 Fangping Gong
Fangping Gong Xiuli Hu
Xiuli Hu Wei Wang
Wei Wang- State Key Laboratory of Wheat and Maize Crop Science, Collaborative Innovation Center of Henan Grain Crops, College of Life Science, Henan Agricultural University, Zhengzhou, China
Introduction
Approximately 80% of human food is composed of crops, which are dominated by cereals that collectively make up 50% of global food production (Langridge and Fleury, 2011). Among cereal crops, rice, wheat, and maize provide approximately half of the calories consumed worldwide. Nevertheless, crop production is seriously hampered by influential abiotic stresses like drought, climate fluctuations, and salinity. It is estimated that up to 50–70% decline in crop productivity is attributed to abiotic stress (Mittler, 2006). Therefore, to ensure the security of global food production, it is essential to produce sustainable crop varieties that can adapt to climate variability, and to develop a broad spectrum of abiotic stress tolerant crops (Tester and Langridge, 2010). This has driven much research into the study of crop responses to abiotic stresses.
Proteomics has been successfully used to study abiotic stress responses in a wide range of crops (Abreu et al., 2013; Barkla et al., 2013; Ngara and Ndimba, 2014), especially rice (Kim et al., 2014), wheat (Komatsu et al., 2014), and maize (Benesova et al., 2012; Gong et al., 2014). It is generally envisioned that at this stage, proteomic-based discoveries in rice are likely to be translated into improving other crop plants against ever-changing environmental factors (Kim et al., 2014).
Despite the potential role of proteomics to advance the study of stress tolerance in crops, thus far little useful information has been made available for crop improvement and breeding, even with the numerous proteomics studies undertaken in recent years. In our opinion, crop stress proteomics should be better focused on the following aspects: dissecting cell specific stress response (especially initial stress responses), identification of stress proteins, and the analysis of post translational modifications (PTMs) of proteins (Figure 1).
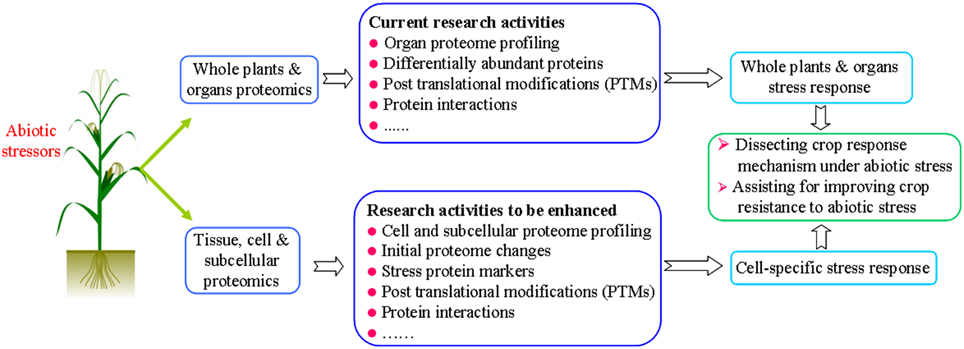
Figure 1. A graphic summary on current research and future research in proteomic analysis of crop plants under abiotic stress conditions.
Dissecting Cell or Tissue Specific Stress Response
Understanding how plant cells sense and respond to abiotic stress is not only fundamental to our understanding of stress tolerance, but has the potential to yield novel approaches to improve crop productivity. Cellular proteomics plays an essential role in determining the functions of cellular compartments and the mechanisms underlying protein/gene targeting and trafficking.
Currently, numerous organ-specific proteomic analyses of abiotic stress in crops have contributed to our understanding of the response mechanisms of crops to abiotic stresses (Komatsu and Hossain, 2013). Obviously, the specifics of proteomic response to abiotic stress vary from tissue to tissue within a plant. Therefore, the crop stress response should be analyzed at a cellular or subcellular level, integrated with studies on whole plants, organs or tissues, to discriminate the specific responses of different cell types to abiotic stress. At present, cell or subcellular proteomic studies focus on relatively abundant, or easily isolated homogenous compartments (e.g., plastids, mitochondria, peroxisomes, and nuclei) mainly in Arabidopsis (Tanz et al., 2013), but also in rice, wheat, barley, maize (Huang et al., 2013; Millar and Taylor, 2014; Hu et al., 2015).
