 Slávka BekešováGeorge Komis
Slávka BekešováGeorge Komis Pavel KřenekPetra Vyplelová
Pavel KřenekPetra Vyplelová Miroslav Ovečka
Miroslav Ovečka Ivan Luptovčiak
Ivan Luptovčiak Peter IllésAnna Kuchařová
Peter IllésAnna Kuchařová Jozef Šamaj*
Jozef Šamaj*- Department of Cell Biology, Centre of the Region Haná for Biotechnological and Agricultural Research, Palacký University Olomouc, Olomouc, Czech Republic
The aim of the present study is to rationalize acrylamide pendant Phos-Tag™ in-gel discrimination of phosphorylated and non-phosphorylated plant protein species with standard immunoblot analysis, and optimize sample preparation, efficient electrophoretic separation and transfer. We tested variants of the method including extraction buffers suitable for preservation of phosphorylated protein species in crude extracts from plants and we addressed the importance of the cation (Mn2+ or Zn2+) used in the gel recipe for efficient transfer to PVDF membranes for further immunoblot analysis. We demonstrate the monitoring of Medicago sativa stress-induced mitogen activated protein kinase (SIMK) in stress-treated wild type plants and transgenic SIMKK RNAi line. We further show the hyperosmotically-induced phosphorylation of the previously uncharacterized HvMPK4 of barley. The method is validated using inducible phosphorylation of barley and wheat α-tubulin and of Arabidopsis MPK6. Acrylamide pendant Phos-Tag™offers a flexible tool for studying protein phosphorylation in crops and Arabidopsis circumventing radioactive labeling and the use of phosphorylation specific antibodies.
Introduction
Protein post-translational modifications (PTMs) and particularly reversible protein phosphorylation are versatile switches regulating protein structure, interactions, function, and subcellular localization (Müller et al., 2010; Nishi et al., 2013, 2014; Offringa and Huang, 2013; Smékalová et al., 2014) under conditional signaling (Ovečka et al., 2014) or during developmental processes (Beck et al., 2010; Smékalová et al., 2014). The inducible phosphorylation of few or even a single protein species might have global consequences on cell physiology, if these proteins—e.g., transcription factors—are involved in central processes such as transactivation of gene expression (Ishihama and Yoshioka, 2012; Meng et al., 2013). Protein phosphorylation is catalyzed in a targeted manner by a large family of enzymes called protein kinases (Dissmeyer and Schnittger, 2011). Protein kinases are attracted by protein substrates via specific and complementary motifs present on both interaction partners as thoroughly studied, for example, in the case of mitogen activated protein kinases (MAPKs; Salazar and Höfer, 2009; Šamajová et al., 2013; Pitzschke, 2015).
The efficiency, the complexity, the spatiotemporal coordination and finally the physiological importance of reversible protein phosphorylation nourished the development of quantitative experimental procedures to address site-specificity, kinetics, and functional consequences of protein phosphorylation (Liu and Chance, 2014).
At large scale, phosphopeptides may be identified by mass spectrometric analyses following appropriate phosphopeptide enrichment strategies (St-Denis and Gingras, 2012) and by subtractive motif- or drug-based approaches, while global phosphoproteomic analyses may provide information on kinase-specific phosphoproteome landscapes (Ruse et al., 2008; Boesger et al., 2012).
Small scale approaches incorporating radioactive metabolic labeling followed by in gel (Cuenda, 2000) or in solution (Dickson, 2008) assays, may provide highly sensitive quantitative and kinetic monitoring of either kinase activity, or the phosphorylation process of a kinase substrate. However, such approaches mandate severe safety precautions, specific training of personnel, and cost-demanding laboratory setup, while inevitably producing undesirable radioactive waste. In addition, these are in vitro assays, presenting the potential of protein phosphorylation but not the actual phosphorylation state within the cell.
Protein phosphorylation in a small scale may be also addressed by phosphorylation-specific antibodies raised against either the protein of interest, or more generally against phosphorylated aminoacids. In the latter case, the lack of specificity of antibodies can be a problem, since phospho-tyrosine monoclonal antibodies have adequate affinity for phosphorylated Tyr residues, but monoclonal antibodies against phospho-serine or phospho-threonine are unpopular because their affinities and specificities are not optimal. An excellent non-radioactive alternative is the separation of protein species by means of one dimensional isoelectric focusing (1D IEF) which can be then coupled with western blot analysis of the proteins of interest in in vivo assays, representing the status quo of the cell (Anderson and Peck, 2008, 2014).
Herein we use an approach somewhat less demanding than 1D IEF in terms of both equipment and reagents since it can be carried out using standard SDS PAGE minigel setup. This method relies on the differential electrophoretic migration of phosphoprotein species compared to their non-phosphorylated counterparts by means of complexing protein phosphogroups with transition metal cations immobilized in-gel using a covalently incorporated chelating agent trademarked as Phos-Tag™(Kinoshita et al., 2006; Kinoshita and Kinoshita-Kikuta, 2011; Kinoshita-Kikuta et al., 2014).
We provide a thorough protocol for acrylamide pendant Phos-Tag™ separation of phosphorylated and non-phosphorylated proteins in four plant species (three crops; Medicago sativa, alfalfa; Hordeum vulgare, barley; and Triticum turgidum, wheat and one genetic model; Arabidopsis thaliana). This methodology aims to identify single or multiple phosphorylation events of proteins of interest, provided that specific antibodies for the protein under study are available. The method requires prior knowledge that the protein of interest is or might be conditionally phosphorylated but it cannot identify the phosphorylated aminoacid per se. This problem can be circumvented if prior to electrophoretic separation the protein is selectively immunoprecipitated and subsequently probed with anti-phospho-Ser/Thr or Tyr antibodies. Importantly, Phos-Tag™ protocols have been developed for monitoring aspartate phosphorylation (Barbieri and Stock, 2008), thiophosphorylation (Kinoshita et al., 2014), and histidine phosphorylation (Ishii et al., 2013). In the present protocol examples of protein phosphorylation elucidated by acrylamide pendant Phos-Tag™ and subsequent western blot analysis, include the Medicago sativa MAPK called SIMK (stress-induced MAPK homologous to AtMPK6 probed with the same antibody as AtMPK6), Arabidopsis thaliana MAPKs AtMPK3 and AtMPK6, Hordeum vulgare MAPK HvMPK4 and the structural protein alpha tubulin in Hordeum vulgare and Triticum turgidum as it was recently identified as a target of hyperosmotically induced protein phosphorylation in rice and Arabidopsis thaliana (Ban et al., 2013; Fujita et al., 2013). The protocol is adapted to crude protein extracts from diverse plant material. Our laboratory routinely employs Phos-Tag™ technology to decipher phosphorylation and activation of MAPKs (Beck et al., 2010) or MAPK substrates (Panteris et al., 2010; Smékalová et al., 2014) in the model plant Arabidopsis thaliana. Importantly we employed a transgenic line of alfalfa with silenced SIMKK (SIMKK RNAi) which is the upstream regulator of SIMK and also studied the hyperosmotic activation of the barley ortholog of MPK4 by means of a custom raised monospecific antibody.
Materials and Methods
Chemicals
All common chemicals were of defined purity (pro analysis) and purchased from Sigma (www.sigma-aldrich.com) and Duchefa (www.duchefa-biochemie.com). Phos-Tag™ was from Wako Pure Chemical Industries Ltd (www.wako-chem.co.jp). Protease and phosphatase inhibitor cocktails (Complete™, EDTA-free, and PhosStop™) were from Roche (lifescience.roche.com). λ-Phosphatase was from New England Biolabs (www.neb.com). The following antibodies were used: anti-AtMPK6 affinity purified rabbit polyclonal IgGs (Sigma), anti-pTEpY rabbit polyclonal IgGs (Cell Signaling; www.cellsignal.com), anti-HvMPK4 rabbit polyclonal IgGs (designated as JK4 raised against the peptide 288-PRQDFRLRFRNM-299 derived from the predicted sequence in Genbank under the accession number AK366765; custom produced by Genscript, www.genscript.com), mouse monoclonal anti-alpha tubulin (clone DM1a; Abcam; www.abcam.com) and horseradish peroxidase conjugated goat anti-rabbit IgGs and rabbit anti-mouse IgGs (both from Santa Cruz Biotech; www.scbt.com). Polyclonal serum against MPK6 was raised against the carboxylterminal residues 384–395 of Arabidopsis thaliana MPK6 and was peptide affinity purified. The anti-pTEpY polyclonal serum was generated using a peptide surrounding the respective motif of mammalian ERK1/2 (extracellular signal related protein kinase 1 and 2). The anti-pTEpY polyclonal serum was affinity purified against the phospho-peptide used for immunization and against the same peptide in its unphosphorylated form. Monoclonal anti-α tubulin antibody, clone DM1a, was generated from hybridomas of mice injected with chick brain tubulin and corresponds to the C-terminus of α tubulin (residues 426–430). The peptide used for JK4 production was synthesized, coupled to keyhole limpet hemocyanin and injected to New Zealand rabbits according to the protocol of the producer. Bradford reagent, acrylamide/bis-acrylamide mixture (30% w/v solution, 37.5:1 ratio acrylamide to bis-acrylamide), pre-stained molecular ladder and enhanced chemiluminescence (ECL; Clarity™) reagent were all from Bio-Rad (www.bio-rad.com).
