 Alberto de Marcos1‡
Alberto de Marcos1‡ Magdalena Triviño1‡
Magdalena Triviño1‡ María Luisa Pérez-Bueno2
María Luisa Pérez-Bueno2 Isabel Ballesteros1†
Isabel Ballesteros1† Matilde Barón2
Matilde Barón2 Montaña Mena1*
Montaña Mena1* Carmen Fenoll1*
Carmen Fenoll1*- 1Facultad de Ciencias Ambientales y Bioquímica, Universidad de Castilla-la Mancha, Toledo, Spain
- 2Departamento de Bioquímica, Biología Celular y Molecular de Plantas, Estación Experimental del Zaidín, Granada, Spain
Loss of function of the positive stomata development regulators SPCH or MUTE in Arabidopsis thaliana renders stomataless plants; spch-3 and mute-3 mutants are extreme dwarfs, but produce cotyledons and tiny leaves, providing a system to interrogate plant life in the absence of stomata. To this end, we compared their cotyledon transcriptomes with that of wild-type plants. K-means clustering of differentially expressed genes generated four clusters: clusters 1 and 2 grouped genes commonly regulated in the mutants, while clusters 3 and 4 contained genes distinctively regulated in mute-3. Classification in functional categories and metabolic pathways of genes in clusters 1 and 2 suggested that both mutants had depressed secondary, nitrogen and sulfur metabolisms, while only a few photosynthesis-related genes were down-regulated. In situ quenching analysis of chlorophyll fluorescence revealed limited inhibition of photosynthesis. This and other fluorescence measurements matched the mutant transcriptomic features. Differential transcriptomes of both mutants were enriched in growth-related genes, including known stomata development regulators, which paralleled their epidermal phenotypes. Analysis of cluster 3 was not informative for developmental aspects of mute-3. Cluster 4 comprised genes differentially up−regulated in mute−3, 35% of which were direct targets for SPCH and may relate to the unique cell types of mute−3. A screen of T-DNA insertion lines in genes differentially expressed in the mutants identified a gene putatively involved in stomata development. A collection of lines for conditional overexpression of transcription factors differentially expressed in the mutants rendered distinct epidermal phenotypes, suggesting that these proteins may be novel stomatal development regulators. Thus, our transcriptome analysis represents a useful source of new genes for the study of stomata development and for characterizing physiology and growth in the absence of stomata.
Introduction
Stomata regulate CO2 uptake and water loss, and are essential for cooling down plant leaves, and for pumping water and nutrients from roots to shoots through transpiration streams. They also play a pivotal role in global carbon and water cycles (Hetherington and Woodward, 2003). Although environmental conditions regulate the extent of stomata aperture, stomata operate within relatively narrow margins, which are set for their optimal function and are rarely wide open (reviewed in Dow and Bergmann, 2014; Dow et al., 2014a). Therefore, their abundance and distribution patterns are key for determining the maximum area available for gas exchange, thus impinging on plant survival and reproduction.
The adoption of Arabidopsis thaliana (Arabidopsis hereafter) as a model for stomatal development has produced a wealth of information regarding the genetic control of this process (Dong and Bergmann, 2010; Pillitteri and Dong, 2013). Stomata differentiation takes place gradually during organ development (Geisler and Sack, 2002) via the interplay of a genetic programme with environmental cues and results in different stomatal numbers under different conditions (Casson and Gray, 2008; Casson et al., 2009; Kang et al., 2009; Xie et al., 2010; Delgado et al., 2012; Tricker et al., 2012; Casson and Hetherington, 2014; Kumari et al., 2014). To date, the mechanisms that link environmental cues to the gene circuits that regulate stomata development remain largely unknown. Recently, the developmental response to elevated atmospheric CO2 was analyzed (Engineer et al., 2014). The analysis involved two carbonic anhydrases and an extracellular protease, which promotes accumulation of the signaling peptide EPF2, a repressor of stomatal development. Evidence exists for a broad intraspecific natural variation in stomatal abundance (Woodward et al., 2002). A detailed study unveiled accessions with extremely high or low values, suggesting that genetically determined stomatal abundance may have an adaptive value in natural environments (Delgado et al., 2011). In the past years, a combination of genetic, genomic, and biochemical approaches have contributed to dissecting gene circuits regulating stomata development, their abundance and patterns (Lau and Bergmann, 2012). While these studies have identified only a few positive regulators of stomata development, they describe a wealth of genes that ensure correct stomata patterns. Amongst these negative regulators of stomatal development are membrane receptors and receptor kinases (TMM and the ERECTA family), signaling peptides, and specific members of MAP kinase cascades, among others (Pillitteri and Torii, 2012; Torii, 2012; Wengier and Bergmann, 2012).
This research efforts have generated mutant or engineered Arabidopsis genotypes with various stomatal abundances and patterns, establishing correlations between reduced stomatal densities and reduced transpiration/increased water-use efficiency (Von Groll et al., 2002; Yoo et al., 2010; Chakravorty et al., 2011; Xing et al., 2011; Franks et al., 2015). Recent work has analyzed a range of genotypes with distinct stomatal abundances and patterns, determining how epidermal phenotypes impinge on physiological parameters such as stomatal conductance or CO2 assimilation. These studies have also highlighted the relevance of proper stomata spacing and have revealed the physiological consequences of different stomatal densities (Dow et al., 2014a,b). Microarray analysis of various genotypes has also proven useful for describing developmental aspects and for identifying new genes (Hachez et al., 2011; Pillitteri et al., 2011). Other genome-wide approaches have identified in vivo chromatin binding sites for SPCH (Lau et al., 2014), providing a suite of putative target genes for this transcription factor, which acts as a positive regulator of stomata development.
As stomata are crucial for land plants, species lacking stomata are very rare (Woodward, 1998). However, loss-of-function of positive stomatal development regulators gives rise to stomataless phenotypes. Among these positive regulators are three related basic helix-loop-helix (bHLH) transcription factors that drive entry into stomatal lineage (SPEECHLESS, SPCH; Macalister et al., 2007), transit from meristemoid to guard mother cell (GMC) (MUTE; Pillitteri et al., 2007) and terminal differentiation of GMC into paired GCs (FAMA; Ohashi-Ito and Bergmann, 2006) (Figure 1A). Loss-of-function mutations in any of these genes give rise to stomataless plants (Figure 1; reviewed by Dong and Bergmann, 2010). spch mutants produce only pavement cells (Macalister et al., 2007); mute mutants produce arrested stomatal lineages (Pillitteri et al., 2007; Triviño et al., 2013) and fama mutants produce caterpillar-like GMC tumors instead of forming stomata (Ohashi-Ito and Bergmann, 2006).
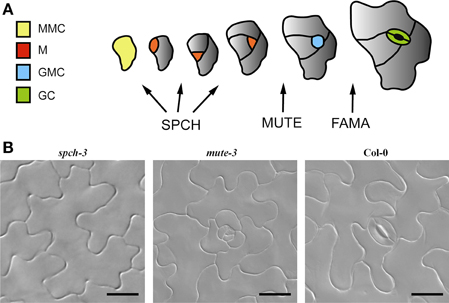
Figure 1. Stomatal development in Arabidopsis. (A) Arabidopsis stomatal development from a protodermal (Meristemoid Mother cell, MMC; yellow) involves sequential cell division and differentiation events that can be grouped into three main stages, regulated by three bHLH transcription factors: SPEECHLESS (SPCH), MUTE, and FAMA. SPCH is required for up to three asymmetric divisions that generate the cells in the stomatal lineage: meristemoids (M, red) and stomatal lineage ground cells (gray) that will eventually differentiate into pavement cells. MUTE is essential for the transition of the youngest meristemoid to a guard mother cell (GMC, blue) and FAMA controls the symmetric division of the GMC, which produces the guard cell pair (GC, green). (B) DIC micrographs of adaxial cotyledons showing the epidermal phenotypes of the genotypes used in this work. Loss-of-function mutations in SPCH (spch-3) prevent the initiation of stomatal lineages, while mutations in MUTE (mute-3) allow lineages to initiate and develop, but they arrest prior to stomata differentiation. Col-0 is a wild-type accession. Bar, 20 μm.
Although mutants lacking stomata are extreme dwarfs, their cotyledons are amenable to molecular, physiological and developmental analysis. Accessing their genome-wide transcriptional complement will provide unbiased clues as to which genes and pathways are active under the conditions imposed by their genotypes. In this work, we exploit the transcriptomes of spch-3, mute-3, and Col-0 cotyledons to identify the molecular trends of stomataless plants (genes similarly expressed in both mutants that had distinct epidermal cell types) and of mute-3 (genes differentially expressed only in this mutant and potentially related to developing stomatal lineages). We made predictions from transcriptome analysis related to physiology or development and tested them functionally by in vivo fluorescence imaging, as well as by phenotyping loss-of-function mutants and transgenic conditional overexpressing lines.
Materials and Methods
Plant Material and Growth Conditions
Arabidopsis thaliana L. (Heyn) ecotype Columbia-0 and the mutant line spch-3 (Macalister et al., 2007) were obtained from NASC (accessions N1092 and SAIL_36_B06, respectively); mute-3 is described in Triviño et al. (2013). Transgenic TRANSPLANTA lines are described in Coego et al. (2014). T-DNA insertion lines were purchased from NASC and homozygous lines were generated and genotyped (Table S1). All lines were in a Col-0 background.
For in vitro growth, sterile seeds were sown in 1X MS + 1% sucrose plates and kept at 4°C in the dark for 48 h. Seedlings were grown in chambers with a 16 h photoperiod, 21°C, 60% relative humidity and 70 μmol photon cm-2 s-1, as described in Delgado et al. (2011).
