 Valmor J. Bianchi
Valmor J. Bianchi Manuel Rubio
Manuel Rubio Livio Trainotti
Livio Trainotti Ignazio Verde
Ignazio Verde Claudio Bonghi
Claudio Bonghi Pedro Martínez-Gómez
Pedro Martínez-Gómez- 1Department of Plant Physiology, Instituto de Biologia, Universidade Federal de Pelotas, Pelotas-RS, Brazil
- 2Department of Plant Breeding, Centro de Edafología y Biología Aplicada del Segura, Consejo Superior de Investigaciones Científicas, Murcia, Spain
- 3Department of Biology, University of Padua, Padova, Italy
- 4Consiglio per la ricerca in agricoltura e l'analisi dell'economia agraria (CRA) - Centro di ricerca per la frutticoltura, Roma, Italy
- 5Department of Agronomy, Food, Natural Resources, and Environment (DAFNAE). University of Padua, Padova, Italy
Many plant processes depend on differential gene expression, which is generally controlled by complex proteins called transcription factors (TFs). In peach, 1533 TFs have been identified, accounting for about 5.5% of the 27,852 protein-coding genes. These TFs are the reference for the rest of the Prunus species. TF studies in Prunus have been performed on the gene expression analysis of different agronomic traits, including control of the flowering process, fruit quality, and biotic and abiotic stress resistance. These studies, using quantitative RT-PCR, have mainly been performed in peach, and to a lesser extent in other species, including almond, apricot, black cherry, Fuji cherry, Japanese apricot, plum, and sour and sweet cherry. Other tools have also been used in TF studies, including cDNA-AFLP, LC-ESI-MS, RNA, and DNA blotting or mapping. More recently, new tools assayed include microarray and high-throughput DNA sequencing (DNA-Seq) and RNA sequencing (RNA-Seq). New functional genomics opportunities include genome resequencing and the well-known synteny among Prunus genomes and transcriptomes. These new functional studies should be applied in breeding programs in the development of molecular markers. With the genome sequences available, some strategies that have been used in model systems (such as SNP genotyping assays and genotyping-by-sequencing) may be applicable in the functional analysis of Prunus TFs as well. In addition, the knowledge of the gene functions and position in the peach reference genome of the TFs represents an additional advantage. These facts could greatly facilitate the isolation of genes via QTL (quantitative trait loci) map-based cloning in the different Prunus species, following the association of these TFs with the identified QTLs using the peach reference genome.
Introduction
Transcription is a complex process in which a DNA strand provides the information for the synthesis of an RNA strand, which transfers the genetic information required for protein synthesis (Watson et al., 2014).
RNA molecules include coding and non-coding RNA. Protein-coding RNA is also called messenger RNA (mRNA) and makes up around 5% of the total RNA in plants. Non-coding RNA includes non-regulatory RNA and is composed of ribosomal RNA (rRNA, up to 85%) and transfer RNA (tRNA, around 15%). In addition, non-coding RNA includes regulatory RNA (less than 5%) with the group of small RNAs (sRNAs); small nuclear RNAs (snRNAs) involved in mRNA and tRNA processing; small interfering RNA (siRNA) and micro RNA involved in mRNA translation; and small cytoplasmic RNA (scRNA) and piwi-interacting RNA (piRNA), with a variable and uncertain function (Figure 1) (Atkins et al., 2011; Watson et al., 2014).
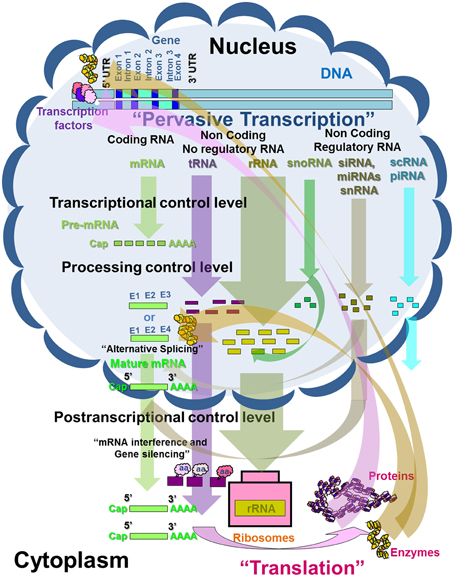
Figure 1. Schematic representation of the transcription control in eukaryotes and Prunus (adapted from Atkins et al., 2011; Watson et al., 2014).
The coding and noncoding-regulatory RNAs are the main molecules involved in the transcription process. This molecule occurs in a highly selective process in which individual genes (monocistronic transcription) are transcribed only when their products, the respective proteins, are required for a cell, a group of cells, or an organ, as a result of spatial and temporal plant growth and development control. The enzymes responsible for transcription in living organisms, including plants, are called RNA polymerases (RNAPs). Plants contain the following four distinct RNA polymerase enzymes, each responsible for synthesizing a different RNA molecule: RNA polymerase I (larger rRNAs); RNA polymerase II (pre mRNAs, snoRNAs -small nucleolar RNAs-, snRNAs, miRNAs); RNA polymerase III (scRNAs, tRNAs, smaller rRNAs); and RNA polymerase IV (siRNAs), which is specific to plants (Kornberg, 2007; Krishnamurthy and Hampsey, 2008). The point on the DNA to which an RNA polymerase enzyme binds prior to initiating transcription is called the promoter. Yet this enzyme is not capable of recognizing promoter regions and requires the help of a large variety of accessory proteins called transcription factors (TFs) (Karp, 2008; Krishnamurthy and Hampsey, 2008).
TFs are proteins that bind a specific DNA sequence and thereby regulate the expression of target genes (Krishnamurthy and Hampsey, 2008). TF/RNAP interaction is thus necessary to form what is also known as the pre-initiation complex to start the transcription process. The same TFs can be involved in the transcription process as co-activators, acting in chromatin remodeling, histone acetylation and nucleic acid methylation, thus up- and down-regulating gene expression (Kornberg, 2007; Watson et al., 2014).
TFs are crucial for the action of the RNAPs, but they have mainly been studied in the case of mRNAs and RNA polymerase II. All major processes of life depend on differential gene expression, which is generally controlled by these TFs (Kornberg, 2007; Karp, 2008; Atkins et al., 2011). The first TFs were described in plants in the 1980s, yet only around 1400 scientific articles about TFs had been published by the year 2000. In the last 14 years, however, with the newly available strategies and tools for molecular studies, TF studies have increased exponentially. Indeed, more than 13,000 articles have been published in plants during this time period. In the case of Prunus species, TF studies have also increased exponentially since 2001. This is indicative of how much remains to be done in order to discover and better understand the real function of TFs and how they influence the main characteristics of agronomical importance in Prunus spp. (Figure 2).
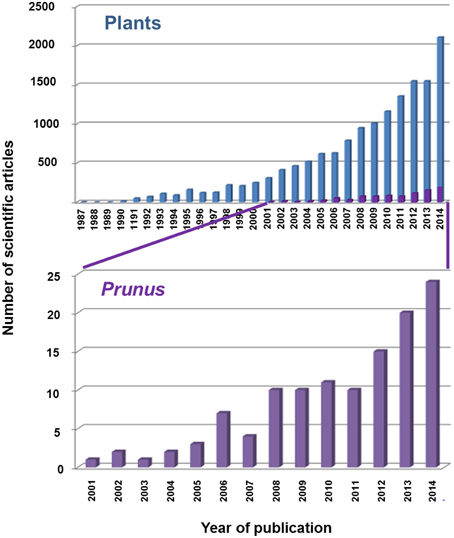
Figure 2. Number of scientific articles related to Prunus transcription factors published in the WOK database (Web of Knowledge, http://apps.webofknowledge.com).
Two main plant TF databases are currently available online: the Plant Transcription Factor Database v3.0 (PlnTFDB) (http://plntfdb.bio.uni-potsdam.de/v3.0/) of the University of Potsdam (Germany) (Pérez-Rodríguez et al., 2009) and the Plant Transcription Factor Database v3.0 (PlantTFDB) (http://planttfdb.cbi.pku.edu.cn/) of the Centre for Bioinformatics of Peking University (China) (Jin et al., 2014). In general, the information and terminology is similar in both databases, although there are some discrepancies, mainly involving the nomenclature of the different TF families. Information regarding Prunus TFs, however, is only available in the PlantTFDB database. According to this database, TFs encoded by the different plant genomes can be classified into 57 major multigene families, including 123,497 different TFs identified (Table 1). The largest families are the basic helix-loop-helix (bHLH) family, the ERF (mTERF) family, the MYB family and the NAC family, all of which have more than 8000 members in this database. The members/genes of these four super-families of TFs are involved in a wide range of biological processes, like the control of mtDNA replication, embryo development, flower and fruit development, fruit dehiscence, meristem determinacy, cell proliferation and differentiation, among others (Littlewood and Evan, 1995; Souer et al., 1996; Roberti et al., 2009) (Table 1).
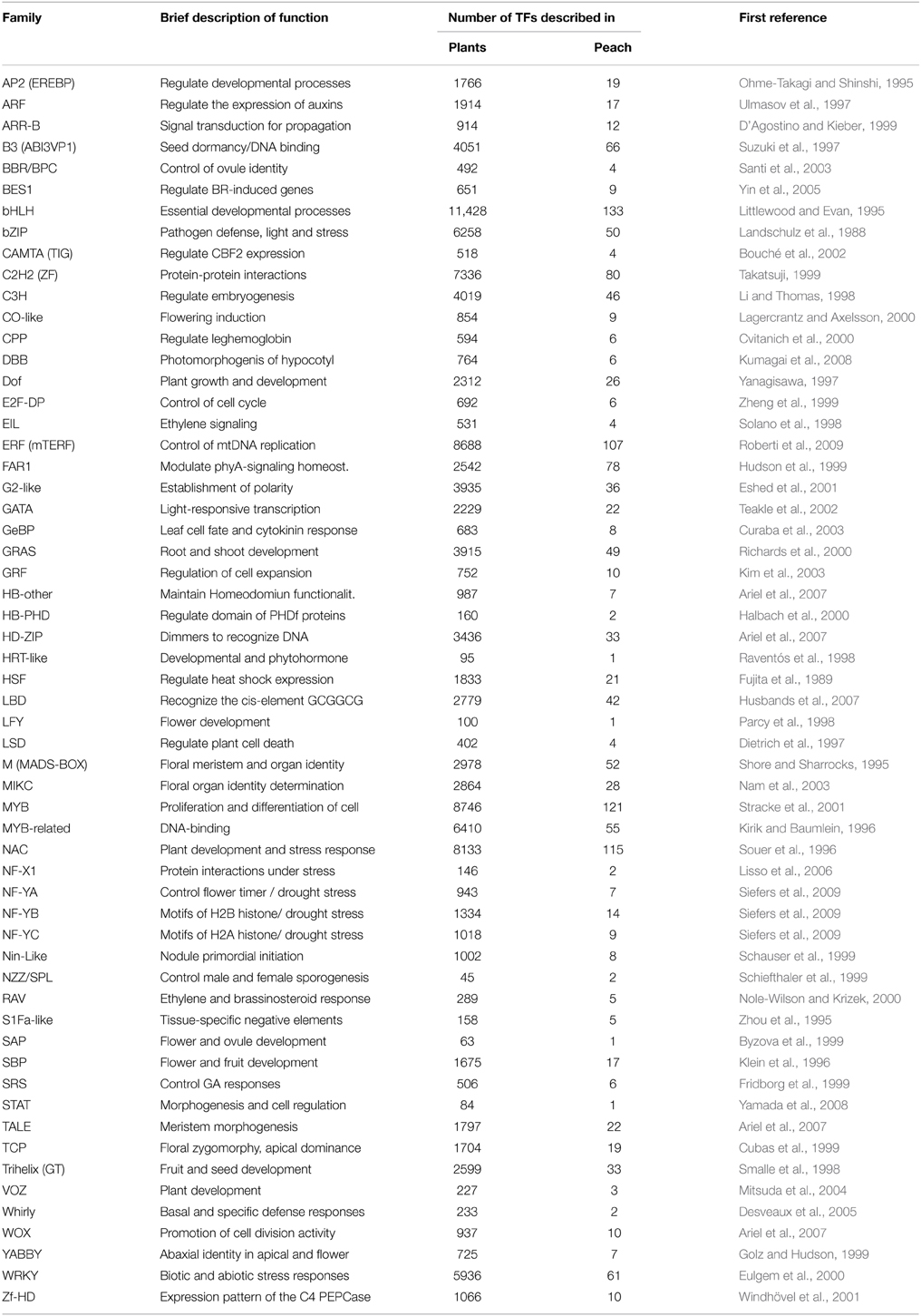
Table 1. Transcription factor (TF) families identified in plants and peach available in the PlantTFDB database (http://planttfdb.cbi.pku.edu.cn/).
The purpose of this study was to summarize the information available from the TF studies in Prunus spp., based on a review of the bibliography. The availability of the peach genome sequence (Verde et al., 2013) made it possible to make an inventory of peach TFs at the whole genome level. This paper also includes a discussion of the main implications of this information for the breeding and development of marker-assisted selection strategies, with particular focus on characteristics of direct agronomical interest.
Transcription Factors Identified in Prunus
The Prunus genus inside the Rosaceae family and the Rosales order is widely grown around the world and includes about 230 species, many of which produce edible fruits and seeds of economic interest (Potter, 2012). Inside this genus, peach [Prunus persica (L.) Batsch] presents several physiological and molecular advantages, including self-compatibility, a short juvenile phase and a small genome size (227.3 Mb). These characteristics make peach a suitable model species within the Prunus genus and even within the Rosaceae family (Arús et al., 2012). The complete peach genome sequence (Peach v1.0) was recently published (Verde et al., 2013) and is now the reference genome in these species. Within this genome, 1529 TFs have been identified to date in the PlantTFDB database, accounting for about 5.3% of the 27,864 protein-coding genes identified in peach. This proportion is similar to that described in Arabidopsis, where Riechmann (2006) estimated that of the 26,000 protein-coding genes, 6.4% were TFs. Among the TF families encoded by the different plant genomes, around 30 families have more than 1000 member genes identified in the PlantTFDB database. With respect to the peach genome, only four families of these TFs [bHLH; ERF (mTERF); MYB; and NAC] have more than 100 identified members, and just 10 of these TFs have 50 or more member genes per family (Table 1). As regards the family size comparison, members of FAR1 are more abundant than members in the Arabidopsis and poplar genomes, while ARF, SBP, ARR-B, CO-like, NF-YA, SRS, BBR/BPC, and LSD families are smaller (Table 1; Supplementary Material, Table S1).
The information about TFs contained in the PlantTFDB database was checked in the GDR database (www.rosecae.org), revealing a wide distribution of the 1529 TFs identified in peach, with a higher number of TFs (transcripts) on pseudomolecule 1 (312 TFs) (Table 2; Supplementary Material, Table S2).
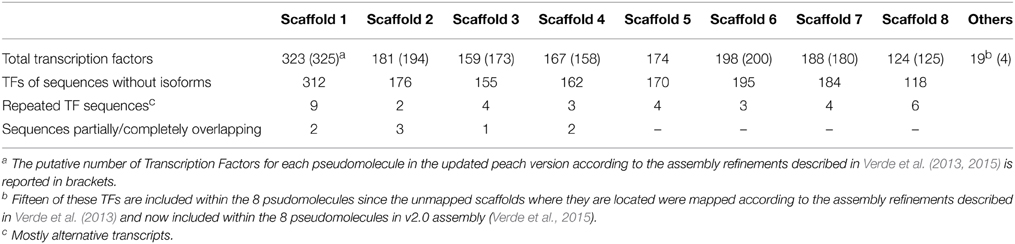
Table 2. Number and distribution of Transcription Factors (TFs) in each pseudomolecule (Scaffold1-8) of the Peach v1.0 genome sequence.