To increase the probability of identifying stress proteins (genes) from specific cells or tissues, an appropriate sampling method needs to be first developed to obtain relatively pure subcellular fractions from this material. A promising sampling method is laser capture microdissection (LCM), which can isolate specific cell types of interest from sectioned specimens of heterogeneous tissues under direct microscopic visualization with the assistance of a laser beam (Longuespée et al., 2014). LCM has been successfully used in transcriptome and microarray studies in maize (Nakazono et al., 2003; Rajhi et al., 2011) and rice (Suwabe et al., 2008; Kubo et al., 2013). Hopefully, combined with more sensitive protein staining technologies and more advanced mass spectrometers, LCM has the potential to promote crop stress proteomics at a cellular level. Another promising technique is free flow electrophoresis (FFE), which can isolate much purer membrane fractions and/or organelles. The FFE technique has been successfully applied in plant proteomics to the isolate tonoplast, mitochondria, plasma membranes, and Golgi apparatus (Barkla et al., 2013).
New approaches are beginning to enable the proteome to be analyzed at cell specific resolution. Not only are these studies leading to a better definition of the specialized proteomic behavior of certain cell types, but they also illustrate that information about proteome levels and responses gained from whole plants or organs can be misleading. This will surely help us to better understand the processes of crop stress acclimation and stress tolerance acquisition.
In addition, the studies on initial stress response of crops should be enhanced. Plant stress response consists of multiple phases, including an initial shock phase, an acclimation phase, a maintenance phase, an exhaustion phase, and/or a recovery phase (Kosová et al., 2011). Each phase of stress response is characterized by its unique transcriptome, proteome and metabolome changes. At 15 and 30 min after the onset of stresses (e.g., UV-B light, drought, and cold), the initial transcriptional changes in Arabidopsis are significant (Kilian et al., 2007). Currently, crop proteomic changes are often analyzed after several hours, even days after a stress onset (e.g., Meng et al., 2014). Thus, the initial proteome changes of crops under abiotic conditions should be further analyzed. These types of studies will give important insights in the signaling cascade activated immediately in crops in response to abiotic stresses. Sensitive proteomic approaches are capable of identifying low-abundance proteins (especially transcription factors and regulatory proteins) involved in the initial stress response in crops.
Identification of Stress Proteins
The sequencing of major crops, especially rice, maize, and wheat represented a major breakthrough in crop proteomic research. For example, stress proteomic studies in maize have increased exponentially since 2009, after the release of the maize genome sequence (Schnable et al., 2009). Because knowledge of the genomic sequence alone does not indicate how a plant interacts with the environment, and not all open reading frames correspond to a functional gene (Ribeiro et al., 2013), proteomic approaches are critical to understand plant mechanisms of stress tolerance. A main aim of stress proteomics in crops is to identify stress proteins which can potentially be used for crop improvement and breeding.
The stress-tolerant phenotype in crops is a result of differential expression of unique proteins in resistant cultivars to protect them during stress periods. To develop better crop plants for sustainable food production, proteomic discovery of these unique stress proteins to further understand the stress-tolerance mechanisms at the molecular level is very important. We can potentially modify these key stress proteins to enhance a crops abiotic stress tolerance. A potential role for crop stress proteomic studies could be the identification and further characterization of key proteins underlying crop tolerance to a given abiotic stress, which can then be used as protein biomarker of a given stress. In such abiotic stress studies, it is common to analyze proteomes by contrasting stressed crop plants against control ones, attempting to correlate changes in protein accumulation with the phenotypic response (Abreu et al., 2013).
