 Shedrack R. Kitimu1†
Shedrack R. Kitimu1† Julian Taylor2
Julian Taylor2 Timothy J. March3
Timothy J. March3 Fred Tairo4
Fred Tairo4 Mike J. Wilkinson1
Mike J. Wilkinson1 Carlos M. Rodríguez López1*
Carlos M. Rodríguez López1*- 1Plant Research Centre, School of Agriculture Food and Wine, Faculty of Sciences, University of Adelaide, Adelaide, SA, Australia
- 2Biometry Hub, School of Agriculture Food and Wine, Faculty of Sciences, University of Adelaide, Adelaide, SA, Australia
- 3School of Agriculture Food and Wine, Faculty of Sciences, University of Adelaide, Adelaide, SA, Australia
- 4Mikocheni Agricultural Research Institute, Dar es Salaam, Tanzania
There is great interest in the phenotypic, genetic and epigenetic changes associated with plant in vitro culture known as somaclonal variation. In vitro propagation systems that are based on the use of microcuttings or meristem cultures are considered analogous to clonal cuttings and so widely viewed to be largely free from such somaclonal effects. In this study, we surveyed for epigenetic changes during propagation by meristem culture and by field cuttings in five cassava (Manihot esculenta) cultivars. Principal Co-ordinate Analysis of profiles generated by methylation-sensitive amplified polymorphism revealed clear divergence between samples taken from field-grown cuttings and those recovered from meristem culture. There was also good separation between the tissues of field samples but this effect was less distinct among the meristem culture materials. Application of methylation-sensitive Genotype by sequencing identified 105 candidate epimarks that distinguish between field cutting and meristem culture samples. Cross referencing the sequences of these epimarks to the draft cassava genome revealed 102 sites associated with genes whose homologs have been implicated in a range of fundamental biological processes including cell differentiation, development, sugar metabolism, DNA methylation, stress response, photosynthesis, and transposon activation. We explore the relevance of these findings for the selection of micropropagation systems for use on this and other crops.
Introduction
Epigenetic control of gene expression plays an important role in development (Meyer et al., 2013). Indeed, normal development in complex higher organisms is dependent upon both spatial and temporal control of gene expression (Zhang et al., 2010), much of which is facilitated by dynamic operation of various epigenetic regulatory systems (Morgan et al., 2005). DNA methylation, and more specifically cytosine methylation (i.e., the incorporation of a methyl group to carbon 5 of the cytosine pyrimidine ring to form 5-methylcytosine) is present across many eukaryotic phyla, including plants, mammals, birds, fish, and invertebrates and provides an important source of epigenetic control for gene expression (Su et al., 2011). In plants, cytosine methylation can occur in three motif contexts (CG, CHG, or CHH, where H = a nucleotide other than G; Rodríguez López and Wilkinson, 2015). DNA methylation occurring within promoters or coding regions typically act to repress gene transcription and can be evoked by small interfering RNA-directed DNA Methylation (RdDM; see Matzke et al., 2007; Verdel et al., 2009). De novo DNA methylation directed by RdDM has been implicated in various types of plant stress responses (e.g., Agorio and Vera, 2007; Tricker et al., 2012, 2013) and developmental progression (e.g., Ruiz-García et al., 2005; Kinoshita et al., 2007).
The vegetative multiplication of elite genotypes to generate the large numbers of plants necessary for commercial production is an essential element for the commercial cultivation of all clonal crops and also of many perennial seed crops. The deployment of in vitro propagation methods rather than more traditional propagation approaches (such as field cuttings) can greatly increase the clonal multiplication throughput (Robert et al., 1992; Quiroz-Figueroa et al., 2006) and so accelerate the time to production. However, some in vitro multiplication techniques are associated with high levels of (usually) unwanted variability; known collectively as ‘somaclonal variation’ (e.g., Peraza-Echeverria et al., 2001). These aberrant regenerated plants can arise from both genetic and/or epigenetic-mediated alterations to gene expression and have sometimes led to significant economic losses. For example, around 5% of commercial oil palm (Elaeis oleifera) plants regenerated via somatic embryogenesis bore somaclonal abnormalities that included the mantled inflorescence syndrome (Jaligot et al., 2000). The appearance of these off-types was later associated with changes to their global DNA methylation status (Matthes et al., 2001) and linked to the use of specific plant hormones, growth regulators and nutrients in the culture media (Varga et al., 1988; Morcillo et al., 2006). The nature of the in vitro propagation system used to produce regenerated plants can have a profound effect on the likelihood of producing significant quantities of somaclonal variant plants. In vitro propagation systems that pass through an intermediate callus phase (such as in somatic embryogenesis) and so rely on a two stage process to generate new plants, are especially prone to evoking genetic and epigenetic change among the regenerated plants (Miguel and Marum, 2011). First, cells from the explant material must de-differentiate to form unspecialized callus cells. Second, some of these callus cells must re-differentiate in a manner that allows for the creation of the specialized cells needed to form tissues and organs. It appears that in some cases at least, one or both of these processes is incomplete. Certainly, Rodríguez López et al. (2010a) showed that the C-methylation profiles of leaves from plants recovered from somatic embryogenesis in cocoa (Theobroma cacao) retained many of the features of the explant tissue (staminoids) as well as only some of those found in the leaves of the mother plant. This finding suggests that at least the epigenetic DNA methylation landscape (and therefore the global gene regulation patterns) had not been entirely wiped (de-differentiated) in the callus cells prior to the formation of new adventitious plant tissues.