In fruit trees, including Prunus, understanding how morphological and phenological traits (flowering timing, bud dormancy, bud and fruit development, cultivar acclimation, chilling requirement, among others) behave in different and changing environments is very important in the search to identify genotypes with better fruit quality, productivity and growth potential to be used in breeding programs. Accordingly, TF studies in Prunus have been performed at the gene expression level for several agronomic traits, such as control of the flowering process, tree shape, fruit quality, and drought and disease resistance, which are found not only in peach, but also in other Prunus species. These TF studies have mainly been performed in peach (Table 3) and to a lesser extent in other species, including almond [P. amygdalus (Batsch) syn. P. dulcis (Miller) Webb], apricot (P. armeniaca L.), black cherry (P. serotina Ehrh), fuji cherry (P. incisa Thunb.), Japanese apricot (P. mume Sieb. Et Zucc.), Japanese plum (P. salicina Lindl), and sour (P. cerasus L.) and sweet (P. avium L.) cherry (Table 4). TF analyses have mainly been performed using quantitative RT-PCR to amplify the known sequences of these TFs. Other tools assayed include cDNA-AFLP, LC-ESI-MS, and RNA and DNA blotting or mapping. More recently, new tools assayed include microarray and high-throughput DNA (DNA-Seq) and RNA (RNA-Seq) sequencing (Tables 3, 4).
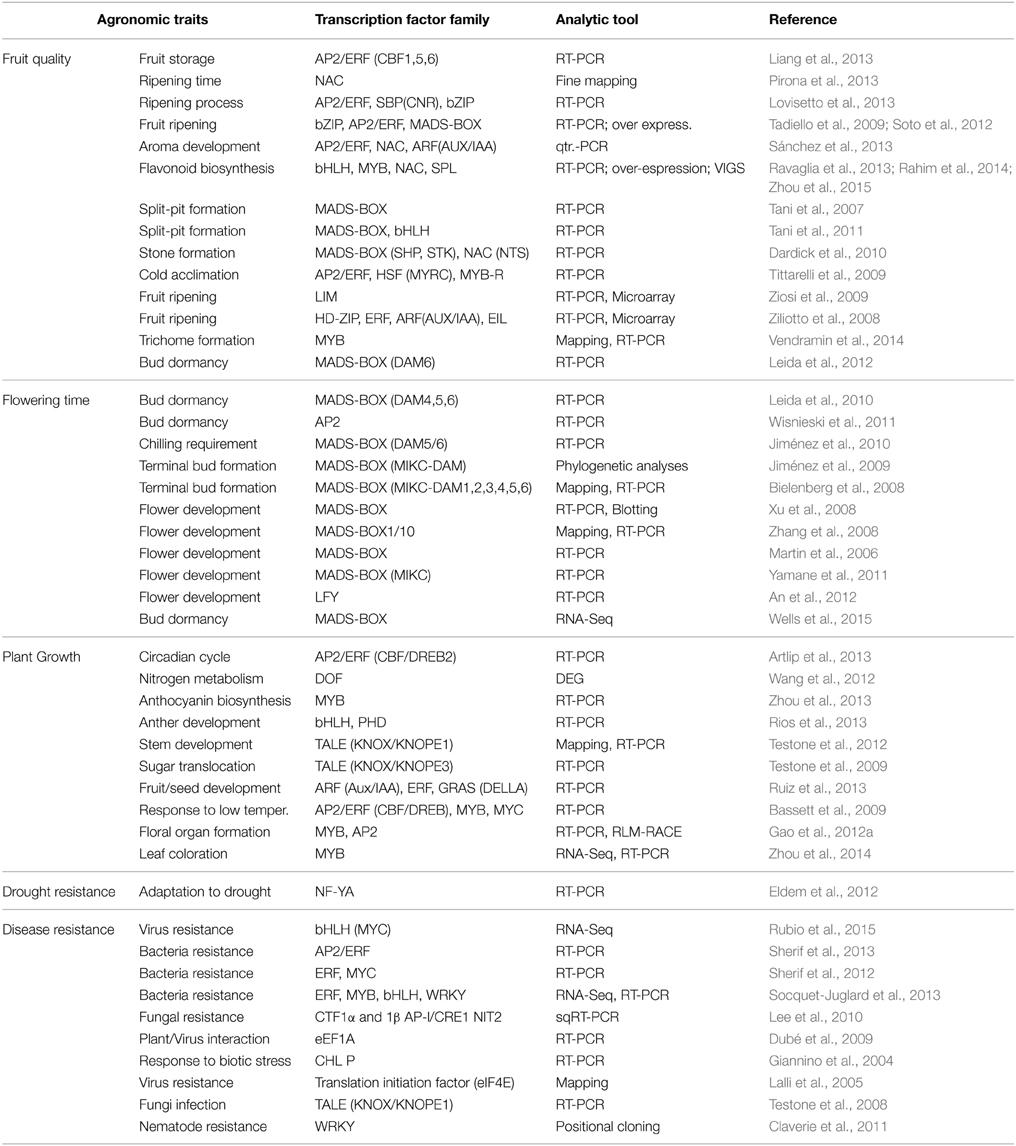
Table 3. Transcription factors (TFs) assayed in peach in the study of different agronomic traits.
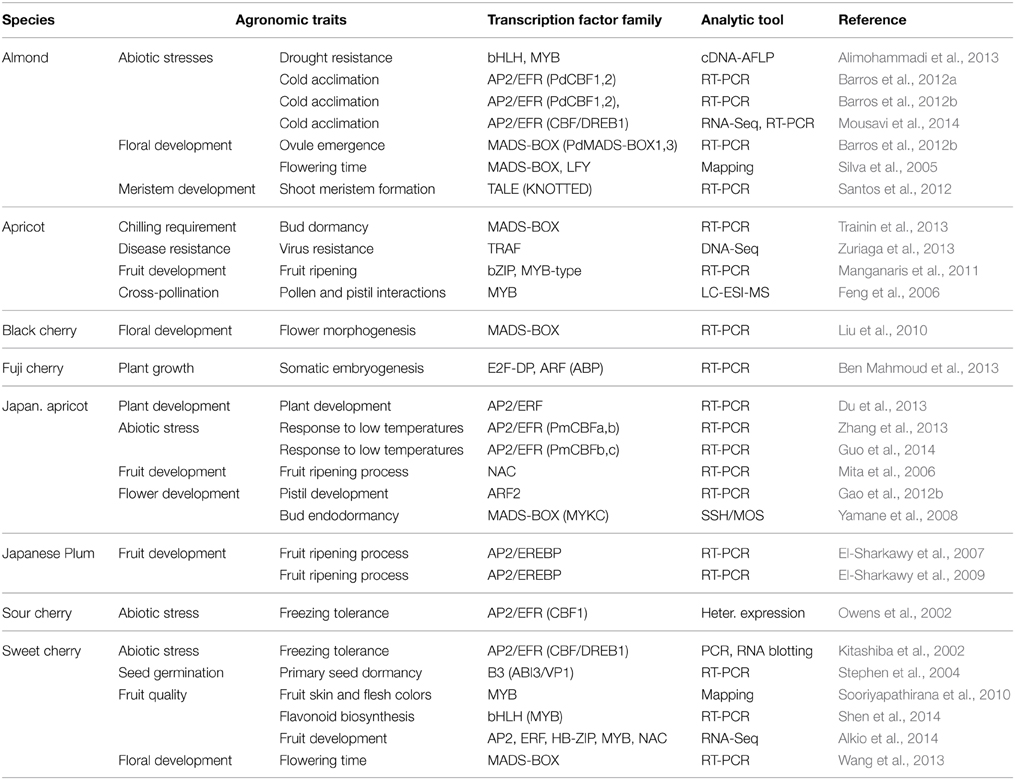
Table 4. Transcription factors (TFs) assayed in almond, apricot, black cherry, sweet cherry, Japanese apricot, and Plum in the study of different agronomic traits.
Flowering Date Control
Late flowering is an important agronomic trait for avoiding spring frost in Prunus species, particularly in the case of the earlier flowering species such as almond. Furthermore, the development of cultivars with early flowering has made Prunus species production a reality in subtropical areas. More knowledge about the factors involved (TFs) in the control of dormancy and flowering date can help in the development of new Prunus genotypes with either later flowering dates and higher chilling requirements to break dormancy to avoid frost or earlier flowering dates and lower chilling requirements to be grown in subtropical areas (peach and Japanese plum) for early production (Wells et al., 2015).
In the case of Prunus and other woody plants of the Rosaceae family, such as apple and pear, dormancy is a mechanism that allows the plants to withstand low temperatures and acclimate to winter conditions. There is usually a relationship between flowering date, bud dormancy, and chilling and heat requirements (Sánchez-Pérez et al., 2012, 2014). In the case of peach, several members of the MADS-BOX TF family (MIKC-DAM 1, 2, 3, 4, 5, and 6) are differentially expressed and have been associated with the control of genes responsible for arresting meristem development, for terminal bud formation and for bud dormancy (Bielenberg et al., 2008; Jiménez et al., 2009). In Arabidopsis, MADS-BOX TFs have been identified as being involved in floral organ identity and in the control of petal, stamen, and carpel development (Parenicová et al., 2003).
In peach, a group of DAM (dormancy-associated) SVP-like (Short Vegetative Phase) MADS-BOX TFs located in the evergrowing (EVG) region has been described as being responsible for the absence of vegetative endodormancy (Li et al., 2009; Jiménez et al., 2010). This MADS-BOX domain (from the founding MCM1, AGAMOUS, DEFICIENS, and SRF TFs) is a conserved DNA-binding region present in a variety of TFs representing a large multigene family in plants. In the peach genome, 79 MADS-BOX TFs have been described, and their annotation has been manually curated (Verde et al., 2013; Wells et al., 2015). Many of the genes of the MADS-BOX family are involved in different steps of flower development, including flowering time determination (Riechmann and Meyerowitz, 1998); bud dormancy (Leida et al., 2010, 2012; Zhong et al., 2013); terminal bud formation (Bielenberg et al., 2008; Jiménez et al., 2009); and flower development (Martin et al., 2006; Xu et al., 2008; Zhang et al., 2008). Yamane et al. (2011) analyzed the expression of PpDAM5 and PpDAM6 during flower bud development in peach cultivars with different chilling requirements, finding that both genes are up-regulated during flower organ differentiation and then down-regulated during flower organ enlargement. Similar patterns of expression for PmDAM5 and PmDAM6 genes were observed in P. mume by Zhong et al. (2013), suggesting that these genes might contribute significantly to terminal bud set and dormancy induction and that their transcript levels could thus provide some sort of measurement of the specific chilling requirements for dormancy release.
It has recently been reported that the expression of DAM 5 and 6 peach genes can be controlled by chromatin remodeling and modification factors [e.g., a putative SWI3C-like element of the SWITCH/SUCROSE NONFERMENTING (SWI/SNF) remodeling complex (ppa001566m); an HDA2-like histone deacetylase (ppa006590m); and a HAM2-like histone acetyltransferase (ppa005747m)] that are co-localized in the same quantitative trait locus (QTL) (Romeu et al., 2014). It is worthy to note that DAM 6 from peach is regulated at the chromatin level by demethylation of H3K4, trimethylation of H3K27 and acetylation of H3 following chill accumulation (Leida et al., 2012).
In Japanese apricot, different ARF-related TFs appear to play an important role during the four stages of seasonal bud dormancy by regulating (both as inducers and repressors) the transcription of auxin-related genes and, thus, the responsiveness to auxin. The interaction between ethylene, ABA, and JA in the transition among the different dormancy phases is worthy of note (Zhong et al., 2013). In particular, Zhong et al. (2013) reported that the JA Carboxy 1 Methyltransferase, EFR1 and ERF5 genes were down-regulated in endodormancy compared with the paradormancy stage, suggesting a strong interaction between JA and ethylene in the establishment of dormancy. On the contrary, two ABA-related genes (ppa006696m and ppa008716m) were up-regulated in endodormancy and presented lower expression levels in the paradormancy stage.
Another class of TFs belonging to the CBF family has been well-documented as being related to cold response and acclimation in peach, almond, apricot, cherry, and Japanese apricot (Kitashiba et al., 2002; Owens et al., 2002; Tittarelli et al., 2009; Li et al., 2009; Barros et al., 2012a,b; Trainin et al., 2013; Zhang et al., 2013; Guo et al., 2014). CBF proteins belong to the CBF/DRE binding (DREB) sub-family of the Apetala2-ethylene responsive factor (AP2/ERF) (Nakano et al., 2006). AP2/ERF or AP2/EREBD (Ethylene Responsive Element Binding Factor) is a multigene superfamily of TFs that act under different growth and developmental mechanisms used by plants to respond to several biological processes and to several types of biotic and abiotic stresses. This TF family is large but unique to plants, and the identity of sequences among different AP2/ERF genes has been estimated to be as low as 13% (Riechmann and Meyerowitz, 1998; Sakuma et al., 2002). This TF family is divided into three subfamilies: the AP2 family proteins that contain two repeated AP2/ERF domains; the EREBP genes with a single AP2/ERF domain (Shigyo et al., 2006); and the RAV family proteins that contain a B3 domain, which is a DNA-binding domain conserved in other plant-specific TFs, in addition to the single AP2/ERF domain (Nakano et al., 2006). Proteins of the AP2/ERF family have been shown to participate in the regulation of developmental processes, like flower development, spikelet meristem determinacy, leaf epidermal cell identity, and embryo development.
Zhebentyayeva et al. (2014) developed a comprehensive program to identify genetic pathways and potential epigenetic mechanisms involved in the control of chilling requirement and flowering time in peach. In almond, integrating genomic and transcriptomic approaches, Silva et al. (2005) described several QTLs (Quantitative Trait Loci) linked to flowering time in an interspecific F2 almond × peach progeny using a Candidate Gene approach (CG) including LFY and MADS-BOX TFs. More recently, two C-repeated binding factor genes in almond (PdCBF1 and PdCBF2) were analyzed in flower buds and shoot internodes, showing that PdCBF2 increased in transcript abundance during cold acclimation, while PdCBF1 was expressed during the summer. Similarly, in P. mume, Guo et al. (2014) found that the PmCBFa, PmCBFb, and PmCBFc genes were cold induced, and the mRNA content was higher in the plants after 168 h of low temperature exposure than at 0 h. However, the mRNA content of PmCBFa and PmCBFb was higher than that of PmCBFc, especially after 168 h, suggesting fewer transcripts of this gene in the flower buds of P. mume in late winter. These results were attributed to the great variation among CBF genes, which can explain the variation in cold tolerance among P. mume populations. Interestingly, CBF-specific CTR/DRE cis elements in promoters of peach PpDAM 5 and PpDAM 6 genes were also found, suggesting their association with a CBF-regulon (Barros et al., 2012b).
In addition to the findings described above, TERMINAL FLOWER1 (TFL1) and FLOWERING LOCUS T (FT) have been identified as being key regulators of flowering time and inflorescence development, but with antagonistic functions (Sánchez-Pérez et al., 2014). In black cherry (Prunus serotina Ehrh.), Wang and Pijut (2013) cloned two TFL1 homologous genes that presented high expression levels in shoot tips and vegetative buds, acting as repressors of floral genes and in the maintenance of vegetative growth. Furthermore, it has been suggested that TFL1 interacts with the bZIP transcription factor FD, repressing the transcription of the FD-dependent genes AP1 and AG, while FT has an activation effect under AP1 and AG. Nevertheless, these authors observed that FT activity was more important in the timing of flowering than TFL1, suggesting that FT and TFL1 have opposite functions in regulating flowering time.
