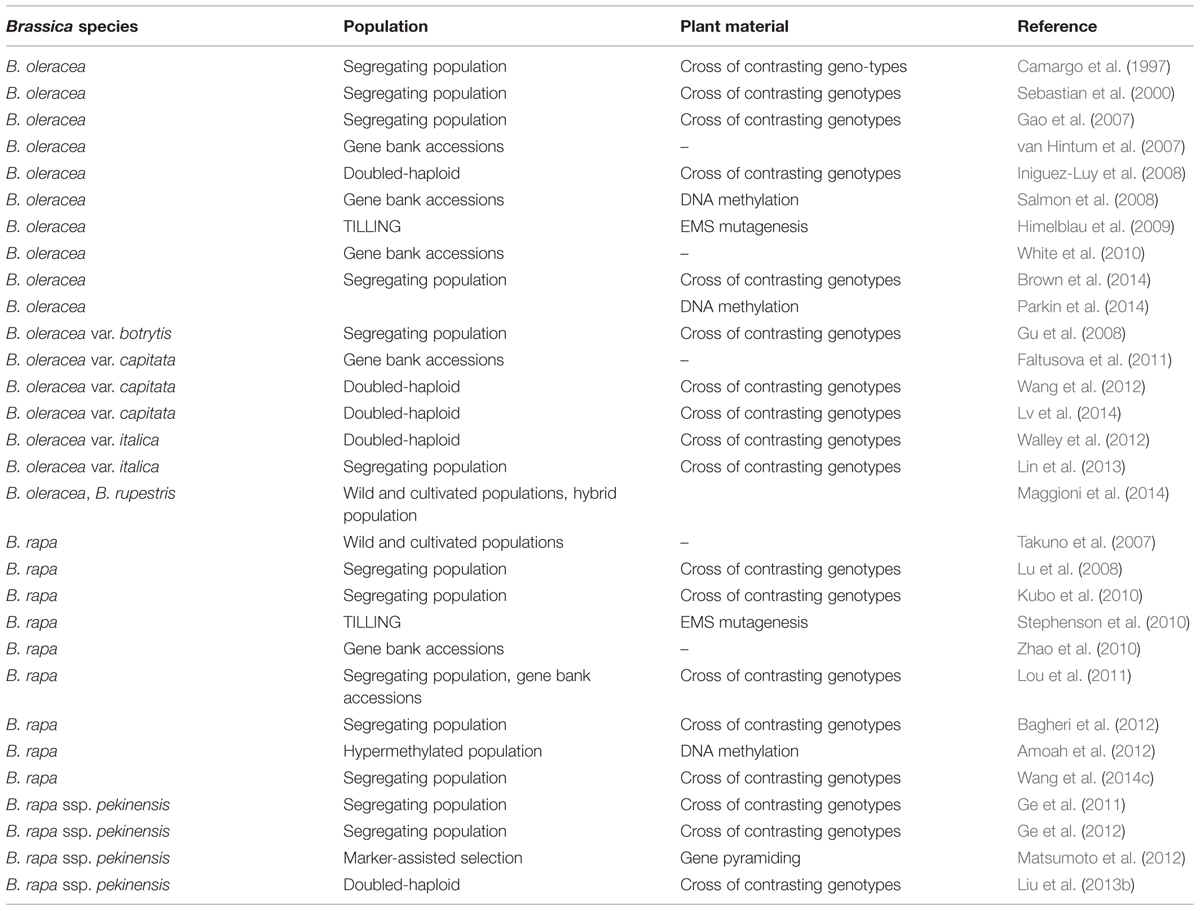
TABLE 1. Published genomics analyses in the brassicaceous vegetables.
Advances in next generation sequencing technologies enabled surveying genotype-phenotype-relationships with the highest resolution to date (Wei et al., 2013; Varshney et al., 2014). A high-density linkage map was derived from the whole genome shotgun sequence of B. oleracea var. capitata, based on 1,227 genetic markers (Wang et al., 2012). The availability of B. rapa genome sequence aided in the generation of a linkage map for a recombinant inbred line descending from a vegetable leafy and an yellow sarson oilseed genotype of B. rapa (Bagheri et al., 2012). Recently, genomes of all three sequences Brassicas were compared to develop SSR markers and a total of 115,869, 185,662, and 356,522 primer pairs were designed from B. rapa, B. oleracea, and B. napus, respectively (Shi et al., 2014). EST markers were developed based on the sequenced transcriptomes of two cabbage lines susceptible or tolerant to black rot disease, demonstrating the feasibility especially for species without a reference genome sequence (Izzah et al., 2014).
One of the main strategies in reverse genetics is Targeting Induced Local Lesions IN Genomes (TILLING) and numerous applications are shown in functional genomics of model plants and crops (Kurowska et al., 2011; Chen et al., 2014). Random point mutations are generated by chemical mutagenesis and high-throughput screening for SNPs in the target gene isolates mutants with loss-of-function or gain-of-function phenotypes. Currently, two TILLING populations are established for brassicaceous vegetables, B. oleracea1 (Himelblau et al., 2009) and B. rapa2 (Stephenson et al., 2010) and both are freely accessible.
Next generation sequencing and AFLP mapping of methylation polymorphism allowed determining the DNA cytosine methylation status and, hence, enabled tackling the epigenome to understand gene expression variation (Niederhuth and Schmitz, 2014). The level of genome methylation was assessed in 30 B. oleracea populations and lines, and then related to phenotypic variability (Salmon et al., 2008). The B. oleracea genome was correlated to the leaf transcriptome and methylome and provided insights into polyplody events (Parkin et al., 2014). A chemically induced hypermethylated B. rapa population was developed recently and can serve for epiallele discovery (Amoah et al., 2012).
Transcriptomics
Transcriptomics is based on the characterization and quantification of RNAs present in a given plant, organ, tissue, or cell. A particular attraction of assaying transcription is that it presages gene expression and so has the potential to provide a link between genotype and phenotype. Rapid developments in the relevant technologies now allow for the design of very large-scaled experiments which can in principle capture and enumerate every transcript present in a given biological sample (summarized in Table 2). The early transcriptomic platforms were built around the concept of immobilizing transcripts in an array, which was used to capture the mRNA content of a sample on the basis of nucleotide sequence complementarity. A more sophisticated form of microarray replaced transcripts with 20–70 nt long oligonucleotides designed to target known gene sequences (Knudsen, 2004). In both cases, the protocol requires bathing the array in a solution of labeled RNA extracted from the test sample. Hybridization of the probe at a given site on the array is detected by the fluorescence emitted by the labeled probe. Typically, two parallel experiments need to be run, in which one sample is derived from a plant exposed to a certain treatment while the other is derived from a plant which has not been exposed (control). Alternatively, genetic contrasts (such as wild type vs. a mutant, wild type vs. a transgenic) can replace the treatment contrast. Many of such experiments have been reported in a range of plant species (for example, Rabbani et al. (2003) in rice, Yu and Setter (2003) in maize, Lee and Yun (2006) in pepper). The quantity of such data collected from the brassicaceous vegetable species is relatively limited, partly perhaps because their full genome sequences have only very recently been acquired. The close phylogenetic relationship between Brassica spp. and A. thaliana has encouraged a number of attempts to use an A. thaliana microarray to analyze Brassica spp. transcriptomes. With the recent acquisition of Brassica spp. genome sequences, a number of Brassica-specific microarrays are finally becoming available – these include the B. rapa 24 K oligo microarray and the Agilent Brassica microarray, which is able to detect the transcription of >27,000 unigenes in a range of Brassica spp. The genetic closeness of Brassica spp. and A. thaliana has allowed large numbers of Brassica unigenes to be assigned a function, but nevertheless, substantial numbers appear to encode either Brassica-specific proteins or represent non-coding RNA (Trick et al., 2009).

TABLE 2. Published transcriptomic analyses in the brassicaceous vegetables.
A cDNA microarray constructed from a Chinese cabbage (B. rapa ssp. pekinensis) pistil-specific cDNA library approach was used by Kwun et al. (2004) to investigate the effect of CO2 on self-incompatibility, and a similar strategy was followed by Yang et al. (2005) to identify genes up-regulated by low temperature stress. Meanwhile Lee et al. (2008) synthesized an oligo-based microarray from the sequences of 24,000 B. rapa unigenes to characterize the response to low temperature, salinity and drought. Based on the identification of candidate genes using a 2 × 104 k Brassica microarray to compare the gene expression of untreated and methyl jasmonate treated pak choi sprouts, Wiesner et al. (2013a, 2014) were able to identify the homologues genes involved in the synthesis of 1-methoxyindole-3-ylmethyl glucosinolate.
The major alternative to microarray technology relies on amplification rather than on hybridization. The most commonly used method, termed RNA-seq, is designed to generate a de novo assembly of the transcriptome. This has largely replaced the earlier approach called “serial analysis of gene expression” (SAGE). In general, a population of RNA (total or fractionated) is converted to a library of cDNA fragments with adaptors attached to one or both ends. Each molecule, with or without amplification, is then sequenced in a high-throughput manner to obtain short sequences from one end (single-end sequencing) or both ends (pair-end sequencing). The reads are typically 300–400 bp, depending on the DNA-sequencing technology used (Wang et al., 2009). In principle, any high-throughput sequencing technology can be used for RNA-Seq. The data generated from RNA-Seq experiments are in the form of an absolute frequency of each transcript identified, which obviates the need for using a reference sequence. For this reason, it can provide a greater level of insight and accuracy than microarray analysis can (Marioni et al., 2008; T’Hoen et al., 2008). In broccoli, the RNA-Seq approach has been taken to describe the transcription of genes involved in glucosinolate metabolism (Gao et al., 2014). Of the nearly 20 k unigenes recovered, more than 2,500 appeared to be differentially transcribed in a contrast between the seed and the seedling, and a large proportion of these were putative transcription factors. Curiously, the up-regulation of candidate glucosinolate synthesis genes was negatively correlated with the glucosinolate content of the germinating seedling; a possible explanation offered by the authors was that the high transcript abundance of an TGG2 orthologue (encoding a myrosinase) had the effect of degrading aliphatic glucosinolates (Gao et al., 2014).
Small RNAs, which range in length from 19 to 25 nt (most are either 21 nt or 24 nt long), are ubiquitous in eucaryotic cells. Extensive transcriptome sequencing has revealed the presence of a large number of such RNAs, which cannot be translated into a protein, but rather act as an important class of regulators, especially in the context of plant development, signal transduction, metabolism and the response to biotic and abiotic stress (Ellendorff et al., 2009; Sun, 2012). The abundance of a particular species of a class of small RNAs, referred to as microRNAs (miRNAs), varies from plant species to plant species, from developmental stage to developmental stage and from tissue to tissue (Carrington and Ambros, 2003; Bartel, 2004; Kenan-Eichler et al., 2011). The transcriptional response of Chinese cabbage to infection by Erwinia carotovora ssp. carotovora included a marked alteration in the abundance of certain small RNAs (Sun, 2014). In turnip (B. rapa ssp. rapa), it appeared that various miRNAs are involved in the regulation of plant growth, development and differentiation in the absence of light (Zhou et al., 2014). A number of Brassica species are polyploids, the genomes of which have been markedly altered by the polyploidization event (Kenan-Eichler et al., 2011). Some of these changes have been associated with the activity of small RNAs. Kim et al. (2012) identified 412 distinct miRNAs in B. rapa, of which 216 were novel. The same study identified 29 novel miRNAs which were only found in the flower.