RNA Extraction and Microarray Hybridization
RNA was extracted from 30 cotyledons collected 21 days post germination (dpg). Mutant genotypes were maintained in heterozygous stocks and segregating homozygous seedlings were identified by microscopic inspection. For each mutant seedling, one cotyledon was phenotyped and the other frozen in liquid nitrogen. Nine independent samples/genotype were collected and pooled into three biological replicates for RNA extraction.
RNA was extracted with Trizol/RNeasy as described in Triviño et al. (2013). RNA quality was tested with an Agilent 2100 Bioanalyzer (Brolingen). Sample preparation and hybridization with ATH1 GeneChip (Affymetrix) was performed at the Centro Nacional de Biotecnología (Madrid). Three independent biological samples were used to hybridize each of the three slides used. The original hybridization results were deposited in ArrayExpress (accession number E-MTAB-3416).
Transcriptome Analyses
Background correction, normalization and expression data averaging following optical reading of GeneChips with a 3000 7G scanner (Affymetrix), as well as conversion to.CEL files, was performed by robust multichip analysis (RMA) (Irizarry et al., 2003). Gene expression values were adjusted to a linear model to apply a contrast by Student's t-test using an empirical Bayes analysis (LIMMA, Smyth, 2004) and to obtain p-values. Benjamini and Hochberg's (1995) method was used to correct for false discovery rate (FDR). FDR adjusted p-values < 0.05 and fold changes between samples > ± 2 were considered significant. Transcripts absent in all genotypes were eliminated according to the MAS5 algorithm (Hubbell et al., 2002). Transcripts detected in at least two of the three replicates were considered “present.”
Genes differentially expressed (DE) among genotypes were obtained using the VENNY software program (Oliveros, 2007). Functional classification of Venn diagram intersections was performed with MapMan 3.0.0 (Thimm et al., 2004) for Figure 5. Gene Ontology-based software AgriGO (Du et al., 2010) was used for Supplementary Figures S1, S2 and S3 using TAIR 10. Clustering was made using a k-means clustering algorithm (k = 4; covariance as a distance method) with Genesis software (Sturn et al., 2002).
Real Time PCR
Expression levels were estimated by quantitative RT-PCR, using cDNA obtained from the RNA samples used for microarray hybridization, with the High-Capacity cDNA Archive Kit (Applied Biosystems); qPCR was performed on a LightCycler® 480 II Real-Time PCR instrument (Roche), using the Maxima SYBR Green qPCR Master Mix (Thermo Scientific) and the primer sets listed in Table S2. Three biological replicates were analyzed per genotype. CT values and relative expression changes were obtained with the LightCycler® 480 software version 1.5 (Roche) and determined by the efficiency method, where fold change is calculated as ECtT(C)−CtT(S)T × ECtR(S)−CtR(C)R. UBQ10 (At4g05320) and ACT2 (At3g18780) were the reference genes.
β-estradiol Treatments
Phenotypic effects of TF overexpression in the TRANSPLANTA lines (Coego et al., 2014) were tested by germinating seeds on MS containing 10 μM 17-β-estradiol (E8875, Sigma) and epidermal inspection 6 days later. At least five plants per line were examined. Lines showing phenotypes were tested at least twice.
Microscopy
Epidermal phenotypes were determined by DIC microscopy, as described in Delgado et al. (2011) and Triviño et al. (2013).
Imaging of Chlorophyll-fluorescence Kinetics
Fluorescence kinetics were recorded according to Granum et al. (2015) using an Open FluorCam FC 800-O (Photon Systems Instruments, Brno, Czech Republic). After 30 min of dark-adaptation, plants were illuminated at 450 μmol photon cm−2 s−1 for 10 min, followed by a 10 min relaxation period in the dark. Saturating pulses of 2000 μmol photon cm−2 s−1 and 1 s long were given 10 s after the beginning of each period, and then every 2 min. After each excitation and relaxation period, a steady-state was reached.
Maximum quantum efficiency of PSII was calculated as FV/FM (1 – F0/FM) where F0 and FM were the minimum and maximum fluorescence in the dark-adapted state, respectively. The quantum yield of PSII (ΦPSII) was calculated as 1 – FS/FM', where FS and FM' were the fluorescence before and during saturating pulses in the light-adapted state, respectively. Non-photochemical quenching (NPQ) was calculated as FM/FM' – 1. Each experiment was repeated four times with similar results; a total of 10–20 plants/genotypes were sampled per experiment.
Multicolor Fluorescence Imaging
Multicolor fluorescence imaging was carried out on the adaxial side of the leaves using an Open FluorCam FC 800-O (Photon Systems Instruments, Brno, Czech Republic). The imaging in blue (F440), green (F520), and red (F680) regions of the spectrum was acquired sequentially for each sample, as described by Granum et al. (2015). Experiments were repeated four times with similar results; a total of 10–20 plants/genotype were sampled per experiment.
Pigment Composition
The content on chlorophylls and xanthophylls plus carotenoids was determined in 90% methanol extracts, as per Lichtenthaler and Buschmann (2001).
Results and Discussion
Transcriptomes of Stomataless Mutants
To investigate gene expression profiles associated with the developmental and physiological features of stomataless plants, a transcriptomic analysis was performed for two mutants lacking stomata, i.e., spch-3 and mute-3 and the wild-type, stomata-bearing Col-0 accession. RNA from 21 dpg cotyledons was used to hybridize Affymetrix ATH1 microarrays (see Materials and Methods).
Transcriptomes were first analyzed for present/absent transcripts, using the MAS5.0 algorithm to set the detection threshold in the Affymetrix chip (Hubbell et al., 2002) and selecting transcripts present in at least two biological replicates. Approximately 14,000 genes were expressed in each genotype (Col-0: 13,985; mute-3: 14,307; spch-3: 13,833), most of which (ca. 13,000) were common to all genotypes. For almost 2000 genes, transcripts were present in only one or two genotypes. Genes exclusively expressed in one genotype were 216 in spch-3, 383 in mute-3 and 382 in Col-0 (Figure 2A).
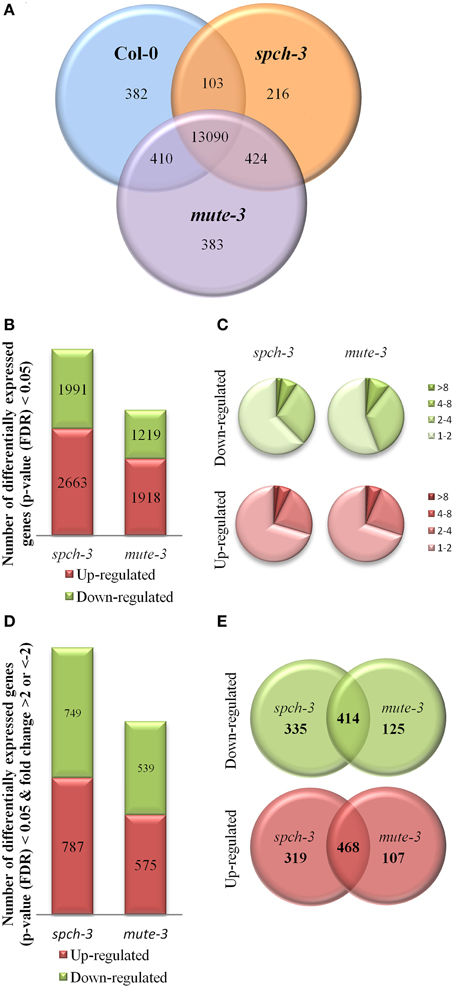
Figure 2. Comparative transcriptomic analysis of spch-3, mute-3, and Col-0 cotyledons. (A) Venn diagram showing the intersection of all genes expressed in each genotype (MAS5-present call in at least two biological replicates in the array). (B) Genes differentially expressed in each mutant with respect to Col-0 (LIMMA FDR < 0.05) and (C) their distribution by fold-change difference. (D) Genes differentially expressed with a fold-change >2 or <–2 in each mutant, compared to Col-0 and (E) Venn diagrams showing their intersections. Transcripts correspond to three independent replicates of RNA extracted from 21 dpg cotyledons.
To determine if the experimental design allowed for detecting genotype-characteristic transcripts, we examined the expression of genes previously involved in stomatal development. Col-0 cotyledons had only pavement cells and stomata, mute-3 showed pavement cells and developing stomatal lineages, including arrested meristemoids and spch-3 had exclusively pavement cells. All genotypes accumulated transcripts for ERECTA, ERL1, YDA, MKK4/5, MPK3/6, STOMAGEN and SCREAM/ICE1, and SCREAM2, whose expression is not exclusive of developing stomatal lineages or specific cell types. In contrast, the genotypes differed in transcripts specific for particular stomatal lineage cell types. For instance, only mute-3 expressed the immature lineage markers MUTE, EPF2, and TMM, and only Col-0 expressed the guard cell marker gene FAMA. As expected, transcripts for all these genes were absent in spch-3. Thus, the observed transcriptional profiles matched the epidermal phenotypes, even for genes expressed in cell types or stages with a very small contribution to all cells represented in the samples. These results strongly support the experimental approach, as it discriminates genotype-specific, low-abundance transcripts. Consequently, these transcriptomes should also contain transcripts from undescribed genes expressed preferentially/exclusively in each of the three genotypes.
Next, pairwise comparisons of gene expression levels were conducted using LIMMA (Smyth, 2004) for a FDR < 0.05 (Figure 2B). The results showed that the number of differentially expressed (DE) genes with respect to Col-0 was higher in spch-3 than in mute-3. The amplitude of DE (fold-change values) with Col-0 ranged between −32 and 53 for mute-3 (corresponding to a plant-specific protein of unknown function encoded by At1g64360 and the transcription factor PISTILLATA), and between −63 and 209 for spch-3 (for transcripts encoding the cell wall protein ECS1 and PISTILLATA, respectively). In both mutants, approximately 8% of the DE genes had absolute fold-change values >4 and around 27% presented values between 2 and 4 (Figure 2C). Genes DE, with an absolute fold-change >2, were 1114 in mute-3 and 1536 in spch-3 (Figure 2D and Table S3); only a fraction of these genes were regulated in a similar manner in the two mutants (Figure 2E).