Fruit and Seed Development
One of the main objectives of all Prunus breeding programs has traditionally been to obtain new genotypes with improved fruit quality according to consumer demand production costs and processes and distribution logistics. Fruit quality involves an important group of traits that determine the success of a new cultivar, such as aroma, solid soluble content (SSC), titratable acidity, health attributes, and both skin and flesh color, among other characteristics (Infante et al., 2011). The study of the implications of the different TF families in the processes related to fruit quality in the different Prunus species provides new opportunities for the marker-assisted breeding of genotypes with more extensive maturity date and the potential to preserve fruit quality after harvesting. Other opportunities include the identification of Prunus rootstocks whose endocarp shows less physical resistance to cracking by natural seed power during the germination process, which is a desirable characteristic.
Phenolic compounds are the precursors of anthocyanins, flavones and proanthocyanidin biosynthesis in the flavonoid pathway (D'Archivio et al., 2007), and these compounds also play a central role as determinants of fruit quality. The most important phenolic compounds are the antioxidant components in fresh fruit (Vinson et al., 2001). The accumulation of these compounds in fruit provides essential cultivar differentiation for consumers and represents an important factor for marketability (Andreotti et al., 2008). TFs of distinct families have been identified as regulating the transcription control of the flavonoid pathway. In this process, R2R3-MYB and basic Helix-Loop-Helix (bHLH) TFs form a complex with WD40 proteins (termed the MBW complex) to activate the anthocyanin and proanthocyanidin biosynthetic genes (reviewed in Petroni and Tonelli, 2011). The MBW complex usually regulates groups of flavonoid biosynthetic genes, and this regulation is via specific binding to motifs in the promoters of the pathway genes (Hartmann et al., 2005). In apple, Espley et al. (2007) have demonstrated that the efficient induction of anthocyanin production during ripening depends upon the co-expression of MYB TFs (MdMYB10) and two bHLH TFs (MdbHLH3 and MdbHLH33). Similarly in peach, the anthocyanin production occurring at ripening, mainly in the peel and in the mesocarp around the stone, is regulated by the coordinated action of MYB10-like and bHLH TFs (Rahim et al., 2014). Three highly similar MYB10-like genes (named MYB10.1, 2, and 3) form a small cluster on chromosome 3 and are closely associated with Ag (anther color), a trait responsible for pigment accumulation in anthers. Transactivation experiments identified PpMYB10.1 and PpbHLH3 as the best partners for the induction of anthocyanin production both in tobacco leaves and the peach mesocarp, thus indicating that the corresponding genes are good targets for genetic improvements (Rahim et al., 2014). Moreover, ppa018744, named MYB10.4, was associated with leaf red coloration in peach (Zhou et al., 2014). Furthermore, it must be noted that a major QTL for skin and flesh color has been mapped in the syntenic region of sweet cherry (Sooriyapathirana et al., 2010).
In peach (and other Prunus species), color formation due to anthocyanin accumulation is also important in flower petals. Pigment accumulation is also regulated by an MYB TF in the petals, although this MYB TF belongs to a different group of MYB10s (Uematsu et al., 2014). Nevertheless, other MYB10-like genes (ppa024617m and ppa010069m) that remain uncharacterized might be important for anthocyanin accumulation either in petals or aging leaves (Rahim et al., 2014). Moreover, ppa018744, named MYB10.4, was associated with leaf red coloration in peach (Zhou et al., 2014). This fact reveals the complexity of the regulation of anthocyanin synthesis, but at the same time it adds more possibilities for the genetic manipulation of this process.
In sweet cherry (Prunus avium L.), Shen et al. (2014) identified the PacMYBA gene, an MYB TF, that was associated with anthocyanin accumulation and that interacted with bHLH TFs to regulate the expression of anthocyanin pathway genes. Although the PacMYBA gene was expressed in several organs and tissues, PacMYBA expression was greatest in the skin of mature fruits and appeared to be directly up-regulated by ABA production. These results indicate that ABA production and PacMYBA expression work together to control anthocyanin synthesis in sweet cherry.
It has been claimed that the MYB TFs play an important role in other plant growth and developmental processes. Vendramin et al. (2014) characterized the gene ppa023142m (PpeMYB25) that encodes an MYB TF, which acts as a positive regulator in trichome formation and is responsible for the fuzzy skin trait in peach. These authors identified an insertion of a Ty1-copia retrotransposon within the PpeMYB25 gene that disrupted the gene, leading to a recessive loss-of-function mutation underlying the nectarine phenotype. The involvement of MYB TFs in the regulation of epidermal cell differentiation and fruit development was also suggested by Alkio et al. (2014). In analysing the exocarp-specific transcripts of sweet cherry fruits, these authors identified the R2R3-MYB Pa_22147 gene, which is also related to anthocyanin biosynthesis. Another three genes (Pa_08841, Pa_02691, and Pa_19618) related to ERF TFs were associated with the regulation of cutin and wax deposition in the exocarp. In addition to these results, the exocarp-specific Pa_05584 gene, an HD-ZIP-related TF, showed high expression levels in later stages (II and III) of sweet cherry fruit development and played a consistent role in cuticular lipid and anthocyanin biosynthesis.
Other processes and characteristics related to fruit quality include fruit development, the potential for fruit storage and the ripening process. Several TFs have been linked to the control of these events, including AP2/ERF; SBP (CNR); bZIP; NAC; HD-ZIP; ARF (and the ARF regulating proteins AUX/IAA); EIL; and LIM (Trainotti et al., 2007; Ziliotto et al., 2008; Ziosi et al., 2008; Soto et al., 2012; Liang et al., 2013; Lovisetto et al., 2013; Pirona et al., 2013). Shigyo et al. (2006) described that the AP2/ERF TFs act under several biological processes and that these TFs may play an important role in fruit growth and development in climacteric fruits (i.e., peaches, nectarines, and Japanese plums), especially in the ethylene signal transduction pathway. ERFs are plant-specific, nucleus-localized proteins. They serve as TFs that bind conserved motifs in promoter regions of target genes (Zhang et al., 2012), providing a route for ethylene signal activation at the level of target gene transcription, suggesting the involvement of ERFs in the ripening process of climacteric fruits (El-Sharkawy et al., 2009). The peach transcript model ppa010982m, similar to the ETHYLENE RESPONSIVE ELEMENT BINDING FACTOR 4 (ERF4) from Arabidopsis, has already been proposed as a candidate gene for fruit maturation date in different climacteric Prunus species (Dirlewanger et al., 2012).
Fruit development results in an increase in size through both cell division and expansion. The SBP genes were first characterized as SQUAMOSA binding proteins (SBPs) that regulate the expression of MADS-BOX genes in early flower development (Klein et al., 1996), and they also play a critical role in regulating flowering in addition to affecting fruit development (Manning et al., 2006). An exhaustive analysis of SBP tomato gene expression revealed that a large proportion of members were ubiquitously and constitutively expressed (from seedling to ripe fruit), while other members showed a more differentiated expression overall (Salinas et al., 2012). In particular, transcripts of SlySBP12b, SlySBP10 and CNR were highly accumulated in ripe fruit. In peach, the gene ppa022739m that codes for a putative TF containing the Squamosa-Promoter Binding Protein (SBP) domain is located in the region in which the major QTL controlling fruit maturation time was mapped (Romeu et al., 2014). On the same locus, the NAC1 ppa008301 has been proposed as a candidate for controlling the harvest date (Pirona et al., 2013).
The involvement of the bZIP gene family in fruit ripening in apricot and peach has also been described by Manganaris et al. (2011). These authors identified different contigs showing homology to the protein phosphatase PP2C family members, such as ABI1 and ABI2, which were only differentially expressed in apricot. ABI1 has been considered as a negative regulator of ABA signaling, and in apricot ripening, the expression of PP2C members was higher than in peach, suggesting a lower sensitivity of apricot to ABA. ABA signaling is linked to the gene Contig298, which, besides being homologous to the TF ATB2/bZIP11 belonging to the bZIP family, is also transcribed more abundantly in ripe peach and immature apricot. In the fleshy fruit of apricot and peach, however, the ATB2/bZIP11 function and the relationship with ABA are not clear. Furthermore, the exogenous application of jasmonates (JAs) during fruit ripening altered the level of transcripts of bZIP contig298, which is associated with developmental regulation (Soto et al., 2012). According to Lovisetto et al. (2013), the dilated ripening and the enhanced metabolism of tomato fruit over-expressing the peach bZIP gene suggests that this gene might participate in ripening regulation, but its molecular action remains unknown.
Fresh fruit quality is closely linked to the control of senescence of the fruit. The NAC TFs were derived from the names of three proteins: NAM (no apical meristem), ATAF1-2, and CUC2 (cup-shaped cotyledon) (Souer et al., 1996). These TFs were initially recognized as factors implicated in various processes of plant development, such as in the response to pathogens and viral infections. More recently, the NAC TFs have been reported to play an essential role in regulating cell division and cell senescence. In the analysis of two populations of peach, segregating for a maturity date locus, Pirona et al. (2013) identified a variant NAC gene (PpNAC1, ppa008301m) on chromosome 4 that was shown to co-segregate with the fruit maturity locus, suggesting this gene as a candidate for controlling ripening time in peach. This gene has been shown to interact with a second NAC, mapped on chromosome 5 and named BLOOD (BL), because it is responsible for the blood-flesh trait (Zhou et al., 2015). The heterodimer BL/PpNAC1 transactivates the expression of the abovementioned PpMYB10.1 gene, thus leading to anthocyanin production in the mesocarp of BL/BL and BL/bl genotypes. The activity of the BL/PpNAC1 heterocomplex is repressed by PpSLP1, an SBP encoded by a gene whose expression ceases at ripening, thus allowing PpMYB10.1 transcription and the resulting anthocyanin accumulation (Zhou et al., 2015). In sweet cherry, Alkio et al. (2014) also identified an NAC gene related to fruit ripening.
The involvement of ARFs and their cognate proteins (Aux/IAA proteins) in peach and apricot fruit ripening has been widely studied (Trainotti et al., 2007; Bonghi et al., 2011; Manganaris et al., 2011). Nonetheless, only four of the 17 ARF genes present in the peach genome have been studied. The role of these TFs in the early phases of fleshy fruit development must therefore be investigated, just as it has recently been deeply examined in tomato (Zouine et al., 2014). In fact, the expression of tomato ARFs has been found to sharply increase upon pollination/fertilization. Given the role of auxin signaling in the fruit set process (De Jong et al., 2009; Devoghalaere et al., 2012), the dynamics of the expression pattern of tomato ARFs is indicative of their putative involvement in mediating auxin responses during the flower-to-fruit transition. Genome-wide expression profiling using RNA-Seq has revealed that tomato ARF genes are regulated by both ethylene and auxin, suggesting the potential contribution of these genes to the convergent mechanism between the signaling pathways of these two hormones. To reinforce this theory of co-operation between auxin and ethylene in the control of fruit ripening, it is worthy to note that the ppa003113m gene, similar to ETHYLENE-INSENSITIVE3-LIKE 3 (EIL3), together with genes related to auxin synthesis and ARFs (ppa002986m, ppa001557m, and ppa002082m), has been located in a region containing a peach QTL associated with fruit ripening time (chromosome 6) (Romeu et al., 2014). It is worth remembering that the expression of softening-related genes, such as Endopolygalacturonase (PpPG) and Expansin3 (PpExp3) in peach fruits, is regulated by ethylene at the transcriptional level (Hayama et al., 2006) and is required for the progression of the fruit softening process. Fruit softening is essential for fruit quality, yet the control of this process is very important for extending the shelf life of post-harvest fruits, especially in peach, nectarine, plum, and apricot.
In peach, the LIM gene has also been associated with changes in firmer flesh that contribute to the regulation of the cell wall structure under signaling by MJ. In peach fruits where JAs were exogenously applied, Ziosi et al. (2008) identified a ripening delay due to a possible interference with ripening and stress-related genes. It was supposed that LIM TF may alter the phenylpropanoid pathway in MJ-treated fruit, leading to an accumulation of lignin precursors, contributing to cell wall strengthening and acting in the process of delaying ripening in peaches. In the case of Prunus species, lignification is a crucial event during fruit development, taking into account that the fruit is a drupe. Endocarp lignification plays a critical role from a practical point of view, because peach varieties showing a phenotype called “split pit,” where the endocarp does not seal along the suture, have seeds that are more vulnerable to pests and disease.
On the other hand, during early phases of peach fruit development, simultaneous activation of the lignin and flavonoid pathways in the mesocarp and endocarp has been detected (Dardick et al., 2010; Hu et al., 2011), while in later phases, high spatial specificity in terms of transcripts, protein and metabolite accumulation has been observed. In fact, in the endocarp, the activation of genes involved in the lignin pathway is accompanied by a repression of genes responsible for flavonoid metabolism, while in the epicarp and mesocarp, these two pathways are regulated in the opposite manner (Dardick and Callahan, 2014). This result suggests that drupe patterning is controlled by a highly coordinated gene network. On the basis of expression profile data, it can be seen that a pivotal role is played in this network by the same TFs that control dehiscence in Brassica species (Dardick et al., 2010). Dardick et al. observed that the expression of the peach homologs of SHATTERPROOF (SHP), SEEDSTICK (STK), and FRUITFUL (FUL), three MADS-BOX genes, was spatially controlled and was restricted to the endocarp for SHP and STK, while FUL transcript accumulation was higher in the mesocarp but constitutively low in the endocarp. This observation is consistent with the theory of a possible role of these genes in delimiting endocarp lignification margins, as demonstrated in the Arabidopsis thaliana silique (Ferraìndiz et al., 2000). Furthermore, Tani et al. (2007) found that SHP expression in a split pit resistant variety was lower during the lignification stage, while FUL expression was significantly elevated in the sensitive variety during later stages of fruit growth.
In the endocarp, expression of SHP and STK is higher until the onset of lignin accumulation. Later on, this event is paralleled by the expression of a peach homolog of NST1 (No Secondary wall Thickening), an NAC TF that rapidly accumulates along with secondary metabolism and cell wall biosynthesis genes, as observed in Arabidopsis (Mitsuda et al., 2005). In addition to these genes, the expression of a peach homolog of SPATULA (SPT), a bHLH TF involved in the control of siliqua valve identity (Groszmann et al., 2011), is consistent with a role in specifying endocarp margins (Tani et al., 2011). Collectively, these data imply that highly similar pathways likely control development in both Prunus and Brassica fruits. Studies of this characteristic raise two contrasting perspectives. On the one hand, the “split pit” phenotype is not wanted because it enhances seed vulnerability, and furthermore, for canning peach cultivars, the “split pit” also creates problems during industrial peach processing. On the other hand, however, while some peach cultivars have a high percentage of germination without mechanically cracking the endocarp, in some peach rootstocks, the endocarp suture is so adhered/lignified that it creates a physical barrier, and seed germination is drastically reduced without mechanical endocarp cracking to release the seeds followed by stratification. To further complicate the situation, the SHP/PLENA peach gene is involved not only in flower development but also in the activation of the ripening process by regulating the expression of ripening-related genes (Tadiello et al., 2009). This is similar to the role played by TAGL1, the tomato ortholog of SHP/PLENA (Vrebalov et al., 2009). Fine-tuning the functions in which this peach gene is involved is therefore a demanding task for breeders.
Resistance to Biotic Stresses
Breeding for pest and disease (biotic stress) resistance is another important breeding objective in Prunus. In this sense, knowledge about the molecular basis of resistance to different pathogens and the role of the different TFs in this process is of critical interest in the development of efficient breeding strategies and markers for selection.
The involvement of the pathogen resistant genes PR1 (Pp-PR1a, Pp-PR1b) and three PR5s (Pp-TLP1, Pp-TLP2, and Pp-TLP3) in the resistance to Xanthomonas arboricola pv. pruni in peach was investigated by Sherif et al. (2012), who verified an induction of PR genes in response to bacterial infection. In this case, the interaction of both signaling molecules and TF MYC2 (JA signaling), ERF (JA/ET signaling), WRKY (SA signaling), and TGA (SA signaling), is determinant in mediating resistance against this pathogen. Lee et al. (2010) investigated the role of the CUTINASE gene (MfCUT1) in wild-type (WT) and MfCUT1-overexpressing transformants of M. fructicola and described several TFs that may be involved in the redox regulation of MfCUT1 expression. The presence of NIT2 in the MfCUT1 promoter region indicates a possible effect of starvation as a form of nitrogen limitation that may regulate MfCUT1 expression, because NIT2 is a nitrogen metabolic regulator and mediates the repression of its target genes when primary nitrogen sources are available. Another TF involved in the response to MfCUT1 expression is an AP-l protein (Activator Protein) that has been linked to signal transduction pathways coupled with oxidative stress.
