 Neha Sharma
Neha Sharma- Plant Molecular Biology Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, India
Plant microRNAs (miRs) have emerged as important regulators of gene expression under normal as well as stressful environments. Rice is an important cereal crop whose productivity is compromised due to various abiotic stress factors such as salt, heat and drought. In the present study, we have investigated the role of rice-specific Osa-miR820, in indica rice cultivars showing contrasting response to salt stress. The dissection of expression patterns indicated that the miR is present in all the tissues but is enriched in the anther tissues. In salinity, the miR levels are up-regulated in the leaf tissues but down-regulated in the root tissues. To map the deregulation under salt stress comprehensive time kinetics of expression was performed in the leaf and root tissues. The reproductive stages were also analyzed under salt stress. It emerged that a common regulatory scheme for Osa-miR820 expression is present in the salt-susceptible Pusa Basmati 1 and salt-tolerant Pokkali varieties, although there is a variation in the levels of the miR and its target transcript, OsDRM2. The regulation of Osa-miR820 and its target were also studied under other abiotic stresses. This study thus captures the window for the miR-target correlation and the putative role of this regulation is discussed. This will help in gaining useful insights on the role of species specific miRs in plant development and abiotic stress response.
Introduction
The sessile nature of plants necessitates a strict and rapid monitoring of gene expression as an important aspect of proper growth and development, thereby delineating a crucial role for the regulatory molecules. MicroRNAs (miRs) are a newly identified class of endogenous non-coding RNA molecules that play a major role in genome dynamics at the molecular level (Voinnet, 2009). The miRs regulate target mRNA expression in a sequence-specific manner, by its cleavage or by translational repression (Llave et al., 2002; Aukerman and Sakai, 2003; Addo-Quaye et al., 2008; Brodersen et al., 2008; Rogers and Chen, 2013). Increasing evidence supports the fact that miRs act as genetic buffers in plant gene regulation providing protection against various abiotic and biotic stress conditions (Sunkar et al., 2007; Sanan-Mishra et al., 2009; Khraiwesh et al., 2012). It is thus important to understand the mechanism of miR mediated regulation of the genetic machinery in plants, under hostile conditions.
The first direct evidence for the involvement of miRs in plant stress responses came from the work of Bartel’s group in Arabidopsis, where they identified several novel miRs, including miR395, which were up-regulated upon sulfate starvation (Jones-Rhoades and Bartel, 2004). Ath-miR395 targets a low-affinity sulfate transporter, AST68 and three ATP sulfurylases (APS1, APS3, and APS4) involved in sulfate assimilation. This was followed by the identification of phosphate deficiency induced expression of Ath-miR399 with a corresponding decrease in its target, Phosphate 2 (PHO2), a ubiquitin-conjugating enzyme (Fujii et al., 2005; Bari et al., 2006; Chiou et al., 2006). miR399 inhibits PHO2 in a dualistic manner, by promoting its transcript decay (Allen et al., 2005; Bari et al., 2006) and also by repressing translation (Bari et al., 2006).
Soil salinity has become a major limiting factor in rice production due to the salt-sensitive nature of most indica cultivars (Lutts et al., 1995; Zeng and Shannon, 2000; Moradi et al., 2003). So, understanding the biology of salt stress associated miRs holds a promising answer for identifying the channels that can reduce yield losses. It was reported that miR396, miR168, miR167, miR165, miR319, miR159, miR394, miR156, miR393, miR171, miR158, and miR169 were up-regulated in salt stress in Arabidopsis (Liu et al., 2008), while Ath-miR398 was down-regulated under salt stress (Jagadeeswaran et al., 2009). Similarly, salt stress in Populus trichocarpa, down-regulated miR530a, miR1445, miR1446a-e, miR1447, and miR171l-n but up-regulated, miR482.2 and miR1450 (Lu et al., 2008). Likewise, in Phaseolus vulgaris, increased accumulation of miRS1 and miR159.2 was observed in response to NaCl addition (Arenas-Huertero et al., 2009). Recently, detailed analysis of the expression levels of individual miRs or a group of miRs has improved our understanding on their role in the stressful environment. The salt stress kinetics revealed the induction of two known miRs (miR171p and miR4416d), and two novel miRs (gly_1, gly_3) in soybean root nodules (Dong et al., 2013). In rice, the time series of salt and alkali treatment was found to de-regulate the expression of Osa-miR393 and Osa-miR396 (Gao et al., 2011). Further thorough profiling showed that, of the six members in Osa-miR396 family, only Osa-miR396c was down-regulated under salt and alkali treatment (Gao et al., 2010). Osa-miR408 showed a differential expression pattern among drought susceptible and tolerant cultivars of rice, where it decreases in the sensitive cultivar but remained at elevated levels in the tolerant cultivar (Mutum et al., 2013). Such studies suggest that comprehensive expression profiling of miRs under stress is important for understanding their precise regulatory roles.
Our group has focussed on identifying miRs that affect growth and yield in rice and we have earlier reported key molecules that exhibit tissue-preferential expression patterns (Mittal et al., 2013). In this study we describe the detailed expression profile of a salt stress deregulated Osa-miR820. It is a rice specific miR, that was first reported from undifferentiated embryogenic callus tissues as miR583 (Luo et al., 2006). Osa-miR820 belongs to a family of three closely related members. These represent the canonical 21 nucleotide (nt) miRs that have been named Osa-miR820a-c and their sequences are available as MI0005263, MI0005264, MI0005265 in mirBase version 21 (Kozomara and Griffiths-Jones, 2014). Later it was proposed to be a member of the transposon derived small RNA family, with its expression being controlled epigenetically at its own locus (Nosaka et al., 2013a,b). Nosaka et al. (2013b) also identified the 24-nt length variants of this family and described two additional family members, Osa-miR820d and Osa-miR820e. Recently, it has been shown to be down-regulated under arsenic stress in two contrasting arsenic responsive rice cultivars (Sharma et al., 2015). Here, we have investigated its role in salt, high temperature and drought stress responses by comparing the expression profiles of Osa-miR820 and its target gene across various tissues of two indica rice cultivars exhibiting a contrasting response to salt stress. The cultivars used include Pusa Basmati 1 (PB1), a breeder variety of basmati which is highly sensitive to salt stress and Pokkali (PK), which is known to thrive well in highly saline soils on salty backwaters of Kerala (Rana et al., 1999; Xie et al., 2000).
Materials and Methods
Plant Material and Growth Conditions
Rice seeds were washed thoroughly with sterile water and placed on germinating sheets. The seeds were grown under controlled conditions at, 28 ± 2°C, 70% relative air humidity and 16/8-h light/dark cycle. For all further analysis, leaf and root tissue samples were harvested from 15 days old seedlings while floral tissues were collected at different stages from rice plants grown in controlled conditions of the green-house. Salt stress was given by replacing nutrient solution with 250 mM NaCl. Heat stress was given by keeping the plants in growth chamber at 42°C. For drought stress, rice seedlings were grown in 100 mM mannitol solution. For time kinetics each stress was provided for the time period of 30 min, 1, 3, 6, 12, and 24 h.
RNA Isolation and Small RNA Library Preparation
Total RNA was extracted from various rice tissues using guanidine isothiocyanate (GITC) based protocol as described previously (Mittal et al., 2013). Briefly, plant tissue is homogenized in liquid nitrogen and GITC buffer is added along with phenol and chloroform. The mixture is allowed to thaw slowly and centrifuged at high speed (∼13000 rpm) for 15 min. The aqueous phase thus obtained is extracted with phenol:chloroform solution twice and kept for precipitation with ethanol. The pellet is washed using 75% DEPC-ethanol twice at 13000 rpm for 15 min each and dried at room temperature. The dried pellet of RNA was dissolved in required amount of DEPC-treated water and stored in –20°C.
For deep sequencing the small RNA libraries were constructed from normal or salt stressed leaf tissues of 15 days old seedlings as described previously (Mittal et al., 2013). Sequencing was performed using GAII sequencer, Illumina.
Sequence Analysis of Osa-miR820 Family in Rice
Sequences of Osa-miR820 precursors were aligned with TIGR version 7 (Ouyang et al., 2007) and 93-11 genome (Zhao et al., 2004) to search all the loci of Osa-miR820 in japonica and indica sequences. Genomic locations of Osa-miR820 family members were identified by using Bowtie 1.0 (Langmead et al., 2009). Exact hits were plotted with the help of MapChart 2.3 (Voorrips, 2002) after converting the chromosome positions to base pair locations in Mbp (Khan et al., 2014). The deep sequencing data was analyzed using in house developed pipelines (Mittal et al., 2013). Digital signatures of Osa-miR820 expression for each dataset was calculated as transcripts per million (TPM) for comparisons across data sets.