Relative expression levels for a panel of genes in the three genotypes was further tested by qPCR, selecting 10 genes that in the microarray comparisons were similarly up- or down-regulated in both mutants, compared to Col-0. The results (Figure 3) showed that for the 10 genes, expression tendencies were the same as estimated by the two independent methods.
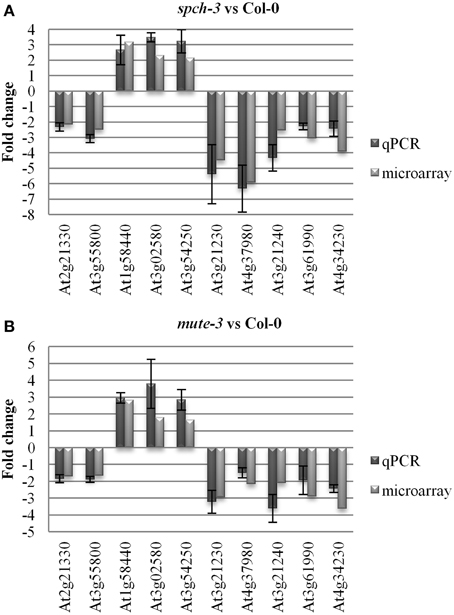
Figure 3. Quantitative PCR for microarray validation. Arrays were validated by quantitative RT-PCR for 10 genes selected from the list in Table S3. UBIQUITIN10 and ACTIN2 served as reference genes. The fold change of the qPCR was determined by the efficiency method (ECtT(C)-CtT(S)T × ECtR(S)-CtR(C)R). The results of qPCR for spch-3 (A) and mute-3 (B) respect to Col-0 were averaged from three independent experiments, with the error bar indicating the standard error of the mean.
A previous microarray study involving spch-3 and other stomatal mutants (Pillitteri et al., 2011) was designed to minimize physiological differences among genotypes, while the present work aimed at stressing such differences. Thus, when compared, these studies attempt to answer different questions and are not comparable. In the present analysis, the high number of DE genes in the mutants might similarly reflect profound differences in growth and physiology among wild-type and stomataless mutant plants, and not merely their epidermal phenotypes, which were different in the two mutants. To our knowledge, the present study is the first to offer a description of a mute mutant transcriptome.
General Transcriptomic Features Common to Stomataless Mutants
We expected genes DE in spch-3 or in mute-3, with respect to Col-0, to include those related to the physiology of stomataless plants and to genotype- or cell type-specific trends. To characterize these gene expression patterns, we performed a k-means cluster analysis, using normalized values of the three biological replicates for each genotype. For the analysis, 1797 genes were selected, based on whether they were significantly (FDR < 0.05) up- (>2 fold) or down-regulated (<–2 fold) in any of the mutants, with respect to either Col-0 or between the two mutants. Z-score transformations of the log2 expression values were used for a k-means clustering (k = 4) (Figure 4). Genes classified in each cluster are listed in Table S4. Clusters 1 and 2 include, respectively, genes with higher or lower expression in the two stomataless genotypes than in Col-0. Clusters 3 and 4 include genes with a lower or higher expression in mute-3, with respect to the other genotypes, thereby representing expression patterns characteristic of mute-3. These genes may relate to epidermal differences of mute-3 with spch-3 and Col-0 (Figure 1B), and they are described in the sections devoted to development.
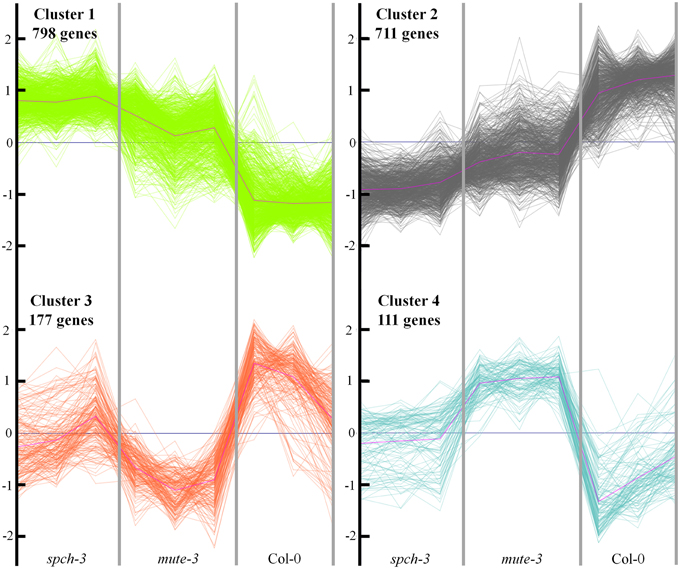
Figure 4. Cluster analysis of expression profiles for differentially expressed genes in the three genotypes. A total of 1797 genes significantly (FDR < 0.05) up- (>2 fold) or down (<–2 fold) regulated in any of the mutants, with respect to Col-0 or between the two mutants, was selected. The normalized expression values were used to perform a k-means clustering for k = 4 (see Methods). The Y-axis shows normalized fold-changes in gene expression and the X-axis indicates each genotype, with the three biological replicates independently represented. Genes classified in each cluster are listed in Table S4.
To identify biological processes similarly altered in both mutants, we applied a gene set enrichment analysis to clusters 1 and 2 (GO database through AgriGO software). Cluster 1 analysis revealed increased transcription of genes classified in several functional categories (Supplementary Figure S1): lipid localization, regulation of cell size and developmental growth. These categories include important growth regulators like PIN3, COBRA, and BRI1. There is also an enriched group of categories involving carbohydrate metabolic processes like polysaccharide and glucan biosynthesis (mostly related to cell wall; see below). In cluster 2, genes with decreased expression in stomataless mutants belonged to three overrepresented categories (Supplementary Figure S2), with cellular nitrogen compound metabolic process appearing as a hub. Genes involved in the biosynthesis of glucosinolates and glycosinolates were mostly depressed, as were the master regulators of these pathways, MYB28, MYB29, and MYB34 (Frerigmann and Gigolashvili, 2014). Down-regulation of genes related to response to stimulus, e.g., chemicals, abiotic cues, or water deprivation was also patent.
We then identified the MapMan metabolic pathways altered in the two mutants based on the gene sets of clusters 1 and 2. Figure 5 represents the common up-regulated (cluster 1) genes in red and in green, common down-regulated (cluster 2) genes (see Figure 4 and Table S4). Almost all DE cell wall-related transcripts cataloged in MapMan were up-regulated in spch-3 and mute-3; they encoded proteins involved in the biosynthesis of precursors, cellulose and hemicelluloses, arabinogalactan and proline-rich wall proteins, extensins, expansins, pectin esterases, and other cell wall-remodeling proteins. These patterns may reflect a potential for growth in the mutants, while Col-0 cotyledons were terminally expanded, according to the biological processes predominant in both mutants (Figures S1, S2). Lipid metabolism-related DE genes were mostly up-regulated. They included 18 genes that encode enzymes involved in fatty-acid synthesis and elongation, e.g., acyl-carrier proteins, acetyl-CoA carboxylases, ketoacyl-ACP synthases and reductases, desaturases, and others. Most lipases were up-regulated, including the membrane-anchored glycerophosphoryl diester phosphodiesterase-like proteins SHAVEN-3 and SHV3-LIKE 1, involved in cell wall deposition and root hair and trichome differentiation (Hayashi et al., 2008). Both genes are expressed during stomatal development, according to the Arabidopsis eFP Browser (Winter et al., 2007); furthermore, a double sha3;slv1 mutant displayed enlarged guard cells (Hayashi et al., 2008), suggesting that both participate in stomatal-lineage cell differentiation. Some transcripts for enzymes related to sphingolipid synthesis were also up-regulated in both mutants. In contrast, the five DE genes in phospholipid synthesis were down-regulated.
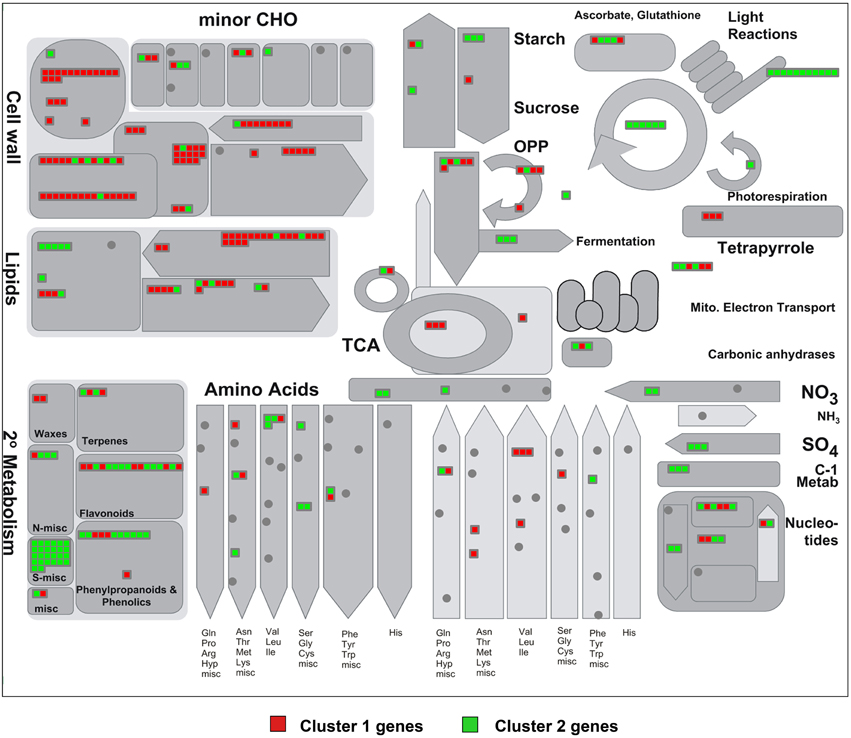
Figure 5. MapMan visualization of the differences in metabolism-related gene expression between stomataless mutants and wild-type plants. Differentially expressed metabolic genes classified in clusters 1 and 2 (Figure 4), and thus similarly regulated in both mutants, were analyzed using MapMan software. Each square represents a gene and displays a qualitative color code: red for genes up-regulated (cluster 1) and green for those down-regulated (cluster 2) in the mutants with respect to Col-0. Note the pathways with predominantly up-regulated (mostly red) and down-regulated (mostly green) genes.