Induced changes to DNA methylation and associated perturbations to gene expression has been reported for genes associated with organogenesis (De-La-Peña et al., 2012) and other developmental processes in plants (Nic-Can et al., 2013). In comparison, in vitro regeneration protocols that preserve meristem anatomy and function, and which generate new plants from the activation of previously dormant meristems are known as micropropagation systems and are widely viewed as being genetically analogous to field cuttings. These systems are thought to generate daughter ramet plants that are morphologically and genetically faithful replicates of the original explant material (Kahn, 2012). To date, however, little is known about the degree to which the epigenetic profiles (and so associated cell regulatory processes) of regenerated plants from micropropagation represent faithful replicates of the original mother plant. In this study we combine methylation-sensitive amplified polymorphism (MSAP) and methylation-sensitive genotype by sequencing (msGBS; Xia et al., 2014) to assess the epigenetic fidelity of meristem micropropagation and to seek specific methylation signatures associated to in vitro propagation in cassava (M. esculenta).
Materials and Methods
Plant Material
Five varieties of cassava (M. esculenta Crantz) namely Kiroba, Kizimbani, Kibandameno, Mfaransa, and Mzungu were used in this study. In vitro micropropagated samples were obtained from the tissue culture facility of the Mikocheni Agriculture Research Institute (MARI) Dar es Salaam, Tanzania and were propagated as described by (Konan et al., 1997). Field samples of the same genotypes were grown at the Sugarcane Research Institute-Kibaha (SRI-KIBAHA), Tanzania. Samples were collected from young leaves (last leaf emerged from bud), newly mature leaves (first fully expanded leaf) and primary root tips from three individual plants representing each cassava variety both from field cutting and meristem culture samples. All samples were kept in dry ice in the field and stored at -80°C until required for DNA extraction.
DNA Isolation
DNA was extracted from all samples at MARI using a DNeasy plant mini kit (Qiagen) according to the manufacturers’ instructions. DNA concentration and quality was estimated using a Nano-Drop 1000 Spectrophotometer (Thermo Scientific). DNA was lyophilised prior to transport to the Plant Research Centre in Adelaide, Australia for use in subsequent MSAP or msGBS analyses. Upon arrival, all DNA samples were re-suspended in nuclease free water (Sigma), and re-quantified using the Thermo Scientific NanoDropTM 1000 Spectrophotometer. DNA concentrations were standardized to produce working solutions of 10 or 20 ng/ul.
Methylation-Sensitive Amplification Polymorphism Procedure
A modification of the MSAP technique (Reyna-Lopez et al., 1997) was used as described by Rodríguez López et al. (2012). In brief, genomic DNA was digested with a combination of the methylation insensitive restriction enzyme EcoRI and one of two isoschizomer enzymes that exhibit differential sensitivity to DNA methylation (HpaII and MspI; Table 1). Adapters were ligated to the digested gDNA and then used as template for the first of two consecutive selective PCR amplifications in which the primers were complementary to the adaptors but possessed unique 3′ overhangs (Table 1). HpaII/MspI selective primers were end labeled using a 6-FAM reporter molecule for fragment detection using capillary electrophoresis. A total of six primer combinations (Table 1) were tested in a pilot study using eight randomly selected DNA samples.