Modified 5′-RLM-RACE for Target Validation
To map the cleavage site of the target mRNA, modified 5′-RLM RACE strategy was employed using Ambion’s 5′-RLM RACE kit. Total RNA was subjected to 5′ adapter ligation using T4 RNA ligase followed by cDNA preparation using oligo(dT) primers. The polymerase chain reactions (PCRs) were performed using 5′ adapter outer forward primer: 5′-GCTGATGGCGATGAATGAACACTG-3′ and target outer reverse primer: 5′-ACCATTTCCTGCTTCCTGTGA-3′. For verification nested PCRs were performed using 5′ adapter inner primer: 5′-CGCGGATCCGAACACTGCGTTTGCTGGCTTT GATG-3’ and target inner primer: 5′-ACCAGAACCATCAGAGTGAGGA-3′. The PCR amplified band was eluted and cloned into pGEMT-easy vector. Ten such clones were sent for sequencing to map the cleavage site.
Stem-Loop and Semi-Quantitative RT PCR
The expression levels of mature miR820 were analyzed using stem-loop end point PCR with slight modifications (Varkonyi-Gasic et al., 2007). Briefly 500 ng total RNA was used to synthesize cDNA using miR-specific stem-loop primer with Superscript reverse transcriptase III (Invitrogen) as per manufacturer’s specifications. A pulsed RT reaction was performed in a thermal cycler as follows: 30 min at 16°C, 60 cycles at 30°C for 30 s, 42°C for 30 s and 50°C for 1 s. RT enzyme was inactivated by incubating the reaction at 85°C for 5 min. 1 μl of direct cDNA was used for PCR using miR specific forward primer (5′-TCGGCCTCGTGGATGG-3′) and universal reverse primer (5′-GTGCAGGGTCCGAGGT-3′) to get a 63 bp amplification products. A 4% agarose gel was used to visualize the RT product. 18S rRNA was used as an endogenous control. Relative abundance was calculated as percentage integrated density values (%IDV) by normalizing the obtained values with 18S rRNA.
For target RT-PCR, 500 ng total RNA was used as starting material to prepare first strand cDNA using Superscript reverse transcriptase II (Invitrogen) with random hexamers as primers. 1 μl direct cDNA was used for PCR using forward primer (5′-GCGAGAAGATTATCGGACGAG-3′) and reverse primer (5′-TCGTCACCAGAACCATCAGA-3′), with 28 cycles of amplification to give 520 bp amplification products. 18S rRNA was used as endogenous control. Spot density measurements were performed using Alpha Imager software. The values were plotted after normalizing with the endogenous control.
Statistical Analysis
All the experiments were repeated atleast three times and biological replicates were employed wherever possible. Mean of three experimental replicate values was plotted and standard deviation is shown as error bars. Paired t-test was performed for all the data using GraphPad Prism version 6.05 for windows (GraphPad, CA, USA). For all samples P < 0.05 was obtained, suggesting that the variations reported here are statistically significant.
Results and Discussion
Mapping miR820 on the Indica Genome
The available information on Osa-miR820 family was used to search for homologs in the indica genome and the results are summarized in Table 1. As miR820 has not been mapped on the indica genome, we attempted to locate all the members on the Beijing indica 93-11 genome based on homology search. Two loci corresponding to pre-miR820b and pre-miR820c were mapped on chromosome 7 and 10, respectively (Figure 1). The locus on chromosome 1 could not be mapped. This may be attributed to the lack of full annotation of the indica genome. The pre-miR820 sequences lie in a repeat rich region of genome flanked by CACTA type DNA retrotransposons on both ends. This is in line with the postulate that young miRs generally originate by segmental duplication events mediated by transposition or recombinational events at the repeat rich regions of genome (Allen et al., 2004; Fahlgren et al., 2010; Cuperus et al., 2011). The Osa-miR820 seems specific to rice, as homologs have not been detected in other plants. The non-conserved nature of this miR also suggests its recent origin.
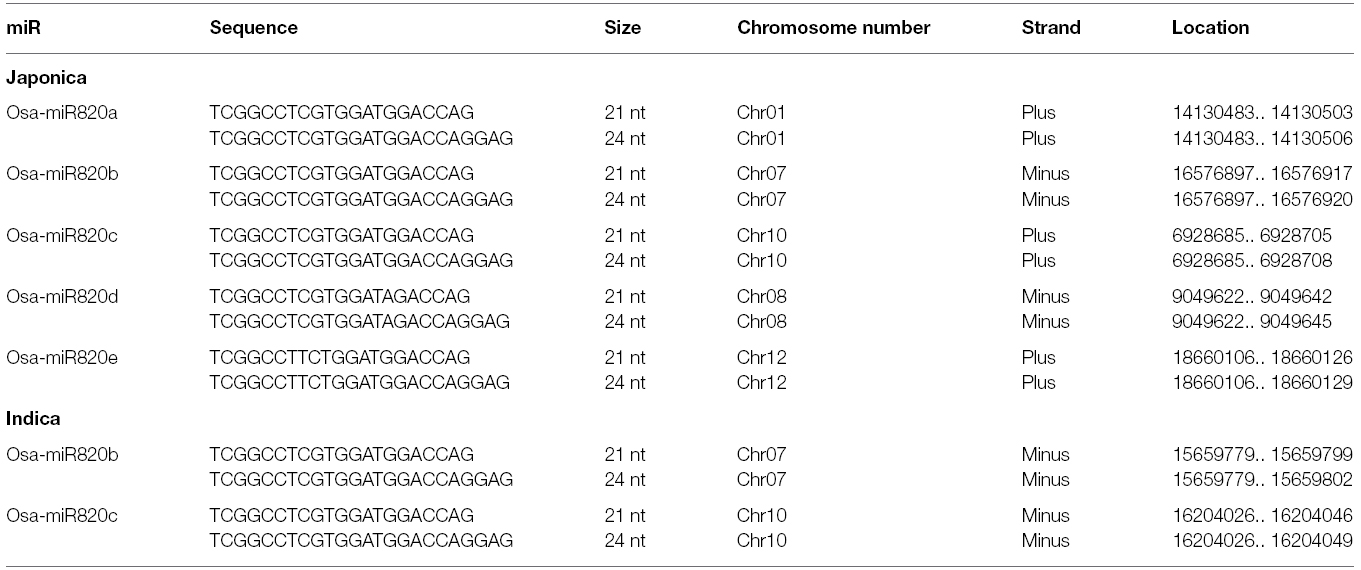
Table 1. List of Osa-miR820 sequences identified in rice.