Overall, the transcriptomes of stomataless mutants suggest a depressed secondary metabolism and dampened mechanisms for stress responses, while maintaining growth-related processes. These trends are hardly surprising, considering that CO2 acquisition is presumably very limited in plants with no stomata. Their extremely slow but sustained growth may as a result monopolize most carbon and energy resources, preventing major investments in non-essential processes.
Genes similarly regulated in both mutants, as compared to Col-0 classified in the remaining metabolic pathways, were mostly down-regulated and are discussed below.
Photosynthesis in Stomataless Mutants
The number of photosynthetic genes DE in both mutants that met the filters of fold-change and statistical significance set for the clustering was surprisingly low. However, most of the down-regulated genes code for crucial proteins, whose depletion should impair photosynthesis. All significant changes in PSII corresponded to repressed genes and included those coding for PsbB, PSBTc, PsbN, PsbQ-LIKE 1, PsbQ-LIKE 2, and PsbP-like protein 2. Other genes repressed with various levels of significance and fold-changes were nuclear or chloroplast genes coding for PSII protein subunits, such as PsbO2 (a regulating subunit in the oxygen-evolving complex), PsbH, PsbA (core protein D1), and PsbC (core chlorophyll-binding protein). The gene encoding the PSI reaction center subunit PsaA was repressed, as were those for five NADH-dehydrogenase subunits (NDH-O, N, D, H, and J). Cytochrome f was down-regulated, as was IMMUTANS, which encodes a chloroplast alternative oxidase (Wu et al., 1999). ATPase subunits ATPB and ATPF transcripts were decreased. Regarding CO2 assimilation, several Calvin cycle genes were repressed, most remarkably the chloroplast gene rbcL encoding the large subunit of RubisCO, but also genes encoding fructose bis-phosphate aldolase (FBA1), several phosphoglycerate kinases and a sedoheptulose bisphosphatase. A photorespiration-related glycolate oxidase was also down-regulated.
This transcriptional profile shared by the two mutants suggests impaired photosynthetic light reactions, low production of reducing power and ATP and depressed carbon assimilation, as expected of plants lacking stomata. However, chloroplasts do not seem dysfunctional in these mutants. Some genes related to tetrapyrrole metabolism, as well as GUN4, which couples nuclear gene transcription to chloroplast status (Larkin et al., 2003), were up-regulated. Several antenna protein subunits were also up-regulated, although with differences not always statistically significant. Therefore, the down-regulation in spch-3 and mute-3 of photosynthesis-related genes seems to be specific and not the result of general chloroplast dysfunctions. It is also possible that many genes in this category did not change their transcription, but that the corresponding proteins were more or less abundant or active in the mutants due to differences in post-transcriptional regulation.
The physiological impact of the observed transcriptional changes for photosynthesis-related genes was examined in vivo by quenching analysis of chlorophyll fluorescence. Kinetic analyses of the red fluorescence emitted by chlorophyll estimated photosynthetic efficiency, as well as mechanisms of energy dissipation indicative of stress. For these experiments, we tested plants grown in vitro for 21 days (as in the microarray experiments) or plants adapted to soil for 4 extra days. Figures 6A,B show that spch-3 and mute-3 displayed a statistically significant decrease in Fv/Fm compared to Col-0, indicating a dysfunction of the photosynthetic machinery. Both mutants also showed low values for PSII quantum yield (ΦPSII) relative to wild-type plants, suggesting an inhibition of photosynthetic electron transport. The decrease in ΦPSII was associated with a larger capacity for energy dissipation (measured as non-photochemical quenching, NPQ) typical of stress conditions and a higher proclivity for photoinhibition, as shown by the higher irreversible NPQ (NPQi). We observed similar trends in 7 dpg cotyledons, although only differences in Fv/Fm were statistically significant (not shown).
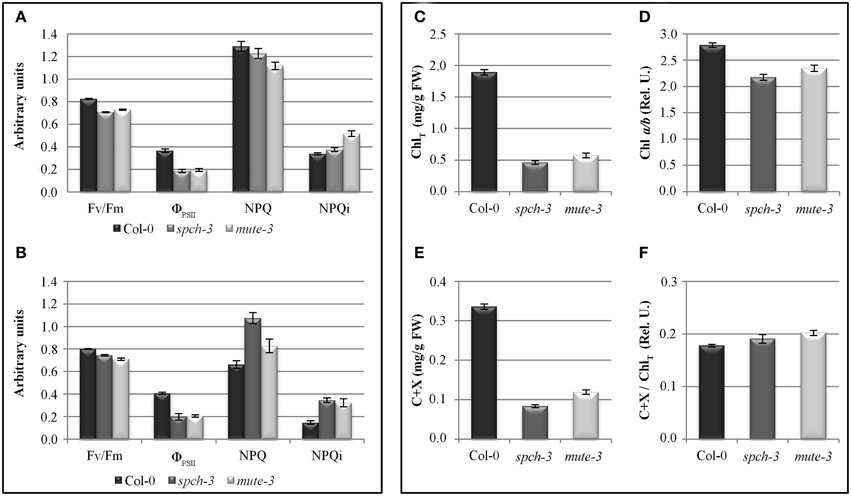
Figure 6. Photosynthetic parameters and pigments in wild-type and stomataless mutants. Left panel: photosynthesis parameters calculated by quenching analysis of chlorophyll fluorescence in 21-day-old plants (A) and after 4 days acclimation to soil (B). Right panel: total chlorophyll content (ChlT (C)), Chl a/b ratio (D), carotenoids and xanthophylls (C+X (E)), and carotenoids and xanthophylls over total chlorophyll content (F) in Col-0, spch-3, and mute-3 plants after adaptation to soil. Error bars are SE. All pairwise differences were statistically significant in a Student t-test (N = 10; T-test p-value < 0.038), except NPQ differences between spch-3 and Col-0 in (A).
These results are consistent with a retroinhibition of the thylakoid electron transport chain, perhaps due to limiting concentrations of internal CO2, which inhibit the Calvin cycle. The strong down-regulation of rbcL transcripts and the repression of genes coding for subunits of PSII and other complexes of the electron transport chain also support this interpretation.
Relative content of chlorophylls (Chl) a and b and accessory pigments (carotenoids and xanthophylls, C + X) were measured in leaf extracts (Figures 6C–F). As expected due to their pale phenotypes, both mutants had a reduced content of all photosynthetic pigments on a fresh weight basis, compared to Col-0. Chl a/b ratios were significantly reduced, while C+X/Chl ratios increased. These results suggest that pigment composition of the photosynthetic complexes I and II, most of which reside in their antenna complexes, is altered in the mutants. The decrease in Chl a/b ratio is compatible with an increase in the proportion of antenna complexes/reaction centers, suggested by the transcriptomic data. DE transcripts in the category antenna proteins had many up-regulated genes (six genes vs. one repressed, all significant and close to the fold-change threshold). The opposite occurred for reaction centres, with five down-regulated genes (all from PSII). The low chlorophyll content in the mutants explains the decrease in the intensity of fluorescence observed in the far-red region (F740) (Figure 7). Alterations in the intensity of red fluorescence (F680) may stem from changes in the Chl a/b ratio in the mutants. Thus, both chlorophyll content and photosynthetic efficiency estimations were those expected from the transcriptomic profiling of the two stomataless mutants and suggested depressed, albeit not null, photosynthetic activity.
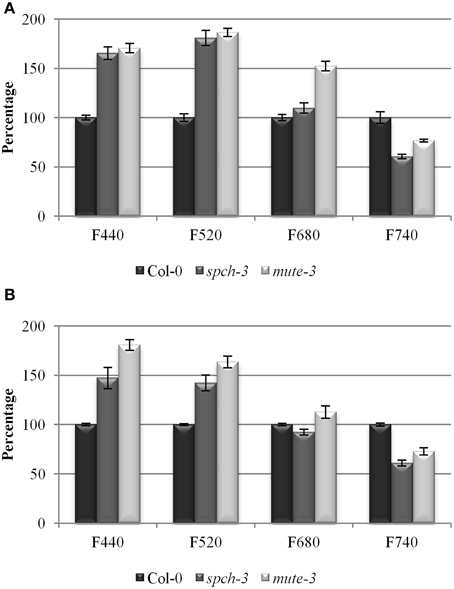
Figure 7. Multicolor fluorescence in the three genotypes. Relative fluorescence as a percentage of Col-0 in the blue (F440), green (F520), red (F680), and far red (F740) regions of the spectrum for 21-day-old plants (A) and after 4 days acclimation to soil (B). Error bars are SE. All pairwise differences were statistically significant in a Student t-test (N = 10; T-test p-value = 3.74 E-07).
In the present work, plants grew in a sucrose-supplemented medium (see Materials and Methods). Sucrose appears to be taken-up and used by the mutants, as suggested by increased transcripts for the phloem unloading sucrose transporter ATSUT2 (Ayre, 2011) and the degradative invertase CINV1 (Barratt et al., 2009). The activation of glycolysis, suggested by the up-regulation of genes coding for plastidic and cytosolic pyruvate kinase subunits, as well as other enzymes from the PPO and TCA cycles may contribute to plant growth. Transcripts for fermentative enzymes were, however, down-regulated. The fact that spch-3 and mute-3 can to some extent grow in soil also indicates that they do perform photosynthesis, perhaps helped by more active carbonic anhydrases, as well as a modified cuticle that might be more permeable to CO2. However, growth was much more extensive (notably for mute-3) in sucrose-containing medium.