Rapid progress in proteome profiling methodologies, such as iTRAQ, DIGE, and high-resolution tandem mass spectrometry has enabled a more accurate comparison of crop stress responses and can detect more differentially abundant proteins than prior analyses. Unfortunately, ascribing a probable function to a newly identified stress responsive protein can be difficult in crops. Unlike Arabidopsis, it is difficult to determine the number of experimentally characterized genes in public databases for many crops. This is mainly due to the lack of high quality functional annotations for many crop genomes. For example, the maize genome sequence contains approximately 40,000 genes (Schnable et al., 2009), but little is known about the function of most genes. A search for maize protein sequences using the keyword “maize” retrieved 262,228 entries at NCBI and 85,389 entries in UniProt (14 May, 2015). This is indicative of the high level of redundancy and repetition, particularly in the NCBI database. In the UniProt database, only 840 maize protein entries have been reviewed (14 May, 2015), with most entries listed as “uncharacterized protein.” Likewise, in rice, only 1% of the protein-coding genes have had a functional annotation based on experimental evidence (Rhee and Mutwil, 2014). Given this situation, the experimental validation of stress proteins and their role in stress tolerance is very important to bridge the gap between proteomic discovery of stress proteins and the selection of potential target proteins for future crop improvements.
More attention should be paid to up-regulated proteins in crop stress proteomics. In plants, translation efficiency can change dramatically in response to abiotic stress, leading to a massive bias in the pool of mRNAs that are actively translated (Mustroph et al., 2009; Juntawong et al., 2014). This might be related to the importance of stress- associated proteins that are required to recalibrate cellular metabolism. Thus, up-regulated proteins are more important for crops to adapt to a stressful environment compared to down-regulated proteins; an important point when considering crop stress tolerance breeding. Of course, down-regulated proteins are also likely to contribute to an acquisition of enhanced plant stress tolerance. For example, some secondary metabolism related proteins affected by stress would likely decrease to conserve energy (Ghosh and Xu, 2014). In addition, due to discordant protein and mRNA expression, especially in plants (Vélez-Bermúdez and Schmidt, 2014), it is essential to identify up-regulated stress proteins rather than mRNA in order to better identify candidates which could be used for crop improvement.
Finally, it is worth noting that much of the stress proteomic research has been performed in laboratories under controlled conditions and relied on screening whole crops for their ability to survive severe stress. The effects of plant growth and gene expression in response to stress can be highly dose-responsive, indicating the existence of sensitive machinery in the plant for assessing the stress level and fine-tuning molecular responses (Claeys et al., 2014). Therefore, many proteomic analysis of abiotic stress in crops can be misleading and may not be useful.
Analysis of PTMs of Proteins
PTMs can affect protein function, interactions with other proteins, subcellular targeting, and stability. In crop stress proteomics, the identification and quantification of PTMs will contribute the detailed functional characterization of a protein and will likely assist in our understanding of crop stress acclimation and stress tolerance acquisition.
Large-scale proteomics studies have revealed that PTMs are more widespread than previously estimated. For example, up to two-thirds of the metabolic proteins in yeast may be affected by protein phosphorylation (Breitkreutz et al., 2010). In Arabidopsis, PTMs include phosphorylation (Heazlewood et al., 2008; Vialaret et al., 2014), N-linked glycosylation (Zielinska et al., 2012), ubiquitination (Kim et al., 2013), methionine oxidation (Marondedze et al., 2013), S-nitrosylation (Fares et al., 2011), and acetylation (Finkemeier et al., 2011).
Few proteomic studies have been performed to specifically characterize PTMs in crops under abiotic stress; this includes the analysis of phosphorylation during salt and water stresses in maize (Zörb et al., 2010; Bonhomme et al., 2012; Hu et al., 2013) and characterization of protein glycosylation in soybean roots under flooding (Mustafa and Komatsu, 2014).
It is expected that in certain cases PTMs play a more important role than protein abundance changes. Thus, the analysis of PTMs of stress-responsive proteins in crops should be strengthened; however the ability to routinely identify and quantify PTMs represents a grand challenge in the field of proteomics (Heazlewood, 2011). In conjunction with improvements in methodological approaches, it would be expected that the study of PTMs will become more common in the area of crop stress proteomics in the future.