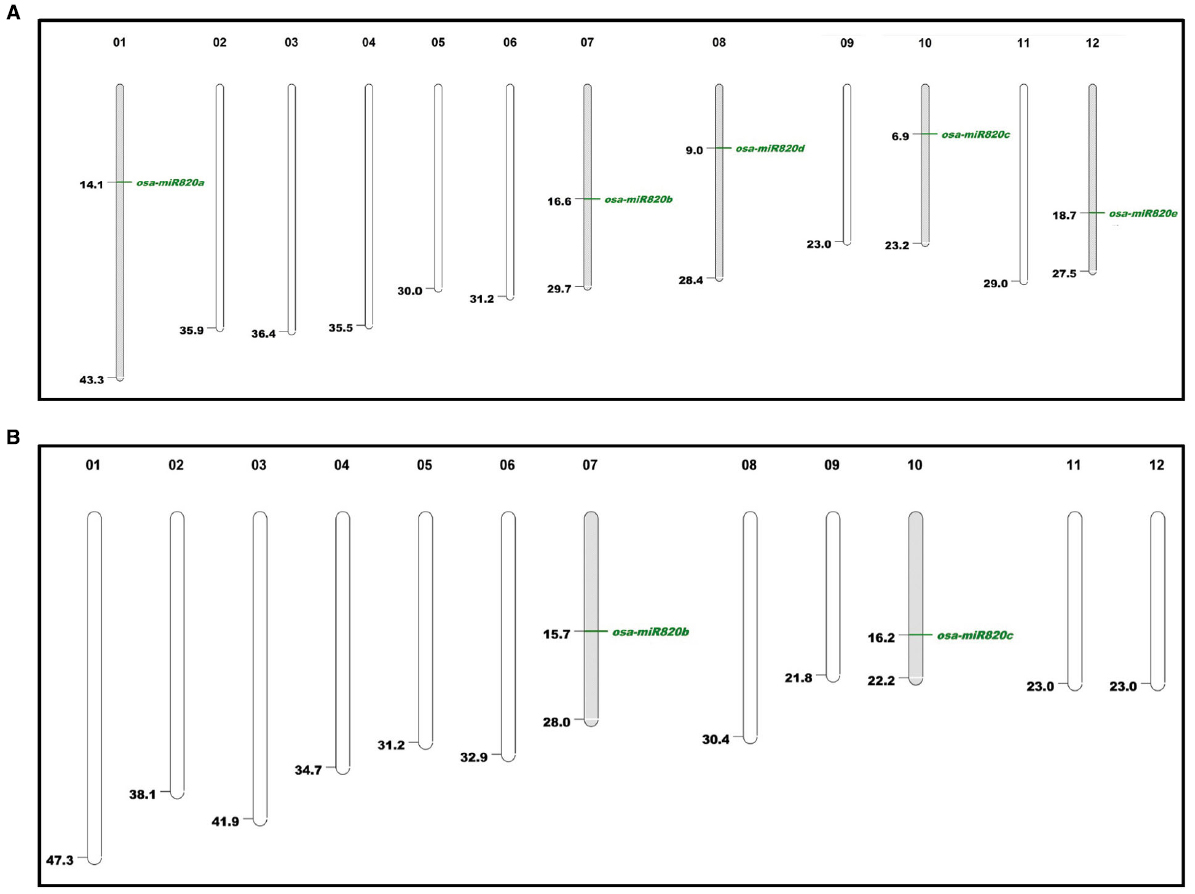
Figure 1. Karyoview of Osa-miR820 family in (A) japonica and (B) indica genomes. Chromosome number is given above each chromosome and mapping positions are shown by green lines.
Both the indica loci contained sequences corresponding to 21-nt and 24-nt length variants of the mature miRs (Nosaka et al., 2013b). It has been established that the canonical 21-nt miRs are generated in a DCL1-dependent pathway and regulate gene expression through post transcriptional gene silencing (PTGS) involving transcript cleavage or translation repression (Vazquez et al., 2008; Zhu et al., 2008; Wu et al., 2010). The 24-nt miRs, commonly referred as long miRs, are produced through the DCL3-mediated pathway and they are supposed to be involved in regulation of gene expression via DNA methylation (Wu et al., 2010). This indicates that the Osa-miR820 family might be involved in a dual mode of target regulation.
Digital Expression Status of Osa-miR820
The digital expression status of Osa-miR820 family, in the leaf and panicle tissues of different indica rice cultivars, was obtained from the Illumina sequencing data available in the lab. The analysis revealed that both the length variants were present in all the libraries and the 24-nt species accumulated to a higher level as compared to the 21-nt species (Figure 2). On comparing the Osa-miR820 levels in the panicle tissues of different indica cultivars, it was observed that the relative concentration of the 21-nt species was higher in the salt-tolerant PK and the heat tolerant Nagina 22 (N22) rice cultivars followed by the salt-sensitive PB1 as compared to the dry season short-duration Satabdi and high yielding drought tolerant Lalat rice cultivars. Whereas the 24-nt species accumulated to very high levels in the PK rice cultivar (Figure 2A). This suggests that the 24-nt length variants may be regulating the genetic machinery to influence the salt-tolerant behavior in PK plants.
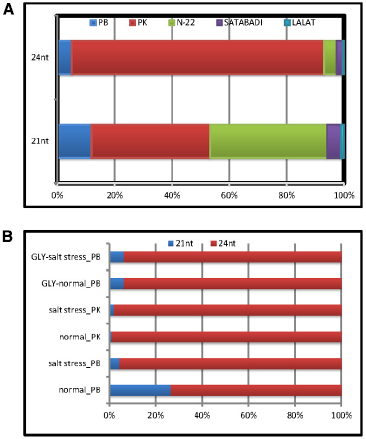
Figure 2. Relative expression patterns of Osa-miR820 length variants. The graph was plotted based on the TPM values obtained from the Illumina sequencing data of different small RNA libraries of rice. The values are presented as percentage to calculate the relative expression. (A) Relative expression in the healthy panicle tissues from different indicia rice cultivars. PB, Pusa Basmati 1; PK, Pokkali; Gly, glyoxalase overexpressing PB1 transgenics; N-22, Nagina 22; Satabdi, and Lalat. (B) Relative expression in the normal (unstressed) and salt stressed leaf tissues from different rice cultivars.
On examining the relative expression patterns of Osa-miR820 length variants in response to salt stress it became apparent that the 21-nt miR was down-regulated in PB1 causing a relative up-regulation of the 24-nt length variant (Figure 2B). In PK leaves the expression of 21-nt miR was up-regulated by more than five-fold but the relative expression levels of the two length variants did not change. A similar observation for Osa-miR820 was made in the salt-tolerant glyoxalase over-expressing transgenics (Singla-Pareek et al., 2008). This suggests that though the expression of Osa-miR820 is deregulated by salt stress and the ratios between the 21-nt and 24-nt length variants may be playing an important role in governing the plants response to salt stress. It also suggests a common scheme of regulation may prevail for its regulation in the salt-tolerant versus the salt-sensitive cultivars. A recent study by Mutum et al. (2013) also indicates such a regulatory layout for the Osa-miR408, which is down-regulated in a drought-susceptible variety and up-regulated in a drought-tolerant variety.
Osa-miR820 Target Prediction and Validation
The miRs are known to regulate gene expression by interacting with their cognate targets in a highly complementary manner. To identify the transcripts that are affected due to changes in expression levels of the Osa-miR820, three different in silico approaches were used. The first approach involved prediction using psRNA target finder, a small RNA target analysis server developed mainly for plants (Dai and Zhao, 2011). Stringent parameters, of no mismatch between 9 and 11 nt, maximum expectation score of 3.0 and Unpaired Energy of 25, predicted seven targets (Table 2). In a second approach the targets were searched in the degradome database StarBase, using the publicly available data (Yang et al., 2011). Degradome sequencing is a high-throughput technique that provides a comprehensive means of analyzing patterns of RNA degradation. Both the techniques identified the DNA methyltransferase as a prominent target (Table 2). This transcript has also been reported as a target in the Zhonghua 11 rice variety (Luo et al., 2006).
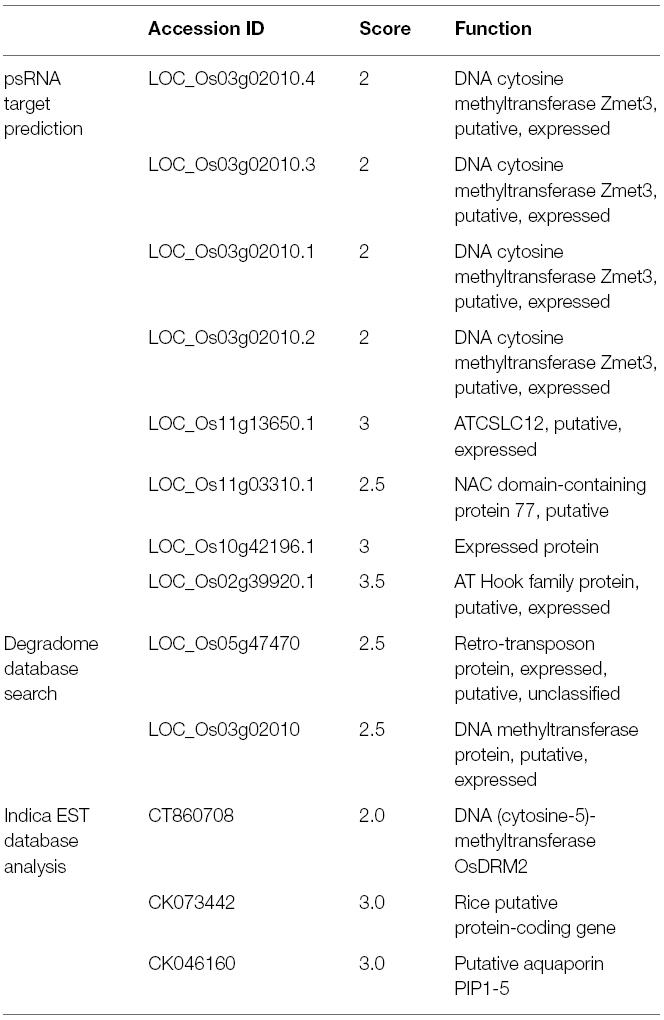
Table 2. List of predicted targets for Osa-miR820 using different approaches.