In peach leaves inoculated with X. arboricola pv. pruni, Sherif et al. (2013) also identified three genes that encode ERF repressors, PpERF12, PpERF3a, and PpERF3b, which showed higher induction in the susceptible peach genotype evaluated than in the resistant one. These results suggest a negative role for these genes in disease resistance. In additional analyses, transgenic Nicotiana tabacum plants overexpressing PpERF3baΔEAR showed less disease symptoms than either plants overexpressing the full-length gene or WT plants, suggesting that the resistance of PpERF3baΔEAR plants is associated with the enhanced induction of pathogenesis-related (PR) genes.
In addition, the transcriptome analysis of peach leaves inoculated with X. arboricola pv pruni revealed a total of six potential ERF TFs, but only one was up-regulated at 2 h post-inoculation (hpi) (Socquet-Juglard et al., 2013). Furthermore, the gene ppa006485m, which is similar to a gene encoding a Mitogen-activated protein kinase kinase kinase (MAPKKK15), was down-regulated, while the genes ppa015973m and ppa018075m, which could putatively belong to the MYB and WRKY family TFs, respectively, were both up-regulated at 12 hpi. These authors also identified three genes similar to bHLH TFs. One of these genes (ppa017640m) was differentially expressed at 2 hpi, and two (ppb012603m and ppa022385m) were differentially expressed at 12 hpi. Furthermore, another four genes (ppa012687m, ppa012737m, ppa012242m, and ppa011359m) belonging to zinc finger families were identified and linked to basal defense against pathogen attacks.
To look at another example, the root-knot nematode (RKN) belonging to the Meloidogyne genus is among the parasites that cause the greatest damage to the roots of Prunus trees around the world. In “Myrobalan plum” rootstocks (P. cerasifera), the Ma gene that confers complete-spectrum resistance to RKN was cloned and characterized by Claverie et al. (2011) as a TNL1 (TIR-NBS-LRR) gene, which contains five post-LRR (PL) exons and a conserved core motif [CG(a)RL(a)Y], similar to the WRKY transcription factor motif (WRKYGQK) from RRS1 identified by Deslandes et al. (2002). The similarity of the PL domains of TNL1 to the WRKY TFs implied that the key targets of the RKN species could be WRKY TFs (Claverie et al., 2011), due to their role as central components of many aspects of the innate immune system of the plant in addition to their basal effects on defense, systemic acquired resistance and plant development (Rushton et al., 2010). The new discoveries about TNL-WRKY protein involvement in plant pathogen resistance open up the possibility of identifying more RKN resistance genes in different Prunus species.
Finally, Plum pox virus (PPV, sharka disease) has been the most studied virus affecting Prunus species. From the genomics point of view, Zuriaga et al. (2013) identified TRAF transcriptional regulators as the genes responsible for resistance in apricot. More recently, Rubio et al. (2015) demonstrated that early PPV infection in peach leaves was associated with an induction of TFs related to pathogen resistance by jasmonic acid (JA). The increase in JA levels leads to a degradation of JAZ proteins and then to the depression of MYC2 (and its redundant homologs MYC3 and MYC4), bHLH TFs that play a central role in JA signaling, resulting in the transcriptional activation of downstream target genes (Katsir et al., 2008). Rodamilans et al. (2014) also described the role of the previously mentioned NBS-LRR genes in the hypersensitive response to PPV in Japanese plum.
Resistance to Abiotic Stresses
Drought, salinity and low temperatures are considered the most important abiotic stresses limiting fruit production and quality in Prunus. Identifying the genes and TFs related to these abiotic stresses, and understanding how gene expression is controlled under these conditions, could represent an important contribution for better managing plants, as well as for reducing the negative impact these stresses cause in fruit production. According to Eldem et al. (2012) the responses to drought stress are regulated at the transcriptional and post-transcriptional levels, and miRNAs have been identified as important gene regulators at post-transcriptional levels. These researchers characterized different miRNAs whose targets were various TF genes [NFYA (miR169) and DRE (miR169)] involved in plant responses to drought. NF-Y TFs are represented by NF-YA, NF-YB, and NF-YC families (Siefers et al., 2009). In Zea mays, Nelson et al. (2007) verified that the overexpression of NF-YB genes enhanced drought resistance. Li et al. (2008) described that NF-YA5 reduces anthocyanin production and stomata aperture, and control of stomata movement is an important mechanism for plants to control loss of water from the leaves during drought stress and to avoid dehydration. Alimohammadi et al. (2013) unraveled the interaction between protein AFC2 kinase and nuclear RNA splicing proteins (including SR45, SR33, SRZ-22, and RSZP21), which are involved in the sugar-mediated signaling pathway as well as in the epigenetic response via histone phosphorylation in the resistance to water deficit in wild almond P. scoparia. Interestingly, promoter analysis showed differentially expressed genes harboring binding sites of MYB1 and MYB TFs, which are involved in the dehydration response through the ABA signaling pathway.
Cold stress causes tissue injury, delay in growth and reduction in photosynthesis. Plants respond to low temperatures by altering the expression of thousands of genes including TFs (Chinnusamy et al., 2007). In almond, Barros et al. (2012b) showed that a progressive increase in the transcript abundance of PdCBF2 (Prunus dulcis C-repeat binding factor) during autumn was closely related to cold acclimation. The AP2 domain is also considered a regulatory element that stimulates transcription in response to low temperatures in plants (Díaz-Martín et al., 2005). The CBF genes belong to the AP2/EREBD multigene superfamily of TFs, and their relationship to cold response and acclimation in Prunus species is well-documented (Tittarelli et al., 2009; Barros et al., 2012a,b; Trainin et al., 2013). CBF genes are considered key regulators of cold acclimation, and the overexpression of CBF 1, 2, or 3 is capable of improving freezing tolerance in A. thaliana plants (Owens et al., 2002). They cloned a CBF1-ortholog gene of Fragaria × ananassa (FaCBF1) and P. cerasus (PcCBF1), and the mRNA levels were up-regulated in the leaves of both crops following exposure to 4°C for a period of between 15 min up to 24 h. In the receptacles of two CaMV35S-CBF1-transgenic lines of Fragaria × ananassa “Honeoye,” no significant changes in freezing tolerance were observed in comparison to wild-type plants. Nevertheless, the temperatures at which 50% electrolyte leakage occurred in detached leaf discs from the two transgenic lines were −8.2°C and −10.3°C, respectively, suggesting the influence of the FaCBF1 gene in cold acclimation. Kitashiba et al. (2002) isolated three DREB1/CBF-like genes from P. avium L. (sweet cherry), but only the expression of the D2 genes was found to be induced at low temperature. In Prunus mume, Zhang et al. (2013) also identified two CBF genes (PmCBFa and PmCBFb), homologs of the sweet cherry PaDREB gene, which were induced at low temperature. Mousavi et al. (2014) observed that the CBF/DREB1 TF was highly expressed in the ovaries of P. dulcis under freezing conditions, while no significant alteration in expression was observed in anthers, reinforcing the regulatory involvement of this TF family in cold acclimation.
Just as WRKY TFs have been linked to biotic stress responses as described above, they have also been associated with abiotic stress responses, like high salt or heat levels, osmotic stress, high CO2 levels, high ozone concentrations, and cold or drought. When plants are exposed to these abiotic stress situations, WRKY TFs form part of the signaling processes associated with transcriptional reprogramming, acting as negative or positive regulators (Rushton et al., 2010; Chen et al., 2012). Several WRKY proteins have been shown to be involved in plant drought and salinity stress responses (Golldack et al., 2011). In rice, the overexpression of the OsWRKY11 gene under the control of the HSP101 promoter has been shown to lead to enhanced drought tolerance and to increase the survival rate of green plant parts (Wu et al., 2009). In A. thaliana, the WRKY25 and WRKY33 genes have been shown to be responsive to both osmotic and oxidative stress. The down-stream regulated target genes of WRKY33 include transcripts with a role in ROS detoxification, such as peroxidases and glutathione-S-transferases (Jiang and Deyholos, 2009), suggesting that WRKY factors play a role as key regulators in both osmotic and oxidative stress adaptation (Golldack et al., 2011). Interaction between the WRKY TFs and an ethylene response transcriptional co-activator (ERTCA) has also been identified. This interaction was specifically induced during a combination of drought and heat shock in tobacco (Rizhsky et al., 2002), which suggests that this combination is accompanied by the activation of a unique genetic program that differs from the programs activated in plants during either drought or heat shock alone.
TFs and miRNAs are Coordinated in the Regulation of Target Genes Involved in Organ Development and Response to Abiotic Stresses
Studies on miRNAs have demonstrated that TFs are one of the main targets of these genes (Molesini et al., 2012). Different miRNAs target transcripts encoding TFs controlling plant development and are involved in the abiotic stress response (Xia et al., 2012).
Computational studies indicate that miRNAs and TFs appear to form a complex regulatory network with their target genes. These two regulatory circuits are strongly related, allowing for the coordination between the transcriptional and post-transcriptional control of their target genes (Cui et al., 2007). In fact, genes with more TF-binding sites have a higher probability of being targeted by miRNAs and have more miRNA-binding sites on average. In this context, the identification of miRNA targets via high-throughput degradome library sequencing, in addition to the identification of transcription factor binding sites (TFBSs) in the promoter region of target genes, can contribute to our understanding of developmental processes. In the case of the peach model species, this approach is feasible due to the availability of a high quality genome sequence, which makes extensive study of promoter regions and miRNA targets obtained from experimental procedures feasible. Using a degradome approach, Luo et al. (2013) identified 259 miRNA targets in peach, among which about 35% were TFs. It is worthy to note that MiR156 and MiR157, two conserved miRNAs, not only targeted SBP TF, but also targeted genes encoding protein associated with energy metabolism, glucose metabolism, redox status, and ion transport. The expression of many peach miRNAs is tissue-specific or developmental stage-specific (Gao et al., 2012a; Luo et al., 2013), suggesting coordination with TFs in the regulation of miRNA target expression, as observed in mammalian cells (Tan et al., 2008). Zhu et al. (2012) identified in peach three miRNAs that collectively target 49 MYBs, 19 of which are known to regulate phenylpropanoid metabolism, a key pathway associated with stone hardening and fruit color development, highlighting a critical role for miRNAs in the regulation of fruit development and ripening.
miRNAs and TFs have been claimed to be responsible for the high fluctuation in the expression profile of protein-coding genes in response to drought at the transcriptional and post-transcriptional levels (Sunkar et al., 2012; Nakashima et al., 2014). A genome-wide identification of miRNAs associated with drought in peach has made it possible to identify miRNAs targeting mainly TFs and transporters that are differently expressed in leaves and roots subjected to water stress (Eldem et al., 2012). These results reinforce the fact that the miRNA-TF regulatory network can differ among tissues. A similar approach has been used to identify miRNAs associated with the chilling response (Barakat et al., 2012). Several of the miRNAs identified in this case were induced in winter buds and co-localized with QTLs for chilling requirement and bloom date, thus making their gene targets potential candidates for mediating plant responses to cold stress.
New Breeding Opportunities
The post-genomic era in Prunus species, as well as in other plant species, is characterized by two elements that can cause a paradigmatic shift in the existing approaches: the development of complete reference genomes and the introduction of new methods of high-throughput sequencing of both DNA (DNA-Seq) and RNA (RNA-Seq) (Martínez-Gómez et al., 2012).
At this moment, only two complete reference genomes have been developed in Prunus. The IPGI (International Peach Genome Initiative) has released the complete peach genome sequence [peach genome (v1.0)], consisting of eight pseudomolecules (scaffold_1 to 8) representing the eight peach chromosomes accounting for up to 96% of the peach sequences (227.3 Mb) (Verde et al., 2013). This species presents important agronomical and molecular advantages, including self-compatibility, a short juvenile phase and a small genome size, which make it suitable as a model within the Prunus genus and the Rosaceae family (Jung et al., 2012; Verde et al., 2013). In addition, Zhang et al. (2012) assembled a 280 Mbp genome of Japanese apricot, anchoring 83.9% of the scaffolds to eight chromosomes.
The availability of these complete reference genomes (mainly the peach reference genome) presents one of the most interesting molecular opportunities for the identification of candidate genes linked to agronomic traits and for promoter identification from genomic data. It is now possible to locate the closest markers or candidate genes (including TFs) identified as associated with different QTLs linked to agronomic traits in the reference genome. These new opportunities are of particular interest in the case of Prunus, where knowledge concerning the link between genes and agronomic traits remains limited (Salazar et al., 2014).
With the genome sequences available, some strategies that could be used in the functional analysis of Prunus TFs include SNP genotyping assays and Genotyping by Sequencing. High-throughput SNP tools have recently been developed in Prunus species. In peach a 9K SNP array was developed using only exonic SNPs (Verde et al., 2012), while both exonic and intronic SNPs were used to construct the 6K cherry SNP array (Peace et al., 2012). In apricot a first approach to developing SNP markers combining RNA sequencing and SNPlex™ high-throughput genotyping technology has been recently described, and a significant decrease in the time and cost of genotyping has been estimated (Salazar et al., 2015). Some of these SNPs have already been located inside TF sequences. Due to their high abundance, SNP markers allow us to cover a large proportion of the genome and are ideal for mapping.
The selected genes and TFs can be blasted against the genomic sequences of peach and A. thaliana in the Phytozome database (http://www.phytozome.net/) to determine the corresponding orthologous genes/sequences in these genomes. The 1500 bp upstream of the transcriptional start point of the corresponding genes in peach and Arabidopsis genomes can then be extracted and considered as promoters. The upstream regions of the selected genes can be analyzed using PLANTPAN (http://plantpan.mbc.nctu.edu.tw) to predict TFs that can activate the selected genes. PLANTPAN finds the TFBs (regulatory elements) on the promoter regions of genes, and, based on the shared TFBs, predicts which TFs might bind/activate all or a majority of the considered genes. The location of the TFs in the peach reference genomes represents an additional advantage, because the gene functions are known. This fact could greatly facilitate the isolation of genes via QTL map-based cloning in the different Prunus species following the association of these TFs with the identified QTLs (Salazar et al., 2014) using peach as model species (Verde et al., 2013).
On the other hand, the high level of performance of new methodologies (“high-throughput” or “next generation” NGS) for DNA sequencing (DNA-Seq, in 2005) and the generation of cDNA from RNA (RNA-Seq, in 2008) have also been causing a revolution in biological research. In this context, the functional domains of TF genes can be used for developing informative genic microsatellite markers, such as those obtained in tomato and pepper (Yu et al., 2010) and chickpea (Kujur et al., 2013). These markers, designed transcription factor gene-derived microsatellite (TFGMS) and transcription factor functional domain-associated microsatellite (TFFDMS) markers, can be used in the high-throughput genotyping of new Prunus accessions. Moreover, DNA-Seq technology allows for easier resequencing of genotypes (Jackson et al., 2011), assuming a reference-like genome, in the identification of new TFs in different species. TF gene-derived markers are already a reality in peach. They have been used, for example, for the selection of nectarine- or peach-type fruits on the basis of the MYB25 sequence (Vendramin et al., 2014) for which a co-dominant functional diagnostic marker (indelG) has been proposed; for the selection of fruit maturity date using the NAC1 sequence variants (Pirona et al., 2013); and for red flesh color, on the basis of the marker linked to the BL allele (Zhou et al., 2015) (Figure 3). Moreover, other TFs are very good candidates for being the genetic determinants of other traits, such as an SBP and DAMS in QTLs controlling fruit maturation (on LG4, Romeu et al., 2014) and bud dormancy (LG1, Fan et al., 2010), respectively.