The dwarf spch-3 and mute-3 phenotypes, when growing on sucrose, may reflect other physiological constraints for growth without stomata. These possible constraints include, e.g., limited O2, depressed transpiration hindering nutrient and water root uptake, as well as evaporative cooling or the accumulation of volatile hormones or metabolites. In spch-3, there are very limited epidermal cell divisions (as stomatal lineages that contribute to epidermal cell numbers are absent). This fact might impose another constraint for growth and explain why dwarfism is more drastic in spch-3 than in mute-3.
Nitrogen, Sulfur, and Secondary Metabolisms
DE genes related to the assimilation of sulfur (such as those coding for one APS and two APS-Kinases) and nitrogen (those coding for GLU and GLN-synthases and two nitrate transporters) were all down-regulated in spch-3 and mute-3. Both mutants displayed repression of all 26 DE genes involved in glucosinolate metabolism, 19 of which are in the biosynthetic pathway. Glucosinolates are nitrogen and sulfur-rich secondary metabolites, whose accumulation depends on sulfur status (Falk et al., 2007). Previous metabolite and transcript profiling revealed coordinated repression of most glucosinolate-related genes in response to sulfate limitation (Hirai et al., 2005). Also repressed were three out of the four DE genes related to N-containing alkaloids. Given the depressed photosynthetic capacity, which might limit reducing power and ATP needed for N and S metabolisms, this behavior was not surprising. In contrast, EXORDIUM-LIKE (EXL) genes EXO, EXL1, EXL3, EXL4, and EXL5, which promote growth during low C and energy supply (Schroder et al., 2011) were up-regulated. Also induced was ASN1, whose overexpression increases plant fitness under N-limiting conditions (Lam et al., 2003) and is triggered by sugar starvation (Baena-Gonzalez et al., 2007) and photosynthesis inhibitors (Fujiki et al., 2001). These trends suggest acclimation of stomataless mutants to low carbon, reduced intermediate metabolites and energy-limiting growth conditions. DE genes related to starch metabolism (two degradative amylases and one starch synthase) were down-regulated; sucrose synthesis was also depressed, while CINV1, a cytosolic invertase related to sucrose degradation, was induced.
Most DE genes in the phenylpropanoid biosynthetic pathway such as those involved in lignin and lignane synthesis (4CL5, 4CL2, ELI3-1, OMTF3 y CAD5) were repressed, with the exception of the laccase-encoding gene LAC8. Flavonoid-related genes showed heterogeneous behavior. While most genes related to secondary metabolism were repressed in the mutants, some were up-regulated, notably some involved in the biosynthesis of chalcones, isoflavones, or anthocianins. For instance, a putative isoflavone reductase (At1g19540) showed strong induction. Many sterol/brassinosteroids biosynthetic genes, such as SQE1, SMT3, and DWF7 were up-regulated. The mevalonate route, involved in carotenoid synthesis, showed induced genes, notably those encoding two mevalonate diphosphate decarboxylases, which promote accumulation of the carotenoid precursor isopentenil pirophosphate. To determine if the mutants were accumulating some end products of these pathways, we performed multicolor fluorescence analysis and found that the two mutants had an increased blue and green autofluorescence (Figure 7).
According to the transcriptomic data, the compounds responsible for this increase may be carotenoids and anthocyanins, and perhaps chalcones. Although phenols also fluoresce in this spectrum, their biosynthetic pathways are down-regulated and as such, they are improbable contributors to the observed fluorescence increases. The notion that the mutants differentially accumulate photoprotective compounds fits with their accessory photosynthetic pigment composition (Figures 6E,F), as well as their high NPQ and NPQi values (Figures 6A,B). An acclimation to the photoinhibitory conditions resulting from the inhibition of photosynthesis in both mutants may explain the accumulation of auxiliary pigments. This accumulation of putative photoprotective pigments and a depressed N and S metabolism are the most notable features of a secondary metabolism in the two stomataless genotypes.
Growth and Development in the Absence of Stomata
In agreement with the analysis of biological processes (Figures S1 and S2), MapMan found that DE genes in clusters 1 and 2, commonly regulated in both mutants (Tables S3 and S4), appeared in several development and growth-related categories, including genes involved in cell division and expansion. For instance, genes coding for the cyclin-dependent kinase CYCP1 and a cyclin-related protein (At2g41830), both with unknown functions, were up-regulated, as was the endoreplication factor FZR2 (FIZZY-RELATED 2). Genes for DNA synthesis encoding a telomere-binding protein, several histones or a putative helicase, as well as most genes encoding cytoskeleton and vesicle transport proteins also showed induction. The transcript with the highest induction in both mutants corresponded to PISTILLATA. Initially described as a flower-specific transcription factor, recent data available at the eFP Browser database (Winter et al., 2007) indicate that PISTILLATA is also expressed in other developmental contexts. These include the quiescent centre, the cellularized seed endosperm, root xylem pole pericycle cells in NPA-treated seedlings, endodermis and columella/root cap under low pH and trichomes of gl3-sst mutants. In 5-day-old spch-3 seedlings, Pillitteri et al. (2011) also found abundant PISTILLATA transcripts. The reasons for its over-accumulation in spch-3 and mute-3 and its possible consequences remain unexplained.
Next, genes in clusters 3 and 4 were analyzed. These were DE in mute-3 respect to Col-0 and spch-3, and may be characteristic of an epidermis containing developing stomatal lineages absent in the two other genotypes. Using gene set enrichment analysis (GO database through AgriGO software) we identified overrepresented biological processes. Inspection of cluster 3 revealed features similar to cluster 2; this was not informative regarding developmental aspects of mute-3. In cluster 4 (Supplementary Figure S3), as expected, very few genes corresponded to metabolic pathways, stress or cell wall, as mute-3 shared most of them with spch-3. Both mutants also shared most of the DE genes classified in the category Development (as described above; Figures S1 and S2), with some notable exceptions: TMM, a marker specific for developing stomatal lineages (Nadeau and Sack, 2002) was present in cluster 4, as was PDF1, specific for the developing epidermis (Abe et al., 1999). Several RNA-related genes were present in this cluster, including SCZ, a meristemoid-specific transcription factor in aerial organs (Pillitteri et al., 2011). Previous work (Pillitteri et al., 2011) using a severe “meristemoid-only” double mutant (scrm-D;mute), which had one cell type similar to those present in mute-3, overlapped only in 24 transcripts with cluster 4. In addition to the important epidermal differences between mute-3 and scrm-D;mute, growth conditions and age differed between Pillitteri et al. (2011) and the present work; hence, the limited overlap of the two transcriptomes was not surprising. We also investigated whether cluster 4 might include genes previously identified as putatively regulated by SPCH (Lau et al., 2014), since SPCH is necessary for the development of the stomatal lineages present in mute-3. For doing so, we compared the 1274 high-confidence SPCH targets represented in the ATH1 array with the 111 genes classified in cluster 4. Results identified 36.9% (41) of the genes characteristic of mute-3 as putative SPCH targets, a proportion much higher than the expected 5.6% by random hits. Therefore, cluster 4 showed highly significant enrichment in genes that might be under direct SPCH regulation.
In this analysis, we also found genes not previously involved in stomatal development. Some examples are BAM2, encoding a CLAVATA1-related receptor kinase-like protein, which is needed for cell fate specification in meristems (Deyoung and Clark, 2008), as well as CLE17, which encodes a putative extracellular peptide similar to CLV3 (Meng and Feldman, 2010). Also induced in mute-3 were TCP3, involved in heterochronic leaf development (Koyama et al., 2007) and NAC35/LOV1 (LONG VEGETATIVE PHASE1). We found that LOV1 co-expressed with the GC genes FAMA, KAT2, and MYB60 (in ATTED, Obayashi et al., 2007), and was expressed in GCs (Leonhardt et al., 2004). EDA17, which will be described below, was found in this cluster. These data enforce the potential of cluster 4 as a source of novel genes involved in stomatal lineage development.
Loss-of-function of Genes Differentially Expressed in the Mutants
We selected a set of 24 DE genes in spch-3 and/or mute-3 compared to Col-0 to inspect the phenotypic effects of their loss-of-function (Table S1). For three genes (LOV1, SOL1, and PI), promoter::reporter fusions were also tested in wild-type plants. Cotyledons of 5 and 10 dpg seedlings from homozygous T-DNA insertion lines were inspected for stomatal and pavement cell morphology, size, and spatial pattern. Only one of these mutants, carrying a T-DNA insertion in EDA17, had an epidermal phenotype; its stomatal index (proportion of epidermal cells that are stomata) was reduced to half that of Col-0 (Supplementary Figure S4). EDA17 was originally named HOTHEAD (HTH) by Krolikowski et al. (2003), because loss-of function mutants in this locus showed floral organ fusion. The line we used (SALK_024611) carried an insertion in the middle of the predicted third exon (Supplementary Figure S4) and was reported by Peng et al. (2006) as displaying the hth phenotype; therefore, it is considered a loss-of-function mutant. In our seed stock, we observed the reported floral organ fusion phenotype and the reduced fertility that characterizes hth mutants.
As EDA17 transcripts accumulate specifically in mute-3 and not in Col-0 or spch-3, EDA17 may be associated with developing stomatal lineage cell types. This hypothesis is supported by differential accumulation of EDA17 transcripts in the “only meristemoids” scrm-D;mute mutant (Pillitteri et al., 2011). EDA17 is involved in the biosynthesis of cuticular, very long-chain fatty acids (Kurdyukov et al., 2006). Since other cuticle- and epidermal wax-defective mutants display stomata pattern defects (Holroyd et al., 2002), further work on EDA17 may strengthen the intriguing connection between cuticle composition and stomata development.