Proteomics
The acquisition of the complete genome sequence of a growing number of plant species3 along with their transcriptomes continues apace. However, much of the physiology of the cell is determined by gene products (particularly, but not exclusively, proteins) rather than by nucleic acid. A proteomic analysis seeks to characterize the full protein complement present in a particular organism, organ, tissue, or cell. Initial attempts to derive this were based on two dimensional gel electrophoresis (2-DE), as described by O’Farrell (1975). Besides 2-DE, current proteomics platforms exploit more versatile LC (liquid chromatography)-based methods (Rabilloud et al., 2010; Matros et al., 2011), coupled with mass spectrometry (MS). Complicating the analysis, and unlike the nucleic acids, protein molecules are subject to a range of functional modification, such as phosphorylation, glycosylation, and acetylation (Cox and Mann, 2011). Nevertheless, proteomic approaches have been successfully applied to a number of plant species to study various developmental processes and environmental adaptation (see reviews by Barkla et al., 2013; Vanderschuren et al., 2013; Zhuang et al., 2014). Kehr and Buhtz (2011) have provided a summary of the literature regarding proteomic analyses in Brassica spp., a list dominated by experiments based on oilseed rape. In the following, we give emphasis to reports on how proteomic techniques are applied to catalog and characterize special, developmental-driven or environmentally induced alterations in the proteome of brassicaceous vegetables (summarized in Table 3).
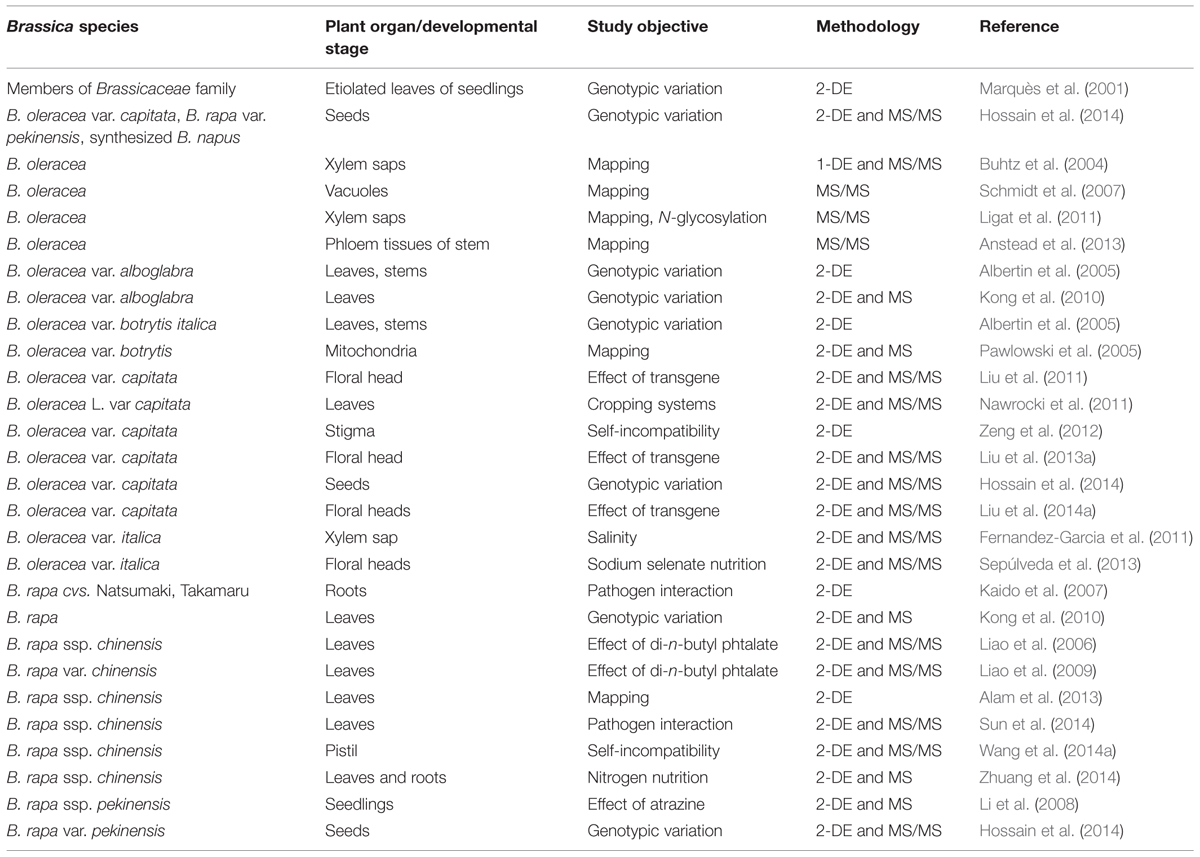
TABLE 3. Published proteomic analyses in the brassicaceous vegetables.
Using a conventional 2-DE approach, Alam et al. (2013) succeeded in detecting about 1,300 distinct low abundance leaf proteins in Chinese cabbage. A characterization of the proteomic content of the xylem sap of broccoli and oilseed rape (and some non- brassicaceous) undertaken by Buhtz et al. (2004) showed little evidence of any species specificity, while De Bernonville et al. (2014) were able to define the proteomic impact in the xylem sap induced by infection of B. oleracea with the pathogen Xanthomonas campestris pv. campestris. Ligat et al. (2011) expanded the B. oleracea data set by subjecting the xylem sap proteome to LC-MS/MS and the N-glycoproteome by prior enrichment with concanavalin A affinity chromatography and LC-MS/MS. Most of the ∼200 proteins identified proved to be involved in cell wall-related carbohydrate metabolism, although a number of oxido-reductases and proteases were also revealed. A proteomic analysis of broccoli tissue enriched with phloem allowed the identification of 379 proteins, some of which were structural and others associated with the biotic and/or abiotic stress response (Anstead et al., 2013). In cauliflower, an analysis of isolated tonoplast membranes revealed 102 tonoplast integral and 214 peripheral proteins (Schmidt et al., 2007). A further study in this species was able to show that its mitochondrial proteome was highly similar to that of A. thaliana, and included at least 51 proteins involved in the generation of ATP, protein folding and protein transport (Pawlowski et al., 2005).
The host-pathogen interaction is important in brassicaceous vegetables, as in all crops. A root proteomic comparison between a pair of turnip cultivars showing a contrasting reaction to the causal agent of clubroot disease was able to demonstrate the presence of certain proteins in the resistant cultivar which were absent from the profile of the sensitive one (Kaido et al., 2007). Sun et al. (2014) tracked the proteomic response of Chinese cabbage resulting from infection with downy mildew (Hyaloperonospora parasitica), and were able to identify some 91 proteins which altered in their abundance; some of these were involved in ethylene signaling. When the abundances of specific transcripts and their gene products were compared, a correlation could be established for only about half of the 33 genes investigated; a finding which was interpreted as implying that many of the early events in the host’s resistance response involved post-translational modification.
Abiotic stress is a further major production constraint. Analysis of the effect of the photosystem II inhibitor atrazine on the chloroplast proteome of Chinese cabbage has identified at least nine proteins as being differentially expressed; some of these were related to isoprene or protein synthesis (Li et al., 2008). Investigations of the impact of the industrial soil pollutant phthalate ester on either pak choi (Liao et al., 2006) or Chinese cabbage (Liao et al., 2009) have shown that the pollutant was responsible for a different set of up- or down-regulated proteins, even though the two species are so closely related to one another. Among the effects of salinity on broccoli are the down-regulation of several proteins involved in cell wall metabolism and the up-regulation of known plant defense-related proteins (Fernandez-Garcia et al., 2011). A third common source of abiotic stress is that imposed by nutrient supply. Wang et al. (2014b) have characterized the response of hydroponically grown pak choi to the supply of nitrogen in different forms. The plants responded to glycine supply by altering the expression of proteins involved in protein processing, amino acid metabolism and redox homeostasis. A similar experiment involving broccoli investigated the effect of supplying the plants with a non-limiting concentration of the essential trace element selenium (Sepúlveda et al., 2013), resulting in an enhanced accumulation in the inflorescence of a number of proteins associated with the general stress response, while that of pathogen defense-related proteins was reduced. The effect of an organic farming regime on the cabbage proteome has shown that the cropping system had a measurable effect on the accumulation of proteins involved in glycolysis, the Krebs cycle, amino acid metabolism and various detoxification processes (Nawrocki et al., 2011).
A plants protein composition is a key determinant of a species’ and a cultivar’s identity, and differences in specific plant features are better reflected by their proteome than by either their genome or transcriptome. Thus proteome variation has been suggested as providing a viable basis for the design of molecular markers able to accelerate breeding (Witzel et al., 2011). Marquès et al. (2001) made comparisons between the 2-DE profiles of various Brassica species in an attempt to establish the nature of the genetic relationship between them. PCR-based markers for the genomes A (B. rapa) and B (B. oleracea) were obtained by Kong et al. (2010) by aligning the proteomic profiles of young leaves. From some of those differentially expressed proteins, PCR-based markers were developed successfully. The effect of the polyploidization process on the proteome was assessed by Albertin et al. (2005), who made comparisons between the leaf and stem proteomes of haploid, diploid and tetraploid sprouting broccoli, and Chinese kale. The analysis concluded that the two different tissues were responsible for 40% of the proteomic variation observed, with only 10% ascribed to ploidy level differences. With the aim to identify proteins responsible for an improved vernalisation trait, the seed proteome of a somatic hybrid line was compared with those of its parent lines B. oleracea (cabbage) and B. rapa (Chinese cabbage) that showed low or high seed production during warm winter conditions, respectively (Hossain et al., 2014).
Understanding the basis of self-incompatibility has been a long-standing problem in the Brassica genus. A proteomic approach addressing this issue in Chinese cabbage was taken by Wang et al. (2014a), who analyzed the protein complement of the pistils from self-incompatible and compatible plants; in some cases, protein abundance was consistent with that of the relevant transcript, particularly with respect to those involved in energy metabolism, protein synthesis and the defense against stress. Recently, for a more cell-type specific proteomics approach related to self-incompatibility, a protocol for the analysis of cabbage stigma proteins was established (Zeng et al., 2012).