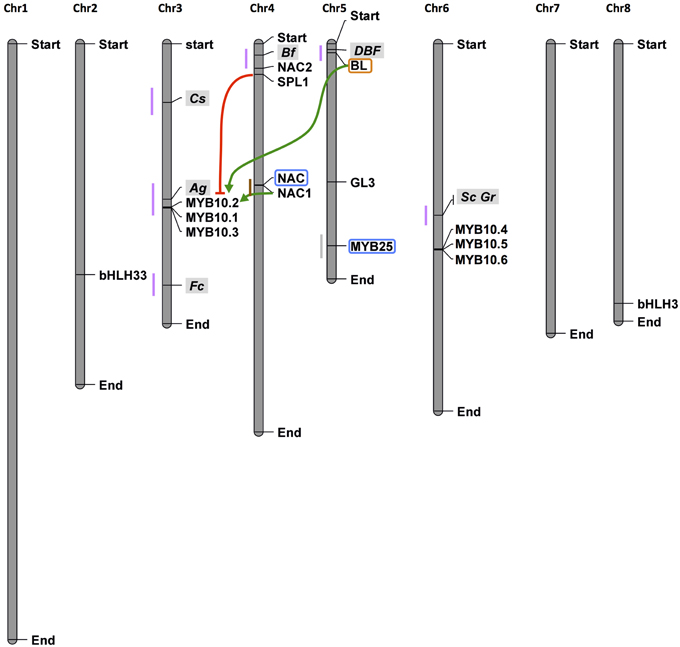
Figure 3. Localization on the peach genome map of loci and transcription factors controlling fruit traits. Fruit color is controlled by several loci within some of which TFs have been demonstrated to be the genetic determinant of the trait such as for BL in the DBF locus on top of chr5. BL interacts with NAC1 to positively (green arrows) regulate MYB10.1 The BL/NAC1 complex is repressed by SPL1 (red line) thus blocking MYB10.1 transcription. MYB10.1, forming a complex with bHLH3 and 33, positively regulate the transcription of structural genes in the flavonoid/anthocyanin pathway, determining pigments accumulation. Closed to NAC1, an additional NAC TF has been shown to contribute to the control of maturity date. On chr5 is located MYB25 that controls the peach/nectarine trait. Genetic markers developed on, or closely to the TF sequences are highlighted by the blue and orange boxes, respectively. These markers have been used to demonstrate that the genes are under the traits, thus will be used for breeding. Similarly, other TFs known be directly involved in the biological process responsible for a trait could be used to develop new genetic markers. Vertical gray bars represent the eight peach chromosomes. Small colored bars represent loci, which names are highlighted in gray, controlling color (purple), maturation date (brown), and peach/nectarine (light gray) traits. Loci for color are: Cs, flesh color around the stone; Ag, anther color; Fc, flower color; Bf, blood flesh; DBF, dominant blood flesh; Sc, fruit skin color; Gr, leaf color.
Finally, the well-known synteny among Prunus and Rosaceae genomes (Jung et al., 2009) and transcriptomes (Martínez-Gómez et al., 2011) offers additional molecular opportunities for the analysis of TFs linked to agronomic traits. We can consider the Prunus genus as a single gene pool (Jung et al., 2009). In this regard, it is important to note the transferability of molecular information about TFs identified in the different Prunus species. This synteny has already been studied in Prunus in relation to other genera inside the Rosaceae family (Jung et al., 2012). This synteny can result in homologous TFs from a common ancestral DNA including orthologous genes from different species or paralogous genes involving new functions (Shulaev et al., 2008).
Author Contributions
VB, IV, and PM participated in the coordination of the study. MR collected and revised the information about resistance biotic stress. IV collected and revised the information about flowering and bud dormancy. VB and PM collected and revised the information about abiotic stress resistance. LT and CB collected and revised the information about fruit and seed development and miRNAs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study has been supported by the projects AGL2010-16335 and AGL2013-43550-R from the Spanish Ministry of Economy and Competiveness to MR and PMG and in part by the Ministero delle Politiche Agricole Alimentari e Forestali–Italy (MiPAAF www.politicheagricole.it) through the project “DRUPOMICS” (grant DM14999/7303/08) to CB, LT, and IV. The authors would like to thank the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES)—Ministério da Educação (Brazil) for the fellowship (BEX9514/13-9) supporting the stay of VB in Spain. Dr. Md Abdur Rahim is thanked for his help in drawing Figure 3.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00443/abstract
Table S1. Plant transcription factor (TF) families of peach, Arabidopsis, and poplar described to date in the PlantTFDB database (http://planttfdb.cbi.pku.edu.cn/).
Table S2. Distribution of identified transcription factors in the peach v1.0 reference genome (http://www.rosaceae.org/species/prunus/prunus_persica). In each scaffold bold letters indicate alternative transcripts of the same protein coding gene. In addition, the correct v2.0 (http://www.rosaceae.org/gb/gbrowse/prunus_persica_v2.1) pseudomolecules where these genes are located, according to the assembly refinements described in Verde et al. (2013, 2015), are indicated in brackets.
References
Alimohammadi, A., Shiran, B., Martínez-Gómez, P., and Ebrahimie, E. (2013). Identification of water-deficit resistance genes in wild almond Prunus scoparia using cDNA-AFLP. Sci. Hort. 159, 19–28. doi: 10.1016/j.scienta.2013.04.023
Alkio, M., Jonas, U., Declercq, M., Van Nocker, S., and Knoche, M. (2014). Transcriptional dynamics of the developing sweet cherry (Prunus avium L.) fruit: sequencing, annotation and expression profiling of exocarp-associated genes. Hort. Res. 1:11. doi: 10.1038/hortres.2014.11
An, L., Lei, H., Shen, X., and Li, T. (2012). Identification and characterization of PpLFL, a homolog of FLORICAULA/LEAFY in peach. Plant Mol. Biol. Rep. 30, 1488–1495. doi: 10.1007/s11105-012-0459-x
Andreotti, C., Ravaglia, D., Ragaini, A., and Costa, G. (2008). Phenolic compounds in peach (Prunus persica) cultivars at harvest and during fruit maturation. Ann. Appl. Biol. 153, 11–23. doi: 10.1111/j.1744-7348.2008.00234.x
Ariel, F. D., Manavella, P. A., Dezar, C. A., and Chan, R. L. (2007). The true story of the HD-Zip family. Trends Plant Sci. 12, 419–426. doi: 10.1016/j.tplants.2007.08.003
Artlip, T. S., Wisniewski, M. E., Bassett, C. L., and Norelli, J. L. (2013). CBF gene expression in peach leaf and bark tissues is gated by a circadian clock. Tree Physiol. 33, 866–877. doi: 10.1093/treephys/tpt056
Arús, P., Verde, I., Sosinski, B., Zhebentyayeva, T., and Abbott, A. G. (2012). The peach genome. Tree Genet. Genomes 8, 531–547. doi: 10.1007/s11295-012-0493-8
Atkins, J. F., Gesteland, R. F., and Cech, T. R. (2011). RNA Worlds: From Life's to Diversity in Gene Regulation. New York, NY: Cold Spring Harbor; Cold Spring Harbor Laboratory Press.
Barakat, A., Sriram, A., Park, J., Zhebentyayeva, T., Main, D., and Abbott, A. (2012). Genome wide identification of chilling responsive microRNAs in Prunus persica. BMC Genomics 13:481. doi: 10.1186/1471-2164-13-481
Barros, P. M., Goncalves, N., Saibo, N. J. M., and Oliveira, M. M. (2012b). Cold acclimation and floral development in almond bud break: insights into the regulatory pathways. J. Exp. Bot. 63, 4585–4596. doi: 10.1093/jxb/ers144
Barros, P. M., Gonçalves, N., Saibo, N. J. M., and Oliveira, M. M. (2012a). Functional characterization of two almond C-repeat-binding factors involved in cold response. Tree Physiol. 32, 1113–1128. doi: 10.1093/treephys/tps067
Bassett, C. L., Wisniewski, M. E., Artlip, T. S., Richart, G., Norelli, J. L., Renaut, J., et al. (2009). Comparative expression and transcript initiation of three peach dehydrin genes. Planta 230, 107–118. doi: 10.1007/s00425-009-0927-1
Ben Mahmoud, K., Delporte, F., Muhovski, Y., Elloumi, N., Jemmali, A., and Druart, P. (2013). Expression of PiABP19, Picdc2 and PiSERK3 during induction of somatic embryogenesis in leaflets of Prunus incisa (Thunb.). Plant Mol. Biol. Rep. 40, 1569–1577. doi: 10.1007/s11033-012-2205-8
Bielenberg, D. G., Wang, Y., Li, Z. G., Zhebentyayeva, T., Fan, S. H., Reighard, G. L., et al. (2008). Sequencing and annotation of the evergrowing locus in peach [Prunus persica (L.) Batsch] reveals a cluster of six MADS-BOX transcription factors as candidate genes for regulation of terminal bud formation Tree Genet. Genomes 4, 495–507. doi: 10.1007/s11295-007-0126-9
Bonghi, C., Trainotti, L., Botton, A., Tadiello, A., Rasori, A., Ziliotto, F., et al. (2011). A microarray approach to identify genes involved in seed-pericarp cross-talk and development in peach. BMC Plant Biol. 11:107. doi: 10.1186/1471-2229-11-107
Bouché, N., Scharlat, A., Snedden, W., Bouchez, D., and Fromm, H. (2002). A novel family of calmodulin-binding transcription activators in multicellular organisms. J. Biol. Chem. 277, 21851–21861. doi: 10.1074/jbc.M200268200
Byzova, M. V., Franken, J., Aarts, M. G., de Almeida-Engler, J., Engler, G., Mariani, C., et al. (1999). Arabidopsis STERILE APETALA, a multifunctional gene regulating inflorescence, flower, and ovule development. Genes Dev. 13, 1002–1014. doi: 10.1101/gad.13.8.1002
Chen, L., Song, Y., Li, S., Zhang, L., Zou, C., and Yu, D. (2012). The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta 1819, 120–128. doi: 10.1016/j.bbagrm.2011.09.002
Chinnusamy, V., Zhu, J., and Zhu, J. K. (2007). Cold stress regulation of gene expression in plants. Trends Plant Sci. 12, 444–451. doi: 10.1016/j.tplants.2007.07.002
Claverie, M., Dirlewanger, E., Bosselut, N., Van Ghelder, C., Voisin, R., Kleinhentz, M., et al. (2011). The Ma gene for complete-spectrum resistance to meloidogyne species in Prunus is a TNL with a huge repeated C-terminal post-LRR region. Plant Physiol. 156, 779–792. doi: 10.1104/pp.111.176230
Cubas, P., Lauter, N., Doebley, J., and Coen, E. (1999). The TCP domain: a motif found in proteins regulating plant growth and development. Plant J. 18, 215–222. doi: 10.1046/j.1365-313X.1999.00444.x
Cui, Q., Yu, Z., Pan, Y., Purisima, E. O., and Wang, E. (2007). MicroRNAs preferentially target the genes with high transcriptional regulation complexity. Biochem. Biophys. Res. Commun. 352, 733–738. doi: 10.1016/j.bbrc.2006.11.080
Curaba, J., Herzog, M., and Vachon, G. (2003). GeBP, the first member of a new gene family in Arabidopsis, encodes a nuclear protein with DNA-binding activity and is regulated by KNAT1. Plant J. 33, 305–317. doi: 10.1046/j.1365-313X.2003.01622.x
Cvitanich, C., Pallisgaard, N., Nielsen, K. A., Hansen, A. C., Larsen, K., Pihakaski-Maunsbach, K., et al. (2000). CPP1, a DNA-binding protein involved in the expression of a soybean leghemoglobin c3 gene. Proc. Natl. Acad. Sci. U.S.A. 97, 8163–8168. doi: 10.1073/pnas.090468497
D'Agostino, I. B., and Kieber, J. J. (1999). Phosphorelay signal transduction: the emerging family of plant response regulators. Trends Biochem. Sci. 24, 452–456.
D'Archivio, M., Filesi, C., Di Benedetto, R., Gargiulo, R., Giovannini, C., and Masella, R. (2007). Polyphenols, dietary sources and bioavailability. Ann. Ist. Super. Sanita 43, 348–361.