Conditional Overexpression of Genes Differentially Expressed in spch-3 and/or mute-3
The suit of genes DE in spch-3 and/or mute-3 includes 158 transcription factors (TFs) listed in the Agris and/or PlantTFDB databases (Yilmaz et al., 2011; Jin et al., 2014), which are candidates for playing key roles in the developmental and metabolic phenotypes of the mutants. Several of the T-DNA lines we inspected corresponded to TFs; however, their loss-of-function rendered no epidermal phenotypes. Arabidopsis TFs often belong to gene families with partially redundant members; it is therefore common that loss-of-function alleles fail to provide informative phenotypes and their ectopic overexpression is used instead to hint at their putative roles. A drawback of this strategy is that it often renders deleterious phenotypes, including early lethality, which conditional overexpression systems can overcome. To examine the possible involvement of TF that were DE in the mutants, we inspected 322 transgenic lines that conditionally overexpressed 128 different TFs (Table S5). Among them, 194 lines corresponded to 76 TFs that were DE in spch-3 and/or mute-3, while the remaining were their paralogues, or showed a tendency to differential expression close to statistical significance. In these lines, β-estradiol induced TF overexpression (Coego et al., 2014). Figure 8A shows a diagram of the construct used for obtaining these lines, which we termed iTFoe (inducible TF overexpressor).
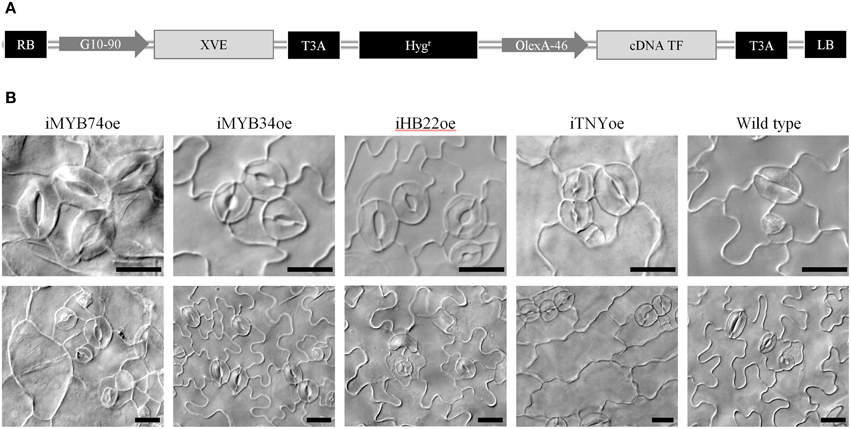
Figure 8. Conditional overexpression of transcription factors differentially expressed in spch-3 or mute-3 transcriptomes. (A) Diagram of the construct on pMDC7 used for generating the TRANSPLANTA lines for β-estradiol-inducible overexpression. LB, RB: left and right T-DNA borders. G10-90: artificial constitutive promoter. XVE: chimeric transcription factor activated by β-estradiol. OlexA-46: promoter recognized by activated XVE. cDNA TF: coding sequence for the different transcription factors. T3A: sequence for gateway cloning. For more details, see Coego et al. (2014). (B) Phenotypic effects of overexpression for selected TF. DIC micrographs of cotyledon abaxial epidermis in seedlings of the indicated genotypes (iTFoe and wild-type) grown for 6 days in β-estradiol plates. For each genotype, two representative fields are shown. Bar, 20 μm.
For each TF-coding gene, seeds from 4 to 1 independent lines were plated on a β-estradiol-containing medium and epidermal phenotypes were inspected in 6 dpg seedlings. iFAMAoe and iMUTEoe control plants rendered the expected phenotypes (Triviño et al., 2013) with all epidermal cells transformed in guard cells or in stomata, respectively. Overexpression of 19 TFs lead to abnormal epidermal phenotypes in all or some of the independent lines. The most recurrent effects were on stomatal density (number of stomata per area unit) and spacing (number of non-stomatal cells between nearest stomata), and in non-stomatal cell shape/size (Table S5).
Figure 8B shows a representative selection of these phenotypes. Overexpression of two R2R3 MYB proteins, MYB74 and MYB34, related to ABA responses (Xin et al., 2005) and IAA synthesis (Celenza et al., 2005), respectively, produced stomatal clusters in otherwise normal seedlings. iMYB74oe also had abnormal non-stomatal cells, with frequent giant rounded cells and small-cell patches in rosette arrays. Recently, ABA was identified as being involved in stomata development (Tanaka et al., 2013), a link that our results with iMYB74oe support. MYB34/ATR regulates the tryptophan pathway and modulates IAA levels, and its transcription is induced by cytokinins (Jones et al., 2010). As it is also involved in indol-glucosinolates homeostasis, MYB34/ATR1 impinges in both primary and secondary metabolism (Malitsky et al., 2008), and stomatal clusters in iMYB34oe might relate to any of these processes. Overexpression of ATHB22, a homeobox-zip TF of unknown function expressed in seedlings and siliques, triggered a strong epidermal phenotype with occasional clusters, frequent unpaired GCs and atypical pavement cell divisions; seedling morphology was normal. The extra cell divisions were not observed if overexpression was induced at 4 dpg, but stomata clusters still formed (not shown), indicating that ATHB22 produces distinct effects in the protodermis and in the developing epidermis. TNY is a NAC-domain protein, whose overexpression renders dwarf plants that accumulate transcripts from DRE- and ERE-driven genes (Sun et al., 2008). A gain-of-function allele with increased TNY expression also showed abnormal epidermal cells (Wilson et al., 1996). β-estradiol treatment of iTNYoe induced a very strong phenotype that included severe dwarfism and a very disturbed epidermis, with stomatal clusters and elongated, un-lobed non-stomatal cells.
These and similar results with other TRANSPLANTA overexpressing lines are the first indication that TFs selected by their differential expression in stomataless mutants may be novel players in stomatal and epidermal development gene circuits.
Conclusions
1. Transcriptomic analysis of expanded cotyledons from two Arabidopsis mutants lacking stomata identified sets of genes commonly regulated in both mutants compared to the wild-type, as well as genotype-specific DE genes. Each genotype expressed diagnostic genes related to stomata development and/or restricted to specific cell stages that matched its distinctive epidermal phenotype. The microarray data (confirmed by qPCR) showed that low abundance, genotype-specific transcripts were detectable in our samples. Hence, our unbiased transcriptomic analysis should also identify novel genes with the potential for explaining the developmental and physiological features of stomataless plants.
2. A set of genes commonly down-regulated in spch-3 and mute-3 suggest that cotyledons lacking stomata had depressed S, N, and secondary metabolisms. In contrast, only a few photosynthesis-related genes showed significant down-regulation, though they coded for crucial proteins. Chlorophyll fluorescence imaging revealed that the mutants maintained a depressed but not negligible photosynthesis. This, as well as the transcriptomic data suggestive of chloroplast functionality, indicates that stomataless plants can indeed perform some photosynthesis, which may be related to their (limited) capacity for growth in soil.
3. Both mutants expressed genes indicative of potential for growth at a time (21 dpg) when Col-0 cotyledons did not. Several genes differentially up-regulated in mute-3 were also identified; these included some high-confidence SPCH targets, as well as development-related genes exclusive to this mutant, which are new candidates for exploring stomatal lineage cell types absent in the other two genotypes.