The market value of vegetables is greatly affected by their visual appearance and their post-harvest shelf life. As part of a transgenic-based attempt to prolong the marketability of broccoli, 2-DE was used to compare the proteomes of the inflorescences of wild type and transgenic lines produced to express delayed senescence (Liu et al., 2011). The conclusion was that a number of stress response related proteins and chaperones accumulated in the transgenic lines, while the build-up of 1-aminocyclopropane-1-carboxylate oxidase was only noted in the wild type. When the effect of exogenously supplying cytokinin (N6-benzylaminopurine) was studied at the proteomic level, Liu et al. (2013a) detected only a minor degree of overlap with the transgenic contrast, an observation which was taken to suggest that the molecular basis of the delayed post-harvest senescence induced by exogenous cytokinin differed from that induced by endogenous cytokinin. Of the up-regulated stress response related proteins detected in the transgenic material, 17 proved to be heat-stable following cooking and were therefore considered to represent potential allergens (Liu et al., 2014a).
Metabolomics
Metabolomics, defined as the systematic study of the by-products of cellular processes, is an ambitious field, as the number of chemically distinct molecules involved in a typical plant sample is estimated to be at least 100,000 (Wink, 1988), each varying with respect to their abundance (De Vos et al., 2007). A number of these compounds are of physiological importance and/or represent dietary sources of antioxidants and other health enhancing compounds; most importantly with respect to vegetable crops, they make a major contribution to product color and flavor. Often these latter quality aspects rely not on individual compounds, but rather on specific mixtures (Ferguson and Schlothauer, 2012). The state-of-the-art analytical platforms employed for metabolomic analyses are a sophisticated combination of nuclear magnetic resonance spectroscopy (NMR), MS, gas chromatography (GC), LC, and capillary electrophoresis (CE). Changes in the primary and secondary metabolite pool have been characterized using a GC-MS approach (Hummel et al., 2013), while CE-MS is more suitable for characterizing the products associated with the central metabolism (Yang et al., 2012). NMR is appropriate for the targeting of phenolic compounds, carbohydrates, organic acids and amino acids, and of particular interest in the context of brassicaceous vegetables, glucosinolates (Table 4). Electrospray ionization (ESI), conducted either in positive (resulting in protonated species) or in negative (deprotonated species) mode is used for the analysis of semi-polar metabolites. Together, these technologies can identify and quantify a wide range of primary and secondary metabolites. For LC-MS and NMR applications, samples are typically extracted in alcohol and not derivatized. In contrast, GC-MS separations require a derivatization step (Kopka et al., 2005). Particularly high mass resolution can be achieved by the use of time-of-flight (TOF)-MS devices, and additional information can be obtained from the MS/MS fragmentation patterns and UV-VIS spectra. Recent software developments have improved the capacity to recognize different metabolites. Profiles based on mass, retention time and signal amplitude provide the data required for filtering biomarkers. Typically a data processing pipeline can be divided into two steps: data processing (filtering, feature detection, alignment, and normalization) and data analysis (algorithm selection, training, evaluation, and model examination; further explanation see Boccard et al., 2010).
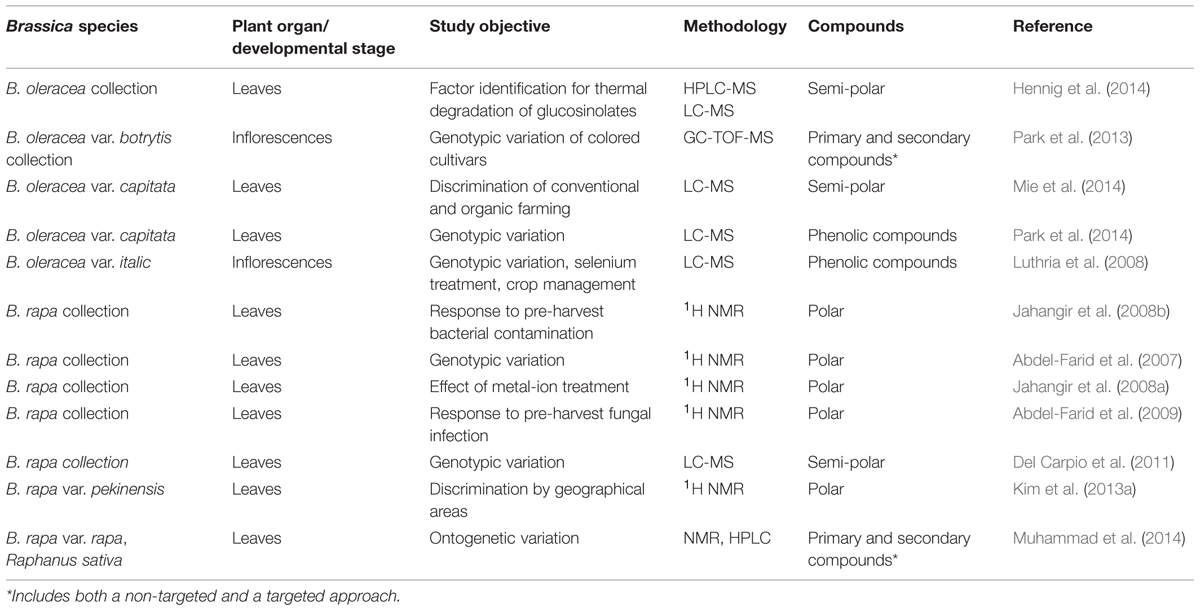
TABLE 4. Published non-targeted metabolomic analyses in the brassicaceous vegetables.
Nevertheless, metabolite identification in non-targeted approaches in the absence of authentic reference compounds remains difficult. Various platforms and spectral databases are available online4.
The metabolome of Brassica spp. as that of all plants, is influenced by both genotype (Luthria et al., 2008) and developmental stage (Abdel-Farid et al., 2007), as well as by the plant’s growing environment (Hall, 2006; Weimer and Slupsky, 2013). Metabolomic analyses in the brassicaceous vegetables have included both non-targeted (Table 4) and targeted (Table 5) approaches (Cevallos-Cevallos et al., 2009). The former have concern a range of both secondary (polyphenols and carotenoids) and primary metabolites (Hall, 2006), while the latter focused heavily on the identification and quantification of glucosinolates (Ramautar et al., 2006). The glucosinolates are a unique component of the brassicaceous vegetable metabolome (Verkerk et al., 2009). These compounds contain a sulfur-linked β-D-glucopyranose moiety and an amino acid-derived side chain, and fall into three definable classes: the aliphatic, indole and aromatic glucosinolates. They are structurally highly diverse, and within the Brassicaceae are mostly dominated by the aliphatic glucosinolates (Halkier and Gershenzon, 2006). Subsequently, aliphatic glucosinolates can be subdivided into straight- or branched-chain alk(en)yl glucosinolates with or without a hydroxy group and into a large group of those that contain an additional sulfur atom with various oxidation levels in the side chain (see for detailed structural overview, Hanschen et al., 2014). The glucosinolates are not known to be bioactive, but their hydrolysis products provide deterrence against feeding (Textor and Gershenzon, 2009), and their presence in the human diet has been associated with both health benefits (Vig et al., 2009) and taste (Beck et al., 2014).

TABLE 5. Published targeted metabolomic analyses in the brassicaceous vegetables, featuring glucosinolates and their break-down products.
Gas chromatography/TOF-MS has been used to characterize the phytochemical diversity of the florets in cauliflower cultivars varying in inflorescence pigmentation (Park et al., 2013). A 1H-NMR platform proved informative for discriminating between a set of Chinese cabbage cultivars from China and Korea: differences with respect to both primary and secondary metabolites were uncovered, although insufficient attention has yet to be given to the effect of the growing environment (Kim et al., 2013a). A non-targeted strategy was chosen by Mie et al. (2014) to reveal the impact of organic farming on the metabolome of cabbage, and led to the conclusion that this approach has potential for the authentication of organic products. The exposure of B. carinata seedlings to lithium ions induced a marked effect on the plant’s lipid and phenolic content: sinapic acid esters and chloroplast lipids were replaced by benzoate derivatives, resveratrol and oxylipins (Li et al., 2009). When GC/TOF-MS was used to discriminate between cabbage cultivars varying with respect to their resistance to feeding by diamondback moth (Plutella xylostella) caterpillars, Kim et al. (2013b) were able to show that the levels of glycolic acid, quinic acid, inositol, fumaric acid, glyceric acid, trehalose, shikimic acid, and aspartic acid were all very different.
Non-targeted metabolic approaches have also been applied to the study of processing quality in brassicaceous vegetables. Changes in the content of certain flavonols have been associated with the thermal degradation of glucosinolates in B. oleracea (Hennig et al., 2014). Several analytical techniques have been used to assess the impact of industrial processing on the phytochemical composition of vegetable purées. In broccoli, most of the metabolites present in the purée were degraded by heating, including various health-related and flavor compounds, vitamins, carotenoids, flavonoids, glucosinolates, and oxylipins (Lopez-Sanchez et al., 2015). Post-harvest storage temperature also has a profound effect on the metabolome. For example, in radish (Raphanus sativus), the content of phenylpropanoids, flavonoids, and glucosinolates decreased with storage time (Jahangir, 2010).
Microbiomics
Microbiomics refers to the application of ‘omics technologies to the microbial community associated with the plant spermosphere, endosphere, rhizosphere, and phyllosphere. The plant microbiome has attracted the focus of several researchers in recent years (reviewed in Vorholt, 2012; Bakker et al., 2013; Bulgarelli et al., 2013), as it has an intimate interaction with the plant, affecting both its metabolism and physiology. Studies of the plant microbiome have relied to date either on the in vitro culture of the relevant micro-organisms or on the recognition of species identity from PCR amplicons derived from variable regions of their genome (such as the 16S rDNA locus) followed by sequencing, microarray analysis or various electrophoretic procedures (Rastogi et al., 2013; Turner et al., 2013; Muller and Ruppel, 2014). Metaproteomics approaches can complement such data sets to provide functional information relevant to the microbiome (Knief et al., 2012). As many vegetable are consumed as a fresh product, their associated microbiome will enter the human digestive system. Exemplarily, the lettuce (Hunter et al., 2010; Rastogi et al., 2012; Jackson et al., 2013; Williams and Marco, 2014) and spinach (Leff and Fierer, 2013; Lopez-Velasco et al., 2013) microbiomes are dependent on the host genotype, the cultivation conditions and the tissue sampled. The impact of commensal microbial diversity on crop disease has been investigated in a range of oilseed rape cultivars infected with the fungal pathogen Verticillium longisporum (Graner et al., 2003). Bacterial strains isolated from tolerant or susceptible plant cultivars showed biocontrol activity against the fungus and imply a possible application in crop protection. Meanwhile De Campos et al. (2013) used next generation sequencing to show how the components of the oilseed rape microbiome depended on the developmental stage of the plant. Links et al. (2014) demonstrated that the seeds of B. juncea, B. napus, and B. rapa, were associated with a core seed microbial population of geographically and ecologically different crops. The structure of the Brassica species -specific microbiome has been shown to be sensitive to the composition of the host’s secondary metabolites (Ruppel et al., 2008). Hunter et al. (2014) established a close relationship between the composition of the root exudate and the structure of the rhizosphere microbiome. Certain microbial strains had a significant impact on the capacity of the rhizosphere to solubilize phosphorus (Schilling et al., 1998; Deubel et al., 2000). Determining the functional interaction between the microbiome’s composition and plant metabolic activities is still at an early stage of development (Bakker et al., 2013).