Plant Material and Treatments
In the present study we used seedlings of Medicago sativa cv. Regen SY (RSY), Hordeum vulgare cv. Golden Promise, Triticum turgidum cv. Athos and Arabidopsis thaliana ecotype Columbia (Col-0). Before plating, seeds of Arabidopsis thaliana and Medicago sativa and caryopses of Hordeum vulgare and Triticum turgidum, were appropriately surface sterilized first with 70% v/v aqueous ethanol for 2 min and subsequently with 10% v/v aqueous bleach solution supplemented with 0.01% v/v Tween-20 for 10 min. Following washing with sterile, distilled water, all plant material was plated to ½Murashige Skoog nutrient medium solidified with 0.8% w/v Phytagel™ and allowed to grow in controlled environmental chambers with standard light:dark cycle (16:8 h)/ temperature (21°C day/18°C night)/ humidity (60% day/70% night) settings. For treatments, 10 day old Arabidopsis thaliana seedlings were treated with 15 mM H2O2 in liquid ½MS for 30 min. 3–4 day old seedlings of Hordeum vulgare and Triticum turgidum were treated with 0.8 M sorbitol in liquid ½MS for 30 min. Wild type seedlings of Medicago sativa were treated with 250 mM NaCl, 15 mM H2O2, and 0.8 M sorbitol in liquid ½MS for 30 min and RNAi expressing Medicago sativa plants growing in soil pots were treated by wounding by lightly scoring leaves with a sharp razor blade and collected in liquid nitrogen 5 min post-wounding.
Cloning of SIMKK RNAi and Transformation of M. sativa
To obtain SIMKK RNAi line of M. sativa, leaves of mature plants were transformed with A. tumefaciens carrying pHellsgate12-SIMKKi expression plasmid. Construction of pHellsgate12-SIMKKi expression plasmid was performed by Gateway® recombination cloning, using pDONR™207 (Life Technologies) donor vector and pHellsgate12 destination vector (obtained from CSIRO Plant Industry, Australia). In the first step, 366 bp PCR fragment was synthesized using SIMKKiFor primer (GGGGACAAGTTTGTACAAAAAAGCAGGCTTCTTAAGGATATATGGAGTTTAGGCGTGAG), SIMKKiRev primer (GGGGACCACTTTGTACAAGAAAGCTGGGTATCTTGGTGGTGGAGGAAGTAAC) and total cDNA of M. sativa as template. Primers were designed according to standards for Gateway® cloning. In addition, restriction site for AflIII (CTTAAG) was inserted between attB1 site and template-complementary site of forward primer, enabling further verification of correct orientation of fragments in expression vector. Consequently, BP recombination reaction (recombination between attB and attP sites) was performed to obtain pDONR207-SIMKKi entry vector by cloning of synthesized PCR fragment into pDONR™207 vector, using Gateway® BP Clonase® II Enzyme Mix (Life Technologies) according to standard protocol of manufacturer. Construction of final expression plasmid pHellsgate12-SIMKKi was performed by recombination of prepared pDONR207-SIMKKi entry vector and pHellsgate12 destination vector (LR reaction; recombination between attL and attR sites), using Gateway® LR Clonase® II Enzyme Mix (Life Technologies) following protocol of manufacturer.
Agrobacterium tumefaciens strain GV3101::pMP90 was transformed with SIMKK-RNAi construct and used for stable transformation of Medicago sativa. Young leaves from in vitro growing plants of the cultivar RSY were selected as explant source. The transformation was performed by cocultivation method (Samac and Austin-Phillips, 2006). Induction of callogenesis from leaf explants, production of somatic embryos from calli, development of shoots and their rooting were performed under selective conditions. Regenerated plants (Austin et al., 1995) that grew on media with selective kanamycin marker were further tested by genotyping. To confirm the presence of the construct in transgenic Medicago plants, two sets of primers for verification of presence of the hairpin were tested. Genomic DNA from control non transformed regenerated plants and SIMKK-RNAi-transformed plants was isolated using CTAB method (Semagn, 2014). Primers were tested and after optimization of PCR conditions, all of them were functional.
Assessment of Transcripts in M. sativa SIMKK RNAi Line by RT-qPCR
Transcript levels of SIMKK in the RNAi and the control Medicago sativa lines were quantified by RT qPCR of total RNA isolated from respective seedlings with Trizol® reagent (Life Technologies, www.lifetechnologies.com) using the following primer sets for SIMKK: CACCAGAAGCTCCAACGACTG (forward) and CGACGGGTCTCTCTGCAAAC (reverse), and the product was normalized against ACTIN2 transcript levels (GGATAAGAGGTGAGATCGGAGGG; forward and GCAACCAACCTACAGACATCCAG; reverse).
Protein Extraction and λ-Phosphatase Treatment
In all cases plant material was harvested immediately before protein isolation and was flash frozen in liquid nitrogen. Material was then crashed into fine powder, collected in prechilled and preweighted microfuge tubes and stored under liquid nitrogen until further processing. Protein extracts were prepared with the following extraction solutions:
RIPA (radio-immunoprecipitation assay) buffer: 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% v/v Triton X-100, 0.5% w/v sodium deoxycholate, 0.1% w/v SDS.
Buffer E: 50 mM HEPES (pH 7.5), 75 mM NaCl, 1 mM EGTA, 1 mM MgCl2, 1 mM NaF, 10% v/v glycerol.
Buffer B: 20 mM Na-phosphate solution (pH 7.4) (mixture of 2 ml of NaH2PO4 with 8 ml Na2HPO4 from 200 mM stock solutions), 50 mM ß-glycerophosphate, 100 μM Na3VO4.
All mentioned buffers were appropriately supplemented with Complete™ EDTA-free and PhosSTOP™.
For the extraction, liquid nitrogen powders of plant material were extracted on ice for 30 min with 1–2 volumes of appropriate extraction buffer. RIPA was the buffer of choice for Arabidopsis thaliana seedlings, buffer E and buffer B worked equally well for Medicago sativa and buffer B was exclusively used for extracts of Hordeum vulgare and Triticum turgidum as mentioned by Ban et al. (2013). Subsequently extracts were cleared at 23000× g, 10 min, 2°C and the resulting supernate was used as crude protein extract. Protein content in the original extract was quantified by Bradford assay and it was further cleaned by repeated rounds of quantitative cold acetone precipitation (80% v/v; Komis et al., 2014). By acetone precipitation proteins are selectively precipitated leaving salts and other soluble impurities in solution. This is quite critical since even minimal amounts of phosphates, especially in the case of buffer B, will interfere with the assay.
In some instances phosphorylation discriminated in western blots following acrylamide pendant Phos-Tag™ SDS-PAGE was verified by treatment of protein extracts with λ PPase. In this case, protein extracts corresponding to a total of 100 μg were successively precipitated with 80% v/v acetone (at least 3 times) at −20°C for at least 1 h each time in order to remove phosphatase inhibitor contaminants from the protein mixture. The final protein pellet was briefly dried and finally resuspended to λ PPase reaction buffer (50 mM HEPES pH 7.5, 10 mM NaCl, 2 mM DTT, 0.01% (v/v) Brij® 35 (supplied prepared in λ PPase kit of New England Biolabs). The reaction mixture was then supplemented with 200–400 units of λ PPase and the reaction was allowed to proceed for 30 min at 30°C according to manufacturer's instructions. At the end of the reaction, the product was precipitated by adding 4 volumes of acetone and kept overnight at −20°C. The resulting pellet was precipitated at 23000× g, 10 min, and 2°C and allowed to slightly dry before dissolving in 1× Laemmli sample buffer and heat denature accordingly.