Dardick, C. D., and Callahan, A. M. (2014). Evolution of the fruit endocarp: molecular mechanism underlying adaptions in seed protection and dispersal strategies. Front. Plant Sci. 5:284. doi: 10.3389/fpls.2014.00284
Dardick, C. D., Callahan, A. M., Chiozzotto, R., Schaffer, R. J., Piagnani, M. C., and Scorza, R. (2010). Stone formation in peach fruit exhibits spatial coordination of the lignin and flavonoid pathways and similarity to Arabidopsis dehiscence. BMC Biol. 8:13. doi: 10.1186/1741-7007-8-13
De Jong, M., Mariani, C., and Vriezen, W. H. (2009). The role of auxin and gibberellin in tomato fruit set. J. Exp. Bot. 60, 1523–1532. doi: 10.1093/jxb/erp094
Deslandes, L., Olivier, J., Theulieres, F., Hirsch, J., Feng, D. X., Bittner-Eddy, P., et al. (2002). Resistance to Ralstonia solanacearum in Arabidopsis thaliana is conferred by the recessive RRS1-R gene, a member of a novel family of resistance genes. Proc. Natl. Acad. Sci. U.S.A. 99, 2404–2409. doi: 10.1073/pnas.032485099
Desveaux, D., Marechal, A., and Brisson, N. (2005). Whirly transcription factors: defence gene regulation and beyond. Trends Plant Sci. 10, 95–102. doi: 10.1016/j.tplants.2004.12.008
Devoghalaere, F., Doucen, T., Guitton, B., Keeling, J., Payne, W., Ling, T. J., et al. (2012). A genomics approach to understanding the role of auxin in apple (Malus × domestica) fruit size control. BMC Plant Biol. 12:7. doi: 10.1186/1471-2229-12-7
Díaz-Martín, J., Almoguera, C., Prieto-Dapena, P., Espinosa, J. M., and Jordano, J. (2005). Functional interaction between two transcription factors involved in the developmental regulation of a small heat stress protein gene promoter. Plant Physiol. 139, 1483–1494. doi: 10.1104/pp.105.069963
Dietrich, R. A., Richberg, M. H., Schmidt, R., Dean, C., and Dangl, J. L. (1997). A novel zinc finger protein is encoded by the Arabidopsis LSD1 gene and functions as a negative regulator of plant cell death. Cell 88, 685–694. doi: 10.1016/S0092-8674(00)81911-X
Dirlewanger, E., Quero-García, J., Le Dantec, L., Lambert, P., Ruiz, D., Dondini, L., et al. (2012). Comparison of the genetic determinism of two key phenological traits, flowering and maturity dates, in three Prunus species: peach, apricot and sweet cherry. Heredity 109, 280–292. doi: 10.1038/hdy.2012.38
Du, D. L., Hao, R. J., Cheng, T. R., Pan, H. T., Yang, W. R., Wang, J., et al. (2013). Genome-wide analysis of the AP2/ERF gene family in Prunus mume. Plant Mol. Biol. Rep. 31, 741–750. doi: 10.1007/s11105-012-0531-6
Dubé, A., Bisaillon, M., and Perreault, J.-P. (2009). Identification of proteins from Prunus persica that interact with peach latent mosaic viroid. J. Virol. 83, 12057–12067. doi: 10.1128/JVI.01151-09
Eldem, V., Akcay, U. C., Ozhuner, E., Bakir, Y., Uranbey, S., and Unver, T. (2012). Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PLoS ONE 7:e50298. doi: 10.1371/journal.pone.0050298
El-Sharkawy, I., Kim, W. S., El-Kereamy, A., Jayasankar, S., Svircev, A. M., and Brown, D. C. W. (2007). Isolation and characterization of four ethylene signal transduction elements in plums (Prunus salicina L.). J. Exp. Bot. 58, 3631–3643. doi: 10.1093/jxb/erm213
El-Sharkawy, I., Sherif, S., Mila, I., Bouzayen, M., and Jayasankar, S. (2009). Molecular characterization of seven genes encoding ethylene-responsive transcriptional factors during plum fruit development and ripening. J. Exp. Bot. 60, 907–922. doi: 10.1093/jxb/ern354
Eshed, Y., Baum, S., Perea, J. V., and Bowman, J. L. (2001). Establishment of polarity in lateral organs of plants. Curr. Biol. 11, 1251–1260. doi: 10.1016/S0960-9822(01)00392-X
Espley, R. V., Hellens, R. P., Putterill, J., Stevenson, D. E., Kutty-Amma, S., and Allan, A. C. (2007). Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 49, 414–427. doi: 10.1111/j.1365-313X.2006.02964.x
Eulgem, T., Rushton, P. J., Robatzek, S., and Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206. doi: 10.1016/S1360-1385(00)01600-9
Fan, S., Bielenberg, D. G., Zhebentyanyeva, T. N., Reighard, G. L., Okie, W. R., Holland, D., et al. (2010). Mapping quantitative trait loci associated with chilling requirement, heat requirement and bloom date in peach (Prunus persica). New Phytol. 185, 917–930. doi: 10.1111/j.1469-8137.2009.03119.x
Feng, J. R., Chen, X. S., Yuan, Z. H., He, T. M., Zhang, L. J., Wu, Y., et al. (2006). Proteome comparison following self- and across-pollination in self-incompatible apricot (Prunus armeniaca L.). Protein J. 25, 328–335. doi: 10.1007/s10930-006-9018-3
Ferraìndiz, C., Liljegren, S. J., and Yanofsky, M. F. (2000). Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science 289, 436–438. doi: 10.1126/science.289.5478.436
Fridborg, I., Kuusk, S., Moritz, T., and Sundberg, E. (1999). The Arabidopsis dwarf mutant shi exhibits reduced gibberellin responses conferred by overexpression of a new putative zinc finger protein. Plant Cell 6, 1019–1032. doi: 10.1105/tpc.11.6.1019
Fujita, A., Kikuchi, Y., Kuhara, S., Misumi, Y., Matsumoto, S., and Kobayashi, H. (1989). Domains of the SFL1 protein of yeasts are homologous to Myc oncoproteins or yeast heat-shock transcription factor. Gene 85, 321–328. doi: 10.1016/0378-1119(89)90424-1
Gao, Z. H., Luo, X. Y., Shi, T., Cai, B., Zhang, Z., Cheng, Z. M., et al. (2012a). Identification and validation of potential conserved microRNAs and their targets in peach (Prunus persica). Mol. Cells 34, 239–249. doi: 10.1007/s10059-012-0004-7
Gao, Z. H., Shi, T., Luo, X. Y., Zhang, Z., Zhuang, W. B., and Wang, L. J. (2012b). High-throughput sequencing of small RNAs and analysis of differentially expressed microRNAs associated with pistil development in Japanese apricot. BMC Genomics 13:371. doi: 10.1186/1471-2164-13-371
Giannino, D., Condello, E., Bruno, L., Testone, G., Tartarini, A., Cozza, R., et al. (2004). The gene geranylgeranyl reductase of peach (Prunus persica [L.] Batsch) is regulated during leaf development and responds differentially to distinct stress factors. J. Exp. Bot. 55, 2063–2073. doi: 10.1093/jxb/erh217
Golldack, D., Lüking, I., and Yang, O. (2011). Plant tolerance to drought and salinity: stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep. 30, 1383–1391. doi: 10.1007/s00299-011-1068-0
Golz, J. F., and Hudson, A. (1999). Plant development: YABBYs claw to the fore. Curr. Biol. 9, 861–863. doi: 10.1016/S0960-9822(00)80047-0
Groszmann, M., Paicu, T., Alvarez, J. P., Swain, S. M., and Smyth, D. R. (2011). SPATULA and ALCATRAZ, are partially redundant, functionally diverging bHLH genes required for Arabidopsis gynoecium and fruit development. Plant J. 68, 816–829. doi: 10.1111/j.1365-313X.2011.04732.x
Guo, C., Zhang, J. Q., Peng, T., Bao, M. Z., and Zhang, J. W. (2014). Structural and expression analyses of three PmCBFs from Prunus mume. Biol. Plant. 58, 247–255. doi: 10.1007/s10535-014-0393-x
Halbach, T., Scheer, N., and Werr, W. (2000). Transcriptional activation by the PHD finger is inhibited through an adjacent leucine zipper that binds 14-3-3 proteins. Nucleic Acids Res. 28, 3542–3550. doi: 10.1093/nar/28.18.3542
Hartmann, U., Sagasser, M., Mehrtens, F., Stracke, R., and Weisshaar, B. (2005). Differential combinatorial interactions of cisacting elements recognized by R2R3-MYB, BZIP, and BHLH factors control light-responsive and tissue-specific activation of phenylpropanoid biosynthesis genes. Plant Mol. Biol. 57, 155–171. doi: 10.1007/s11103-004-6910-0
Hayama, H., Shimada, T., Fujii, H., Ito, A., and Kashimura, Y. (2006). Ethylene-regulation of fruit softening and softening-related genes in peach. J. Exp. Bot. 57, 4071–4077. doi: 10.1093/jxb/erl178
Hu, H., Liu, Y., Shi, G. L., Liu, Y. P., Wu, R. J., Yang, A. Z., et al. (2011). Proteomic analysis of peach endocarp and mesocarp during early fruit development. Physiol. Plant. 142, 390–406. doi: 10.1111/j.1399-3054.2011.01479.x
Hudson, M., Ringli, C., Boylan, M. T., and Quail, P. H. (1999). The FAR1 locus encodes a novel nuclear protein specific to phytochrome A signalling. Genes Dev. 13, 2017–2027. doi: 10.1101/gad.13.15.2017
Husbands, A., Bell, E. M., Shuai, B., Smith, H. M. S., and Springer, P. S. (2007). LATERAL ORGAN BOUNDARIES defines a new family of DNA-binding transcription factors and can interact with specific bHLH proteins. Nucleic Acids Res. 35, 6663–6671. doi: 10.1093/nar/gkm775
Infante, R., Martínez-Gómez, P., and Predieri, S. (2011). “Breeding for fruit quality in Prunus,” in Breeding for Fruit Quality, eds M. A. Jenks and P. J. Bebeli (New York, NY: Wiley & Blackwel), 201–229.
Jackson, S. A., Iwata, A., Lee, S. H., Schmutz, J., and Shoemaker, R. (2011). Sequencing crop genomes: approaches and applications. New Phytol. 191, 915–925. doi: 10.1111/j.1469-8137.2011.03804.x
Jiang, Y., and Deyholos, M. K. (2009). Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 69, 91–105. doi: 10.1007/s11103-008-9408-3
Jiménez, S., Lawton-Rauh, A. L., Reighard, G. L., Abbott, A. G., and Bielenberg, D. G. (2009). Phylogenetic analysis and molecular evolution of the dormancy associated MADS-BOX genes from peach. BMC Plant Biol. 9:81. doi: 10.1186/1471-2229-9-81
Jiménez, S., Reighard, G. L., and Bielenberg, D. G. (2010). Gene expression of DAM5 and DAM6 is suppressed by chilling temperatures and inversely correlated with bud break rate. Plant Mol. Biol. 73, 157–167. doi: 10.1007/s11103-010-9608-5
Jin, J. P., Zhang, H., Kong, L., Gao, G., and Luo, J. C. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Jung, S., Cestaro, A., Troggio, M., Main, D., Zheng, P., Cho, I., et al. (2012). Whole genome comparisons of Fragaria, Prunus and Malus reveal different modes of evolution between Rosaceous subfamilies. BMC Genomics 13:129. doi: 10.1186/1471-2164-13-129
Jung, S., Jiwan, D., Cho, I., Abbott, A., Tomkins, J., and Main, D. (2009). Synteny of Prunus and other model plant species. BMC Genomics 10:76. doi: 10.1186/1471-2164-10-76
Katsir, L., Schilmiller, A. L., Staswick, P. E., He, S. Y., and Howe, G. A. (2008). COI1 is a critical component of a receptor for jasmonate and the bacterial virulence factor coronatine. Proc. Natl. Acad. Sci. U.S.A. 105, 7100–7105. doi: 10.1073/pnas.0802332105
Kim, J. H., Choi, D., and Kende, H. (2003). The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 36, 94–104. doi: 10.1046/j.1365-313X.2003.01862.x
Kirik, V., and Baumlein, H. (1996). A novel leaf-specific myb-related protein with a single binding repeat. Gene 183, 109–113. doi: 10.1016/S0378-1119(96)00521-5
Kitashiba, H., Matsuda, N., Ishizaka, T., Nakano, H., and Suzuki, T. (2002). Isolation of genes similar to DREB1/CBF from sweet cherry (Prunus avium L.). J. Jpn. Soc. Hort. Sci. 71, 651–657. doi: 10.2503/jjshs.71.651
Klein, J., Saedler, H., and Huijser, P. (1996). A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol. Gen. Genet. 250, 7–16.
Kornberg, R. D. (2007). The molecular basis of eukaryotic transcription. Proc. Natl. Acad. Sci. U.S.A. 104, 12955–12961. doi: 10.1073/pnas.0704138104
Krishnamurthy, S., and Hampsey, M. (2008). Eukaryotic transcription initiation. Curr. Biol. 19, R153–R156. doi: 10.1016/j.cub.2008.11.052
Kujur, A., Bajaj, D., Saxena, M. S., Tripathi, S., Upadhyaya, H. D., Gowda, C. L. L., et al. (2013). Functionally relevant microsatellite markers from chickpea transcription factor genes for efficient genotyping applications and trait association mapping. DNA Res. 20, 355–374. doi: 10.1093/dnares/dst015
Kumagai, T., Ito, S., Nakamichi, N., Niwa, Y., Murakami, M., Yamashino, T., et al. (2008). The common function of a novel subfamily of B-Box zinc finger proteins with reference to circadian-associated events in Arabidopsis thaliana. Biosci. Biotechnol. Biochem. 72, 1539–1549. doi: 10.1271/bbb.80041
Lagercrantz, U., and Axelsson, T. (2000). Rapid evolution of the family of CONSTANS LIKE genes in plants. Mol. Biol. Evol. 17, 1499–1507. doi: 10.1093/oxfordjournals.molbev.a026249
Lalli, D. A., Decroocq, V., Blenda, A. V., Schurdi-Levraud, V., Garay, L., Le Gall, O., et al. (2005). Identification and mapping of resistance gene analogs (RGAs) in Prunus: a resistance map for Prunus. Theor. Appl. Genet. 111, 1504–1513. doi: 10.1007/s00122-005-0079-z
Landschulz, W. H., Johnson, P. F., and McKnight, S. L. (1988). The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 240, 1759–1764. doi: 10.1126/science.3289117
Lee, H., Yoo, S. J., Lee, J. H., Kim, W., Yoo, S. K., Fitzgerald, H., et al. (2010). Genetic framework for flowering-time regulation by ambient temperature-responsive miRNAs in Arabidopsis. Nucleic Acids Res. 38, 3081–3093. doi: 10.1093/nar/gkp1240
Leida, C., Conesa, A., Llacer, G., Badenes, M. L., and Rios, G. (2012). Histone modifications and expression of DAM6 gene in peach are modulated during bud dormancy release in a cultivar-dependent manner. New Phytol. 193, 67–80. doi: 10.1111/j.1469-8137.2011.03863.x
Leida, C., Terol, J., Marti, G., Agusti, M., Llacer, G., Badenes, M. L., et al. (2010). Identification of genes associated with bud dormancy release in Prunus persica by suppression subtractive hybridization. Tree Physiol. 30, 655–666. doi: 10.1093/treephys/tpq008
Li, W.-X., Oono, Y., Zhu, J., He, X.-J., Wu, J.-M., Iida, K., et al. (2008). The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20, 2238–2251. doi: 10.1105/tpc.108.059444
Li, Z., and Thomas, T. L. (1998). PEI1, an embryo-specific zinc finger protein gene required for heart-stage embryo formation in Arabidopsis. Plant Cell 10, 383–398. doi: 10.2307/3870596
Li, Z., Reighard, G. L., Abbott, A. G., and Bielenberg, D. G. (2009). Dormancy associated MADS-BOX genes from the EVG locus of peach [Prunuspersica (L.) Batsch] have distinct seasonal and photoperiodic expression patterns. J. Exp. Bot. 60, 3521–3530. doi: 10.1093/jxb/erp195
Liang, L., Zhang, B., Yin, X. R., Xu, C. J., Sun, C. D., and Chen, K. S. (2013). Differential expression of the CBF gene family during postharvest cold storage and subsequent shelf-life of peach fruit. Plant Mol. Biol. Rep. 31, 1358–1367. doi: 10.1007/s11105-013-0600-5
Lisso, J., Altmann, T., and Mussig, C. (2006). The AtNFXL1 gene encodes a NF-X1 type zinc finger protein required for growth under salt stress. FEBS Lett. 580, 4851–4856. doi: 10.1016/j.febslet.2006.07.079
Littlewood, T. D., and Evan, G. I. (1995). Transcription factors 2: helix-loop-helix. Protein Profile 2, 621–702.