Concluding Remarks
Many refinements in all the ‘omics technologies can be expected to come on-stream over the next few years, and these will provide exciting new avenues of research in the brassicaceous vegetables. Some of the topics likely to feature in the near future are flux analysis, the identification of informative breeding markers for the selection of beneficial bioactives and enhanced responses to both abiotic and biotic stress (Figure 2). The increasing volume of transcriptomic data acquired continues to provide a wealth of information relevant to obtaining a detailed picture of key regulatory mechanisms and pathways active in the plant. Improving the capacity to identify the components of the proteome is a fast moving area of technology development. So far, proteomic analysis in the brassicaceous vegetables has relied almost exclusively on 2-DE separation, even though the more sophisticated chromatography and MS platforms have been quite widely used in other (particularly model) plant species; these technologies are of particular value in illuminating post-translational modifications. The acquisition of full genome sequences for some (or all) of the brassicaceous vegetables can only aid in facilitating proteomic analysis in this groups of crop plants. Gaining some control over the composition of metabolites will surely provide a powerful means of varietal improvement and the optimization of post-harvest technology.

FIGURE 2. Potential applications of ‘omics technologies for brassicaceous vegetables improvement.
The marriage of ‘omics technology and plant breeding should trigger a major efficiency breakthrough in crop improvement, and also generate novel opportunities in the fields of nutrigenomics (systems approach to understand the relationship between diet and health) and microbiomics. Although substantial progress has already been made both in the data acquisition and its interpretation, many technical and scientific challenges remain before the level of understanding of the workings of a complex biological system such as a plant rises above the current state. Global information of different hierarchies (genomics, transcriptomics, proteomics, and metabolomics) need to be integrated using mathematical and statistical methods to refine existing knowledge and make new discoveries.
There are successful studies were systems perspectives revealed correlations that had not been suggested by classical approaches in Arabidopsis, (Mochida and Shinozaki, 2011), tomato (Enfissi et al., 2010), maize (Casati et al., 2011), and others. Elucidating principles that govern relationships between biological instances will help to improve plant functions also in brassicaceous vegetables; i.e., stress-resistant plants and high- yield cultivars.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ http://www3.botany.ubc.ca/can-till
- ^ http://revgenuk.jic.ac.uk
- ^ phytozome.jgi.doe.gov/
- ^ www.metabolomicsociety.org
References
Abdel-Farid, B. B., Kim, H. K., Choi, Y. H., and Verpoorte, R. (2007). Metabolic characterization of Brassica rapa leaves by NMR spectroscopy. J. Agric. Food Chem. 55, 7936–7943. doi: 10.1021/jf071294b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Abdel-Farid, I. B., Jahangir, M., Mustafa, N. R., Van Dam, N. M., Van Den Hondel, C., Kim, H. K., et al. (2010). Glucosinolate profiling of Brassica rapa cultivars after infection by Leptosphaeria maculans and Fusarium oxysporum. Biochem. Syst. Ecol. 38, 612–620. doi: 10.1016/j.bse.2010.07.008
Abdel-Farid, I. B., Jahangir, M., Van Den Hondel, C., Kim, H. K., Choi, Y. H., and Verpoorte, R. (2009). Fungal infection-induced metabolites in Brassica rapa. Plant Sci. 176, 608–615. doi: 10.1016/j.plantsci.2009.01.017
Alam, I., Sharmin, S. A., Kim, K. H., Kim, Y. G., Lee, J. J., and Lee, B. H. (2013). An improved plant leaf protein extraction method for high resolution two-dimensional polyacrylamide gel electrophoresis and comparative proteomics. Biotech. Histochem. 88, 61–75. doi: 10.3109/10520295.2012.729863
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Albertin, W., Brabant, P., Catrice, O., Eber, F., Jenczewski, E., Chèvre, A.-M., et al. (2005). Autopolyploidy in cabbage (Brassica oleracea L.) does not alter significantly the proteomes of green tissues. Proteomics 5, 2131–2139. doi: 10.1002/pmic.200401092
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amoah, S., Kurup, S., Lopez, C. M. R., Welham, S. J., Powers, S. J., Hopkins, C. J., et al. (2012). A hypomethylated population of Brassica rapa for forward and reverse epi-genetics. BMC Plant Biol. 12:193. doi: 10.1186/1471-2229-12-193
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Anstead, J., Hartson, S., and Thompson, G. (2013). The broccoli (Brassica oleracea) phloem tissue proteome. BMC Genomics 14:764. doi: 10.1186/1471-2164-14-764
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Augustine, R., Arya, G. C., Nambiar, D. M., Kumar, R., and Bisht, N. C. (2014). Translational genomics in Brassica crops: challenges, progress, and future prospects. Plant Biotechnol. Rep. 8, 65–81. doi: 10.1007/s11816-013-0298-8
Ayele, M., Haas, B. J., Kumar, N., Wu, H., Xiao, Y. L., Van Aken, S., et al. (2005). Whole genome shotgun sequencing of Brassica oleracea and its application to gene discovery and annotation in Arabidopsis. Genome Res. 15, 487–495. doi: 10.1101/gr.3176505
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bagheri, H., El-Soda, M., Van Oorschot, I., Hanhart, C., Bonnema, G., Jansen-Van Den Bosch, T., et al. (2012). Genetic analysis of morphological traits in a new, versatile, rapid-cycling Brassica rapa recombinant inbred line population. Front. Plant Sci. 3:183. doi: 10.3389/fpls.2012.00183
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bakker, P. A. H. M., Berendsen, R. L., Doornbos, R. F., Wintermans, P. C. A., and Pieterse, C. M. J. (2013). The rhizosphere revisited: root microbiomics. Front. Plant Sci. 4:165. doi: 10.3389/fpls.2013.00165
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barkla, B. J., Castellanos-Cervantes, T., Diaz De León, J. L., Matros, A., Mock, H.-P., Perez-Alfocea, F., et al. (2013). Elucidation of salt stress defense and tolerance mechanisms of crop plants using proteomics - current achievements and perspectives. Proteomics 13, 1885–1900. doi: 10.1002/pmic.201200399
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Beck, T. K., Jensen, S., Bjoern, G. K., and Kidmose, U. (2014). The masking effect of sucrose on perception of bitter compounds in Brassica vegetables. J. Sens. Stud. 29, 190–200. doi: 10.1111/joss.12094
Bieda, M. (2012). Kepler for ‘Omics Bioinformatics. Procedia Comput. Sci. 9, 1635–1638. doi: 10.1016/j.procs.2012.04.180
Boccard, J., Veuthey, J.-L., and Rudaz, S. (2010). Knowledge discovery in metabolomics: an overview of MS data handling. J. Sep. Sci. 33, 290–304. doi: 10.1002/jssc.200900609
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brown, A. F., Yousef, G. G., Chebrolu, K. K., Byrd, R. W., Everhart, K. W., Thomas, A., et al. (2014). High-density single nucleotide polymorphism (SNP) array mapping in Brassica oleracea: identification of QTL associated with carotenoid variation in broccoli florets. Theor. Appl. Genet. 127, 2051–2064. doi: 10.1007/s00122-014-2360-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buhtz, A., Kolasa, A., Arlt, K., Walz, C., and Kehr, J. (2004). Xylem sap protein composition is conserved among different plant species. Planta 219, 610–618. doi: 10.1007/s00425-004-1259-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bulgarelli, D., Schlaeppi, K., Spaepen, S., Van Themaat, E. V. L., and Schulze-Lefert, P. (2013). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Camargo, L. E. A., Savides, L., Jung, G., Nienhuis, J., and Osborn, T. C. (1997). Location of the self-incompatibility locus in an RFLP and RAPD map of Brassica oleracea. J. Hered. 88, 57–60. doi: 10.1093/oxfordjournals.jhered.a023057
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carrington, J. C., and Ambros, V. (2003). Role of microRNAs in plant and animal development. Science 301, 336–338. doi: 10.1126/science.1085242
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Casati, P., Campi, M., Morrow, D. J., Fernandes, J. F., and Walbot, V. (2011). Transcriptomic, proteomic and metabolomic analysis of UV-B signaling in maize. BMC Genomics 12:321. doi: 10.1186/1471-2164-12-321
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cevallos-Cevallos, J. M., Reyes-De-Corcuera, J. I., Etxeberria, E., Danyluk, M. D., and Rodrick, G. E. (2009). Metabolomic analysis in food science: a review. Trends Food Sci. Technol. 20, 557–566. doi: 10.1016/j.tifs.2009.07.002
Chen, L., Hao, L. G., Parry, M. A. J., Phillips, A. L., and Hu, Y. G. (2014). Progress in TILLING as a tool for functional genomics and improvement of crops. J. Integr. Plant Biol. 56, 425–443. doi: 10.1111/jipb.12192
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cox, J., and Mann, M. (2011). “Quantitative, high-resolution proteomics for data-driven systems biology,” in Annual Review of Biochemistry, Vol. 80, eds R. D. Kornberg, C. R. H. Raetz, J. E. Rothman, and J. W. Thorner (Palo Alto, CA: Annual Reviews), 273–299.