To validate this as a target transcript in the indica genome the miR binding site was searched in the expressed sequence tags (EST) database, as its fully annotated transcriptome is unavailable (Table 2). Again the putative cytosine DNA methyltransferase was identified as a target transcript. Its closest rice homolog corresponds to Domain Rearranged Methyltransferase 2 (OsDRM2; Os03g0110800). These proteins are known to be involved in catalyzing the de novo methylation of the C-5 residue of cytosine along with enzyme, Chromomethylase 3 (CMT3; Chan et al., 2005; Saze et al., 2012; Stroud et al., 2014). This role is implicated in genome defense against transposons and epigenetic activity during reproductive development and stress (Cao and Jacobsen, 2002; Sahu et al., 2013). Interestingly, the 24-nt Osa-miR820 family members were also shown to target and repress the OsDRM2 at the transcriptional level (Nosaka et al., 2013b). Thus, both 21- and 24-nt Osa-miR820 length variants seems to regulate the same target though at different points indicating that the dynamics of the plant genetic machinery may require dual mode of regulation.
The EST analysis also identified a putative NAC domain containing protein 77 (N77) and a putative aquaporin, PIP1-5, as targets of Osa-miR820. The N77 is a member of one of the largest families of plant-specific transcription factors which have multifarious roles such as embryonic and floral development, lateral root formation, auxin signaling and stress response (Olsen et al., 2005). Whereas PIP1-5, also known as plasma membrane intrinsic protein 1–5, is a conserved protein, known to be involved in water channel activity and can thus affect salt stress responses by regulating transport through water channels (Marmagne et al., 2004; Gawwad et al., 2013).
Considering the ubiquitous presence of DNA methyltransferase in the prediction results, LOC_Os03g02010 was validated as a cleavage target by modified 5′-RACE strategy in both PB1 and PK cultivars (Figure 3). This indicates that the 21-nt miRs are regulating the targets at the PTGS level by target cleavage across different rice genotypes. Therefore to understand the functional significance and putative interaction of Osa-miR820 and OsDRM2 during reproductive development and abiotic stress detailed transcript profiling was performed.
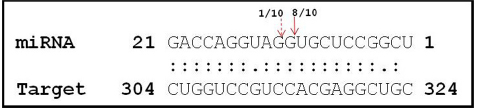
Figure 3. Cleavage site mapping of Osa-miR820 target DNA cytosine-5-methyltransferase (OsDRM2). The 5′ end of the cleaved product determined by 5′ RACE followed by cloning and sequencing. The cleavage site is indicated by an arrow in the miRNA:mRNA base-pairing diagram. The numbers indicate the clones in which the cleavage site was mapped, among the total clones analyzed.
Expression of miR820 and Its Targets in Different Tissues
To delineate the role of Osa-miR820 in the physiology of rice the expression levels of the miR and OsDRM2 transcripts were checked in different tissues of PB1 and PK (Figure 4). It was observed that though Osa-miR820 is present in all the tissues, its expression was abundant in the mature leaf and root tissues (Figure 4A). The OsDRM2 levels were elevated in young leaf and root tissues, however a clear negative correlation could not be observed in the mature tissues (Figure 4A). This ambiguity may be due to overlapping expression domains of individual miR loci as the amplifications provided an overall status of the miR family due to high sequence similarity of the various Osa-miR820 isoforms. Thus it was broadly speculated that the window of transcript regulation might be restricted to the young or developing tissues of rice.
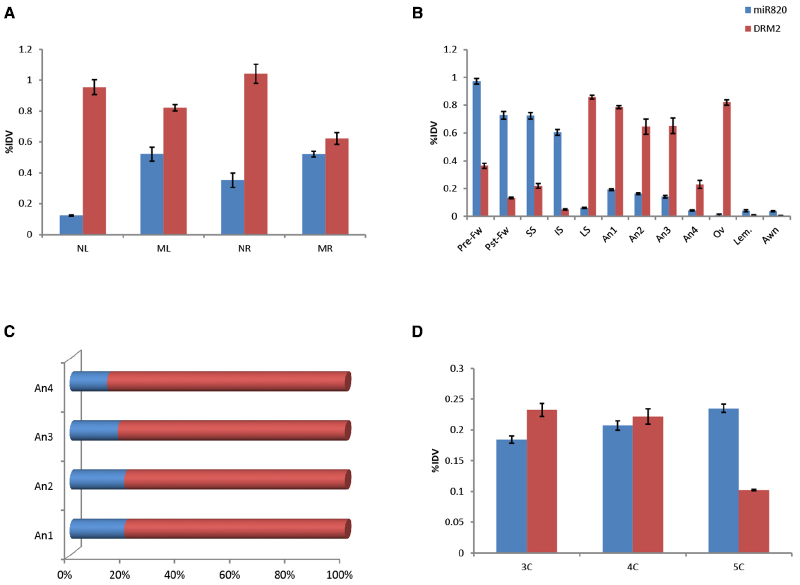
Figure 4. Expression of Osa-miR820 and its target, OsDRM2 in various tissues of Pusa Basmati 1. (A) The vegetative tissues from 15 days old seedlings and mature plant. NL, seedling leaf; ML, mature leaf; NR, seedling root; MR, mature root. (B) The floral tissues from late booting stage. Pre-Fw, pre-pollinated flower; Pst-Fw, post-pollinated flower; SS, superior spikelets; IS, inferior spikelets; LS, leaf sheath covering panicle; An1, anther stage 1; An2, anther stage 2; An3, anther stage 3; An4, anther stage 4; Ov, ovary; Lem, lemma. (C) The ratio of OsmiR820:OsDRM2 at the different stages of the anther. (D) Embryonic callus at different times post initiation. 3C, 3-week old callus; 4C, 4-week old callus; 5C, 5-week old callus. The integrated density values (IDV) were normalized with 18S rRNA and plotted as percentage relative abundance. The error bards indicate the standard deviation.
The expression levels of Osa-miR820 were also checked within the developing floral tissues (Figure 4B). It was observed that at the late booting stage when the panicles were completely encased within the flag leaf sheath (Moldenhauer and Slaton, 2001), the superior spikelets had a higher level of this miR as compared to inferior spikelets. This is in line with the earlier observations (Peng et al., 2011), and negatively correlated with the OsDRM2 transcripts. Nevertheless there was no direct correlation of the target accumulation with the fluctuations in miR expression (Figure 4B). Analysis of individual spikelets showed a two-fold higher accumulation of Osa-miR820 at pre-pollinated stage compared to the post-pollinated stage. The levels of OsDRM2 were reduced in these stages though relatively higher levels of the target transcripts were present in the pre-pollinated spikelets as compared to the post-pollinated spikelets (Figure 4B). This indicated that the miR is preferentially expressed during early reproductive developmental stage in rice and during this time the OsDRM2 transcripts were maintained at low levels.
Within the florets the ovary, lemma and awn had very low levels of Osa-miR820, though the OsDRM2 transcripts accumulated only in the ovary. Relatively higher level of Osa-miR820 was observed in the anthers at stage 1–3 representing the stage before anthesis, after anthesis and at the time of dehiscence, respectively, as compared to stage 4 representing the anthers after dehiscence. The anther stages 1–4 correspond to stage 11–14 as described earlier (Zhang et al., 2011). It was observed that within the anthers the expression of OsDRM2 was high. However, the ratio of Osa-miR820:OsDRM2 was maintained at stages 1 and 2 and decreased slowly in stages 3 and 4 (Figure 4C). This indicates the association of Osa-miR820:OsDRM2 regulation in pollen biology, which may in turn regulate the grain filling and/or grain quality in rice.