Electrophoresis and Western Blot
Two different electrophoresis procedures were followed. One involving the in-gel combination of Phos-Tag™ with Mn2+ and the other involving the combination of Phos-Tag™ with Zn2+. For the Mn2+/Phos-tag™ combination the standard Tris-Cl buffered stacking (4% w/v acrylamide) and separating (8% w/v acrylamide) gel recipes were applied. The Zn2+/Phos-tag™ stacking and separating gels were prepared as follows:
Stacking Zn2+/Phos-tag™ gel buffer: 357 mM Bis-Tris (pH 6.8), 0.1% v/v TEMED, 0.05% w/v ammonium persulfate (APS). Acrylamide, bis-acrylamide and water should be adjusted according to the desired gel pore size. In the present protocol a 4% w/v acrylamide stacking gel was used.
Separating Zn2+/Phos-tag™ gel buffer: 357 mM Bis-Tris (pH 6.8), 25 μM Phos-Tag™, 50 μM Zn(NO3)2, 0.05% v/v TEMED, 0.025% w/v APS. Acrylamide, bis-acrylamide and water should be adjusted according to the desired gel pore size. In the present protocol an 8% w/v acrylamide separating gel was used. The concentration of Phost-Tag™, will impact the separation and the transfer efficiency of the gel. For higher MW proteins that are already difficult to transfer, lower Phos-Tag™ concentrations (e.g., 5 μM) have been used quite efficiently (Kinoshita et al., 2009).
The running buffer for standard and Mn2+/ Phos-Tag™ gels was 25 mM Tris base, 192 mM glycine and 0.1% w/v SDS. For Zn2+/ Phos-Tag™ the running buffer consisted of 100 mM Tris, 100 mM MOPS, and 0.1% (w/v) SDS to which 5 mM of sodium bisulfite were added immediately before electrophoresis.
In all cases, the power supply settings were adjusted for optimal separation of phosphorylated and non-phosphorylated protein species. For this reason, all gels run at 10–15 mA/gel constant current and electrophoresis required 4–6 h for completion.
For transfer of gel separated proteins to PVDF membranes for subsequent immunoblotting of the protein of interest, gels were pretreated with washing in methanol-free transfer buffer (25 mM Tris-Cl pH 7.4, 192 mM glycine, 10 mM EDTA) for 6 times 10 min each in order to remove bivalent cations that would immobilize phosphorylated proteins in the gel not allowing their transfer to PVDF membrane. Finally the gel was equilibrated for 2 × 10 min in methanol-free transfer buffer with 1 mM EDTA. Gels were transferred to PVDF membranes with transfer buffer containing 5% v/v methanol and 1 mM EDTA at 24V overnight. For efficient transfer of Phos-Tag™ gel separated proteins we used wet-tank transfer approach since semi-dry transfer protocols are less efficient and complicate the procedure (Kosako, 2009).
Following electrophoretic transfer, membranes were allowed to completely dry. Subsequently they were reactivated in methanol and immediately stained with 3% w/v Ponceau S in 5% v/v aqueous solution of acetic acid to validate transfer efficiency. Subsequently membranes were thoroughly destained with milliQ water and TBST (10 mM Tris–HCl (pH 7.5), 100 mM NaCl, and 0.10% v/v Tween-20). Non-specific antibody binding was blocked by incubating membranes in 6% w/v BSA in TBST for at least 1 h. Primary antibodies were appropriately diluted in 6% w/v BSA in TBST and incubation of the membranes was carried out overnight at 4°C. Following extensive washing in TBST (6 × 10 min), membranes were incubated with diluted HRP-conjugated secondary antibodies for 1 h. After washing in TBST (6 × 10 min) membranes were incubated with ECL reagent according to manufacturer's instructions. Immunoreactive bands were documented with a BioRad ChemiDoc™ MP System. Western blots of Phos-Tag™ gels cannot be used to make MW assignments. Prestained MW markers migrate anomalously in such gels, whereas all proteins migrate slower compared to standard SDS-PAGE gels. Therefore, protein identity should be based on known immunoreactivity.
Results
As already mentioned Phos-Tag™ identification of phosphorylated proteins following SDS-PAGE separation and western blotting has been well-established in Arabidopsis thaliana (Bethke et al., 2009; Beck et al., 2010; Mao et al., 2011; Fujita et al., 2013; Li et al., 2014; Smékalová et al., 2014; Kang et al., 2015) while few applications included dicotyledonous and monocotyledonous crops (Panteris et al., 2010; Taylor et al., 2012; Ban et al., 2013). Herein we attempt to provide proper conditions for preservation of phosphorylated proteins upon extraction, for their adequate separation in acrylamide pendant Phos-Tag™ SDS-PAGEs and their quantitative transfer to solid membranes for subsequent western blot-based probing of the proteins of interest.
Salt, Oxidative, and Wound-induced Activation of M. sativa SIMK
A prominent MAPK of Medicago sativa is SIMK which can be equally activated by exposure to hyperosmotic salt or sorbitol solutions or by wounding and oxidative stress (Kiegerl et al., 2000; Cardinale et al., 2002). In all cases the strongest activator of SIMK is the MAPKK called SIMKK which can directly activate SIMK (Cardinale et al., 2002). Moreover, SIMK is a highly conserved homolog of AtMPK6 with absolute identity in the carboxylterminal region. Therefore, a commercial antibody generated against the 10 carboxylterminal residues of AtMPK6 can equally detect SIMK (46 kDa; Šamaj et al., 2002) in appropriate western blots (Ovečka et al., 2014; Figure 1A1 bottom panel; arrow). For this reason we monitored the activation of Medicago sativa SIMK following salt, sorbitol and wounding stress in roots of either wild type seedlings or in leaves of wound-inflicted plants stably transformed with a siRNA SIMKK construct using this SIMK antibody. Therefore, this SIMKK silenced M. sativa line was used for further Phos-tag and western blot analyses.
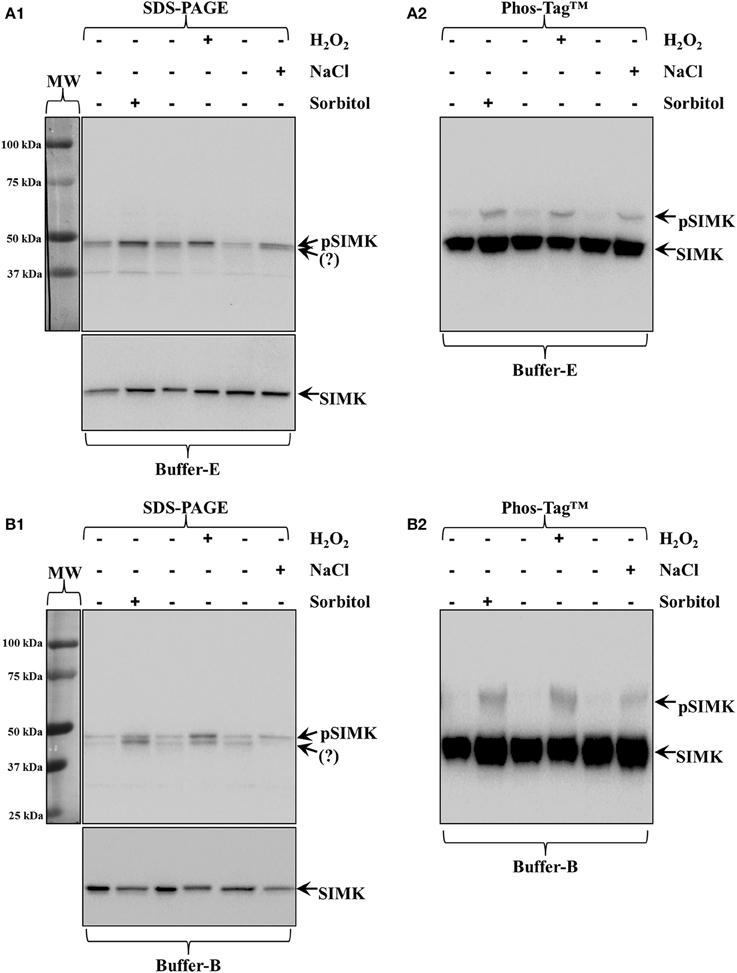
Figure 1. Activation of Medicago sativa SIMK by three different stresses (250 mM NaCl, 15 mM H2O2 and 0.8 M sorbitol, all applied for a total of 30 min). (A1,A2) All stresses induce the accumulation of phosphorylated MAPK species compared to untreated seedlings as recognized by pTEpY antibody in extracts prepared in buffer E (A1) which by Phos-Tag™ (A2) show clear SIMK activation (top arrow, pSIMK; bottom arrow, SIMK). (B1,B2) The same samples extracted with buffer B show better discrimination of pTEpY immunoreactive species in western blots following common SDS-PAGE (B1) and much better separation of pSIMK (top arrow) and SIMK (bottom arrow) in Phos-Tag™ gels (B2). Panels on the left of (A1,B1) correspond to images of the protein ladder following Ponceau S staining of the membrane. Question marks in (A1,B1) denote unknown anti-pTEpY immunoreactive species.