De Bernonville, T., Albenne, C., Arlat, M., Hoffmann, L., Lauber, E., and Jamet, E. (2014). “Xylem sap proteomics,” in Plant Proteomics, eds J. V. Jorrin-Novo, S. Komatsu, W. Weckwerth, and S. Wienkoop (New York, NY: Humana Press), 391–405. doi: 10.1007/978-1-62703-631-3_28
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Campos, S. B., Youn, J. W., Farina, R., Jaenicke, S., Junemann, S., Szczepanowski, R., et al. (2013). Changes in root bacterial communities associated to two different development stages of canola (Brassica napus L. var oleifera) evaluated through next-generation sequencing technology. Microb. Ecol. 65, 593–601. doi: 10.1007/s00248-012-0132-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Vos, R. C. H., Moco, S., Lommen, A., Keurentjes, J. J. B., Bino, R. J., and Hall, R. D. (2007). Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2, 778–791. doi: 10.1038/nprot.2007.95
Del Carpio, D. P., Basnet, R. K., De Vos, R. C. H., Maliepaard, C., Visser, R., and Bonnema, G. (2011). The patterns of population differentiation in a Brassica rapa core collection. Theor. Appl. Genet. 122, 1105–1118. doi: 10.1007/s00122-010-1516-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deubel, A., Gransee, A., and Merbach, W. (2000). Transformation of organic rhizodepositions by rhizosphere bacteria and its influence on the availability of tertiary calcium phosphate. J. Plant Nutr. Soil Sci. 163, 387–392. doi: 10.1002/1522-2624(200008)163:4<387::AID-JPLN387>3.0.CO;2-K
Devisetty, U. K., Covington, M. F., Tat, A. V., Lekkala, S., and Maloof, J. N. (2014). Polymorphism identification and improved genome annotation of Brassica rapa through deep RNA sequencing. G3 (Bethesda) 4, 2065–2078. doi: 10.1534/g3.114.012526
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ellendorff, U., Fradin, E. F., De Jonge, R., and Thomma, B. P. (2009). RNA silencing is required for Arabidopsis defence against Verticillium wilt disease. J. Exp. Bot. 60, 591–602. doi: 10.1093/jxb/ern306
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Enfissi, E. M. A., Barneche, F., Ahmed, I., Lichtlé, C., Gerrish, C., Mcquinn, R. P., et al. (2010). Integrative transcript and metabolite analysis of nutritionally enhanced DE-ETIOLATED1 downregulated tomato fruit. Plant Cell 22, 1190–1215. doi: 10.1105/tpc.110.073866
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fahey, J. W., Zhang, Y. S., and Talalay, P. (1997). Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. U.S.A. 94, 10367–10372. doi: 10.1073/pnas.94.19.10367
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Faltusova, Z., Kucera, L., and Ovesna, J. (2011). Genetic diversity of Brassica oleracea var. capitata gene bank accessions assessed by AFLP. Electr. J. Biotechnol. 14, 10. doi: 10.2225/vol14-issue3-fulltext-4
Ferguson, L. R., and Schlothauer, R. C. (2012). The potential role of nutritional genomics tools in validating high health foods for cancer control: broccoli as example. Mol. Nutr. Food Res. 56, 126–146. doi: 10.1002/mnfr.201100507
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fernandez-Garcia, N., Hernandez, M., Casado-Vela, J., Bru, R., Elortza, F., Hedden, P., et al. (2011). Changes to the proteome and targeted metabolites of xylem sap in Brassica oleracea in response to salt stress. Plant Cell Environ. 34, 821–836. doi: 10.1111/j.1365-3040.2011.02285.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, J. J., Yu, X. X., Ma, F. M., and Li, J. (2014). RNA-Seq analysis of transcriptome and glucosinolate metabolism in seeds and sprouts of broccoli (Brassica oleracea var. italic). PLoS ONE 9:e88804. doi: 10.1371/journal.pone.0088804
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, M. Q., Li, G. Y., Yang, B., Qiu, D., Farnham, M., and Quiros, C. (2007). High-density Brassica oleracea linkage map: identification of useful new linkages. Theor. Appl. Genet. 115, 277–287. doi: 10.1007/s00122-007-0568-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ge, Y., Ramchiary, N., Wang, T., Liang, C., Wang, N., Wang, Z., et al. (2011). Mapping quantitative trait loci for leaf and heading-related traits in Chinese cabbage (Brassica rapa L. ssp pekinesis). Hortic. Environ. Biotechnol. 52, 494–501. doi: 10.1007/s13580-011-0031-x
Ge, Y., Wang, T., Wang, N., Wang, Z., Liang, C., Ramchiary, N., et al. (2012). Genetic mapping and localization of quantitative trait loci for chlorophyll content in Chinese cabbage (Brassica rapa ssp pekinensis). Sci. Hortic. 147, 42–48. doi: 10.1016/j.scienta.2012.09.004
Gehlenborg, N., O’Donoghue, S. I., Baliga, N. S., Goesmann, A., Hibbs, M. A., Kitano, H., et al. (2010). Visualization of omics data for systems biology. Nat. Methods 7, S56–S68. doi: 10.1038/nmeth.1436
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Graner, G., Persson, P., Meijer, J., and Alstrom, S. (2003). A study on microbial diversity in different cultivars of Brassica napus in relation to its wilt pathogen, Verticillium longisporum. FEMS Microbiol. Lett. 224, 269–276. doi: 10.1016/S0378-1097(03)00449-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gu, Y., Zhao, Q. C., Sun, D. L., and Song, W. Q. (2008). A genetic linkage map based on AFLP and NBS markers in cauliflower (Brassica oleracea var. botrytis). Bot. Stud. 49, 93–99.
Halkier, B. A., and Gershenzon, J. (2006). Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 57, 303–333. doi: 10.1146/annurev.arplant.57.032905.105228
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hall, R. D. (2006). Plant metabolomics: from holistic hope, to hype, to hot topic. New Phytol. 169, 453–468. doi: 10.1111/j.1469-8137.2005.01632.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanschen, F. S., Lamy, E., Schreiner, M., and Rohn, S. (2014). Reactivity and stability of glucosinolates and their breakdown products in foods. Angew. Chem. Int. Ed. 53, 11430–11450. doi: 10.1002/anie.201402639
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hennig, K., De Vos, R. C. H., Maliepaard, C., Dekker, M., Verkerk, R., and Bonnema, G. (2014). A metabolomics approach to identify factors influencing glucosinolate thermal degradation rates in Brassica vegetables. Food Chem. 155, 287–297. doi: 10.1016/j.foodchem.2014.01.062
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Higdon, J. V., Delage, B., Williams, D. E., and Dashwood, R. H. (2007). Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol. Res. 55, 224–236. doi: 10.1016/j.phrs.2007.01.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Himelblau, E., Gilchrist, E. J., Buono, K., Bizzell, C., Mentzer, L., Vogelzang, R., et al. (2009). Forward and reverse genetics of rapid-cycling Brassica oleracea. Theor. Appl. Genet. 118, 953–961. doi: 10.1007/s00122-008-0952-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hong, E., Kim, S. J., and Kim, G. H. (2011). Identification and quantitative determination of glucosinolates in seeds and edible parts of Korean Chinese cabbage. Food Chem. 128, 1115–1120. doi: 10.1016/j.foodchem.2010.11.102
Hossain, M. M., Li, X., Evans, I. H., and Rahman, M. A. (2014). A proteomic analysis of seed proteins expressed in a Brassica somatic hybrid and its two parental species. J. Plant Tissue Cult. Biotechnol. 24, 11–26. doi: 10.3329/ptcb.v24i1.19187
Hummel, J., Strehmel, N., Bölling, C., Schmidt, S., Walther, D., and Kopka, J. (2013). “Mass spectral search and analysis using the golm metabolome database,” in The Handbook of Plant Metabolomics, eds W. Weckwerth and G. Kahl (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA), 321–343. doi: 10.1002/9783527669882.ch18
Hunter, P. J., Hand, P., Pink, D., Whipps, J. M., and Bending, G. D. (2010). Both leaf properties and microbe-microbe interactions influence within-species variation in bacterial population diversity and structure in the lettuce (Lactuca species) Phyllosphere. Appl. Environ. Microbiol. 76, 8117–8125. doi: 10.1128/AEM.01321-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hunter, P. J., Teakle, G., and Bending, G. D. (2014). Root traits and microbial community interactions in relation to phosphorus availability and acquisition, with particular reference to Brassica. Front. Plant Sci. 5:27. doi: 10.3389/fpls.2014.00027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iniguez-Luy, F. L., Voort, A. V., and Osborn, T. C. (2008). Development of a set of public SSR markers derived from genomic sequence of a rapid cycling Brassica oleracea L. genotype. Theor. Appl. Genet. 117, 977–985. doi: 10.1007/s00122-008-0837-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Izzah, N. K., Lee, J., Jayakodi, M., Perumal, S., Jin, M., Park, B. S., et al. (2014). Transcriptome sequencing of two parental lines of cabbage (Brassica oleracea L. var. capitata L.) and construction of an EST-based genetic map. BMC Genomics 15:149. doi: 10.1186/1471-2164-15-149
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jackson, C. R., Randolph, K. C., Osborn, S. L., and Tyler, H. L. (2013). Culture dependent and independent analysis of bacterial communities associated with commercial salad leaf vegetables. BMC Microbiol. 13:274. doi: 10.1186/1471-2180-13-274
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jahangir, M. (2010). Stress Response and Health Affecting Compounds in Brassicaceae. Ph.D. thesis, Department of Pharmacognosy and Metabolomics, Institute of Biology (IBL), Faculty of Science, Leiden University, Leiden.
Jahangir, M., Abdel-Farid, I. B., Choi, Y. H., and Verpoorte, R. (2008a). Metal ion-inducing metabolite accumulation in Brassica rapa. J. Plant Physiol. 165, 1429–1437. doi: 10.1016/j.jplph.2008.04.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jahangir, M., Kim, H. K., Choi, Y. H., and Verpoorte, R. (2008b). Metabolomic response of Brassica rapa submitted to pre-harvest bacterial contamination. Food Chem. 107, 362–368. doi: 10.1016/j.foodchem.2007.08.034
Jeffery, E. H., and Araya, M. (2009). Physiological effects of broccoli consumption. Phytochem. Rev. 8, 283–298. doi: 10.1007/s11101-008-9106-4
Kaido, M., Ishikawa, T., and Hori, H. (2007). Comparative proteomic analysis of the resistant response in Brassica rapa root culture to the clubroot disease agent Plasmodiophora brassicae. Bull. Facul. Agric. Niigata Univ. 60, 67–71.
Kang, J., Zhang, G., Bonnema, G., Fang, Z., and Wang, X. (2008). Global analysis of gene expression in flower buds of Ms-cd1 Brassica oleracea conferring male sterility by using an Arabidopsis microarray. Plant Mol. Biol. 66, 177–192. doi: 10.1007/s11103-007-9261-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Katajamaa, M., and Oresic, M. (2007). Data processing for mass spectrometry-based metabolism. J. Chromatogr. A 1158, 318–328. doi: 10.1016/j.chroma.2007.04.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaul, S., Koo, H. L., Jenkins, J., Rizzo, M., Rooney, T., Tallon, L. J., et al. (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kehr, J., and Buhtz, A. (2011). “Brassica proteomics and metabolomics,” in Genetics, Genomics and Breeding of Oilseed Brassicas, eds D. Edwards, J. Batley, I. Parkin, and C. Kole (Lebanon: Science Publishers, Inc), 174–193.