Tissue profiling was extended to the embryogenic callus tissues to check the level of miR and its target transcript (Figure 4D). Higher accumulation of both was observed in calli at 3- and 4-weeks post initiation. The reverse-correlation was captured in calli at 5 weeks post initiation, when the Osa-miR820 levels got enhanced while the OsDRM2 levels decreased. Absence of miR:target correlation at earlier stages could be due to overlapping functional domains of miR and target gene (Mittal et al., 2013). Analyzing the spatio-temporal expression of miRs is pivotal in understanding their role in plant growth and development. Recently, focused research on tissue preferential miRs has helped in understanding organ development and phase transitions in a lucid manner (Rubio-Somoza et al., 2009; Wollmann and Weigel, 2010; Mittal et al., 2013; Pandey et al., 2014). The profiling of Osa-miR820 and its target transcript in the different tissues enabled the identification of its regulatory zones. It was observed that the Osa-miR820 expression was more in the early reproductive phases and late embryogenic callus as compared to the other vegetative tissues. However, as the members of Osa-miR820 family share identical mature sequences it was difficult to predict which locus was active in these regions.
Expression of miR820 and Its Targets in Diverse Rice Cultivars
Considering the observations that Osa-miR820 is expressed during early reproductive developmental stages and is deregulated under salt stress, we investigated its expression profiles in various floral tissues under salt stress conditions, using both salt-susceptible (PB1) and salt-tolerant (PK) cultivars (Figure 5). It was observed that as compared to PB1, in PK superior spikelets accumulated lower levels while the inferior spikelets accumulated higher levels of Osa-miR820 (Figures 5A,B). A similar pattern was observed for OsDRM2 expression levels as well indicating that the 21-nt miR mediated cleavage regulation alone may not be operative in these tissues. Under salt stress, the levels of Osa-miR820 did not change extensively though a reverse pattern of deregulation was obtained in PB1 and PK. It was interesting to note that in salt stressed PK, the OsDRM2 levels decreased in the inferior spikelets but increased in the superior spikelets (Figures 5A,B). Superior spikelets flower earlier than inferior spikelets and contribute more toward grain filling as compared to inferior spikelets which either fill poorly or are sterile, which leads to compromised yield in most cases (Dong et al., 2012). This differential expression of Osa-miR820 in different zones of panicle indicates an important role of Osa-miR820 in controlling rice panicle formation.
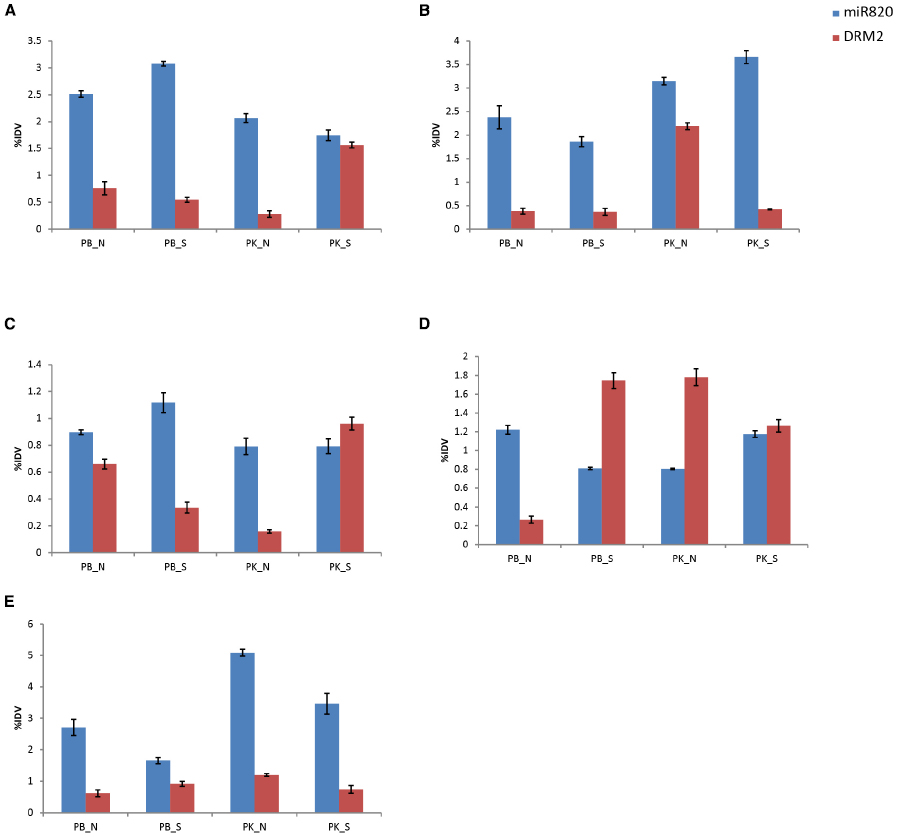
Figure 5. Expression profiling of Osa-miR820 and its target, OsDRM2 in different floral tissues of Pusa Basmati 1 (PB) and Pokkali (PK) under control (N) and salt stressed (S) conditions. (A) superior spikelets, (B) inferior spikelets, (C) pre-pollinated panicle, (D) post-pollinated panicle, (E) flag leaf. The integrated density values (IDV) were normalized with 18S rRNA and plotted as percentage relative abundance. The error bards indicate the standard deviation.
In pre-pollinated floral tissues, salt stress induces the expression of Osa-miR820 in PB1, with corresponding decrease in OsDRM2 expression levels (Figure 5C). But in PK, a mild decrease in Osa-miR820 levels is accompanied by increase in OsDRM2 levels in salt stress. However, at the post-pollinated stage an antagonistic expression pattern is observed. In PB1, Osa-miR820 gets down-regulated leading to increase in OsDRM2 expression levels, while in PK Osa-miR820 gets up-regulated leading to decrease in OsDRM2 expression levels (Figure 5D). This indicates a delicate but controlled regulation of Osa-miR820 under changing developmental stages and time. The levels of Osa-miR820 were similar between salt stressed PB1 and normal PK tissues in pre-and post-pollinated panicles (Figures 5C,D). This indicates that gene regulatory patterns of unstressed PK cultivar are closer to salt challenged PB1 cultivar. Further understanding of this regulation is important as it could have important implications in crop improvement program for stress tolerance.
Flag leaf is the top-most leaf enclosing the emerging panicle. It also helps in assimilating a major portion of incoming solar radiations and thus, contributes to the carbohydrate synthesis for accumulation during grain filling (Li et al., 1998). The expression profiling in flag leaf revealed a greater accumulation of Osa-miR820 in flag leaf of PK as compared to PB1, though the levels declined under salt stress (Figure 5E). The OsDRM2 transcripts were maintained at lower levels though a concomitant fluctuation in its expression was not observed.
Kinetics of miR820 and Its Target Under Salt Stress
The analysis was extended to understand the deregulation of Osa-miR820 and its target under increasing duration of salt stress, in 14-day-old seedlings of PB and PK. A time kinetics of 250 mM salt stress indicated an increase in the Osa-miR820 levels in the leaf (Figure 6A) and a decrease in the root tissues (Figure 6B). It was observed that in the leaves at 30 min of stress there is decrease in Osa-miR820 but it gradually increases by 24 h of stress (Figure 6A). A contrasting pattern was obtained for OsDRM2 indicating regulation by the miR. A similar observation on the salt stress induced deregulation of OsDRM2 transcript was reported from Arabidopsis seedlings (Rae et al., 2014). In both rice and Arabidopsis leaves, the OsDRM2 levels are induced at 30 min of stress and then decrease at 1 h although OsDRM2 is regulated by Osa-miR820 in rice only. This indicates that an overlapping and complicated regulatory network is operative in rice. In roots, low levels of Osa-miR820 were maintained throughout the stress (Figure 6B) but the levels of OsDRM2 begin to increase only at 24 h of stress period. Time course experiments for individual miRs are essential to understand the dynamism between the miR:target pair.