The activation of SIMK upon brief salt, H2O2 and sorbitol treatment of Medicago sativa roots (see Materials and Methods) was confirmed by western blot analysis of protein extracts acquired in buffer E and buffer B using a commercial anti-pTEpY antibody (Figure 1A1, top panel; arrow and Figure 1B1, top panel, top arrow respectively).
In the case of buffer E extracted proteins, the anti-pTEpY antibody detected one specific band at the expected molecular weight for SIMK (Figure 1A1 top panel, top arrow) and a second band corresponding to another unidentified salt-induced MAPK (Figure 1A1 top panel, bottom arrow). The first band detected with the anti-pTEpY antibody was coinciding with a band detected with anti-SIMK antibody (Figure 1A1 bottom panel, arrow) suggesting that it corresponds to phosphorylated SIMK. The same pattern was revealed when buffer B extracted proteins were similarly separated and immunoblotted, although they showed better separation of the pSIMK band when compared to E buffer extracted proteins (Figure 1B1, top arrow) from the band corresponding to the unidentified salt-induced MAPK (Figure 1B1, bottom arrow) as further corroborated with anti-SIMK antibody (Figure 1B1, bottom panel, arrow).
Extracts in buffer E and buffer B were separated in Zn2+/Phos-Tag™ gel and immunoprobed with anti-SIMK antibody resulting in two well discriminated bands in both cases separating unphosphorylated and phosphorylated SIMK (Figures 1A2,B2; bottom arrows, SIMK; top arrows, pSIMK).
Phosphorylation of SIMK was further pursued in wounded Medicago sativa leaves from control plants and from plants expressing SIMKK RNAi construct. The SIMKK RNAi line was selected on the basis of hairpin detections by PCR (Figures S1A,B, arrows) and subsequent SIMKK transcript quantitation by RT qPCR (Figure S1C) showing reduction of SIMKK transcript by 57% compared to the wild type RSY (Figure S1C). Leaf extracts of control M. sativa line RSY as well as extracts from M. sativa SIMKK RNAi line with knock downed SIMKK were probed with anti-pTEpY antibody before and after wounding. In this case, the anti-pTEpY antibody recognized two bands with the top one corresponding to phosphorylated SIMK (Figure 2A, top panel, top arrow, pSIMK) and the bottom one to an unknown wound-inducible MAPK species (Figure 2A, top panel, bottom arrow, question mark). The band corresponding to pSIMK was significantly reduced in the SIMKK RNAi line (Figure 2A, top panel, top arrow, pSIMK). When the same extracts were immunoblotted with anti-SIMK antibody to verify the position of SIMK, it was found that the total levels of SIMK were also reduced in the SIMKK RNAi line (Figure 2A, bottom panel, arrow, SIMK). In western blots following Zn2+/Phos-Tag™ gel of the same extracts as in Figure 2A, the control line showed the separation of two SIMK immunoreactive bands, one corresponding to the phosphorylated SIMK (Figure 2B, top arrow, pSIMK) while the bulk immunoreactivity resided in the lower band corresponding to non-phosphorylated SIMK (Figure 2B, bottom arrow). By contrast, extracts of SIMKK RNAi line tested hereby failed to show the upper band corresponding to phosphorylated SIMK (Figure 2B).
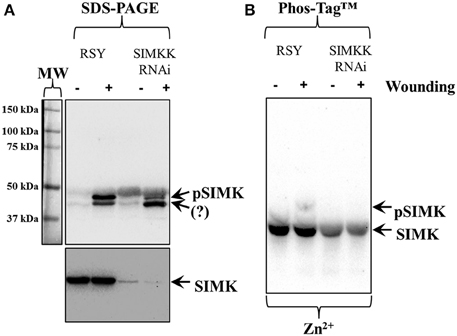
Figure 2. Effects of silencing of SIMKK on inducible activation and total levels of SIMK in SIMKK RNAi expressing Medicago sativa plants following wound induction. (A) extracts control (RSY) and RNAi (SIMKK RNAi) obtained from resting or wounded leaves, where probed against anti-pTEpY antibody recognizing phosphorylated SIMK (A, top panel, top arrow, pSIMK) or with anti-MPK6 antibody recognizing total levels of SIMK (A, bottom panel, arrow, SIMK). The RNAi line shows reduced accumulation of anti-pTEpY immunoreactivity corresponding to SIMK (A, top panel, top arrow, pSIMK). Surprisingly the total levels of SIMK where found reduced in both RNAi lines compared to the controls (A, bottom panel, arrow, SIMK). (B) Identical extracts as those used for Panel (A) where separated in Zn2+/Phos-Tag™ gel and probed with anti-MPK6 antibody, showing the absence of the upper band corresponding to pSIMK (top arrow) in the RNAi line. The bottom arrow denotes the unphosphorylated form of SIMK. Panel on the left of (A) corresponds to image of the protein ladder following Ponceau S staining of the membrane.
Phosphorylation of Monocot Proteins
Next we used Phos-Tag™ electrophoretic separation to document phosphorylation of alpha-tubulin in hyperosmotically-treated Hordeum vulgare and Triticum turgidum seedlings as previously published for Oryza sativa and Arabidopsis thaliana (Ban et al., 2013; Fujita et al., 2013). In the case of Hordeum vulgare, the efficient separation and detection of hyperosmotically-induced protein phosphorylation was further validated in the case of barley MAPK (HvMPK4) detected with a custom raised antibody.
In western blots of barley extracts after sorbitol treatment, an anti-pTEpY antibody recognized one prominent band which was not apparent in extracts of resting plants (Figure 3A1). Alpha-tubulin immunoreactivity was slightly enhanced in extracts from hyperosmotically treated plants (Figure 3A2 right lane cf left lane). Phosphorylation of alpha-tubulin following hyperosmotic treatment was monitored in western blots following both Mn2+/ and Zn2+/Phos-Tag™ gel separation (Figures 3B1,B2 respectively). While in both cases, tubulin immunoreactivity appeared as two bands (right lanes of Figures 3B1,B2) compared to extracts of untreated samples (left lanes of Figures 3B1,B2), the separation of these two bands was clearly better in western blots following Zn2+/Phos-Tag™ gels highlighting the efficiency of Zn2+/Phos-Tag™ gels to separate phosphorylated (pTUA) from non-phosphorylated alpha-tubulin (TUA) compared to Mn2+/Phos-Tag™ gels. Further we studied, as a proof-of-principle, the hyperosmotically induced phosphorylation of a previously uncharacterized barley ortholog of Arabidopsis thaliana MPK4 (HvMPK4) using a custom raised antibody. The specificity of the antibody was corroborated in western blots following standard SDS-PAGE showing a band at ca. 43 kDa which remained roughly conserved between extracts of untreated and hyperosmotically treated plants (Figure 3C1 left and right panel respectively). By Zn2+/Phos-Tag™ analyses, it was found that the HvMPK4 immunoreactivity appeared in two bands in extracts of untreated plants, the lower of which corresponded to unphosphorylated HvMPK4 (Figure 3C2 left lane, bottom arrow, HvMPK4) while the upper one probably corresponded to basally phosphorylated HvMPK4 (Figure 3C2 left lane, top arrow, pHvMPK4). Interestingly, in extracts of hyperosmotically treated plants, the HvMPK4 immunoreactivity breaks down to three bands. Again the lowest corresponding to the non-phosphorylated HvMPK4 (Figure 3C2 right lane, bottom arrow, HvMPK4), while the two upper ones that reside close together probably reflected two different phosphorylation states of HvMPK4 induced after hyperosmotic treatment (Figure 3C2 right lane, top arrows, pHvMPK4).