Concluding Remarks and Outlook
Proteomics has an important role to play in assisting our understating at the molecular level of how crops respond to abiotic stress. In-depth proteomic analysis of crop stress responses will be essential for future crop improvements. Though proteomic characterization of cell and tissue specific stress responses, stress proteins and PTMs is still a difficult undertaking in crops, the development of more sensitive methodologies, particularly for the cell specific analysis of the proteome will be crucial for understanding stress responses at the cellular level. In addition, rapid advances in high-throughput omics technologies (e.g., proteomics, transcriptomics, genomics, and metabolomics) will make it possible to use a systems biology approach to understand crop responses to abiotic stresses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Joshua Heazlewood for copy-editing our manuscript. We acknowledge the National Natural Science Foundation of China (Grant No. 31371543), the Plan for Scientific Innovation Talent of Henan Province (Grant No. 144200510012), and the Program for Innovative Research Team (in Science and Technology) in University of Henan Province (Grant No. 15IRTSTHN015) for financial support.
References
Abreu, I. A., Farinha, A. P., Negrão, S., Gonçalves, N., Fonseca, C., Rodrigues, M., et al. (2013). Coping with abiotic stress: proteome changes for crop improvement. J. Proteomics 93, 145–168. doi: 10.1016/j.jprot.2013.07.014
Barkla, B. J., Vera-Estrella, R., and Pantoja, O. (2013). Progress and challenges for abiotic stress proteomics of crop plants. Proteomics 13, 1801–1815. doi: 10.1002/pmic.201200401
Benesova, M., Hola, D., Fischer, L., Jedelsky, P. L., Hnilicka, F., Wilhelmova, N., et al. (2012). The physiology and proteomics of drought tolerance in maize: early stomatal closure as a cause of lower tolerance to short-term dehydration? PLoS ONE 7: e38017. doi: 10.1371/journal.pone.0038017
Bonhomme, L., Benoît, V., Tardieu, F., and Zivy, M. (2012). Phosphoproteome dynamics upon changes in plant water status reveal early events associated with rapid growth adjustment in maize leaves. Mol. Cell. Proteomics 11, 957–972. doi: 10.1074/mcp.M111.015867
Breitkreutz, A., Choi, H., Sharom, J. R., Boucher, L., Neduva, V., Larsen, B., et al. (2010). A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046. doi: 10.1126/science.1176495
Claeys, H., Van Landeghem, S., Dubois, M., Maleux, K., and Inzé, D. (2014). What is stress? Dose-response effects in commonly used in vitro stress assays. Plant Physiol. 165, 519–527. doi: 10.1104/pp.113.234641
Fares, A., Rossignol, M., and Peltier, J. B. (2011). Proteomics investigation of endogenous S-nitrosylationin Arabidopsis. Biochem. Biophys. Res. Commun. 416, 331–336. doi: 10.1016/j.bbrc.2011.11.036
Finkemeier, I., Laxa, M., Miguet, L., Howden, A. J. M., and Sweetlove, L. J. (2011). Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant Physiol. 155, 1779–1790. doi: 10.1104/pp.110.171595
Ghosh, D., and Xu, J. (2014). Abiotic stress responses in plant roots: a proteomics perspective. Front. Plant Sci. 5:6. doi: 10.3389/fpls.2014.00006
Gong, F. P., Yang, L., Tai, F., Hu, X. L., and Wang, W. (2014). “Omics” of maize stress response for sustainable food production: opportunities and challenges. OMICS 18, 714–732. doi: 10.1089/omi.2014.0125
Heazlewood, J. L. (2011). The green proteome: challenges in plant proteomics. Front. Plant Sci. 2:6. doi: 10.3389/fpls.2011.00006
Heazlewood, J. L., Durek, P., Hummel, J., Selbig, J., Weckwerth, W., Walther, D., et al. (2008). PhosPhAt: a database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucl. Acids Res. 36, D1015–D1021. doi: 10.1093/nar/gkm812