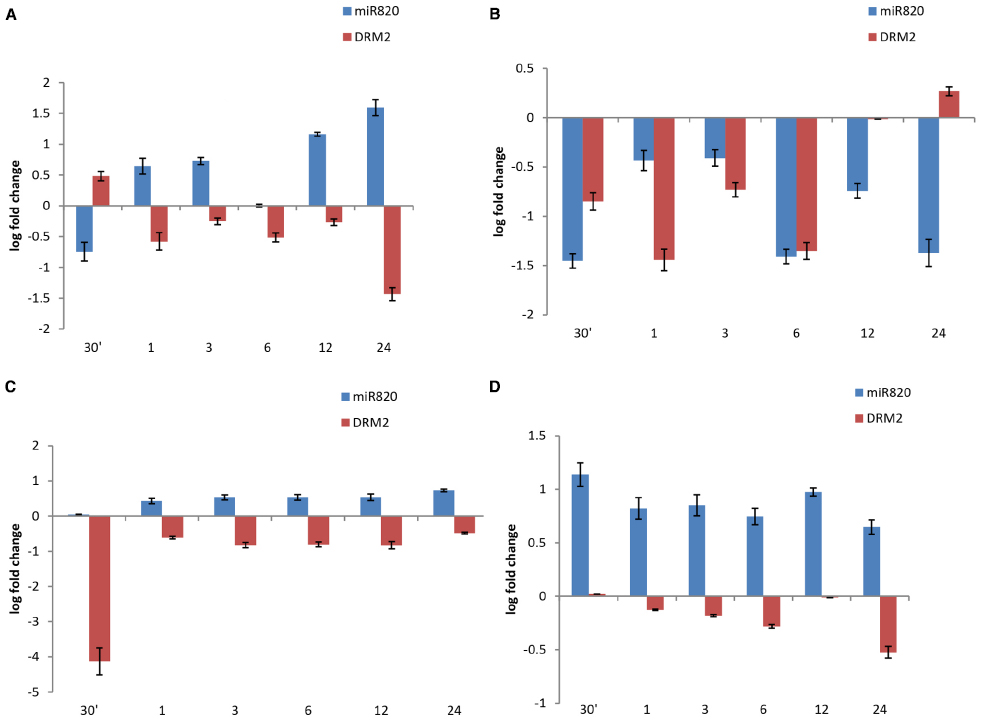
Figure 6. Time kinetics of fold change of Osa-miR820 and its target OsDRM2 under salt stress in leaf and root tissues of Pusa Basmati 1 (PB) and Pokkali (PK). The stress was given to the leaf (L) or root (R) tissues for 30 min (30′); 1 h (1); 3 h (3); 6 h (6); 12 h (12); and 24 h (24). Salt stress was provided by giving 250 mM NaCl to (A) leaf tissues of PB and (B) root tissues of PB, (C) leaf tissues of PK and (D) root tissues of PK. The error bards indicate the standard deviation.
The time kinetics of salt stress revealed an increase in levels of Osa-miR820 in PK leaves similar to the pattern observed in PB1 though there was difference in the level of miR expression (Figure 6C). However, the miR levels were much higher in PK roots as compared to the PB1 roots (Figure 6D). The OsDRM2 transcript levels were reduced in both the tissues. In leaves, a sharp dip in OsDRM2 transcripts was observed after 30 min of stress and relatively low levels were maintained at subsequent time points (Figure 6C) whereas in roots the transcript levels decreased slowly over the stress duration of 24 h (Figure 6D). The results indicate a variation in the behavior of the miR between the salt-tolerant and susceptible varieties. Such variations have been reported earlier as with Osa-miR408 where the time kinetics of drought stress revealed its differential response in drought susceptible and tolerant rice cultivars (Mutum et al., 2013). Similarly, comparative expression analysis identified Osa-miR393 and Osa-miR396c as salt down-regulated miRs and their over-expression conferred salt sensitivity to the transgenic plants (Gao et al., 2010, 2011). These studies will contribute toward understanding the role of miRs in governing the stress responses in plants.
Expression of miR820 and Its Targets Under Other Abiotic Stresses
The analysis was extended to understand the deregulation of Osa-miR820 and its target under high temperature and drought stress. The time kinetics of high temperature stress indicated an increase in the Osa-miR820 levels in the leaves of PB1 (Figure 7A) and PK (Figure 7C). The levels of OsDRM2 transcripts were correspondingly low though the extent of deregulation was more in PK as compared to PB1. In the root tissues of PB1 the expression of Osa-miR820 decreased though the concomitant increase in the levels of OsDRM2 was not observed (Figure 7B). In PK roots a high variation in the levels of Osa-miR820 and OsDRM2 was observed (Figure 7D).
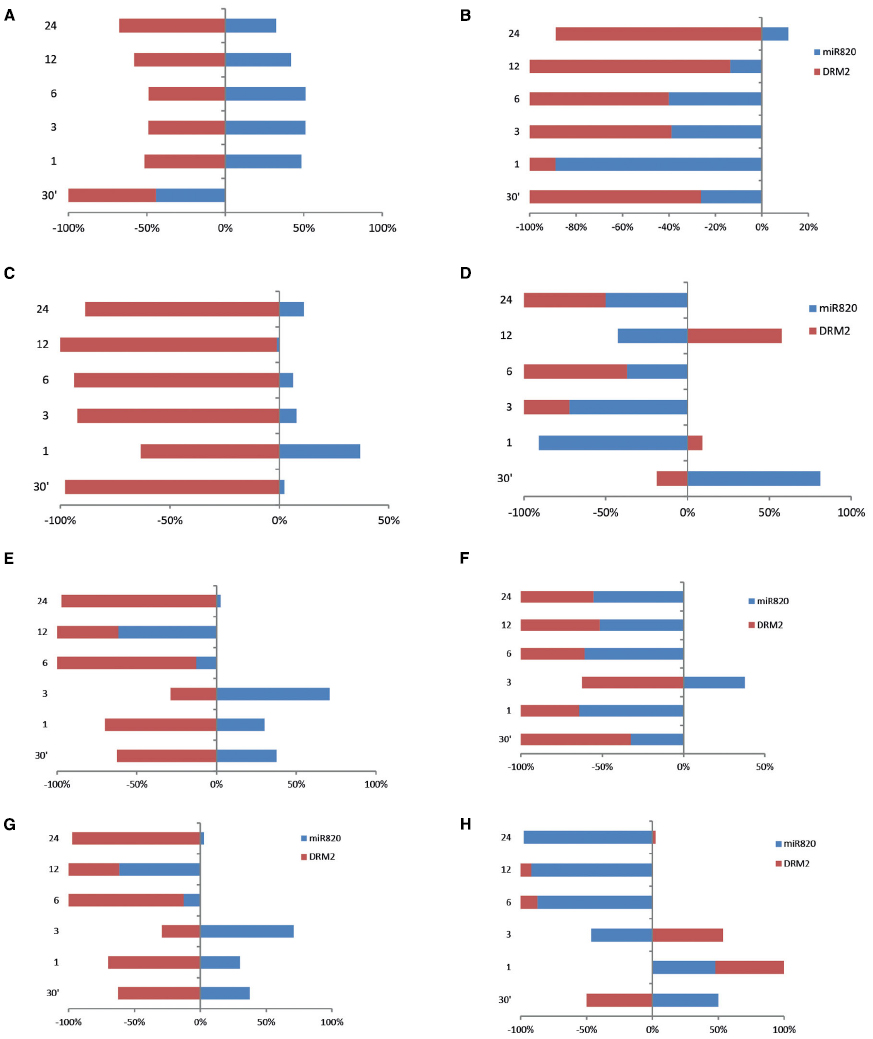
Figure 7. Time kinetics of Osa-miR820 and its target OsDRM2 under high temperature and drought stress. The stress was given to the leaf (L) or root (R) tissues of Pusa Basmati 1 (PB) and Pokkali (PK) for 30 min (30′); 1 h (1); 3 h (3); 6 h (6); 12 h (12); and 24 h (24). High temperature stress was provided by subjecting at 42°C to (A) leaf tissues of PB and (B) root tissues of PB, (C) leaf tissues of PK and (D) root tissues of PK. Physiological drought stress was given by 100 mM mannitol solution to (E) leaf tissues of PB and (F) root tissues of PB, (G) leaf tissues of PK and (H) root tissues of PK. The integrated density values (IDV) were normalized with 18S rRNA and plotted as percentage relative abundance. The error bards indicate the standard deviation.
In the presence of physiological drought conditions, the Osa-miR820 levels were up-regulated in the leaves till 3 h beyond which these reduced drastically. This was accompanied by a corresponding change in the OsDRM2 levels (Figures 7E,G). In PB1 roots, however, both Osa-miR820 and OsDRM2 levels were maintained at low levels under drought stress except at the 3 h time point (Figure 7F). In PK roots, Osa-miR820 levels decreased gradually after 1 h of stress but OsDRM2 transcripts accumulated till 3 h beyond which they decreased drastically (Figure 7H). The general pattern that emerged from the time kinetics studies suggests a stress induced increase in the Osa-miR820 levels in PB1 and PK leaves accompanied by a down-regulation of OsDRM2. In the root tissues both Osa-miR820 and OsDRM2 decreased in PB1 although the Osa-miR820 levels increased during early stages of stress.