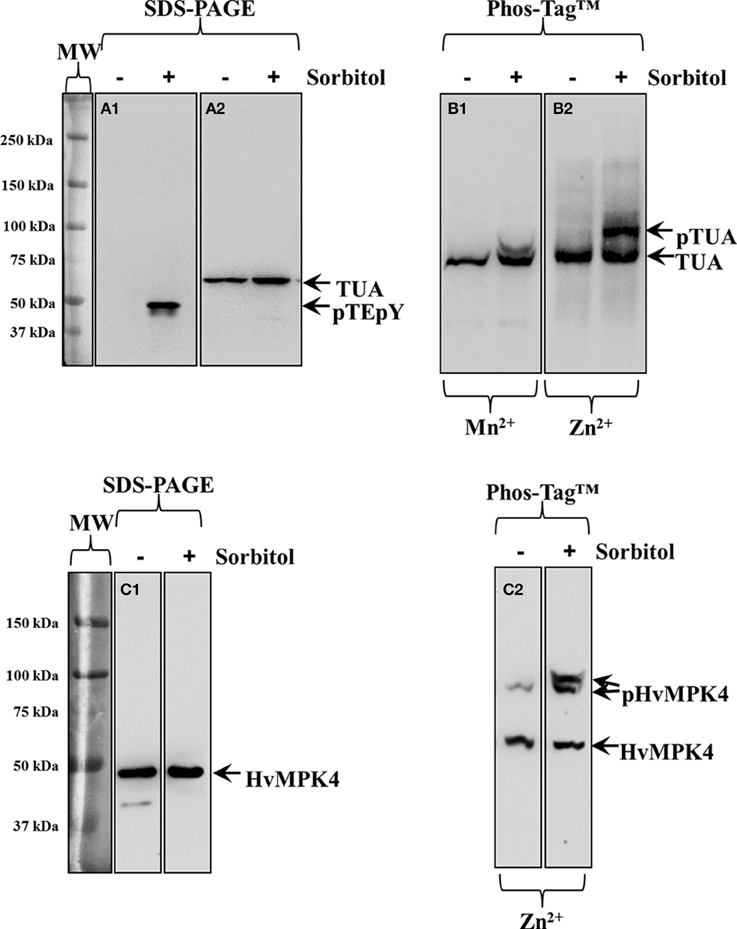
Figure 3. Hyperosmotically induced (0.8 M sorbitol for 30 min) phosphorylation of alpha-tubulin and HvMPK4 as probed in barley root extracts. (A1) Severe, non-ionic hyperosmotic stress induces activation of unknown MAPKs recognized by pTEpY antibody. (A2) Under the same conditions total levels of alpha-tubulin are slightly upregulated as evidenced in immunoblots following standard SDS-PAGE. (B1) Phos-Tag™ gel separates alpha-tubulin to a phosphorylated (top arrow, pTUA) and a non-phosphorylated band (bottom arrow TUA) in the presence of Mn2+. (B2) A wider separation of phosphorylated (top arrow, pTUA) from non-phosphorylated alpha tubulin (bottom arrow, TUA) is seen in the presence of Zn2+ in the Phos-Tag™ gel indicating better efficiency of Zn2+ containing Phos-Tag™ gels to separate phosphorylated tubulin. The panels on the left of (A1) corresponds to image of the protein ladder following Ponceau S staining of the membrane. (C1) Identical extracts as those used in Panel (A) where probed with anti-HvMPK4 antibody. The antibody specifically recognizes HvMPK4 (left arrow) and further shows conservation of total HvMPK4 levels in control and hyperosmotically-treated samples. (C2) Hyperosmotically-induced phosphorylation of HvMPK4 as monitored by Zn2+/Phos-Tag™ gel electrophoresis and western blot. The lower band corresponds to unphosphorylated HvMPK4 (bottom arrow) while the upper bands correspond to different phosphorylation statuses of HvMPK4 after hyperosmotic induction (right lane, top arrows, pHvMPK4). In untreated samples (left lane) only one faint upper band appears probably corresponding to basally phosphorylated HvMPK4. Panels on the left of (A1) and (C1) correspond to images of the protein ladder following Ponceau S staining of the membranes.
Tubulin phosphorylation was also followed in extracts of hyperosmotically-treated wheat seedlings. By standard SDS-PAGE and subsequent western blot, alpha-tubulin immunoreactivity was detected as a single band, showing slight upregulation of alpha-tubulin intensity after the hyperosmotic treatment (Figure 4A, right lane cf left lane corresponding to control). As previously described for barley, Zn2+/Phos-Tag™ gels discriminated tubulin immunoreactivity in two bands, the upper one of which likely corresponded to phosphorylated alpha-tubulin (Figure 4B, top arrow, pTUA) while the lower one corresponded to non-phosphorylated alpha tubulin (Figure 4B, bottom arrow, TUA).
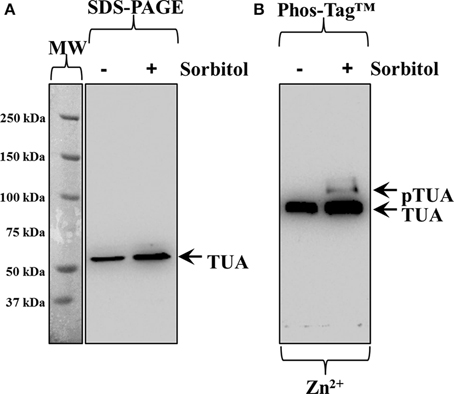
Figure 4. Hyperosmotically induced (0.8 M sorbitol for 30 min) phosphorylation of alpha-tubulin as probed in wheat root extracts. (A) Hyperosmotic stress induces small increase of alpha-tubulin (left arrow, TUA) levels as evidenced in standard SDS-PAGE followed by immunoblot analysis. (B) Zn2+/Phos-Tag™ analysis of the same extract, shows phosphorylation of a fraction of alpha-tubulin (top arrow, pTUA) compared to extracts of control, untreated roots. The bottom arrow denotes unphosphorylated alpha-tubulin. The panel left to (A) corresponds to image of the protein ladder following Ponceau S staining of the membrane.
Validation of the Protocol with Extracts of A. thaliana
When executing the protocol it is best to characterize proteins which are known to undergo conditional phosphorylation. In the case of Arabidopsis thaliana this is best exemplified in the case of the MPK6, representing mitogen activated protein kinase known to be activated by oxidative stress (Wang et al., 2013).
Oxidative stress, performed by treatment of Arabidopsis thaliana seedlings with 15 mM H2O2 for 30 min, is not inducing changes in total MPK6 (Figure 5A, bottom panel, arrow, MPK6) levels but does promote the accumulation of phosphorylated MAPK species probed with phosphospecific antibody (Figure 5A, top panel, top arrow, pMPK6). Depending on the separation efficiency of the gel, the phosphospecific antibody may detect two (Figure 5A, top panel, arrows) bands, the most prominent of which corresponds to MPK6 (Figure 5A, top panel, top arrow, pMPK6) while the lower probably corresponds to phosphorylated MPK3 (Figure 5A, top panel, bottom arrow, pMPK3; see also Willmann et al., 2014). Phos-Tag™ analyses show that the H2O2-induced accumulation of phosphorylated MPK6 can be discriminated by both the Mn2+ and the Zn2+ variants with variable efficiency and depending on the time of electrophoretic separation. The separation of dually-phosphorylated MPK6 (Figures 5B1,C1,C2, top arrows, pMPK6) from its non-phosphorylated counterpart (Figures 5B1,C1,C2, bottom arrows, MPK6) is depending on the transition metal used in Phos-Tag™ gel assembly (Figure 5B1 of Figure 5B2 showing better separation in the presence of Zn2+ compared to Mn2+) but also depends on gel running time allowing better separation after longer times of electrophoresis (Figures 5B1,B2 come from blots of Phos-Tag™ gels separated for 4 h while Figures 5C1,C2 come from gels separated for 6 h).

Figure 5. Overview of the protocol applied on Arabidopsis thaliana root protein extracts following treatment with 15 mM H2O2 for 30 min. (A) Immunoblot detection of MPK6 (bottom panel, arrow, MPK6), and phosphorylated MPK6 (top panel, top arrow, pMPK6) in conventional SDS-PAGE gels by means of anti-pTEpY antibody. In top panel of (A) the fainter band likely corresponds to phosphorylated MPK3 (bottom arrow, pMPK3) which is detectable depending on the separation efficiency of the gel. (B1,B2) Identical immunoblot from Phos-Tag™ separated samples in the presence of Zn2+ (B1) or Mn2+ (B2) immunodetected with antibody against MPK6 (B1,B2). Proteins were allowed to separate in the Phos-Tag™ gel for 4 h. (C1,C2) Identical immunoblots from Phos-Tag™ separated samples in the presence of Zn2+ (C1) or Mn2+ (C2). Proteins were allowed to separate for 6 h in the Phos-Tag™ gel. Panels on the left of (A) correspond to images of the protein ladder following Ponceau S staining of the membrane.
In order to verify that Phos-Tag™ gels truly discriminate between phosphorylated and non-phosphorylated protein species a control experiment should be carried out. In the case of H2O2-induced phosphorylation of MPK6, we eliminated MPK6 phosphorylation (Figure 6 middle lane, top arrow, pMPK6) by λ PPase treatment of the protein extract, leading to the disappearance of phosphorylated MPK6 in the respective Phos-Tag™ gel (Figure 6) allowing the detection of non-phosphorylated MPK6 alone (last lane, bottom arrow, MPK6).