Hu, X. L., Yang, Y. F., Gong, F. P., Zhang, D. Y., Zhang, L., Wu, L. J., et al. (2015). Protein sHSP26 improves chloroplast performance under heat stress by interacting with specific chloroplast proteins in maize (Zea mays). J. Proteomics 115, 81–92. doi: 10.1016/j.jprot.2014.12.009
Hu, Y., Guo, S., Li, X., and Ren, X. (2013). Comparative analysis of salt-responsive phosphoproteins in maize leaves using Ti4+-IMAC enrichment and ESI-Q-TOF MS. Electrophoresis 34, 485–492. doi: 10.1002/elps.201200381
Huang, S., Shingaki-Wells, R. N., Taylor, N. L., and Millar, A. H. (2013). The rice mitochondria proteome and its response during development and to the environment. Front. Plant Sci. 4:16. doi: 10.3389/fpls.2013.00016
Juntawong, P., Girke, T., Bazin, J., and Bailey-Serres, J. (2014). Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111, E203–E212. doi: 10.1073/pnas.1317811111
Kilian, J., Whitehead, D., Horak, J., Wanke, D., Weinl, S., Batistic, O., et al. (2007). The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 50, 347–363. doi: 10.1111/j.1365-313X.2007.03052.x
Kim, D. Y., Scalf, M., Smith, L. M., and Vierstra, R. D. (2013). Advanced proteomic analyses yield a deep catalog of ubiquitylation targets in Arabidopsis. Plant Cell 25, 1523–1540. doi: 10.1105/tpc.112.108613
Kim, S. T., Kim, S. G., Agrawal, G. K., Kikuchi, S., and Rakwa, R. (2014). Rice proteomics: a model system for crop improvement and food security. Proteomics 14, 593–610. doi: 10.1002/pmic.201300388
Komatsu, S., and Hossain, Z. (2013). Organ-specific proteome analysis for identification of abiotic stress response mechanism in crop. Front. Plant Sci. 4: 71. doi: 10.3389/fpls.2013.00071
Komatsu, S., Kamal, A. H. M., and Hossain, Z. (2014). Wheat proteomics: proteome modulation and abiotic stress acclimation. Front. Plant Sci. 5: 684. doi: 10.3389/fpls.2014.00684
Kosová, K., Vítámvás, P., Prášil, I. T., and Renaut, J. (2011). Plant proteome changes under abiotic stress–contribution of proteomics studies to understanding plant stress response. J. Proteomics 74, 1301–1322. doi: 10.1016/j.jprot.2011.02.006
Kubo, T., Fujita, M., Takahashi, H., Nakazono, M., Tsutsumi, N., and Kurata, N. (2013). Transcriptome analysis of developing ovules in rice isolated by laser microdissection. Plant Cell Physiol. 54, 750–765. doi: 10.1093/pcp/pct029
Langridge, P., and Fleury, D. (2011). Making the most of ‘omics’ for crop breeding. Trends Biotechnol. 29, 33–40. doi: 10.1016/j.tibtech.2010.09.006
Longuespée, R., Fléron, M., Pottier, C., Quesada-Calvo, F., Meuwis, M. A., Baiwir, D., et al. (2014). Tissue proteomics for the next decade? Towards a molecular dimension in histology. OMICS 18, 539–552. doi: 10.1089/omi.2014.0033
Marondedze, C., Turek, I., Parrott, B., Thomas, L., Jankovic, B., Lilley, K. S., et al. (2013). Structural and functional characteristics of cGMP-dependent methionine oxidation in Arabidopsis thaliana proteins. Cell Commun. Signal. 11: 1. doi: 10.1186/1478-811X-11-1
Meng, L. B., Chen, Y. B., Lu, T. C., Wang, Y. F., Qian, C. R., Yu, Y., et al. (2014). A systematic proteomic analysis of NaCl-stressed germinating maize seeds. Mol. Biol. Rep. 41, 3431–3443. doi: 10.1007/s11033-014-3205-7
Millar, A. H., and Taylor, N. L. (2014). Subcellular proteomics—where cell biology meets protein chemistry. Front. Plant Sci. 5: 55. doi: 10.3389/fpls.2014.00055
Mittler, R. (2006). Abiotic stress, the field environment and stress combination. Trends Plant Sci. 11, 15–19. doi: 10.1016/j.tplants.2005.11.002
Mustafa, G., and Komatsu, S. (2014). Quantitative proteomics reveals the effect of protein glycosylation in soybean root under flooding stress. Front. Plant Sci. 5: 627. doi: 10.3389/fpls.2014.00627