Kenan-Eichler, M., Leshkowitz, D., Tal, L., Noor, E., Melamed-Bessudo, C., Feldman, M., et al. (2011). Wheat hybridization and polyploidization results in deregulation of small RNAs. Genetics 188, 263–272. doi: 10.1534/genetics.111.128348
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khan, M. A. M., Ulrichs, C., and Mewis, I. (2011). Water stress alters aphid-induced glucosinolate response in Brassica oleracea var. italica differently. Chemoecology 21, 235–242. doi: 10.1007/s00049-011-0084-4
Kim, B., Yu, H. J., Park, S. G., Shin, J. Y., Oh, M., Kim, N., et al. (2012). Identification and profiling of novel microRNAs in the Brassica rapa genome based on small RNA deep sequencing. BMC Plant Biol. 12:218. doi: 10.1186/1471-2229-12-218
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, H. A., Lim, C. J., Kim, S., Choe, J. K., Jo, S. H., Baek, N., et al. (2014). High-throughput sequencing and de novo assembly of Brassica oleracea var. capitata L. for transcriptome analysis. PLoS ONE 9:e92087. doi: 10.1371/journal.pone.0092087
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J., Jung, Y., Song, B., Bong, Y. S., Ryu, D. H., Lee, K. S., et al. (2013a). Discrimination of cabbage (Brassica rapa ssp pekinensis) cultivars grown in different geographical areas using H-1 NMR-based metabolomics. Food Chem. 137, 68–75. doi: 10.1016/j.foodchem.2012.10.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J. K., Choi, S. R., Lee, J., Park, S.-Y., Song, S. Y., Na, J., et al. (2013b). Metabolic differentiation of diamondback moth (Plutella xylostella (L.)) resistance in cabbage (Brassica oleracea L. ssp. capitata). J. Agric. Food Chem. 61, 11222–11230. doi: 10.1021/jf403441t
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Knief, C., Delmotte, N., Chaffron, S., Stark, M., Innerebner, G., Wassmann, R., et al. (2012). Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. Isme J. 6, 1378–1390. doi: 10.1038/ismej.2011.192
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Knudsen, S. (2004). Guide to Analysis of DNA Microarray Data, 2nd Edn. Hoboken, N.J: Wiley-Liss. doi: 10.1002/0471670278
Kohl, M., Megger, D. A., Trippler, M., Meckel, H., Ahrens, M., Bracht, T., et al. (2014). A practical data processing workflow for multi-OMICS projects. Biochim. Biophys. Acta 1844, 52–62. doi: 10.1016/j.bbapap.2013.02.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kong, F., Ge, C., Fang, X., Snowdon, R. J., and Wang, Y. (2010). Characterization of seedling proteomes and development of markers to distinguish the Brassica A and C genomes. J. Genet. Genomics 37, 333–340. doi: 10.1016/S1673-8527(09)60051-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kopka, J., Schauer, N., Krueger, S., Birkemeyer, C., Usadel, B., Bergmüller, E., et al. (2005).R01EQENTQi5EQg==: the Golm Metabolome Database. Bioinformatics 21, 1635–1638. doi: 10.1093/bioinformatics/bti236
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kubo, N., Saito, M., Tsukazaki, H., Kondo, T., Matsumoto, S., and Hirai, M. (2010). Detection of quantitative trait loci controlling morphological traits in Brassica rapa L. Breed. Sci. 60, 164–171. doi: 10.1270/jsbbs.60.164
Kurowska, M., Daszkowska-Golec, A., Gruszka, D., Marzec, M., Szurman, M., Szarejko, I., et al. (2011). TILLING - a shortcut in functional genomics. J. Appl. Genet. 52, 371–390. doi: 10.1007/s13353-011-0061-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kwun, M., Choi, Y., Yoon, H., Park, B. S., Kang, B. J., and Chung, Y. Y. (2004). Expression analysis of the pistil genes in controlling self-incompatibility of Brassica campestris by CO2 gas using microarray. Mol. Cells 18, 94–99.
Lee, J. G., Bonnema, G., Zhang, N. W., Kwak, J. H., De Vos, R. C. H., and Beekwilder, J. (2013). Evaluation of glucosinolate variation in a collection of turnip (Brassica rapa) germplasm by the analysis of intact and desulfo glucosinolates. J. Agric. Food Chem. 61, 3984–3993. doi: 10.1021/jf400890p
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, S. C., Lim, M. H., Kim, J. A., Lee, S. I., Kim, J. S., Jin, M., et al. (2008). Transcriptome analysis in Brassica rapa under the abiotic stresses using Brassica 24K oligo microarray. Mol. Cells 26, 595–605.
Lee, S., and Yun, S. C. (2006). The ozone stress transcriptome of pepper (Capsicum annuum L.). Mol. Cells 21, 197–205.
Leff, J. W., and Fierer, N. (2013). Bacterial communities associated with the surfaces of fresh fruits and vegetables. PLoS ONE 8:e59310. doi: 10.1371/journal.pone.0059310
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, X., Gao, P., Gjetvaj, B., Westcott, N., and Gruber, M. Y. (2009). Analysis of the metabolome and transcriptome of Brassica carinata seedlings after lithium chloride exposure. Plant Sci. 177, 68–80. doi: 10.1016/j.plantsci.2009.03.013
Li, X., Ramchiary, N., Dhandapani, V., Choi, S. U., and Lim, Y. P. (2013). “Omics applications in Brassica species,” in OMICS Applications in Crop Science, ed. D. Barh (Boca Raton, FL: CRC Press), 163–190. doi: 10.1201/b16352-7
Li, X., Zhang, W., Wang, Y., Wang, Z., and Peng, Y. (2008). Changes in chloroplast proteome of Chinese cabbage seedlings induced by PS II inhibiting herbicide Atrazine. Acta Agron. Sin. 34, 238–242. doi: 10.3724/SP.J.1006.2008.00238
Liao, C.-S., Yen, J.-H., and Wang, Y.-S. (2006). Effects of endocrine disruptor di-n-butyl phthalate on the growth of Bok choy (Brassica rapa subsp. chinensis). Chemosphere 65, 1715–1722. doi: 10.1016/j.chemosphere.2006.04.093
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liao, C.-S., Yen, J.-H., and Wang, Y.-S. (2009). Growth inhibition in Chinese cabbage (Brassica rapa var. chinensis) growth exposed to di-n-butyl phthalate. J. Hazard. Mater. 163, 625–631. doi: 10.1016/j.jhazmat.2008.07.025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ligat, L., Lauber, E., Albenne, C., Clemente, H. S., Valot, B., Zivy, M., et al. (2011). Analysis of the xylem sap proteome of Brassica oleracea reveals a high content in secreted proteins. Proteomics 11, 1798–1813. doi: 10.1002/pmic.201000781
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, K. H., Chang, L. C., Lai, C. D., and Lo, H. F. (2013). AFLP mapping of quantitative trait loci influencing seven head-related traits in broccoli (Brassica oleracea var. italica). J. Hortic. Sci. Biotechnol. 88, 257–268.
Links, M. G., Demeke, T., Grafenhan, T., Hill, J. E., Hemmingsen, S. M., and Dumonceaux, T. J. (2014). Simultaneous profiling of seed-associated bacteria and fungi reveals antagonistic interactions between microorganisms within a shared epiphytic microbiome on triticum and Brassica seeds. New Phytol. 202, 542–553. doi: 10.1111/nph.12693
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, M.-S., Ko, M.-H., Li, H.-C., Tsai, S.-J., Lai, Y.-M., Chang, Y.-M., et al. (2014a). Compositional and proteomic analyses of genetically modified broccoli (Brassica oleracea var. italica) harboring an agrobacterial gene. Int. J. Mol. Sci. 15, 15188–15209. doi: 10.3390/ijms150915188
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, S., Liu, Y., Yang, X., Tong, C., Edwards, D., Parkin, I. a. P., et al. (2014b). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930. doi: 10.1038/ncomms4930
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, M.-S., Li, H.-C., Chang, Y.-M., Wu, M.-T., and Chen, L.-F. O. (2011). Proteomic analysis of stress-related proteins in transgenic broccoli harboring a gene for cytokinin production during postharvest senescence. Plant Sci. 181, 288–299. doi: 10.1016/j.plantsci.2011.06.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, M.-S., Li, H.-C., Lai, Y.-M., Lo, H.-F., and Chen, L.-F. O. (2013a). Proteomics and transcriptomics of broccoli subjected to exogenously supplied and transgenic senescence-induced cytokinin for amelioration of postharvest yellowing. J. Proteom. 93, 133–144. doi: 10.1016/j.jprot.2013.05.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, Y., Zhang, Y., Xing, J. Y., Liu, Z. Y., and Feng, H. (2013b). Mapping quantitative trait loci for yield-related traits in Chinese cabbage (Brassica rapa L. ssp pekinensis). Euphytica 193, 221–234. doi: 10.1007/s10681-013-0931-1
Lopez-Sanchez, P., De Vos, R. C. H., Jonker, H. H., Mumm, R., Hall, R. D., Bialek, L., et al. (2015). Comprehensive metabolomics to evaluate the impact of industrial processing on the phytochemical composition of vegetable purees. Food Chem. 168, 348–355. doi: 10.1016/j.foodchem.2014.07.076
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lopez-Velasco, G., Carder, P. A., Welbaum, G. E., and Ponder, M. A. (2013). Diversity of the spinach (Spinacia oleracea) spermosphere and phyllosphere bacterial communities. FEMS Microbiol. Lett. 346, 146–154. doi: 10.1111/1574-6968.12216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lou, P., Xie, Q., Xu, X., Edwards, C. E., Brock, M. T., Weinig, C., et al. (2011). Genetic architecture of the circadian clock and flowering time in Brassica rapa. Theor. Appl. Genet. 123, 397–409. doi: 10.1007/s00122-011-1592-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lu, G., Cao, J. S., Yu, X. L., Xiang, X., and Chen, H. (2008). Mapping QTLs for root morphological traits in Brassica rapa L. based on AFLP and RAPD markers. J. Appl. Genet. 49, 23–31. doi: 10.1007/BF03195245
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luthria, D. L., Lin, L. Z., Robbins, R. J., Finley, J. W., Banuelos, G. S., and Harnly, J. M. (2008). Discriminating between cultivars and treatments of broccoli using mass spectral fingerprinting and analysis of variance-principal component analysis. J. Agric. Food Chem. 56, 9819–9827. doi: 10.1021/jf801606x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lv, H. H., Wang, Q. B., Zhang, Y. Y., Yang, L. M., Fang, Z. Y., Wang, X. W., et al. (2014). Linkage map construction using InDel and SSR markers and QTL analysis of heading traits in Brassica oleracea var. capitata L. Mol. Breed. 34, 87–98. doi: 10.1007/s11032-014-0019-1
Maggioni, L., Von Bothmer, R., Poulsen, G., Branca, F., and Jorgensen, R. B. (2014). Genetic diversity and population structure of leafy kale and Brassica rupestris Raf. in south Italy. Hereditas 151, 145–158. doi: 10.1111/hrd2.00058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., and Gilad, Y. (2008). RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517. doi: 10.1101/gr.079558.108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marquès, K., Sarazin, B., Chané-Favre, L., Zivy, M., and Thiellement, H. (2001). Comparative proteomics to establish genetic relationships in the Brassicaceae family. Proteomics 1, 1457–1462. doi: 10.1002/1615-9861(200111)1:11<1457::AID-PROT1457>3.0.CO;2-W
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matros, A., Kaspar, S., Witzel, K., and Mock, H.-P. (2011). Recent progress in liquid chromatography-based separation and label-free quantitative plant proteomics. Phytochemistry 72, 963–974. doi: 10.1016/j.phytochem.2010.11.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsumoto, E., Ueno, H., Aruga, D., Sakamoto, K., and Hayashida, N. (2012). Accumulation of three clubroot resistance genes through marker-assisted selection in Chinese cabbage (Brassica rapa ssp pekinensis). J. Jpn. Soc. Hort. Sci. 81, 184–190. doi: 10.2503/jjshs1.81.184
Mewis, I., Schreiner, M., Nguyen, C. N., Krumbein, A., Ulrichs, C., Lohse, M., et al. (2012). UV-B irradiation changes specifically the secondary metabolite profile in broccoli sprouts: induced signaling overlaps with defense response to biotic stressors. Plant Cell Physiol. 53, 1546–1560. doi: 10.1093/pcp/pcs096
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mie, A., Laursen, K. H., Aberg, K. M., Forshed, J., Lindahl, A., Thorup-Kristensen, K., et al. (2014). Discrimination of conventional and organic white cabbage from a long-term field trial study using untargeted LC-MS-based metabolomics. Anal. Bioanal. Chem. 406, 2885–2897. doi: 10.1007/s00216-014-7704-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mochida, K., and Shinozaki, K. (2011). Advances in omics and bioinformatics tools for systems analyses of plant functions. Plant Cell Physiol. 52, 2017–2038. doi: 10.1093/pcp/pcr153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muhammad, J., Abdel-Farid, I. B., Vos, R. C. H. D., Jonker, H. H., Choi, Y., and Verpoorte, R. (2014). Metabolomic variation of Brassica rapa var. rapa (var. raapstelen) and Raphanus sativus L. at different developmental stages. Pak. J. Bot. 46, 1445–1452.