Conclusion
The results indicate that Osa-miR820 expression is precisely regulated in the different tissues of rice. In general, it accumulates more in the aerial and floral tissues as compared to the root tissues. Within the floral tissues, the miR levels were maintained at high levels in the anthers. It was observed that under salt, high temperature and drought stress the miR levels were up-regulated in the leaf tissues whereas the miR levels were down-regulated in the root tissues. A variation in the levels of miR was observed between the salt-susceptible PB1 and salt-tolerant PK varieties, but the overall pattern of deregulation appeared to be similar. The regulatory role of this miR on OsDRM2 transcripts was evident and the narrow windows of transcript regulation were captured. However, it is important to understand that the OsDRM2 transcript levels may also be influenced at the transcriptional level by the 24-nt Osa-miR820 species. Knowledge on the delicate balance in the ratios of the 21- and 24-nt miRs and their influence on the transcript expression and accumulation is required to reveal the intricate genetic reprogramming mediated by the Osa-miR820. It will also be interesting to decipher the expression zones of individual Osa-miR820 loci and understand their deregulation in stress.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors are thankful for the funds received from International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi and Department of Biotechnology (DBT), Government of India. The authors would like to thank Prof. S. K. Sopory and Dr. S. L. S. Pareek for providing the Gly transgenics. NS is thankful to Council of Scientific and Industrial Research (CSIR), India, for providing research fellowship.
References
Addo-Quaye, C., Eshoo, T. W., Bartel, D. P., and Axtell, M. J. (2008). Endogenous siRNA and microRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 18, 758–762. doi: 10.1016/j.cub.2008.04.042
Allen, E., Xie, Z., Gustafson, A. M., and Carrington, J. C. (2005). microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121, 207–221. doi: 10.1016/j.cell.2005.04.004
Allen, E., Xie, Z., Gustafson, A. M., Sung, G.-H., Spatafora, J. W., and Carrington, J. C. (2004). Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat. Genet. 36, 1282–1290. doi: 10.1038/ng1478
Arenas-Huertero, C., Perez, B., Rabanal, F., Blanco-Melo, D., De La Rosa, C., Estrada-Navarrete, G., et al. (2009). Conserved and novel miRNAs in the legume Phaseolus vulgaris in response to stress. Plant Mol. Biol. 70, 385–401. doi: 10.1007/s11103-009-9480-3
Aukerman, M. J., and Sakai, H. (2003). Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15, 2730–2741. doi: 10.1105/tpc.016238
Bari, R., Datt Pant, B., Stitt, M., and Scheible, W.-R. (2006). PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol. 141, 988–999. doi: 10.1104/pp.106.079707
Brodersen, P., Sakvarelidze-Achard, L., Bruun-Rasmussen, M., Dunoyer, P., Yamamoto, Y. Y., Sieburth, L., et al. (2008). Widespread translational inhibition by plant miRNAs and siRNAs. Science 320, 1185–1190. doi: 10.1126/science.1159151
Cao, X., and Jacobsen, S. E. (2002). Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 12, 1138–1144. doi: 10.1016/S0960-9822(02)00925-9
Chan, S. W. L., Henderson, I. R., and Jacobsen, S. E. (2005). Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 6, 351–360. doi: 10.1038/nrg1601
Chiou, T.-J., Aung, K., Lin, S.-I., Wu, C.-C., Chiang, S.-F., and Su, C.-L. (2006). Regulation of phosphate homeostasis by microRNA in Arabidopsis. Plant Cell 18, 412–421. doi: 10.1105/tpc.105.038943
Cuperus, J. T., Fahlgren, N., and Carrington, J. C. (2011). Evolution and functional diversification of MIRNA genes. Plant Cell 23, 431–442. doi: 10.1105/tpc.110.082784
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Dong, M. H., Chen, P. F., Xie, Y. L., Qiao, Z. Y., and Yang, J. C. (2012). Variations in carbohydrate and protein accumulation among spikelets at different positions within a panicle during rice grain filling. Rice Sci. 19, 223–232. doi: 10.1016/S1672-6308(12)60044-4
Dong, Z., Shi, L., Wang, Y., Chen, L., Cai, Z., Wang, Y., et al. (2013). Identification and dynamic regulation of microRNAs involved in salt stress responses in functional soybean nodules by high-throughput sequencing. Int. J. Mol. Sci. 14, 2717–2738. doi: 10.3390/ijms14022717
Fahlgren, N., Jogdeo, S., Kasschau, K. D., Sullivan, C. M., Chapman, E. J., Laubinger, S., et al. (2010). MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 22, 1074–1089. doi: 10.1105/tpc.110.073999
Fujii, H., Chiou, T.-J., Lin, S.-I., Aung, K., and Zhu, J.-K. (2005). A miRNA involved in phosphate-starvation response in Arabidopsis. Curr. Biol. 15, 2038–2043. doi: 10.1016/j.cub.2005.10.016
Gao, P., Bai, X., Yang, L., Lv, D., Li, Y., Cai, H., et al. (2010). Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta 231, 991–1001. doi: 10.1007/s00425-010-1104-2
Gao, P., Bai, X., Yang, L., Lv, D., Pan, X., Li, Y., et al. (2011). osa-MIR393: a salinity-and alkaline stress-related microRNA gene. Mol. Biol. Rep. 38, 237–242. doi: 10.1007/s11033-010-0100-8
Gawwad, M. R. A., Šutković, J., Mataković, L., Musrati, M., and Zhang, L. (2013). Functional interactome of Aquaporin 1 sub-family reveals new physiological functions in Arabidopsis thaliana. Netw. Biol. 3, 87–96.
Jagadeeswaran, G., Saini, A., and Sunkar, R. (2009). Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 229, 1009–1014. doi: 10.1007/s00425-009-0889-3
Jones-Rhoades, M. W., and Bartel, D. P. (2004). Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 14, 787–799. doi: 10.1016/j.molcel.2004.05.027
Khan, Y., Yadav, A., Suresh, V. B., Muthamilarasan, M., Yadav, C. B., and Prasad, M. (2014). Comprehensive genome-wide identification and expression profiling of foxtail millet [Setaria italica (L.)] miRNAs in response to abiotic stress and development of miRNA database. Plant Cell Tissue and Organ Cult. 118, 279–292. doi: 10.1007/s11240-014-0480-x
Khraiwesh, B., Zhu, J.-K., and Zhu, J. (2012). Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 1819, 137–148. doi: 10.1016/j.bbagrm.2011.05.001
Kozomara, A., and Griffiths-Jones, S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. doi: 10.1093/nar/gkt1181
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25
Li, Z., Pinson, S. M., Stansel, J., and Paterson, A. (1998). Genetic dissection of the source-sink relationship affecting fecundity and yield in rice (shape Oryza sativa L.). Mol. Breed. 4, 419–426. doi: 10.1023/a:1009608128785
Liu, H. H., Tian, X., Li, Y. J., Wu, C. A., and Zheng, C. C. (2008). Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14, 836–843. doi: 10.1261/rna.895308
Llave, C., Xie, Z., Kasschau, K. D., and Carrington, J. C. (2002). Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297, 2053–2056. doi: 10.1126/science.1076311
Lu, S., Sun, Y. H., and Chiang, V. L. (2008). Stress-responsive microRNAs in Populus. Plant J. 55, 131–151. doi: 10.1111/j.1365-313X.2008.03497.x
Luo, Y.-C., Zhou, H., Li, Y., Chen, J.-Y., Yang, J.-H., Chen, Y.-Q., et al. (2006). Rice embryogenic calli express a unique set of microRNAs, suggesting regulatory roles of microRNAs in plant post-embryogenic development. FEBS Lett. 580, 5111–5116. doi: 10.1016/j.febslet.2006.08.046
Lutts, S., Kinet, J. M., and Bouharmont, J. (1995). Changes in plant response to NaCl during development of rice (Oryza sativa L.) varieties differing in salinity resistance. J. Exp. Bot. 46, 1843–1852. doi: 10.1093/jxb/46.12.1843
Marmagne, A., Rouet, M.-A., Ferro, M., Rolland, N., Alcon, C., Joyard, J., et al. (2004). Identification of new intrinsic proteins in Arabidopsis plasma membrane proteome. Mol. Cell. Proteomics 3, 675–691. doi: 10.1074/mcp.M400001-MCP200
Mittal, D., Mukherjee, S. K., Vasudevan, M., and Mishra, N. S. (2013). Identification of tissue-preferential expression patterns of rice miRNAs. J. Cell. Biochem. 114, 2071–2081. doi: 10.1002/jcb.24552
Moldenhauer, K., and Slaton, N. (2001). “Rice growth and development,” in Rice Production Handbook, eds N. A. Slaton, L. B. Ford, J. L. Bernhardt, R. D. Cartwright, D. Gardisser, J. Gibbons, et al. (Lawrence: Cooperative Extension Service, Division of Agriculture, University of Arkansas), 7–14.