4. A selection of development-related genes DE in one or the two mutants was examined for epidermal phenotypes associated with their loss-of-function, using T-DNA insertion lines. Of the 24 insertion mutant genes examined, one rendered an epidermal phenotype, leading to the identification of EDA17 as a new candidate gene involved in stomata development. Conditional overexpression of 19 transcription factors that were DE in the mutants induced altered stomatal phenotypes, suggesting functional roles for these novel transcription factors in stomatal or epidermal differentiation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Ana Rapp for technical assistance and NASC for providing Col-0, spch-3 and T-DNA lines. This work was supported by the Spanish Government (BIO2012-33952 and CSD2007-00057 to CF and BIO2007-60276 to MM), the Junta de Comunidades de Castilla-la Mancha (PPII10-0194-4164 to MM) and the CICE-Junta de Andalucía (P12-AGR-0370 to MBA).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00456
References
Abe, M., Takahashi, T., and Komeda, Y. (1999). Cloning and characterization of an L1 layer-specific gene in Arabidopsis thaliana. Plant Cell Physiol. 40, 571–580. doi: 10.1093/oxfordjournals.pcp.a029579
Ayre, B. G. (2011). Membrane-transport systems for sucrose in relation to whole-plant carbon partitioning. Mol. Plant 4, 377–394. doi: 10.1093/mp/ssr014
Baena-Gonzalez, E., Rolland, F., Thevelein, J. M., and Sheen, J. (2007). A central integrator of transcription networks in plant stress and energy signalling. Nature 448, 938–942. doi: 10.1038/nature06069
Barratt, D. H. P., Derbyshire, P., Findlay, K., Pike, M., Wellner, N., Lunn, J., et al. (2009). Normal growth of Arabidopsis requires cytosolic invertase but not sucrose synthase. Proc. Natl. Acad. Sci. U.S.A. 106, 13124–13129. doi: 10.1073/pnas.0900689106
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Casson, S. A., Franklin, K. A., Gray, J. E., Grierson, C. S., Whitelam, G. C., and Hetherington, A. M. (2009). phytochrome B and PIF4 regulate stomatal development in response to light quantity. Curr. Biol. 19, 229–234. doi: 10.1016/j.cub.2008.12.046
Casson, S. A., and Hetherington, A. M. (2014). phytochrome B Is required for light-mediated systemic control of stomatal development. Curr. Biol. 24, 1216–1221. doi: 10.1016/j.cub.2014.03.074
Casson, S., and Gray, J. E. (2008). Influence of environmental factors on stomatal development. New Phytol. 178, 9–23. doi: 10.1111/j.1469-8137.2007.02351.x
Celenza, J. L., Quiel, J. A., Smolen, G. A., Merrikh, H., Silvestro, A. R., Normanly, J., et al. (2005). The Arabidopsis ATR1 Myb transcription factor controls indolic glucosinolate homeostasis. Plant Physiol. 137, 253–262. doi: 10.1104/pp.104.054395
Chakravorty, D., Trusov, Y., Zhang, W., Acharya, B. R., Sheahan, M. B., McCurdy, D. W., et al. (2011). An atypical heterotrimeric G-protein gamma-subunit is involved in guard cell K(+)-channel regulation and morphological development in Arabidopsis thaliana. Plant J. 67, 840–851. doi: 10.1111/j.1365-313X.2011.04638.x
Coego, A., Brizuela, E., Castillejo, P., Ruiz, S., Koncz, C., Del Pozo, J. C., et al. (2014). The TRANSPLANTA collection of Arabidopsis lines: a resource for functional analysis of transcription factors based on their conditional overexpression. Plant J. 77, 944–953. doi: 10.1111/tpj.12443
Delgado, D., Alonso-Blanco, C., Fenoll, C., and Mena, M. (2011). Natural variation in stomatal abundance of Arabidopsis thaliana includes cryptic diversity for different developmental processes. Ann. Bot. 107, 1247–1258. doi: 10.1093/aob/mcr060
Delgado, D., Ballesteros, I., Torres-Contreras, J., Mena, M., and Fenoll, C. (2012). Dynamic analysis of epidermal cell divisions identifies specific roles for COP10 in Arabidopsis stomatal lineage development. Planta 236, 447–461. doi: 10.1007/s00425-012-1617-y
Deyoung, B. J., and Clark, S. E. (2008). BAM receptors regulate stem cell specification and organ development through complex interactions with CLAVATA signaling. Genetics 180, 895–904. doi: 10.1534/genetics.108.091108
Dong, J., and Bergmann, D. C. (2010). Stomatal patterning and development. Curr. Top. Dev. Biol. 91, 267–297. doi: 10.1016/s0070-2153(10)91009-0
Dow, G. J., and Bergmann, D. C. (2014). Patterning and processes: how stomatal development defines physiological potential. Curr. Opin. Plant Biol. 21, 67–74. doi: 10.1016/j.pbi.2014.06.007
Dow, G. J., Bergmann, D. C., and Berry, J. A. (2014a). An integrated model of stomatal development and leaf physiology. New Phytol. 201, 1218–1226. doi: 10.1111/nph.12608
Dow, G. J., Berry, J. A., and Bergmann, D. C. (2014b). The physiological importance of developmental mechanisms that enforce proper stomatal spacing in Arabidopsis thaliana. New Phytol. 201, 1205–1217. doi: 10.1111/nph.12586
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Engineer, C. B., Ghassemian, M., Anderson, J. C., Peck, S. G., Hu, H., and Schroeder, J. I. (2014). Carbonic anhydrases, EPF2 and a novel protease mediate CO2 control of stomatal development. Nature 513, 246–250. doi: 10.1038/nature13452
Falk, K. L., Tokuhisa, J. G., and Gershenzon, J. (2007). The effect of sulfur nutrition on plant glucosinolate content: physiology and molecular mechanisms. Plant Biol. 9, 573–581. doi: 10.1055/s-2007-965431
Franks, P. J. W., Doheny-Adams, T., Britton-Harper, Z. J., and Gray, J. E. (2015). Increasing water-use efficiency directly through genetic manipulation of stomatal density. New Phytol. 207, 188–195. doi: 10.1111/nph.13347
Frerigmann, H., and Gigolashvili, T. (2014). Update on the role of R2R3-MYBs in the regulation of glucosinolates upon sulfur deficiency. Front. Plant Sci. 5:626. doi: 10.3389/fpls.2014.00626
Fujiki, Y., Yoshikawa, Y., Sato, T., Inada, N., Ito, M., Nishida, I., et al. (2001). Dark-inducible genes from Arabidopsis thaliana are associated with leaf senescence and repressed by sugars. Physiol. Plant. 111, 345–352. doi: 10.1034/j.1399-3054.2001.1110312.x
Geisler, M. J., and Sack, F. D. (2002). Variable timing of developmental progression in the stomatal pathway in Arabidopsis cotyledons. New Phytol. 153, 469–476. doi: 10.1046/j.0028-646X.2001.00332.x
Granum, E., Pérez-Bueno, M. L., Calderón, C. E., Ramos, C., de Vicente, A., Cazorla, F. M., et al. (2015). Metabolic responses of avocado plants to stress induced by Rosellinia necatrix analysed by fluorescence and thermal imaging. Eur. J. Plant Pathol. 142, 625–632. doi: 10.1007/s10658-015-0640-9
Hachez, C., Ohashi-Ito, K., Dong, J., and Bergmann, D. C. (2011). Differentiation of Arabidopsis guard cells: analysis of the networks incorporating the basic helix-loop-helix transcription factor, FAMA. Plant Physiol. 155, 1458–1472. doi: 10.1104/pp.110.167718
Hayashi, S., Ishii, T., Matsunaga, T., Tominaga, R., Kuromori, T., Wada, T., et al. (2008). The glycerophosphoryl diester phosphodiesterase-like proteins SHV3 and its homologs play important roles in cell wall organization. Plant Cell Physiol. 49, 1522–1535. doi: 10.1093/pcp/pcn120
Hetherington, A. M., and Woodward, F. I. (2003). The role of stomata in sensing and driving environmental change. Nature 424, 901–908. doi: 10.1038/nature01843
Hirai, M. Y., Klein, M., Fujikawa, Y., Yano, M., Goodenowe, D. B., Yamazaki, Y., et al. (2005). Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 280, 25590–25595. doi: 10.1074/jbc.M502332200
Holroyd, G. H., Hetherington, A. M., and Gray, J. E. (2002). A role for the cuticular waxes in the environmental control of stomatal development. New Phytol. 153, 433–439. doi: 10.1046/j.0028-646X.2001.NPH326.doc.x
Hubbell, E., Liu, W. M., and Mei, R. (2002). Robust estimators for expression analysis. Bioinformatics 18, 1585–1592. doi: 10.1093/bioinformatics/18.12.1585
Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U., et al. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. doi: 10.1093/biostatistics/4.2.249
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Jones, B., Gunneras, S. A., Petersson, S. V., Tarkowski, P., Graham, N., May, S., et al. (2010). Cytokinin regulation of auxin synthesis in Arabidopsis involves a homeostatic feedback loop regulated via auxin and cytokinin signal transduction. Plant Cell 22, 2956–2969. doi: 10.1105/tpc.110.074856
Kang, C. Y., Lian, H. L., Wang, F. F., Huang, J. R., and Yang, H. Q. (2009). Cryptochromes, phytochromes, and COP1 regulate light-controlled stomatal development in Arabidopsis. Plant Cell 21, 2624–2641. doi: 10.1105/tpc.109.069765
Koyama, T., Furutani, M., Tasaka, M., and Ohme-Takagi, M. (2007). TCP transcription factors control the morphology of shoot lateral organs via negative regulation of the expression of boundary-specific genes in Arabidopsis. Plant Cell 19, 473–484. doi: 10.1105/tpc.106.044792
Krolikowski, K. A., Victor, J. L., Wagler, T. N., Lolle, S. J., and Pruitt, R. E. (2003). Isolation and characterization of the Arabidopsis organ fusion gene HOTHEAD. Plant J. 35, 501–511. doi: 10.1046/j.1365-313X.2003.01824.x
Kumari, A., Jewaria, P. K., Bergmann, D. C., and Kakimoto, T. (2014). Arabidopsis reduces growth under osmotic stress by decreasing SPEECHLESS protein. Plant Cell Physiol. 55, 2037–2046. doi: 10.1093/pcp/pcu159
Kurdyukov, S., Faust, A., Trenkamp, S., Bar, S., Franke, R., Efremova, N., et al. (2006). Genetic and biochemical evidence for involvement of HOTHEAD in the biosynthesis of long-chain alpha-,omega-dicarboxylic fatty acids and formation of extracellular matrix. Planta 224, 315–329. doi: 10.1007/s00425-005-0215-7
Lam, H. M., Wong, P., Chan, H. K., Yam, K. M., Chen, L., Chow, C. M., et al. (2003). Overexpression of the ASN1 gene enhances nitrogen status in seeds of Arabidopsis. Plant Physiol. 132, 926–935. doi: 10.1104/pp.103.020123
Larkin, R. M., Alonso, J. M., Ecker, J. R., and Chory, J. (2003). GUN4, a regulator of chlorophyll synthesis and intracellular signaling. Science 299, 902–906. doi: 10.1126/science.1079978
Lau, O. S., and Bergmann, D. C. (2012). Stomatal development: a plant's perspective on cell polarity, cell fate transitions and intercellular communication. Development 139, 3683–3692. doi: 10.1242/dev.080523
Lau, O. S., Davies, K. A., Chang, J., Adrian, J., Rowe, M. H., Ballenger, C. E., et al. (2014). Direct roles of SPEECHLESS in the specification of stomatal self-renewing cells. Science 345, 1605–1609. doi: 10.1126/science.1256888
Leonhardt, N., Kwak, J. M., Robert, N., Waner, D., Leonhardt, G., and Schroeder, J. I. (2004). Microarray expression analyses of Arabidopsis guard cells and isolation of a recessive abscisic acid hypersensitive protein phosphatase 2C mutant. Plant Cell 16, 596–615. doi: 10.1105/tpc.019000
Lichtenthaler, H. K., and Buschmann, C. (2001). “Chlorophylls and carotenoids: measurement and characterization by UV-VIS spectroscopy,” in Current Protocols in Food Analytical Chemistry, eds R. E. Wrolstad, T. E. Acree, H. An, E. A. Decker, M. H. Penner, D. S. Reid, S. J. Schwartz, C. F. Shoemaker, and P. Sporns (New York, NY: John Wiley & Sons, Inc.), F4.3.1–F4.3.8.