Mustroph, A., Zanettia, M. E., Jang, C. J. H., Holtan, H. E., Repetti, P. P., Galbraith, D. W., et al. (2009). Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 106, 18843–18848. doi: 10.1073/pnas.0906131106
Nakazono, M., Qiu, F., Borsuk, L. A., and Schnable, P. S. (2003). Laser-capture microdissection, a tool for the global analysis of gene expression in specific plant cell ty pes: identification of genes expressed differentially in epidermal cells or vascular tissues of maize. Plant Cell 15, 583–596. doi: 10.1105/tpc.008102
Ngara, R., and Ndimba, B. K. (2014). Understanding the complex nature of salinity and drought-stress response in cereals using proteomics technologies. Proteomics 14, 611–621. doi: 10.1002/pmic.201300351
Rajhi, I., Yamauchi, T., Takahashi, H., Nishiuchi, S., Shiono, K., Watanabe, R., et al. (2011). Identification of genes expressed in maize root cortical cells during lysigenous aerenchyma formation using laser microdissection and microarray analyses. New Phytol. 190, 351–368. doi: 10.1111/j.1469-8137.2010.03535.x
Rhee, S. Y., and Mutwil, M. (2014). Towards revealing the functions of all genes in plants. Trends Plant Sci. 19, 212–221. doi: 10.1016/j.tplants.2013.10.006
Ribeiro, M., Nunes-Miranda, J. D., Branlard, G., Carrillo, J. M., Rodriguez-Quijano, M., and Igrejas, G. (2013). One hundred years of grain omics: identifying the glutens that feed the world. J. Proteome Res. 12, 4702–4716. doi: 10.1021/pr400663t
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115. doi: 10.1126/science.1178534
Suwabe, K., Suzuki, G., Takahashi, H., Katsuhiro, S., Makoto, E., Kentaro, Y., et al. (2008). Separated transcriptomes of male gametophyte and tapetum in rice: validity of a laser microdissection (LM) microarray. Plant Cell Physiol. 49, 1407–1416. doi: 10.1093/pcp/pcn124
Tanz, S. K., Castleden, I., Hooper, C. M., Vacher, M., Small, I., and Millar, H. A. (2013). SUBA3: a data base for integrating experimentation and prediction to define the SUBcellular location of proteins in Arabidopsis. Nucl. Acids Res. 41, D1185–D1191. doi: 10.1093/nar/gks1151
Tester, M., and Langridge, P. (2010). Breeding technologies to increase crop production in a changing world. Science 327, 818–822. doi: 10.1126/science.1183700
Vélez-Bermúdez, I. C., and Schmidt, W. (2014). The conundrum of discordant protein and mRNA expression. Are plants special? Front. Plant Sci. 5: 619. doi: 10.3389/fpls.2014.00619
Vialaret, J., Di Pietro, M., Hem, S., Maurel, C., Rossignol, M., and Santoni, V. (2014). Phosphorylation dynamics of membrane proteins from Arabidopsis roots submitted to salt stress. Proteomics 14, 1058–1070. doi: 10.1002/pmic.201300443
Zielinska, D. F., Gnad, F., Schropp, K., Wisniewski, J. R., and Mann, M. (2012). Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell 46, 542–548. doi: 10.1016/j.molcel.2012.04.031
Keywords: crop stress proteomics, abiotic stress, subcellular proteome, stress proteins, initial proteome stress response, post-translational modifications
Citation: Gong F, Hu X and Wang W (2015) Proteomic analysis of crop plants under abiotic stress conditions: where to focus our research? Front. Plant Sci. 6:418. doi: 10.3389/fpls.2015.00418
Received: 15 February 2015; Accepted: 24 May 2015;
Published: 05 June 2015.
Edited by:
Silvia Mazzuca, Università della Calabria, ItalyReviewed by:
Dipanjana Ghosh, National University of Singapore, SingaporeKlára Kosová, Crop Research Institute, Czech Republic
Copyright © 2015 Gong, Hu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Wang,d2FuZ3dlaUBoZW5hdS5lZHUuY24=;d2FuZ193QGFsaXl1bi5jb20=