Moradi, F., Ismail, A., Egdane, J., and Gregorio, G. (2003). Salinity tolerance of rice during reproductive development and association with tolerance at the seedling stage. Indian J. Plant Physiol. 8, 276–287.
Mutum, R. D., Balyan, S. C., Kansal, S., Agarwal, P., Kumar, S., Kumar, M., et al. (2013). Evolution of variety-specific regulatory schema for expression of osa-miR408 in indica rice varieties under drought stress. FEBS J. 280, 1717–1730. doi: 10.1111/febs.12186
Nosaka, M., Ishiwata, A., Shimizu-Sato, S., Ono, A., Ishimoto, K., Noda, Y., et al. (2013a). The copy number of rice CACTA DNA transposons carrying MIR820 does not correlate with MIR820 expression. Plant Signal. Behav. 8, e25169. doi: 10.4161/psb.25169
Nosaka, M., Ono, A., Ishiwata, A., Shimizu-Sato, S., Ishimoto, K., Noda, Y., et al. (2013b). Expression of the rice microRNA miR820 is associated with epigenetic modifications at its own locus. Genes Genet. Syst. 88, 105–112. doi: 10.1266/ggs.88.105
Olsen, A. N., Ernst, H. A., Leggio, L. L., and Skriver, K. (2005). NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 10, 79–87. doi: 10.1016/j.tplants.2004.12.010
Ouyang, S., Zhu, W., Hamilton, J., Lin, H., Campbell, M., Childs, K., et al. (2007). The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res. 35, D883–D887. doi: 10.1093/nar/gkl976
Pandey, R., Joshi, G., Bhardwaj, A. R., Agarwal, M., and Katiyar-Agarwal, S. (2014). A comprehensive genome-wide study on tissue-specific and abiotic stress-specific miRNAs in Triticum aestivum. PLoS ONE 9:e95800. doi: 10.1371/journal.pone.0095800
Peng, T., Lv, Q., Zhang, J., Li, J., Du, Y., and Zhao, Q. (2011). Differential expression of the microRNAs in superior and inferior spikelets in rice (Oryza sativa). J. Exp. Bot. 62, 4943–4954. doi: 10.1093/jxb/err205
Rae, G. M., Uversky, V. N., David, K., and Wood, M. (2014). DRM1 and DRM2 expression regulation: potential role of splice variants in response to stress and environmental factors in Arabidopsis. Mol. Genet. Genomics 289, 317–332. doi: 10.1007/s00438-013-0804-2
Rana, P., Nainawatee, H., Sindhu, A., Jain, R., and Chowdhury, J. (1999). RAPD-based assessment of diversity in indica rice genotypes. Indian J. Exp. Biol. 37, 1209–1212.
Rogers, K., and Chen, X. (2013). Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 25, 2383–2399. doi: 10.1105/tpc.113.113159
Rubio-Somoza, I., Cuperus, J. T., Weigel, D., and Carrington, J. C. (2009). Regulation and functional specialization of small RNA-target nodes during plant development. Curr. Opin. Plant Biol. 12, 622–627. doi: 10.1016/j.pbi.2009.07.003
Sahu, P., Pandey, G., Sharma, N., Puranik, S., Muthamilarasan, M., and Prasad, M. (2013). Epigenetic mechanisms of plant stress responses and adaptation. Plant Cell Rep. 32, 1151–1159. doi: 10.1007/s00299-013-1462-x
Sanan-Mishra, N., Kumar, V., Sopory, S. K., and Mukherjee, S. K. (2009). Cloning and validation of novel miRNA from basmati rice indicates cross talk between abiotic and biotic stresses. Mol. Genet. Genomics 282, 463–474. doi: 10.1007/s00438-009-0478-y
Saze, H., Tsugane, K., Kanno, T., and Nishimura, T. (2012). DNA methylation in plants: relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol. 53, 766–784. doi: 10.1093/pcp/pcs008
Sharma, D., Tiwari, M., Lakhwani, D., Tripathi, R. D., and Trivedi, P. K. (2015). Differential expression of microRNAs by arsenate and arsenite stress in natural accessions of rice. Metallomics 7, 174–187. doi: 10.1039/c4mt00264d
Singla-Pareek, S., Yadav, S., Pareek, A., Reddy, M. K., and Sopory, S. K. (2008). Enhancing salt tolerance in a crop plant by overexpression of glyoxalase II. Transgenic Res. 17, 171–180. doi: 10.1007/s11248-007-9082-2
Stroud, H., Do, T., Du, J., Zhong, X., Feng, S., Johnson, L., et al. (2014). Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 21, 64–72. doi: 10.1038/nsmb.2735
Sunkar, R., Chinnusamy, V., Zhu, J., and Zhu, J. K. (2007). Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 12, 301–309. doi: 10.1016/j.tplants.2007.05.001
Varkonyi-Gasic, E., Wu, R., Wood, M., Walton, E. F., and Hellens, R. P. (2007). Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3, 12–12. doi: 10.1186/1746-4811-3-12
Vazquez, F., Blevins, T., Ailhas, J., Boller, T., and Meins, F. (2008). Evolution of Arabidopsis MIR genes generates novel microRNA classes. Nucleic Acids Res. 36, 6429–6438. doi: 10.1093/nar/gkn670
Voinnet, O. (2009). Origin, biogenesis, and activity of plant microRNAs. Cell 136, 669–687. doi: 10.1016/j.cell.2009.01.046
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Wollmann, H., and Weigel, D. (2010). Small RNAs in flower development. Eur. J. Cell Biol. 89, 250–257. doi: 10.1016/j.ejcb.2009.11.004
Wu, L., Zhou, H., Zhang, Q., Zhang, J., Ni, F., Liu, C., et al. (2010). DNA methylation mediated by a microRNA pathway. Mol. Cell 38, 465–475. doi: 10.1016/j.molcel.2010.03.008
Xie, J., Zapata-Arias, F., Shen, M., and Afza, R. (2000). Salinity tolerant performance and genetic diversity of four rice varieties. Euphytica 116, 105–110. doi: 10.1023/A:1004041900101
Yang, J.-H., Li, J.-H., Shao, P., Zhou, H., Chen, Y.-Q., and Qu, L.-H. (2011). starBase: a database for exploring microRNA–mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 39, D202–D209. doi: 10.1093/nar/gkq1056
Zeng, L., and Shannon, M. C. (2000). Salinity effects on seedling growth and yield components of rice. Crop Sci. 40, 996–1003. doi: 10.2135/cropsci2000.404996x
Zhang, D., Luo, X., and Zhu, L. (2011). Cytological analysis and genetic control of rice anther development. J. Genet. Genomics 38, 379–390. doi: 10.1016/j.jgg.2011.08.001
Zhao, W., Wang, J., He, X., Huang, X., Jiao, Y., Dai, M., et al. (2004). BGI-RIS: an integrated information resource and comparative analysis workbench for rice genomics. Nucleic Acids Res. 32, D377–D382. doi: 10.1093/nar/gkh085
Keywords: rice, salt stress, microRNA, expression patterns, panicles
Citation: Sharma N, Tripathi A and Sanan-Mishra N (2015) Profiling the expression domains of a rice-specific microRNA under stress. Front. Plant Sci. 6:333. doi: 10.3389/fpls.2015.00333
Received: 23 February 2015; Accepted: 27 April 2015;
Published: 13 May 2015.
Edited by:
Laurent Deluc, Oregon State University, USAReviewed by:
Chenglin Chai, Louisiana State University, USARobert Henry, The University of Queensland, Australia
Copyright © 2015 Sharma, Tripathi and Sanan-Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neeti Sanan-Mishra, Plant Molecular Biology Group, International Centre for Genetic Engineering and Biotechnology, Aruna Asaf Ali Marg, New Delhi 110067, India,bmVldGlAaWNnZWIucmVzLmlu