Liu, X. M., Anderson, J. M., and Pijut, P. M. (2010). Cloning and characterization of Prunus serotina AGAMOUS, a putative flower homeotic gene. Plant Mol. Biol. Rep. 28, 193–203. doi: 10.1007/s11105-009-0140-1
Lovisetto, A., Guzzo, F., Tadiello, A., Confortin, E., Pavanello, A., Botton, A., et al. (2013). Characterization of a bZIP gene highly expressed during ripening of the peach fruit. Plant Physiol. Biochem. 70, 462–470. doi: 10.1016/j.plaphy.2013.06.014
Luo, X., Gao, Z., Shi, T., Cheng, Z., Zhang, Z., and Ni, Z. (2013). Identification of miRNAs and their target genes in peach (Prunus persica L.) using high-throughput sequencing and degradome analysis. PLoS ONE 8:e79090. doi: 10.1371/journal.pone.0079090
Manganaris, G. A., Rasori, A., Bassi, D., Geuna, F., Ramina, A., Tonutti, P., et al. (2011). Comparative transcript profiling of apricot (Prunus armeniaca L.) fruit development and on-tree ripening. Tree Genet. Genomes 7, 609–616. doi: 10.1007/s11295-010-0360-4
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Martin, T., Hu, M., Labbe, H., McHugh, S., Svircev, A., and Miki, B. (2006). PpAG1, a homolog of AGAMOUS, expressed in developing peach flowers and fruit. Can. J. Bot. 84, 767–776. doi: 10.1139/b06-031
Martínez-Gómez, P., Crisosto, C., Bonghi, C., and Rubio, M. (2011). New approaches to Prunus transcriptome analysis. Genetica 139, 755–769. doi: 10.1007/s10709-011-9580-2
Martínez-Gómez, P., Sánchez-Pérez, R., and Rubio, M. (2012). Clarifying omics concepts, challenges and opportunities for Prunus breeding in the post-genomic era. OMICS 16, 268–283. doi: 10.1089/omi.2011.0133
Mita, S., Nagai, Y., and Asai, T. (2006). Isolation of cDNA clones corresponding to genes differentially expressed in pericarp of mume (Prunus mume) in response to ripening, ethylene and wounding signals. Physiol. Plant. 128, 531–545. doi: 10.1111/j.1399-3054.2006.00749.x
Mitsuda, N., Hisabori, T., Takeyasu, K., and Sato, M. H. (2004). VOZ: isolation and characterization of novel vascular plant transcription factors with a one-zinc finger from Arabidopsis thaliana. Plant Cell Physiol. 45, 845–854. doi: 10.1093/pcp/pch101
Mitsuda, N., Seki, M., Shinozaki, K., and Ohme-Takagi, M. (2005). The NAC transcription factors NST1 and NST2 of Arabidopsis regulate secondary wall thickenings and are required for anther dehiscence. Plant Cell 17, 2993–3006. doi: 10.1105/tpc.105.036004
Molesini, B., Pii, Y., and Pandolfini, T. (2012). Fruit improvement using intragenesis and artificial microRNA. Trends Biotechnol. 30, 80–88. doi: 10.1016/j.tibtech.2011.07.005
Mousavi, S., Alisoltani, A., Shiran, B., Fallahi, H., Ebrahimie, E., Imani, A., et al. (2014). De novo transcriptome assembly and comparative analysis of differentially expressed genes in Prunus dulcis Mill. In response to freezing stress. PLoS ONE 9:e104541. doi: 10.1371/journal.pone.0104541
Nakano, T., Suzuki, K., Fujimura, T., and Shinshi, H. (2006). Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 140, 411–432. doi: 10.1104/pp.105.073783
Nakashima, K., Yamaguchi-Shinozaki, K., and Shinozaki, K. (2014). The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 5:170. doi: 10.3389/fpls.2014.00170
Nam, J., de Pamphilis, C. W., Ma, H., and Nei, M. (2003). Antiquity and evolution of the MADS-box gene family controlling flower development in plants. Mol. Biol. Evol. 20, 1435–1447. doi: 10.1093/molbev/msg152
Nelson, D. E., Repetti, P. P., Adams, T. R., Creelman, R. A., Wu, J., Warner, D. C., et al. (2007). Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved corn yields on water-limited acres. Proc. Natl. Acad. Sci. U.S.A. 104, 16450–16455. doi: 10.1073/pnas.0707193104
Nole-Wilson, S., and Krizek, B. A. (2000). DNA binding properties of the Arabidopsis floral development protein AINTEGUMENTA. Nucleic Acids Res. 28, 4076–4082. doi: 10.1093/nar/28.21.4076
Ohme-Takagi, M., and Shinshi, H. (1995). Ethylene-inducible DNA binding proteins that interact with an ethylene-responsive element. Plant Cell 7, 173–182. doi: 10.1105/tpc.7.2.173
Owens, C. L., Thomashow, M. F., Hancock, J. F., and Iezzoni, A. F. (2002). CBF1 orthologs in sour cherry and strawberry and the heterologous expression of CBF1 in strawberry. J. Amer. Soc. Hort. Sci. 127, 489–494.
Parcy, F., Nilsson, O., Busch, M. A., Lee, I., and Weigel, D. (1998). A genetic framework for floral patterning. Nature 395, 561–566. doi: 10.1038/26903
Parenicová, L., Folter, S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and phylogenetic analyses of the complete MADS-BOX transcription factor family in Arabidopsis: new openings to the MADS-BOX world. Plant Cell 15, 1538–1551. doi: 10.1105/tpc.011544
Peace, C., Bassil, N., Main, D., Ficklin, S., Rosyara, U. R., Stegmeir, T., et al. (2012). Development and evaluation of a genome-wide 6K SNP array for diploid sweet cherry and tetraploid sour cherry. PLoS ONE 7:e48305. doi: 10.1371/journal.pone.0048305
Pérez-Rodríguez, P., Riano-Pachon, D. M., Guedes Correa, L. G., Rensing, S. A., Kersten, B., and Mueller-Roeber, B. (2009). PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, D822–D827. doi: 10.1093/nar/gkp805
Petroni, K., and Tonelli, C. (2011). Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 181, 219–229. doi: 10.1016/j.plantsci.2011.05.009
Pirona, R., Eduardo, I., Pacheco, I., Linge, C. S., Miculan, M., Verde, I., et al. (2013). Fine mapping and identification of a candidate gene for a major locus controlling maturity date in peach. BMC Plant Biol. 13:166. doi: 10.1186/1471-2229-13-166
Potter, D. (2012). “Basic information on the stone fruit crops,” in Genetics, Genomics and Breeding of Stone Fruits, eds C. Kole, and A. G. Abbott (New York, NY: CRC Press), 1–21.
Rahim, M. A., Busatto, N., and Trainotti, L. (2014). Regulation of anthocyanin biosynthesis in peach fruits. Planta 240, 913–929. doi: 10.1007/s00425-014-2078-2
Ravaglia, D., Espley, R. V., Henry-Kirk, R. A., Andreotti, C., Ziosi, V., Hellens, R. P., et al. (2013). Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biol. 13:68. doi: 10.1186/1471-2229-13-68
Raventós, D., Skriver, K., Schlein, M., Karnahl, K., Rogers, S. W., Rogers, J. C., et al. (1998). HRT, a novel zinc finger, transcriptional repressor from barley. J. Biol. Chem. 273, 23313–23320. doi: 10.1074/jbc.273.36.23313
Richards, D. E., Peng, J., and Harberd, N. P. (2000). Plant GRAS and metazoan STATs: one family? Bioessays 22, 573–577. doi: 10.1002/(SICI)1521-1878(200006)22:6<573::AID-BIES10>3.0.CO;2-H
Riechmann, J. L. (2006). Transcription factors of Arabidopsis and rice: a genomic prespective. Annu. Plant Rev. 29, 28–53. doi: 10.1002/9780470988886.ch2
Riechmann, J. L., and Meyerowitz, E. M. (1998). The AP2/EREBP family of plant transcription factors. Biol. Chem. 379, 633–646.
Rios, G., Tadeo, F. R., Leida, C., and Badenes, M. L. (2013). Prediction of components of the sporopollenin synthesis pathway in peach by genomic and expression analyses. BMC Genomics 14:40. doi: 10.1186/1471-2164-14-40
Rizhsky, L., Liang, H., and Mittler, R. (2002). The combined effect of drought stress and heat shock on gene expression in tobacco. Plant Physiol. 130, 1143–1151. doi: 10.1104/pp.006858
Roberti, M., Polosa, P. L., Bruni, F., Manzari, C., Deceglie, S., Gadaleta, M. N., et al. (2009). The MTERF family proteins: mitochondrial transcription regulators and beyond. Biochim. Biophys. Acta 1787, 303–3011. doi: 10.1016/j.bbabio.2009.01.013
Rodamilans, B., San León, D., Mühlberger, L., Candresse, T., Neumüller, M., Oliveros, J. C., et al. (2014). Transcriptomic analysis of Prunus domestica undergoing hypersensitive response to plum pox virus infection. PLoS ONE 9:e100477. doi: 10.1371/journal.pone.0100477
Romeu, J. F., Monforte, A. J., Sánchez, G., Granell, A., García-Brunton, J., Badenes, M. L., et al. (2014). Quantitative trait loci affecting reproductive phenology in peach. BMC Plant Biol. 14:52. doi: 10.1186/1471-2229-14-52
Rubio, M., Rodríguez-Moreno, L., Ballester, A. R., Castro de Moura, M., Bonghi, C., Candresse, T., et al. (2015). Analysis of gene expression changes in peach leaves in response to Plum pox virus infection using RNA-Seq. Mol. Plant Pathol. 16, 164–176. doi: 10.1111/mpp.12169
Ruiz, K. B., Trainotti, L., Bonghi, C., Ziosi, V., Costa, G., and Torrigiani, P. (2013). Early methyl jasmonate application to peach delays fruit/seed development by altering the expression of multiple hormone-related genes. J. Plant Growth Regul. 32, 852–864. doi: 10.1007/s00344-013-9351-7
Rushton, P. J., Somssich, I. E., Ringler, P., and Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15, 247–258. doi: 10.1016/j.tplants.2010.02.006
Sakuma, Y., Liu, Q., Dubouzet, J. G., Abe, H., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2002). DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 290, 998–1009. doi: 10.1006/bbrc.2001.6299
Salazar, J. A., Rubio, M., Ruiz, D., Tartarini, S., Martínez-Gómez, P., and Dondini, L. (2015). SNP development for genetic diversity analysis in apricot. Tree Genet. Genomes 11, 15. doi: 10.1007/s11295-015-0845-2
Salazar, J. A., Ruiz, D., Campoy, J. A., Sánchez-Pérez, R., Crisosto, C. H., Martínez-García, P. J., et al. (2014). Quantitative Trait Loci (QTL) and Mendelian Trait Loci (MTL) analysis in Prunus: a breeding perspective and beyond. Plant Mol. Biol. Rep. 32, 1–18. doi: 10.1007/s11105-013-0643-7
Salinas, M., Xing, S., Höhmann, S., Berndtgen, R., and Huijser, P. (2012). Genomic organization, phylogenetic comparison and differential expression of the SBP-box family of transcription factors in tomato. Planta 235, 1171–1184. doi: 10.1007/s00425-011-1565-y
Sánchez, G., Venegas-Calerón, M., Salas, J. J., Monforte, A., Badenes, M. L., and Granell, A. (2013). An integrative “omics” approach identifies new candidate genes to impact aroma volatiles in peach fruit. BMC Genomics 14:343. doi: 10.1186/1471-2164-14-343
Sánchez-Pérez, R., Del Cueto, J., Dicenta, F., and Martínez-Gómez, P. (2014). Recent advancements to study flowering time in almond and other Prunus species. Front. Plant Sci. 5:334. doi: 10.3389/fpls.2014.00334
Sánchez-Pérez, R., Dicenta, F., and Martínez-Gómez, P. (2012). Inheritance of chilling and heat requirements for flowering in almond and QTL analysis. Tree Genet. Genomes 8, 379–389. doi: 10.1007/s11295-011-0448-5
Santi, L., Wang, Y., Stile, M. R., Berendzen, K., Wanke, D., Roig, C., et al. (2003). The GA octodinucleotide repeat binding factor BBR participates in the transcriptional regulation of the homeobox gene Bkn3. Plant J. 34, 813–826. doi: 10.1046/j.1365-313X.2003.01767.x
Santos, A. M., Oliver, M. J., Sanchez, A. M., and Oliveira, M. M. (2012). Expression of almond Knotted1 homologue (PdKn1) anticipates adventitious shoot initiation. In Vitro Cell. Dev. Biol. Plant 48, 4–49. doi: 10.1007/s11627-011-9388-x
Schauser, L., Roussis, A., Stiller, J., and Stougaard, J. (1999). A plant regulator controlling development of symbiotic root nodules. Nature 402, 191–195. doi: 10.1038/46058
Schiefthaler, U., Balasubramanian, S., Sieber, P., Chevalier, D., Wisman, E., and Schneitz, K. (1999). Molecular analysis of NOZZLE, a gene involved in pattern formation and early sporogenesis during sex organ development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 96, 11664–11669. doi: 10.1073/pnas.96.20.11664
Shen, X., Zhao, K., Liu, L., Zhang, K., Yuan, H., Liao, X., et al. (2014). A role for PacMYBA in ABA-regulated anthocyanin biosynthesis in red-colored sweet cherry cv. Hong Deng (Prunus avium L.). Plant Cell Physiol. 55, 862–880. doi: 10.1093/pcp/pcu013
Sherif, S., El-Sharkawy, I., Paliyath, G., and Jayasankar, S. (2013). PpERF3b, a transcriptional repressor from peach, contributes to disease susceptibility and side branching in EAR-dependent and -independent fashions. Plant Cell Rep. 32, 1111–1124. doi: 10.1007/s00299-013-1405-6
Sherif, S., Paliyath, G., and Jayasankar, S. (2012). Molecular characterization of peach PR genes and their induction kinetics in response to bacterial infection and signaling molecules. Plant Cell Rep. 31, 697–711. doi: 10.1007/s00299-011-1188-6
Shigyo, M., Hasebe, M., and Ito, M. (2006). Molecular evolution of the AP2 subfamily. Gene 366, 256–265. doi: 10.1016/j.gene.2005.08.009
Shore, P., and Sharrocks, A. D. (1995). The MADS-BOX family of transcription factors. Eur. J. Biochem. 229, 1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x
Shulaev, V., Korban, S. S., Sosinski, B., Abbott, A. G., Aldwinckle, H. S., Folta, K. M., et al. (2008). Multiple models for Rosaceae genomics. Plant Physiol. 147, 985–1003. doi: 10.1104/pp.107.115618
Siefers, N., Dang, K. K., Kumimoto, R. W., Bynum, W. E., Tayrose, G., and Holt, B. F. (2009). Tissue-specific expression patterns of Arabidopsis NF-Y transcription factors suggest potential for extensive combinatorial complexity. Plant Physiol. 149, 625–641. doi: 10.1104/pp.108.130591
Silva, C., García-Mas, J., Sánchez, A. M., Arús, P., and Oliveira, M. M. (2005). Looking into flowering time in almond (Prunus dulcis (Mill) D.A. Webb): the candidate gene approach. Theor. Appl. Genet. 110, 959–968. doi: 10.1007/s00122-004-1918-z
Smalle, J., Kurepa, J., Haegman, M., Gielen, J., Van Montagu, M., and Van Der Straeten, D. (1998). The trihelix DNA-binding motif in higher plants is not restricted to the transcription factors GT-1 and GT-2. Proc. Natl. Acad. Sci. U.S.A. 95, 3318–3322. doi: 10.1073/pnas.95.6.3318
Socquet-Juglard, D., Kamber, T., Pothier, J. F., Christen, D., Gessler, G., Duffy, B., et al. (2013). Comparative RNA-Seq analysis of early-infected peach leaves by the invasive phytopathogen Xanthomonas arboricola pv. Pruni. PLoS ONE 8:e54196. doi: 10.1371/journal.pone.0054196
Solano, R., Stepanova, A., Chao, Q., and Ecker, J. R. (1998). Nuclear events in ethylene signaling: a transcriptional cascade mediated by ETHYLENE-INSENSITIVE3 and ETHYLENE-RESPONSE-FACTOR1. Genes Dev. 12, 3703–3714. doi: 10.1101/gad.12.23.3703
Sooriyapathirana, S. S., Khan, A., Sebolt, A. M., Wang, D. C., Bushakra, J. M., Lin-Wang, K., et al. (2010). QTL analysis and candidate gene mapping for skin and flesh color in sweet cherry fruit (Prunus avium L.). Tree Genet. Genomes 6, 821–832. doi: 10.1007/s11295-010-0294-x
Soto, A., Ruiz, K. B., Ziosi, V., Costa, G., and Torrigiani, P. (2012). Ethylene and auxin biosynthesis and signaling are impaired by methyl jasmonate leading to a transient slowing down of ripening in peach fruit. J. Plant Physiol. 169, 1858–1865. doi: 10.1016/j.jplph.2012.07.007