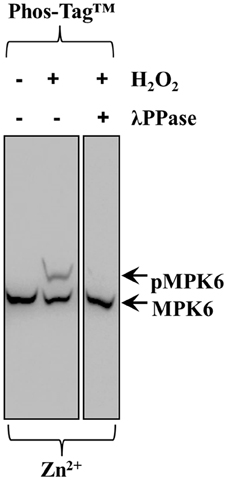
Figure 6. Effects of λ PPase treatment on the ability of Zn2+/Phos-Tag™ gel to discriminate phosphorylated MPK6 (pMPK6) in root extracts of Arabidopsis thaliana seedlings treated with 15 mM H2O2 for 30 min. Extracts untreated with λ PPase show an upper band corresponding to phosphorylated MPK6 (top arrow, pMPK6) and after treatment with λ PPase the upper band is eliminated. The lower band (arrow, MPK6) corresponds to the unphosphorylated MPK6.
Discussion
Development of the Protocol
There are two principal strategies for implementing acrylamide pendant Phos-Tag™ separation of phosphorylated proteins from their non-phosphorylated counterparts and these differ on the transition metal cation used. The original method utilized Mn2+ on a standard Tris-based SDS PAGE (Kinoshita et al., 2006) and in our earlier studies we used it as such (Beck et al., 2010; Panteris et al., 2010). Later, improved electrophoretic transfer of Phos-Tag™ separated proteins of variable Mr was reported using Zn2+ instead, incorporated to a NuPAGE setup with Bis-Tris based gels and MOPS-Tris-based electrophoresis buffer (Kinoshita and Kinoshita-Kikuta, 2011; Kinoshita et al., 2012; Kinoshita-Kikuta et al., 2014), which we appropriately adopted and modified for the separation of phosphorylated proteins from whole extracts of Arabidopsis thaliana seedlings or roots (Komis et al., 2014; Smékalová et al., 2014).
Importance of Divalent Cation in Acrylamide Pendant Phos-Tag™ Separation of Phosphorylated Proteins
We found that both Phos-Tag™ variants are useful for the separation of phosphorylated proteins and their non-phosphorylated counterparts, provided careful management of electrophoresis running time and thorough post-treatment of the Phos-Tag™ gel to ensure quantitative protein transfer to PVDF membrane. Thus, as we showed in the case of H2O2-induced phosphorylation of Arabidopsis thaliana MPK6, the separation of the phospho- from the non-phosphorylated form is more efficient in the Zn2+/Phos-Tag™ regime and after a gel running time of 6 h instead of 4 h (Figures 5B1,C1). Ultimately, the running time will depend on the size of the protein of interest, but for smaller proteins than those detected herein, higher acrylamide percentages may be used to prevent the elution of such proteins from the gel during prolonged electrophoresis.
Importance of the Extraction Buffer in the Preservation of Phosphorylated Groups
Preservation of phosphorylated groups in crude protein extracts is of paramount importance for the successful detection of phosphorylated proteins by Phos-Tag™, since such proteins frequently occur as a minute fraction of the total protein pool. Therefore, it was crucial to establish extraction conditions to preserve phosphorylation status of the proteins under study. From three common extraction buffers used for protein isolation, we found that buffer E and especially buffer B, were superior to RIPA buffer. We believe that the presence of phosphates in buffer B, further prevents spontaneous protein dephosphorylation which might occur during prolonged storage of protein extracts, even in the presence of phosphatase inhibitors.
Importance of Post-Electrophoretic Processing of the Gel
Phosphorylated proteins remain bound in Phos-Tag™ gels by transition metal bridges. In order to ensure their quantitative transfer to solid supports like PVDF membranes, divalent cations have to be efficiently removed. This is normally done by washing gels in solutions of chelating agents such as EDTA. The original protocol for post-electrophoretic treatment of Phos-Tag™ gels suggested 10 min incubation time in transfer buffer containing 1 mM EDTA (Kinoshita et al., 2006). We and others (Ban et al., 2013; Smékalová et al., 2014), found this step insufficient for quantitative transfer of proteins from the Phos-Tag™ gel to the PVDF membrane and thus we increased the concentration of EDTA to 10 mM while increasing the incubation time to 1 h with as many as six changes of the washing solution. It is critical, however, to omit methanol from washing steps as it may fix proteins to the gel and prolong the washing as much as possible and with as many changes as possible until protein diffusion might become a problem.
Applications of the Method
We have used acrylamide pendant Phos-Tag™ to delineate the phosphorylation status of the microtubule associated protein 65-1 (MAP65-1) in four MAPK-related mutants (anp2 × anp3, mpk4, yda, and ΔNyda) of Arabidopsis thaliana and also showed phosphorylation of AtMPK4 under conditions of hyperosmotic stress in wild type Arabidopsis thaliana seedlings (Beck et al., 2010; Smékalová et al., 2014). Moreover, we showed phosphorylation of MAP65-1 ortholog of Vigna sinensis following hyperosmotic stress (Panteris et al., 2010).
In the plant science, acrylamide pendant Phos-Tag™ had been further used to demonstrate alpha-tubulin phosphorylation in Arabidopsis thaliana and rice following hyperosmotic and salt stress or antimicrotubular drug treatments associated with the function of an atypical kinase originally identified as phosphatase (Ban et al., 2013; Fujita et al., 2013). Phos-Tag™ was also used to follow the pathogen elicited phosphorylation of either plant or pathogen proteins during the development of innate immune responses (Mao et al., 2011; Taylor et al., 2012; Li et al., 2014) and the MPK6-mediated phosphorylation of ethylene responsive factor ERF104 (Bethke et al., 2009) and the BES1 (BRI1-EMS suppressor 1) transcription factor (Kang et al., 2015) among others.
Herein we provide a survey of acrylamide pendant Phos-Tag™ extending its application to discriminate phosphorylated from non-phosphorylated protein species in two monocots (barley and wheat) and one dicot crop (alfalfa). By means of Zn2+/Phos-Tag™ we showed the hyperosmotically induced phosphorylation of alpha-tubulin as previously described for rice and Arabidopsis thaliana (Ban et al., 2013; Fujita et al., 2013). It is likely that tubulin phosphorylation detected herein, corresponds to Thr-349 as previously reported (Ban et al., 2013). However, α-tubulin might be also targeted for Tyr phosphorylation regulating microtubule sensitivity to low temperatures (Sheremet et al., 2012a), growth and morphogenesis of Arabidopsis root cells (Yemets et al., 2008) and mitotic progression in dividing tobacco BY-2 suspension culture cells (Sheremet et al., 2012b). Although, the identity of phosphorylated residues in tubulin (and any other protein) cannot be revealed by Phos-Tag™, the method can be readily combined with immunoprecipitation of the protein of interest and subsequent probing of the resulting western blot with phospho-amino acid specific antibodies. Moreover, we demonstrated the hyperosmotically induced accumulation of phosphorylated HvMPK4 in appropriately treated barley plants. It was of interest to see that by the use of an otherwise monospecific custom raised antibody, the phosphorylated form of HvMPK4 was further discriminated in two bands. During MAPK activation the TXY motif within the activation loop becomes simultaneously dually phosphorylated and indeed in the case of Arabidopsis thaliana MAPKs (e.g., MPK4 and MPK6) separated by Phos-Tag™ we only detected one upper band corresponding to pMPK6 (Beck et al., 2010; Smékalová et al., 2014). The appearance of two upper bands probably reflects to two different phosphorylation states of HvMPK4, one probably corresponding to the dually phosphorylated TEY motif while the second one might correspond to another phosphorylation site that will require further attention.
On the other hand Phos-Tag™ helped to corroborate the previously published robust reaction of Medicago sativa SIMK in root extracts exposed to H2O2, salt and hyperosmotic stress (Kiegerl et al., 2000; Ovečka et al., 2014) without a phospho-specific antibody. Additionally, we aimed to extend the application of Phos-Tag™ in order to characterize a transgenic Medicago sativa line with downregulated SIMKK, an upstream regulator of SIMK (Cardinale et al., 2002; Ovečka et al., 2014), by using siRNA approach. The SIMKK RNAi line used herein showed decreased accumulation of pSIMK in western blots after standard SDS-PAGE probed with anti-pTEpY antibody and no separation of pSIMK from SIMK following Zn2+/Phos-Tag™. The reduced anti-pTEpY immunoreactivity of SIMK and the absence of a band corresponding to pSIMK from western blot that followed Zn2+/Phos-Tag™, may be explained by the reduced levels of SIMK upstream regulator, SIMKK (Figure S1B) and the severe downregulation of endogenous SIMK in the RNAi line even if equal loading was verified by Ponceau S staining. This rather unexpected finding will merit further attention.