Muller, T., and Ruppel, S. (2014). Progress in cultivation-independent phyllosphere microbiology. FEMS Microbiol. Ecol. 87, 2–17. doi: 10.1111/1574-6941.12198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nawrocki, A., Thorup-Kristensen, K., and Jensen, O. N. (2011). Quantitative proteomics by 2DE and MALDI MS/MS uncover the effects of organic and conventional cropping methods on vegetable products. J. Proteom. 74, 2810–2825. doi: 10.1016/j.jprot.2011.06.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Niederhuth, C. E., and Schmitz, R. J. (2014). Covering your bases: inheritance of DNA methylation in plant genomes. Mol. Plant 7, 472–480. doi: 10.1093/mp/sst165
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
O’Farrell, P. H. (1975). High-resolution 2-dimensional electrophoresis of proteins. J. Biol. Chem. 250, 4007–4021.
Okazaki, K., Shinano, T., Oka, N., and Takebe, M. (2012). Metabolite profiling of Komatsuna (Brassica rapa L.) field-grown under different soil organic amendment and fertilization regimes. Soil Sci. Plant Nutr. 58, 696–706. doi: 10.1080/00380768.2012.733924
Park, S., Arasu, M. V., Jiang, N., Choi, S. H., Lim, Y. P., Park, J. T., et al. (2014). Metabolite profiling of phenolics, anthocyanins and flavonols in cabbage (Brassica oleracea var. capitata). Ind. Crops Prod. 60, 8–14. doi: 10.1016/j.indcrop.2014.05.037
Park, S. Y., Lim, S. H., Ha, S. H., Yeo, Y., Park, W. T., Kwon, D. Y., et al. (2013). Metabolite profiling approach reveals the interface of primary and secondary metabolism in colored cauliflowers (Brassica oleracea L. ssp botrytis). J. Agric. Food Chem. 61, 6999–7007. doi: 10.1021/jf401330e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parkin, I. A. P., Koh, C., Tang, H. B., Robinson, S. J., Kagale, S., Clarke, W. E., et al. (2014). Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15, 18. doi: 10.1186/gb-2014-15-6-r77
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pawlowski, T., Rurek, M., Janicka, S., Raczynska, K. D., and Augustyniak, H. (2005). Preliminary analysis of the cauliflower mitochondrial proteome. Acta Physiol. Plant. 27, 275–281. doi: 10.1007/s11738-005-0003-9
Pink, D., Bailey, L., Mcclement, S., Hand, P., Mathas, E., Buchanan-Wollaston, V., et al. (2008). Double haploids, markers and QTL analysis in vegetable Brassicas. Euphytica 164, 509–514. doi: 10.1007/s10681-008-9742-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rabbani, M. A., Maruyama, K., Abe, H., Khan, M. A., Katsura, K., Ito, Y., et al. (2003). Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol. 133, 1755–1767. doi: 10.1104/pp.103.025742
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rabilloud, T., Chevallet, M., Luche, S., and Lelong, C. (2010). Two-dimensional gel electrophoresis in proteomics: past, present and future. J. Proteom. 73, 2064–2077. doi: 10.1016/j.jprot.2010.05.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramautar, R., Demirci, A., and De Jong, G. J. (2006). Capillary electrophoresis in metabolomics. Trends Analyt. Chem. 25, 455–466. doi: 10.1016/j.trac.2006.02.004
Rastogi, G., Coaker, G. L., and Leveau, J. H. J. (2013). New insights into the structure and function of phyllosphere microbiota through high-throughput molecular approaches. FEMS Microbiol. Lett. 348, 1–10. doi: 10.1111/1574-6968.12225
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rastogi, G., Sbodio, A., Tech, J. J., Suslow, T. V., Coaker, G. L., and Leveau, J. H. J. (2012). Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. ISME J. 6, 1812–1822. doi: 10.1038/ismej.2012.32
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reichelt, M., Brown, P. D., Schneider, B., Oldham, N. J., Stauber, E., Tokuhisa, J., et al. (2002). Benzoic acid glucosinolate esters and other glucosinolates from Arabidopsis thaliana. Phytochemistry 59, 663–671. doi: 10.1016/S0031-9422(02)00014-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rochfort, S. J., Trenerry, V. C., Imsic, M., Panozzo, J., and Jones, R. (2008). Class targeted metabolomics: ESI ion trap screening methods for glucosinolates based on MSn fragmentation. Phytochemistry 69, 1671–1679. doi: 10.1016/j.phytochem.2008.02.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ruppel, S., Krumbein, A., and Schreiner, M. (2008). Composition of the phyllospheric microbial populations on vegetable plants with different glucosinolate and carotenoid compositions. Microb. Ecol. 56, 364–372. doi: 10.1007/s00248-007-9354-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Salmon, A., Clotault, J., Jenczewski, E., Chable, V., and Manzanares-Dauleux, M. J. (2008). Brassica oleracea displays a high level of DNA methylation polymorphism. Plant Sci. 174, 61–70. doi: 10.1016/j.plantsci.2007.09.012
Schilling, G., Gransee, A., Deubel, A., Lezovic, G., and Ruppel, S. (1998). Phosphorus availability, root exudates, and microbial activity in the rhizosphere. J. Plant Nutr. Soil Sci. 161, 465–478. doi: 10.1002/jpln.1998.3581610413
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmidt, U. G., Endler, A., Schelbert, S., Brunner, A., Schnell, M., Neuhaus, H. E., et al. (2007). Novel tonoplast transporters identified using a proteomic approach with vacuoles isolated from cauliflower buds. Plant Physiol. 145, 216–229. doi: 10.1104/pp.107.096917
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schonhof, I., Blankenburg, D., Muller, S., and Krumbein, A. (2007a). Sulfur and nitrogen supply influence growth, product appearance, and glucosinolate concentration of broccoli. J. Plant Nutr. Soil Sci. 170, 65–72. doi: 10.1002/jpln.200620639
Schonhof, I., Klaring, H. P., Krumbein, A., and Schreiner, M. (2007b). Interaction between atmospheric CO2 and glucosinolates in broccoli. J. Chem. Ecol. 33, 105–114. doi: 10.1007/s10886-006-9202-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schreiner, M., Beyene, B., Krumbein, A., and Stutzel, H. (2009). Ontogenetic changes of 2-propenyl and 3-indolylmethyl glucosinolates in Brassica carinata leaves as affected by water supply. J. Agric. Food Chem. 57, 7259–7263. doi: 10.1021/jf901076h
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schreiner, M., Krumbein, A., Knorr, D., and Smetanska, I. (2011). Enhanced glucosinolates in root exudates of Brassica rapa ssp. rapa mediated by salicylic acid and methyl jasmonate. J. Agric. Food Chem. 59, 1400–1405. doi: 10.1021/jf103585s
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schumacher, A., Rujan, T., and Hoefkens, J. (2014). A collaborative approach to develop a multi-omics data analytics platform for translational research. Appl. Transl. Genomics 3, 105–108. doi: 10.1016/j.atg.2014.09.010
Sebastian, R. L., Howell, E. C., King, G. J., Marshall, D. F., and Kearsey, M. J. (2000). An integrated AFLP and RFLP Brassica oleracea linkage map from two morphologically distinct doubled-haploid mapping populations. Theor. Appl. Genet. 100, 75–81. doi: 10.1007/s001220050011
Sepúlveda, I., Barrientos, H., Mahn, A., and Moenne, A. (2013). Changes in SeMSC, glucosinolates and sulforaphane levels, and in proteome profile in broccoli (Brassica oleracea var. Italica) fertilized with sodium selenate. Molecules 18, 5221–5234. doi: 10.3390/molecules18055221
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shapiro, T. A., Fahey, J. W., Dinkova-Kostova, A. T., Holtzclaw, W., Stephenson, K. K., Wade, K. L., et al. (2006). Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: a clinical phase I study. Nutr. Cancer 55, 53–62. doi: 10.1207/s15327914nc5501_7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shapiro, T. A., Fahey, J. W., Wade, K. L., Stephenson, K. K., and Talalay, P. (2001). Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: metabolism and excretion in humans. Cancer Epidemiol. Biomarkers Prev. 10, 501–508.