Souer, E., van Houwelingen, A., Kloos, D., Mol, J., and Koes, R. (1996). The No Apical Meristem gene of petunia is required for pattern formation in embryos and flowers and is expressed as meristem and primordia boundaries. Cell 85, 159–170. doi: 10.1016/S0092-8674(00)81093-4
Stephen, J. R., Dent, K. C., and Finch-Savage, W. E. (2004). Molecular responses of Prunus avium (wild cherry) embryonic axes to temperatures affecting dormancy. New Phytol. 161, 401–413. doi: 10.1046/j.1469-8137.2003.00927.x
Stracke, R., Werber, M., and Weisshaar, B. (2001). The R2R3-MYB geen family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 4, 447–456. doi: 10.1016/S1369-5266(00)00199-0
Sunkar, R., Li, Y. F., and Jagadeeswaran, G. (2012). Functions of microRNAs in plant stress responses. Trends Plant Sci. 17, 196–203. doi: 10.1016/j.tplants.2012.01.010
Suzuki, M., Kao, C. Y., and McCarty, D. R. (1997). The conserved B3 domain of VIPAROUS1 has a cooperative DNA binding activity. Plant Cell 9, 799–807. doi: 10.1105/tpc.9.5.799
Tadiello, A., Pavanello, A., Zanin, D., Caporali, E., Colombo, L., Rotino, G. L., et al. (2009). A PLENA-like gene of peach is involved in carpel formation and subsequent transformation into a fleshy fruit. J. Exp. Bot. 60, 651–661. doi: 10.1093/jxb/ern313
Takatsuji, H. (1999). Zinc-finger proteins: the classical zinc finger emerges in contemporary plant science. Plant Mol. Biol. 39, 1073–1078. doi: 10.1023/A:1006184519697
Tan, K., Tegner, J., and Ravasi, T. (2008). Integrated approaches to uncovering transcription regulatory networks in mammalian cells. Genomics 91, 219–231. doi: 10.1016/j.ygeno.2007.11.005
Tani, E., Polidoros, A. N., and Tsaftaris, A. S. (2007). Characterization and expression analysis of FRUITFULL- and SHATTERPROOF-like genes from peach (Prunus persica) and their role in split-pit formation. Tree Physiol. 27, 649–659. doi: 10.1093/treephys/27.5.649
Tani, E., Tsaballa, A., Stedel, C., Kalloniati, C., Papaefthimiou, D., Polidoros, A., et al. (2011). The study of a SPATULA-like bHLH transcription factor expressed during peach (Prunus persica) fruit development. Plant Physiol. Biochem. 49, 654–663. doi: 10.1016/j.plaphy.2011.01.020
Teakle, G. R., Manfield, I. W., Graham, J. F., and Gilmartin, P. M. (2002). Arabidopsis thaliana GATA factors: organization, expression and DNA-binding characteristics. Plant Mol. Biol. 50, 43–57. doi: 10.1023/A:1016062325584
Testone, G., Bruno, L., Condello, E., Chiappetta, A., Bruno, A., Mele, G., et al. (2008). Peach [Prunus persica (L.) Batsch] KNOPE1, a class 1 KNOX orthologue to Arabidopsis BREVIPEDICELLUS/KNAT1, is misexpressed during hyperplasia of leaf curl disease. J. Exp. Bot. 59, 389–402. doi: 10.1093/jxb/erm317
Testone, G., Condello, E., Verde, I., Caboni, E., Iannelli, M. A., Bruno, L., et al. (2009). The peach (Prunus persica [L.] Batsch) homeobox gene KNOPE3, which encodes a class 2 knotted-like transcription factor, is regulated during leaf development and triggered by sugars. Mol. Genet. Genomics 282, 47–64. doi: 10.1007/s00438-009-0445-7
Testone, G., Condello, E., Verde, I., Nicolodi, C., Caboni, E., Dettori, M. T., et al. (2012). The peach (Prunus persica L. Batsch) genome harbours 10 KNOX genes, which are differentially expressed in stem development, and the class 1 KNOPE1 regulates elongation and lignification during primary growth. J. Exp. Bot. 63, 5417–5435. doi: 10.1093/jxb/ers194
Tittarelli, A., Santiago, M., Morales, A., Meisel, L. A., and Silva, H. (2009). Isolation and functional characterization of cold-regulated promoters, by digitally identifying peach fruit cold-induced genes from a large EST dataset. BMC Plant Biol. 9:121. doi: 10.1186/1471-2229-9-121
Trainin, T., Bar-Ya'akov, I., and Holland, D. (2013). ParSOC1, a MADS-BOX gene closely related to Arabidopsis AGL20/SOC1, is expressed in apricot leaves in a diurnal manner and is linked with chilling requirements for dormancy break. Tree Genet. Genomes 9, 753–766. doi: 10.1007/s11295-012-0590-8
Trainotti, L., Tadiello, A., and Casadoro, G. (2007). The involvement of auxin in the ripening of climacteric fruits comes of age: the hormone plays a role of its own and has an intense interplay with ethylene in ripening peaches. J. Exp. Bot. 58, 3299–3308. doi: 10.1093/jxb/erm178
Uematsu, C., Katayama, H., Makino, I., Inagaki, A., Arakawa, O., and Martin, C. (2014). Peace, a MYB-like transcription factor, regulates petal pigmentation in flowering peach “Genpei” bearing variegated and fully pigmented flowers. J. Exp. Bot. 65, 1081–1094. doi: 10.1093/jxb/ert456
Ulmasov, T., Hagen, G., and Guilfoyle, T. J. (1997). ARF1, a transcription factor that binds to auxin response elements. Science 276, 1865–1868. doi: 10.1126/science.276.5320.1865
Vendramin, E., Pea, G., Dondini, L., Pacheco, I., Dettori, M. T., Gazza, L., et al. (2014). A unique mutation in a MYB gene cosegregates with the nectarine phenotype in peach. PLoS ONE 9:e90574. doi: 10.1371/journal.pone.0090574
Verde, I., Abbott, A. G., Scalabrin, S., Jung, S., Shu, S., Marroni, F., et al. (2013). The high-quality draft of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494. doi: 10.1038/ng.2586
Verde, I., Bassil, N., Scalabrin, S., Gilmore, B., Lawley, C. T., Gasic, K., et al. (2012). Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS ONE 7:e35668. doi: 10.1371/journal.pone.0035668
Verde, I., Shu, S., Jenkins, J., Zuccolo, A., Dettori, M. T., Dardick, C., et al. (2015). “The peach v2.0 release: an improved genome sequence for bridging the gap between genomics and breeding in Prunus,” in Plant & Animal Genome XXIII Conference (San Diego, CA).
Vinson, J., Su, X., Zubik, L., and Bose, P. (2001). Phenol antioxidant quantity and quality in foods: fruits. J. Agric. Food Chem. 49, 5315–5321. doi: 10.1021/jf0009293
Vrebalov, J., Pan, I. L., Arroyo, A. J. M., McQuinn, R., Chung, M., Poole, M., et al. (2009). Fleshy fruit expansion and ripening are regulated by the tomato SHATTERPROOF gene TAGL1. Plant Cell 21, 3041–3062. doi: 10.1105/tpc.109.066936
Wang, J., Zhang, X., Yan, G., Zhou, Y., and Zhang, K. (2013). Over-expression of the PaAP1 gene from sweet cherry (Prunus avium L.) causes early flowering in Arabidopsis thaliana. J. Plant Physiol. 170, 315–320. doi: 10.1016/j.jplph.2012.09.015
Wang, X. L., Peng, F. T., Yang, L., Li, M. J., and Zhang, S. S. (2012). Expression of genes involved in nitrate signaling and metabolism in peach roots in response to elevated levels of nitrate. Plant Mol. Biol. Rep. 30, 1450–1460. doi: 10.1007/s11105-012-0462-2
Wang, Y., and Pijut, P. M. (2013). Isolation and characterization of a TERMINAL FLOWER 1 homolog from Prunus serotina Ehrh. Tree Physiol. 33, 855–865. doi: 10.1093/treephys/tpt051
Watson, J. D., Gann, A., Baker, T. A., Levine, M., Bell, S. P., and Harrison, S. C. (2014). Molecular Biology of the Gene. New York, NY: Cold Spring Harbor; Cold Spring Harbor Laboratory Press.
Wells, C. E., Vendramin, E., Jiménez-Tarodo, S., Verde, I., and Bielenberg, D. G. (2015). A genome-wide analysis of MADS-BOX genes in peach [Prunus persica (L.) Batsch]. BMC Plant Biol. 15:41. doi: 10.1186/s12870-015-0436-2
Windhövel, A., Hein, I., Dabrowa, R., and Stockhaus, J. (2001). Characterization of a novel class of plant homeodomain proteins that bind to the C4 phosphoenolpyruvate carboxylase gene of Flaveria trinervia. Plant Mol. Biol. 45, 201–214. doi: 10.1023/A:1006450005648
Wisnieski, M., Norelli, J., Bassete, C., Artlip, T., and Macarisin, D. (2011). Ectopic expression of a novel peach (Prunus persica) CBF transcription factor in apple results in short-day induced dormancy and increased hardiness. Planta 233, 971–983. doi: 10.1007/s00425-011-1358-3
Wu, X., Shiroto, Y., Kishitani, S., Ito, Y., and Toriyama, K. (2009). Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep. 28, 21–30. doi: 10.1007/s00299-008-0614-x
Xia, R., Zhu, H., An, Y., Beers, E. P., and Liu, Z. (2012). Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 13:R47. doi: 10.1186/gb-2012-13-6-r47
Xu, Y., Zhang, L., Xie, H., Zhang, Y. Q., Oliveira, M. M., and Ma, R. C. (2008). Expression analysis and genetic mapping of three SEPALLATA-like genes from peach (Prunus persica (L.) Batsch). Tree Genet. Genomes 4, 693–703. doi: 10.1007/s11295-008-0143-3
Yamada, Y., Wang, H. Y., Fukuzawa, M., Barton, G. J., and Williams, J. G. (2008). A new family of transcription factors. Development 135, 3093–3101. doi: 10.1242/dev.026377
Yamane, H., Kashiwa, Y., Ooka, T., Tao, R., and Yonemori, K. (2008). Suppression subtractive hybridization and differential screening reveals endodormancy-associated expression of an SVP/AGL24-type MADS-BOX gene in lateral vegetative buds of japanese apricot. J. Amer. Soc. Hort. Sci. 133, 708–716.
Yamane, H., Ooka, T., Jotatsu, H., Sasaki, R., and Tao, R. (2011). Expression analysis of PpDAM5 and PpDAM6 during flower bud development in peach (Prunus persica). Sci. Hort. 129, 844–848. doi: 10.1016/j.scienta.2011.05.013
Yanagisawa, S. (1997). Dof DNA-binding domains of plant transcription factors contribute to multiple protein-protein interactions. Eur. J. Biochem. 250, 403–410. doi: 10.1111/j.1432-1033.1997.0403a.x
Yin, Y., Vafeados, D., Tao, Y., Yoshida, S., Asami, T., and Chory, J. (2005). A new class of transcription factors mediates brassinosteroid-regulated gene expression in Arabidopsis. Cell 120, 249–259. doi: 10.1016/j.cell.2004.11.044
Yu, J. K., Paik, H., Choi, J. P., Han, J. H., Choe, J. K., and Hur, C. G. (2010). Functional domain marker (FDM): an in silico demonstration in Solanaceae using simple sequence repeats (SSRs). Plant Mol. Biol. Rep. 28, 352–356. doi: 10.1007/s11105-009-0154-8
Zhang, J., Yang, W. R., Cheng, T. R., Pan, H. T., and Zhang, Q. X. (2013). Functional and evolutionary analysis of two CBF genes in Prunus mume. Can. J. Plant Sci. 93, 455–464. doi: 10.4141/cjps2012-193
Zhang, L., Xu, Y., and Ma, R. C. (2008). Molecular cloning, identification, and chromosomal localization of two MADS-BOX box genes in peach (Prunus persica). J. Genet. Genomics 35, 365–372. doi: 10.1016/S1673-8527(08)60053-3
Zhang, Q., Chen, W., Sun, L., Zhao, F., Huang, B., Yang, W., et al. (2012). The genome of Prunus mume. Nat. Commun. 3, 1318. doi: 10.1038/ncomms2290
Zhebentyayeva, T. N., Fan, S., Chandra, A., Bielenberg, D. G., Reighard, G. L., Okie, W. R., et al. (2014). Dissection of chilling requirement and bloom date QTLs in peach using a whole genome sequencing of sibling trees from an F2 mapping population. Tree Genet. Genomes 10, 35–51. doi: 10.1007/s11295-013-0660-6
Zheng, N., Fraenkel, E., Pabo, C. O., and Pavletich, N. P. (1999). Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev. 13, 666–674. doi: 10.1101/gad.13.6.666
Zhong, W., Gao, Z., Zhuang, W., Shi, T., Zhang, Z., and Ni, Z. (2013). Genome-wide expression profiles of seasonal bud dormancy at four critical stages in Japanese apricot. Plant Mol. Biol. 83, 247–264. doi: 10.1007/s11103-013-0086-4
Zhou, D. X., Bisanz-Seyer, C., and Mache, R. (1995). Molecular cloning of a small DNA binding protein with specificity for a tissue-specific negative element within the rps1 promoter. Nucleic Acids Res. 23, 1165–1169. doi: 10.1093/nar/23.7.1165
Zhou, H., Lin-Wang, K., Wang, H., Gu, C., Dare, A. P., Espley, R. V., et al. (2015). Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 82, 105–121. doi: 10.1111/tpj.12792
Zhou, Y., Guo, D., Li, J., Cheng, J., Zhou, H., Gu, C., et al. (2013). Coordinated regulation of anthocyanin biosynthesis through photorespiration and temperature in peach (Prunus persica f. atropurpurea). Tree Genet. Genomes 9, 265–278. doi: 10.1007/s11295-012-0552-1
Zhou, Y., Zhou, H., Lin-Wand, K., Vimolmangkang, S., Esplet, R. V., Wang, L., et al. (2014). Transcriptome analysis and transient transformation suggest an ancient duplicated MYB transcription factor as candidate gene for leaf red coloration in peach. BMC Plant Biol. 14:375. doi: 10.1186/s12870-014-0388-y
Zhu, H., Xia, R., Zhao, B. Y., An, Y. Q., Dardick, C. D., Callahan, A. M., et al. (2012). Unique expression, processing regulation, and regulatory network of peach (Prunus persica) miRNAs. BMC Plant Biol. 12:149. doi: 10.1186/1471-2229-12-149
Ziliotto, F., Begheldo, M., Rasori, A., Bonghi, C., and Tonutti, P. (2008). Transcriptome profiling of ripening nectarine (Prunus persica L. Batsch) fruit treated with 1-MCP. J. Exp. Bot. 59, 2781–2791. doi: 10.1093/jxb/ern136
Ziosi, V., Bregoli, A. M., Fregola, F., Costa, G., and Torrigiani, P. (2009). Jasmonate-induced ripening delay is associated with up-regulation of polyamine levels in peach fruit. J. Plant Physiol. 166, 938–946. doi: 10.1016/j.jplph.2008.11.014
Ziosi, V., Noferini, M., Fiori, G., Tadiello, A., Trainotti, L., Casadoro, G., et al. (2008). A new index based on vis spectroscopy to characterize the progression of ripening in peach fruit. Postharvest Biol. Technol. 49, 319–329. doi: 10.1016/j.postharvbio.2008.01.017
Zouine, M., Fu, Y., Chateigner-Boutin, A.-L., Mila, I., Frasse, P., Wang, H., et al. (2014). Characterization of the tomato ARF gene family uncovers a multi-levels post-transcriptional regulation including alternative splicing. PLoS ONE 9:e84203. doi: 10.1371/journal.pone.0084203
Keywords: Prunus spp., breeding, gene regulation, transcription factors, flowering time, fruit quality, abiotic stress, biotic stress
Citation: Bianchi VJ, Rubio M, Trainotti L, Verde I, Bonghi C and Martínez-Gómez P (2015) Prunus transcription factors: breeding perspectives. Front. Plant Sci. 6:443. doi: 10.3389/fpls.2015.00443
Received: 09 March 2015; Accepted: 29 May 2015;
Published: 12 June 2015.
Edited by:
Ariel Orellana, Universidad Andres Bello, ChileReviewed by:
Swarup Kumar Parida, Jawaharlal Nehru University, IndiaShichen Wang, Kansas State University, USA
Andrea Miyasaka Almeida, Universidad Andrés Bello, Chile
Copyright © 2015 Bianchi, Rubio, Trainotti, Verde, Bonghi and Martínez-Gómez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Martínez-Gómez, Department of Plant Breeding, Centro de Edafología y Biología Aplicada del Segura, Consejo Superior de Investigaciones Científicas, Campus Universitario de Espinardo, Building 25, E-30100 Espinardo, Murcia, Spain,cG1hcnRpbmV6QGNlYmFzLmNzaWMuZXM=