Comparison with Other Methods
Acrylamide pendant Phos-Tag™ identification and monitoring of protein phosphorylation is a small scale method and thus will be compared to similar approaches. The identification of phosphorylated protein species and their subsequent quantitative analysis requires their efficient separation from their non-phosphorylated counterparts. In small scale experiments, phosphorylation can be monitored by metabolic labeling of phosphorylated proteins with radioactive ATP (γ32P-ATP; Cuenda, 2000; Dickson, 2008). Radioactive phosphorus is a beta emitter and exhibits the highest emission energy compared to other biologically used radioisotopes. For this reason γ32P-ATP handling requires dedicated laboratory space, equipment and special training of the personnel, adhering to institutional rules for storage, handling and disposal of radioactive waste. Although γ32P-ATP comes at affordable prices by many vendors, the health risks and the cost for establishing appropriate laboratory space and training of handling personnel make the use of γ32P-ATP prohibitive especially for research groups that only occasionally study protein phosphorylation.
Alternatively, protein phosphorylation may be semi quantitatively addressed on standard immunoblots following denaturing SDS PAGE with phosphorylation specific antibodies (Willmann et al., 2014). Although some commonly used phospho-specific antibodies are commercially available most will have to be custom ordered at a significant cost.
A last important alternative to Phos-Tag is the one dimensional electrophoretic separation of phosphorylated proteins by means of isoelectric focusing coupled to western blot analysis (Anderson and Peck, 2008, 2014). Although there are no principal differences with Phos-Tag™ one must take into account the necessity of specialized chemicals, high voltage power supplies and a setup which is more complicated and expensive than the acrylamide pendant Phos-Tag approach presented hereby.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by two grants P501/11/1764 and P501/12/P455 from Czech Science Foundation (GACR), and by grant No LO1204 (Sustainable development of research in the Centre of the Region Haná) from the National Program of Sustainability I, MEYS. We gratefully acknowledge expert technical assistance by Katarína Takáčová in transformation of Medicago sativa somatic embryos, plant regeneration, and caretaking.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00336/abstract
References
Anderson, J. C., and Peck, S. C. (2008). A simple and rapid technique for detecting protein phosphorylation using one-dimensional isoelectric focusing gels and immunoblot analysis. Plant J. 55, 881–885. doi: 10.1111/j.1365-313X.2008.03550.x
Anderson, J. C., and Peck, S. C. (2014). Detection of protein phosphorylation and charge isoforms using vertical one-dimensional isoelectric focusing gels. Methods Mol. Biol. 1171, 39–46. doi: 10.1007/978-1-4939-0922-3_4
Austin, S., Bingham, E. T., Mathews, D. E., Shahan, M. N., Will, J., and Burgess, R. R. (1995). Production and field performance of transgenic alfalfa (Medicago sativa L.) expressing alpha amylase and manganese-dependent lignin peroxidase. Euphytica 85, 381–393. doi: 10.1007/BF00023971
Ban, Y., Kobayashi, Y., Hara, T., Hamada, T., Hashimoto, T., Takeda, S., et al. (2013). α-tubulin is rapidly phosphorylated in response to hyperosmotic stress in rice and Arabidopsis. Plant Cell Physiol. 54, 848–858. doi: 10.1093/pcp/pct065
Barbieri, C. M., and Stock, A. M. (2008). Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal Biochem. 376, 73–82. doi: 10.1016/j.ab.2008.02.004
Beck, M., Komis, G., Müller, J., Menzel, D., and Šamaj, J. (2010). Arabidopsis homologs of nucleus- and phragmoplast-localized kinase 2 and 3 and mitogen-activated protein kinase 4 are essential for microtubule organization. Plant Cell. 22, 755–771. doi: 10.1105/tpc.109.071746
Bethke, G., Unthan, T., Uhrig, J. F., Pöschl, Y., Gust, A. A., Scheel, D., et al. (2009). Flg22 regulates the release of an ethylene response factor substrate from MAP kinase 6 in Arabidopsis thaliana via ethylene signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 8067–8072. doi: 10.1073/pnas.0810206106
Boesger, J., Wagner, V., Weisheit, W., and Mittag, M. (2012). Application of phosphoproteomics to find targets of casein kinase 1 in the flagellum of chlamydomonas. Int. J. Plant Genomics 2012:581460. doi: 10.1155/2012/581460
Cardinale, F., Meskiene, I., Ouaked, F., and Hirt, H. (2002). Convergence and divergence of stress-induced mitogen-activated protein kinase signaling pathways at the level of two distinct mitogen-activated protein kinase kinases. Plant Cell 14, 703–711. doi: 10.1105/tpc.010256
Cuenda, A. (2000). Methods to assay stress-activated protein kinases. Methods Mol. Biol. 99, 127–143. doi: 10.1385/1-59259-054-3:127
Dickson, C. (2008). Protein techniques: immunoprecipitation, in vitro kinase assays, and Western blotting. Methods Mol. Biol. 461, 735–744. doi: 10.1007/978-1-60327-483-8_53
Dissmeyer, N., and Schnittger, A. (2011). The age of protein kinases. Methods Mol. Biol. 779, 7–52. doi: 10.1007/978-1-61779-264-9_2
Fujita, S., Pytela, J., Hotta, T., Kato, T., Hamada, T., Akamatsu, R., et al. (2013). An atypical tubulin kinase mediates stress-induced microtubule depolymerization in Arabidopsis. Curr. Biol. 23, 1969–1978. doi: 10.1016/j.cub.2013.08.006
Ishihama, N., and Yoshioka, H. (2012). Post-translational regulation of WRKY transcription factors in plant immunity. Curr. Opin. Plant Biol. 15, 431–437. doi: 10.1016/j.pbi.2012.02.003
Ishii, E., Eguchi, Y., and Utsumi, R. (2013). Mechanism of activation of PhoQ/PhoP two-component signal transduction by SafA, an auxiliary protein of PhoQ histidine kinase in Escherichia coli. Biosci. Biotechnol. Biochem. 77, 814–819. doi: 10.1271/bbb.120970
Kang, S., Yang, F., Li, L., Chen, H., Chen, S., and Zhang, J. (2015). The Arabidopsis transcription factor BES1 is a direct substrate of MPK6 and regulates immunity. Plant Physiol. 167, 1076–1086. doi: 10.1104/pp.114.250985
Kiegerl, S., Cardinale, F., Siligan, C., Gross, A., Baudouin, E., Liwosz, A., et al. (2000). SIMKK, a mitogen-activated protein kinase (MAPK) kinase, is a specific activator of the salt stress-induced MAPK, SIMK. Plant Cell. 12, 2247–2258. doi: 10.1105/tpc.12.11.2247
Kinoshita, E., and Kinoshita-Kikuta, E. (2011). Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics 11, 319–323. doi: 10.1002/pmic.201000472
Kinoshita, E., Kinoshita-Kikuta, E., and Koike, T. (2012). Phos-tag SDS-PAGE systems for phosphorylation profiling of proteins with a wide range of molecular masses under neutral pH conditions. Proteomics 12, 192–202. doi: 10.1002/pmic.201100524
Kinoshita, E., Kinoshita-Kikuta, E., Ujihara, H., and Koike, T. (2009). Mobility shift detection of phosphorylation on large proteins using a Phos-tag SDS-PAGE gel strengthened with agarose. Proteomics 9, 4098–4101. doi: 10.1002/pmic.200900020
Kinoshita, E., Kinoshita-Kikuta, E., Shiba, A., Edahiro, K., Inoue, Y., Yamamoto, K., et al. (2014). Profiling of protein thiophosphorylation by Phos-tag affinity electrophoresis: evaluation of adenosine 5'-O-(3-thiotriphosphate) as a phosphoryl donor in protein kinase reactions. Proteomics 14, 668–679. doi: 10.1002/pmic.201300533