Macalister, C. A., Ohashi-Ito, K., and Bergmann, D. C. (2007). Transcription factor control of asymmetric cell divisions that establish the stomatal lineage. Nature 445, 537–540. doi: 10.1038/nature05491
Malitsky, S., Blum, E., Less, H., Venger, I., Elbaz, M., Morin, S., et al. (2008). The transcript and metabolite networks affected by the two clades of Arabidopsis glucosinolate biosynthesis regulators. Plant Physiol. 148, 2021–2049. doi: 10.1104/pp.108.124784
Meng, L., and Feldman, L. J. (2010). CLE14/CLE20 peptides may interact with CLAVATA2/CORYNE receptor-like kinases to irreversibly inhibit cell division in the root meristem of Arabidopsis. Planta 232, 1061–1074. doi: 10.1007/s00425-010-1236-4
Nadeau, J. A., and Sack, F. D. (2002). Control of stomatal distribution on the Arabidopsis leaf surface. Science 296, 1697–1700. doi: 10.1126/science.1069596
Obayashi, T., Kinoshita, K., Nakai, K., Shibaoka, M., Hayashi, S., Saeki, M., et al. (2007). ATTED-II: a database of co-expressed genes and cis elements for identifying co-regulated gene groups in Arabidopsis. Nucleic Acids Res. 35, D863–D869. doi: 10.1093/nar/gkl783
Ohashi-Ito, K., and Bergmann, D. C. (2006). Arabidopsis FAMA controls the final proliferation/differentiation switch during stomatal development. Plant Cell 18, 2493–2505. doi: 10.1105/tpc.106.046136
Oliveros, J. (2007). VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Madrid: BioinfoGP-CNB-CSIC.
Peng, P., Chan, S. W. L., Shah, G. A., and Jacobsen, S. E. (2006). Plant genetics: increased outcrossing in hothead mutants. Nature 443, E8. doi: 10.1038/nature05251
Pillitteri, L. J., and Dong, J. (2013). Stomatal development in Arabidopsis. Arabidopsis Book 11:e0162. doi: 10.1199/tab.0162
Pillitteri, L. J., Peterson, K. M., Horst, R. J., and Torii, K. U. (2011). Molecular profiling of stomatal meristemoids reveals new component of asymmetric cell division and commonalities among stem cell populations in Arabidopsis. Plant Cell 23, 3260–3275. doi: 10.1105/tpc.111.088583
Pillitteri, L. J., Sloan, D. B., Bogenschutz, N. L., and Torii, K. U. (2007). Termination of asymmetric cell division and differentiation of stomata. Nature 445, 501–505. doi: 10.1038/nature05467
Pillitteri, L. J., and Torii, K. U. (2012). Mechanisms of stomatal development. Annu. Rev. Plant Biol. 63, 591–614. doi: 10.1146/annurev-arplant-042811-105451
Schroder, F., Lisso, J., and Mussig, C. (2011). EXORDIUM-LIKE1 promotes growth during low carbon availability in Arabidopsis. Plant Physiol. 156, 1620–1630. doi: 10.1104/pp.111.177204
Smyth, G. K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3. doi: 10.2202/1544-6115.1027
Sturn, A., Quackenbush, J., and Trajanoski, Z. (2002). Genesis: cluster analysis of microarray data. Bioinformatics 18, 207–208. doi: 10.1093/bioinformatics/18.1.207
Sun, S., Yu, J. P., Chen, F., Zhao, T. J., Fang, X. H., Li, Y. Q., et al. (2008). TINY, a dehydration-responsive element (DRE)-binding protein-like transcription factor connecting the DRE- and ethylene-responsive element-mediated signaling pathways in Arabidopsis. J. Biol. Chem. 283, 6261–6271. doi: 10.1074/jbc.M706800200
Tanaka, Y., Nose, T., Jikumaru, Y., and Kamiya, Y. (2013). ABA inhibits entry into stomatal-lineage development in Arabidopsis leaves. Plant J. 74, 448–457. doi: 10.1111/tpj.12136
Thimm, O., Blasing, O., Gibon, Y., Nagel, A., Meyer, S., Kruger, P., et al. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939. doi: 10.1111/j.1365-313X.2004.02016.x
Torii, K. U. (2012). Mix-and-match: ligand-receptor pairs in stomatal development and beyond. Trends Plant Sci. 17, 711–719. doi: 10.1016/j.tplants.2012.06.013
Tricker, P. J., Gibbings, J. G., Rodriguez Lopez, C. M., Hadley, P., and Wilkinson, M. J. (2012). Low relative humidity triggers RNA-directed de novo DNA methylation and suppression of genes controlling stomatal development. J. Exp. Bot. 63, 3799–3813. doi: 10.1093/jxb/ers076
Triviño, M., Martin-Trillo, M., Ballesteros, I., Delgado, D., De Marcos, A., Desvoyes, B., et al. (2013). Timely expression of the Arabidopsis stoma-fate master regulator MUTE is required for specification of other epidermal cell types. Plant J. 75, 808–822. doi: 10.1111/tpj.12244
Von Groll, U., Berger, D., and Altmann, T. (2002). The subtilisin-like serine protease SDD1 mediates cell-to-cell signaling during Arabidopsis stomatal development. Plant Cell 14, 1527–1539. doi: 10.1105/tpc.001016
Wengier, D. L., and Bergmann, D. C. (2012). On fate and flexibility in stomatal development. Cold Spring Harb. Symp. Quant. Biol. 77, 53–62. doi: 10.1101/sqb.2013.77.015883
Wilson, K., Long, D., Swinburne, J., and Coupland, G. (1996). A Dissociation insertion causes a semidominant mutation that increases expression of TINY, an Arabidopsis gene related to APETALA2. Plant Cell 8, 659–671. doi: 10.1105/tpc.8.4.659
Winter, D., Vinegar, B., Nahal, H., Ammar, R., Wilson, G. V., and Provart, N. J. (2007). An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2:e718. doi: 10.1371/journal.pone.0000718
Woodward, F. I. (1998). Do plants really need stomata? J. Exp. Bot. 49, 471–480. doi: 10.1093/jxb/49.Special_Issue.471
Woodward, F. I., Lake, J. A., and Quick, W. P. (2002). Stomatal development and CO2: ecological consequences. New Phytol. 153, 477–484. doi: 10.1046/j.0028-646X.2001.00338.x
Wu, D., Wright, D. A., Wetzel, C., Voytas, D. F., and Rodermel, S. (1999). The IMMUTANS variegation locus of Arabidopsis defines a mitochondrial alternative oxidase homolog that functions during early chloroplast biogenesis. Plant Cell 11, 43–55. doi: 10.1105/tpc.11.1.43
Xie, Z., Li, D., Wang, L., Sack, F. D., and Grotewold, E. (2010). Role of the stomatal development regulators FLP/MYB88 in abiotic stress responses. Plant J. 64, 731–739. doi: 10.1111/j.1365-313X.2010.04364.x
Xin, Z., Zhao, Y., and Zheng, Z. L. (2005). Transcriptome analysis reveals specific modulation of abscisic acid signaling by ROP10 small GTPase in Arabidopsis. Plant Physiol. 139, 1350–1365. doi: 10.1104/pp.105.068064
Xing, H. T., Guo, P., Xia, X. L., and Yin, W. L. (2011). PdERECTA, a leucine-rich repeat receptor-like kinase of poplar, confers enhanced water use efficiency in Arabidopsis. Planta 234, 229–241. doi: 10.1007/s00425-011-1389-9
Yilmaz, A., Mejia-Guerra, M. K., Kurz, K., Liang, X., Welch, L., and Grotewold, E. (2011). AGRIS: the Arabidopsis Gene Regulatory Information Server, an update. Nucleic Acids Res. 39, D1118–D1122. doi: 10.1093/nar/gkq1120
Yoo, C. Y., Pence, H. E., Jin, J. B., Miura, K., Gosney, M. J., Hasegawa, P. M., et al. (2010). The Arabidopsis GTL1 transcription factor regulates water use efficiency and drought tolerance by modulating stomatal density via transrepression of SDD1. Plant Cell 22, 4128–4141. doi: 10.1105/tpc.110.078691
Keywords: epidermis development, fluorescence imaging, mute-3, photosynthesis, spch-3, stomata, transcription factor, transcriptome
Citation: de Marcos A, Triviño M, Pérez-Bueno ML, Ballesteros I, Barón M, Mena M and Fenoll C (2015) Transcriptional profiles of Arabidopsis stomataless mutants reveal developmental and physiological features of life in the absence of stomata. Front. Plant Sci. 6:456. doi: 10.3389/fpls.2015.00456
Received: 18 April 2015; Accepted: 08 June 2015;
Published: 23 June 2015.
Edited by:
Stefan De Folter, Centro de Investigación y de Estudios Avanzados - Instituto Politécnico Nacional, MexicoReviewed by:
Mitsuhiro Aida, Nara Institute of Science and Technology, JapanJuan José Ripoll, University of California, San Diego, USA
Copyright © 2015 de Marcos, Triviño, Pérez-Bueno, Ballesteros, Barón, Mena and Fenoll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Montaña Mena and Carmen Fenoll, Facultad de Ciencias Ambientales y Bioquímica, Universidad de Castilla-la Mancha, Avda. Carlos III, s/n, Toledo, 45071, Spain,TW9udGFuYS5NZW5hQHVjbG0uZXM=;Q2FybWVuLkZlbm9sbEB1Y2xtLmVz
†Present Address: Isabel Ballesteros, Departamento de Tecnología de Alimentos, Subdirección General de Investigación y Tecnología-Instituto Nacional de Investigaciones Agrarias, Madrid, Spain
‡These authors have contributed equally to this work.