Shi, J. Q., Huang, S. M., Zhan, J. P., Yu, J. Y., Wang, X. F., Hua, W., et al. (2014). Genome-wide microsatellite characterization and marker development in the sequenced Brassica crop species. DNA Res. 21, 53–68. doi: 10.1093/dnares/dst040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shumin, L., Schonhof, I., Krumbein, A., Long, L., Stuetzel, H., and Schreiner, M. (2007). 771 Glucosinolate concentration in turnip (Brassica rapa spp. rapifera L.) roots as 772 affected by nitrogen and sulfur supply. J. Agric. Food Chem. 55, 8452–8457. doi: 10.1021/jf070816k
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sonderby, I. E., Geu-Flores, F., and Halkier, B. A. (2010). Biosynthesis of glucosinolates - gene discovery and beyond. Trends Plant Sci. 15, 283–290. doi: 10.1016/j.tplants.2010.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Song, X., Liu, G., Duan, W., Liu, T., Huang, Z., Ren, J., et al. (2014). Genome-wide identification, classification and expression analysis of the heat shock transcription factor family in Chinese cabbage. Mol. Genet. Genomics 289, 541–551. doi: 10.1007/s00438-014-0833-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stephenson, P., Baker, D., Girin, T., Perez, A., Amoah, S., King, G. J., et al. (2010). A rich TILLING resource for studying gene function in Brassica rapa. BMC Plant Biol. 10:62. doi: 10.1186/1471-2229-10-62
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sun, C. B. (2014). Characterization of the small RNA transcriptome in plant-microbe (Brassica/Erwinia) interactions by high-throughput sequencing. Biotechnol. Lett. 36, 371–381. doi: 10.1007/s10529-013-1362-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sun, C., Wang, L., Hu, D., Riquicho, A. R. M., Liu, T., Hou, X., et al. (2014). Proteomic analysis of non-heading Chinese cabbage infected with Hyaloperonospora parasitica. J. Proteom. 98, 15–30. doi: 10.1016/j.jprot.2013.11.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sun, G. (2012). MicroRNAs and their diverse functions in plants. Plant Mol. Biol. 80, 17–36. doi: 10.1007/s11103-011-9817-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takuno, S., Kawahara, T., and Ohnishi, O. (2007). Phylogenetic relationships among cultivated types of Brassica rapa L. em. Metzg. as revealed by AFLP analysis. Genet. Resour. Crop Evol. 54, 279–285. doi: 10.1007/s10722-005-4260-7
Textor, S., and Gershenzon, J. (2009). Herbivore induction of the glucosinolate-myrosinase defense system: major trends, biochemical bases and ecological significance. Phytochem. Rev. 8, 149–170. doi: 10.1007/s11101-008-9117-1
T’Hoen, P. A., Ariyurek, Y., Thygesen, H. H., Vreugdenhil, E., Vossen, R. H., De Menezes, R. X., et al. (2008). Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 36, e141. doi: 10.1093/nar/gkn705
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Trick, M., Cheung, F., Drou, N., Fraser, F., Lobenhofer, E. K., Hurban, P., et al. (2009). A newly-developed community microarray resource for transcriptome profiling in Brassica species enables the confirmation of Brassica-specific expressed sequences. BMC Plant Biol. 9:50. doi: 10.1186/1471-2229-9-50
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turner, T. R., James, E. K., and Poole, P. S. (2013). The plant microbiome. Genome Biol. 14, 209. doi: 10.1186/gb-2013-14-6-209
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vanderschuren, H., Lentz, E., Zainuddin, I., and Gruissem, W. (2013). Proteomics of model and crop plant species: status, current limitations and strategic advances for crop improvement. J. Proteom. 93, 5–19. doi: 10.1016/j.jprot.2013.05.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Hintum, T. J. L., De Wiel, C., Visser, D. L., Van Treuren, R., and Vosman, B. (2007). The distribution of genetic diversity in a Brassica oleracea gene bank collection related to the effects on diversity of regeneration, as measured with AFLPs. Theor. Appl. Genet. 114, 777–786. doi: 10.1007/s00122-006-0456-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Varshney, R. K., Terauchi, R., and Mccouch, S. R. (2014). Harvesting the promising fruits of genomics: applying genome sequencing technologies to crop breeding. PLoS Biol. 12:e1001883. doi: 10.1371/journal.pbio.1001883
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verkerk, R., Schreiner, M., Krumbein, A., Ciska, E., Holst, B., Rowland, I., et al. (2009). Glucosinolates in Brassica vegetables: the influence of the food supply chain on intake, bioavailability and human health. Mol. Nutr. Food Res. 53, S219–S265. doi: 10.1002/mnfr.200800065
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vig, A. P., Rampal, G., Thind, T. S., and Arora, S. (2009). Bio-protective effects of glucosinolates - a review. Lwt-Food Sci. Technol. 42, 1561–1572. doi: 10.1016/j.lwt.2009.05.023
Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walley, P., Carder, J., Skipper, E., Mathas, E., Lynn, J., Pink, D., et al. (2012). A new broccoli × broccoli immortal mapping population and framework genetic map: tools for breeders and complex trait analysis. Theor. Appl. Genet. 124, 467–484. doi: 10.1007/s00122-011-1721-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, L., Peng, H., Ge, T., Liu, T., Hou, X., and Li, Y. (2014a). Identification of differentially accumulating pistil proteins associated with self-incompatibility of non-heading Chinese cabbage. Plant Biol. 16, 49–57. doi: 10.1111/plb.12016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, X., Tang, D., and Huang, D. (2014b). Proteomic analysis of pakchoi leaves and roots under glycine–nitrogen conditions. Plant Physiol. Biochem. 75, 96–104. doi: 10.1016/j.plaphy.2013.12.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y. G., Zhang, L., Ji, X. H., Yan, J. F., Liu, Y. T., Lv, X. X., et al. (2014c). Mapping of quantitative trait loci for the bolting trait in Brassica rapa under vernalizing conditions. Genet. Mol. Res. 13, 3927–3939. doi: 10.4238/2014.May.23.3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, W. X., Huang, S. M., Liu, Y. M., Fang, Z. Y., Yang, L. M., Hua, W., et al. (2012). Construction and analysis of a high-density genetic linkage map in cabbage (Brassica oleracea L. var. capitata). BMC Genomics 13:523. doi: 10.1186/1471-2164-13-523
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1083/nrg2484
Wei, L. J., Xiao, M. L., Hayward, A., and Fu, D. H. (2013). Applications and challenges of next-generation sequencing in Brassica species. Planta 238, 1005–1024. doi: 10.1007/s00425-013-1961-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weimer, B. C., and Slupsky, C. (2013). “Metabolomics in food and nutrition,” in Metabolomics in Food and Nutrition, eds B. C. Weimer and C. M. Slupsky (Cambridge: Woodhead), 237.
White, P. J., Hammond, J. P., King, G. J., Bowen, H. C., Hayden, R. M., Meacham, M. C., et al. (2010). Genetic analysis of potassium use efficiency in Brassica oleracea. Ann. Bot. 105, 1199–1210. doi: 10.1093/aob/mcp253
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wiesner, M., Hanschen, F. S., Schreiner, M., Glatt, H., and Zrenner, R. (2013a). Induced production of 1-methoxy-indol-3-ylmethyl glucosinolate by jasmonic acid and methyl jasmonate in sprouts and leaves of pak choi (Brassica rapa ssp. chinensis). Int. J. Mol. Sci. 14, 14996–15016. doi: 10.3390/ijms140714996
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wiesner, M., Zrenner, R., Krumbein, A., Glatt, H., and Schreiner, M. (2013b). Genotypic variation of the glucosinolate profile in pak choi (Brassica rapa ssp. chinensis). J. Agric. Food Chem. 61, 1943–1953. doi: 10.1021/jf303970k
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wiesner, M., Schreiner, M., and Zrenner, R. (2014). Functional identification of genes responsible for the biosynthesis of 1-methoxy-indol-3-ylmethyl-glucosinolate in Brassica rapa ssp. chinensis. BMC Plant Biol. 14:124. doi: 10.1186/1471-2229-14-124
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Williams, T. R., and Marco, M. L. (2014). Phyllosphere microbiota composition and microbial community transplantation on lettuce plants grown indoors. Mbio 5, e01564–14. doi: 10.1128/mBio.01564-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wink, M. (1988). Plant breeding - importance of plant secondary metabolites for protection against pathogens and herbivores. Theor. Appl. Genet. 75, 225–233. doi: 10.1007/BF00303957
Wittstock, U., and Burow, M. (2010). Glucosinolate breakdown in Arabidopsis: mechanism, regulation and biological significance. Arabidopsis Book 8, e0134. doi: 10.1199/tab.0134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Witzel, K., Pietsch, C., Strickert, M., Matros, A., Röder, M., Weschke, W., et al. (2011). Mapping of quantitative trait loci associated with protein expression variation in barley grains. Mol. Breed. 27, 301–314. doi: 10.1007/s11032-010-9432-2
Wu, Q. J., Yang, Y., Vogtmann, E., Wang, J., Han, L. H., Li, H. L., et al. (2013). Cruciferous vegetables intake and the risk of colorectal cancer: a meta-analysis of observational studies. Ann. Oncol. 24, 1079–1087. doi: 10.1093/annonc/mds601
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, K. A., Lim, C. J., Hong, J. K., Jin, Z. L., Hong, J. C., Yun, D. J., et al. (2005). Identification of Chinese cabbage genes up-regulated by prolonged cold by using microarray analysis. Plant Sci. 168, 959–966. doi: 10.1016/j.plantsci.2004.11.011
Yang, Z., Kobayashi, E., Katsuno, T., Asanuma, T., Fujimori, T., Ishikawa, T., et al. (2012). Characterisation of volatile and non-volatile metabolites in etiolated leaves of tea (Camellia sinensis) plants in the dark. Food Chem. 135, 2268–2276. doi: 10.1016/j.foodchem.2012.07.066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, L. X., and Setter, T. L. (2003). Comparative transcriptional profiling of placenta and endosperm in developing maize kernels in response to water deficit. Plant Physiol. 131, 568–582. doi: 10.1104/pp.014365
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zeng, J., Chen, S., Zhu, L., Gao, Q., Wang, X., and Yang, X. (2012). Establishment of two-dimensional electrophoresis system of Brassica oleracea L. var. capitata L. stigma protein. China Vegetables 1, 30–36.
Zhao, J. J., Artemyeva, A., Del Carpio, D. P., Basnet, R. K., Zhang, N. W., Gao, J., et al. (2010). Design of a Brassica rapa core collection for association mapping studies. Genome 53, 884–898.
Zhou, B., Fan, P., and Li, Y. (2014). High-throughput sequence analysis of small RNAs in skotomorphogenic seedlings of Brassica rapa ssp. rapa. Gene 548, 68–74. doi: 10.1016/j.gene.2014.07.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: genomics, transcriptomics, metabolomics, proteomics, crop, microbiomics
Citation: Witzel K, Neugart S, Ruppel S, Schreiner M, Wiesner M and Baldermann S (2015) Recent progress in the use of ‘omics technologies in brassicaceous vegetables. Front. Plant Sci. 6:244. doi: 10.3389/fpls.2015.00244
Received: 30 December 2014; Paper pending published: 22 February 2015;
Accepted: 26 March 2015; Published online: 14 April 2015
Edited by:
Hartmut Stützel, Leibniz Universität Hannover, GermanyReviewed by:
Juan B. Alvarez, Universidad de Córdoba, SpainAbu Hena Mostafa Kamal, National Agriculture and Food Research Organization, Japan
Ziyin Yang, South China Botanical Garden – Chinese Academy of Sciences, China
Copyright © 2015 Witzel, Neugart, Ruppel, Schreiner, Wiesner and Baldermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanne Baldermann, Leibniz-Institute of Vegetable and Ornamental Crops Großbeeren/Erfurt e.V., Theodor-Echtermeyer-Weg 1, 14979 Großbeeren, GermanyYmFsZGVybWFubkBpZ3pldi5kZQ==