Kinoshita, E., Kinoshita-Kikuta, E., Takiyama, K., and Koike, T. (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757. doi: 10.1074/mcp.T500024-MCP200
Kinoshita-Kikuta, E., Kinoshita, E., Matsuda, A., and Koike, T. (2014). Tips on improving the efficiency of electrotransfer of target proteins from Phos-tag SDS-PAGE gel. Proteomics 14, 2437–2442. doi: 10.1002/pmic.201400380
Komis, G., Takáč, T., Bekešová, S., Vadovič, P., and Šamaj, J. (2014). Affinity-based SDS PAGE identification of phosphorylated Arabidopsis MAPKs and substrates by acrylamide pendant Phos-Tag™. Methods Mol. Biol. 1171, 47–63. doi: 10.1007/978-1-4939-0922-3_5
Kosako, H. (2009). Phos-tag Western blotting for detecting stoichiometric protein phosphorylation in cells. Protoc. Exch. doi: 10.1038/nprot.2009.170
Li, R. F., Lu, G. T., Li, L., Su, H. Z., Feng, G. F., Chen, Y., et al. (2014). Identification of a putative cognate sensor kinase for the two-component response regulator HrpG, a key regulator controlling the expression of the hrp genes in Xanthomonas campestris pv. campestris. Environ. Microbiol. 16, 2053–2071. doi: 10.1111/1462-2920.12207
Liu, Y., and Chance, M. R. (2014). Integrating phosphoproteomics in systems biology. Comput. Struct. Biotechnol. J. 10, 90–97. doi: 10.1016/j.csbj.2014.07.003
Mao, G., Meng, X., Liu, Y., Zheng, Z., Chen, Z., and Zhang, S. (2011). Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in Arabidopsis. Plant Cell 23, 1639–1653. doi: 10.1105/tpc.111.084996
Meng, X., Xu, J., He, Y., Yang, K. Y., Mordorski, B., Liu, Y., et al. (2013). Phosphorylation of an ERF transcription factor by Arabidopsis MPK3/MPK6 regulates plant defense gene induction and fungal resistance. Plant Cell 25, 1126–1142. doi: 10.1105/tpc.112.109074
Müller, J., Beck, M., Mettbach, U., Komis, G., Hause, G., Menzel, D., et al. (2010). Arabidopsis MPK6 is involved in cell division plane control during early root development, and localizes to the pre-prophase band, phragmoplast, trans-Golgi network and plasma membrane. Plant J. 61, 234–248. doi: 10.1111/j.1365-313X.2009.04046.x
Nishi, H., Fong, J. H., Chang, C., Teichmann, S. A., and Panchenko, A. R. (2013). Regulation of protein-protein binding by coupling between phosphorylation and intrinsic disorder: analysis of human protein complexes. Mol. Biosyst. 9, 1620–1626. doi: 10.1039/c3mb25514j
Nishi, H., Shaytan, A., and Panchenko, A. R. (2014). Physicochemical mechanisms of protein regulation by phosphorylation. Front. Genet. 5:270. doi: 10.3389/fgene.2014.00270
Offringa, R., and Huang, F. (2013). Phosphorylation-dependent trafficking of plasma membrane proteins in animal and plant cells. J. Integr. Plant Biol. 55, 789–808. doi: 10.1111/jipb.12096
Ovečka, M., Takáč, T., Komis, G., Vadovič, P., Bekešová, S., Doskočilová, A., et al. (2014). Salt-induced subcellular kinase relocation and seedling susceptibility caused by overexpression of Medicago SIMKK in Arabidopsis. J. Exp. Bot. 65, 2335–2350. doi: 10.1093/jxb/eru115
Panteris, E., Komis, G., Adamakis, I. D., Šamaj, J., and Bosabalidis, A. M. (2010). MAP65 in tubulin/colchicine paracrystals of Vigna sinensis root cells: possible role in the assembly and stabilization of atypical tubulin polymers. Cytoskeleton 67, 152–160. doi: 10.1002/cm.20432
Pitzschke, A. (2015). Modes of MAPK substrate recognition and control. Trends Plant Sci. 20, 49–55. doi: 10.1016/j.tplants.2014.09.006
Ruse, C. I., McClatchy, D. B., Lu, B., Cociorva, D., Motoyama, A., Park, S. K., et al. (2008). Motif-specific sampling of phosphoproteomes. J. Proteome Res. 7, 2140–2150. doi: 10.1021/pr800147u
Salazar, C., and Höfer, T. (2009). Multisite protein phosphorylation–from molecular mechanisms to kinetic models. FEBS J. 276, 3177–3198. doi: 10.1111/j.1742-4658.2009.07027.x
Samac, D. A., and Austin-Phillips, S. (2006). Alfalfa (Medicago sativa L.). Methods Mol. Biol. 343, 301–311. doi: 10.1385/1-59745-130-4:301
Šamaj, J., Ovečka, M., Hlavačka, A., Lecourieux, F., Meskiene, I., Lichtscheidl, I., et al. (2002). Involvement of the mitogen-activated protein kinase SIMK in regulation of root hair tip growth. EMBO J. 21, 3296–3306. doi: 10.1093/emboj/cdf349
Šamajová, O., Komis, G., and Šamaj, J. (2013). Emerging topics in the cell biology of mitogen-activated protein kinases. Trends Plant Sci. 18, 140–148. doi: 10.1016/j.tplants.2012.11.004
Semagn, K. (2014). Leaf tissue sampling and DNA extraction protocols. Methods Mol. Biol. 1115, 53–67. doi: 10.1007/978-1-62703-767-9_3
Sheremet, Y. A., Yemets, A. I., Azmi, A., Vissenberg, K., Verbelen, J. P., and Blume, Y. B. (2012b). Effects of tyrosine kinase and phosphatase inhibitors on mitosis progression in synchronized tobacco BY-2 cells. Tsitol. Genet. 46, 3–11. doi: 10.3103/S0095452712050088
Sheremet, Y. A., Yemets, A. I., and Blume, Y. B. (2012a). Inhibitors of tyrosine kinases and phosphatases as a tool for the investigation of microtubule role in plant cold response. Tsitol. Genet. 46, 3–9. doi: 10.3103/S0095452712010112
Smékalová, V., Luptovčiak, I., Komis, G., Šamajová, O., Ovečka, M., Doskočilová, A., et al. (2014). Involvement of YODA and mitogen activated protein kinase 6 in Arabidopsis post-embryogenic root development through auxin up-regulation and cell division plane orientation. New Phytol. 203, 1175–1193. doi: 10.1111/nph.12880
St-Denis, N., and Gingras, A. C. (2012). Mass spectrometric tools for systematic analysis of protein phosphorylation. Prog. Mol. Biol. Transl. Sci. 106, 3–32. doi: 10.1016/B978-0-12-396456-4.00014-6
Taylor, K. W., Kim, J. G., Su, X. B., Aakre, C. D., Roden, J. A., Adams, C. M., et al. (2012). Tomato TFT1 is required for PAMP-triggered immunity and mutations that prevent T3S effector XopN from binding to TFT1 attenuate Xanthomonas virulence. PLoS Pathog. 8:e1002768. doi: 10.1371/journal.ppat.1002768
Wang, P., Du, Y., Zhao, X., Miao, Y., and Song, C. P. (2013). The MPK6-ERF6-ROS-responsive cis-acting Element7/GCC box complex modulates oxidative gene transcription and the oxidative response in Arabidopsis. Plant Physiol. 161, 1392–1408. doi: 10.1104/pp.112.210724
Willmann, R., Haischer, D. J., and Gust, A. A. (2014). Analysis of MAPK activities using MAPK-specific antibodies. Methods Mol. Biol. 1171, 27–37. doi: 10.1007/978-1-4939-0922-3_3
Keywords: protein phosphorylation, SDS-PAGE Phos-TagTM, Arabidopsis thaliana, Hordeum vulgare, Medicago sativa, Triticum turgidum, mitogen activated protein kinase
Citation: Bekešová S, Komis G, Křenek P, Vyplelová P, Ovečka M, Luptovčiak I, Illés P, Kuchařová A and Šamaj J (2015) Monitoring protein phosphorylation by acrylamide pendant Phos-Tag™ in various plants. Front. Plant Sci. 6:336. doi: 10.3389/fpls.2015.00336
Received: 10 March 2015; Accepted: 28 April 2015;
Published: 13 May 2015.
Edited by:
Roger Deal, Emory University, USAReviewed by:
Yaroslav B. Blume, Institute of Food Biotechnology and Genomics, UkrainePolydefkis Hatzopoulos, Agricultural University of Athens, Greece
Copyright © 2015 Bekešová, Komis, Křenek, Vyplelová, Ovečka, Luptovčiak, Illés, Kuchařová and Šamaj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jozef Šamaj, Department of Cell Biology, Centre of the Region Haná for Biotechnological and Agricultural Research, Palacký University Olomouc, Šlechtitelů 27, Olomouc 783 71, Czech Republic,am96ZWYuc2FtYWpAdXBvbC5jeg==