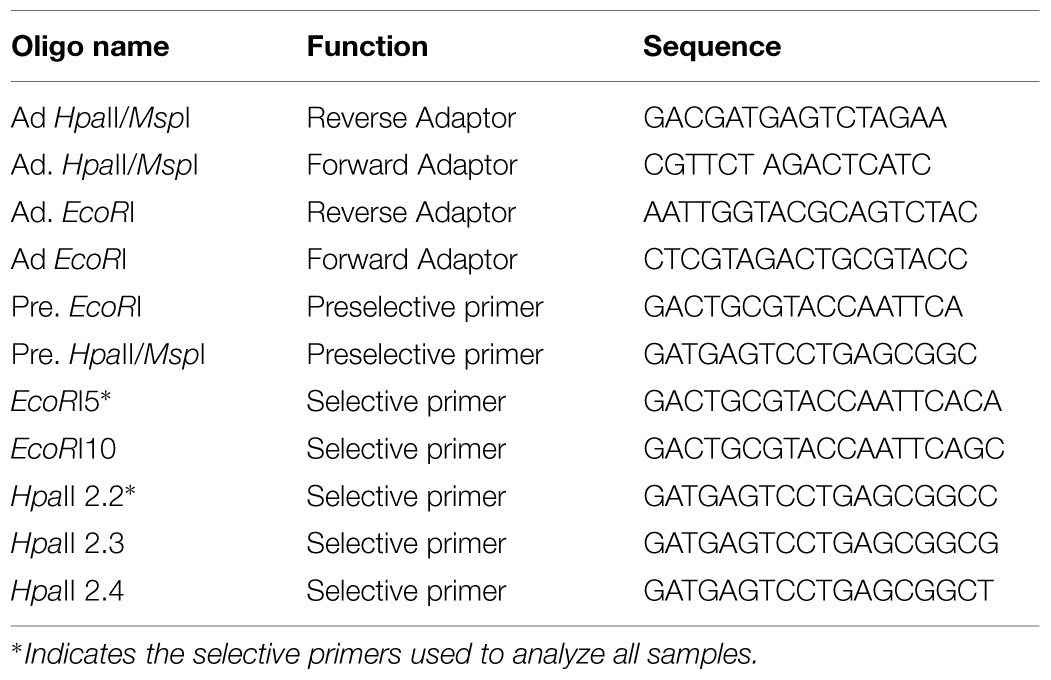
TABLE 1. Primer sequences used for MSAP.
Sample Fractionation by Capillary Electrophoresis
Single base resolution separation of the MSAP products was achieved by capillary electrophoresis on an ABI PRISM 3130 (Applied Biosystems, Foster City, CA, USA) housed at the Australian Genome Research Facility Ltd, Adelaide South Australia. Sample fractionation was performed as follows: 2 μl of the labeled MSAP products were combined with 15 μl of HiDi formamide (Applied Biosystems, Foster City, CA, USA) and mixed with 0.5 μl of GeneScanTM 500 ROXTM Size Standard (Applied Biosystems, Foster City, CA). Samples were heat-denatured at 95°C for 5 min and snap-cooled on ice for 5 min. Samples were fractionated at 15 kV for 6 s and at 15 kV for 33 min at 66°C.
Methylation Sensitive Genotyping by Sequencing
We performed the methylation-sensitive modification of the genotype by sequencing (GBS) technique (Poland et al., 2012) as described by Xia et al. (2014). In brief, a two-enzyme approach was used to generate restriction products. In this experiment, only one enzyme combination was used (MspI with EcoRI). The selected enzyme combination was based on the results obtained using the MSAP approach. Two hundred nanogram of genomic DNA from each of the 86 selected samples [comprising three replicate per tissue/variety and growing condition (i.e., in vitro or field) see Supplementary Table S1] were used in a reaction volume of 20 μl containing 2 μl of NEB Smart cut buffer, 8 U of HF-EcoRI (High-Fidelity) and 8 U of MspI (New England BioLabs Inc., Ipswich, MA, USA). Reactions were prepared in a 96 well plate containing 87 reactions (86 DNA samples plus one Negative control water sample) and conducted on a BioRad 100 thermocycler at 37°C for 2 h and then 65°C for 20 min for enzyme inactivation. A set of 96 barcoded adapters with an MspI overhang and a common Y adapter with an EcoRI overhang were designed for the ligation reaction using barcode script made by Thomas P. van Gurp1. Adapters were annealed prior to ligation as described by Poland et al. (2012). A full list of adapters for MspI (with corresponding barcodes and cassava samples) and EcoRI is listed in Supplementary Table S1. The ligation reaction (40 μl) was carried out on the same PCR plate adding to the restriction products T4 Ligase (200 U) and T4 Ligase buffer (NEB T4 DNA Ligase #M0202), 0.1 pmol of the respective barcoded MspI adapter and 15 pmol of the common Y-adapter. Ligation was completed at 22°C for 2 h followed by an enzyme inactivation step of 20 min at 65°C. Five micro liter from each ligation reaction were pooled into a single tube and then divided into two equal volumes for column clean-up using PureLink® PCR Purification Kit (Life Technologies) according to manufacturer’s instructions. Samples were re-suspended in 60 μl of nanopure water. Both clean-ups were then combined and divided again into eight samples for PCR amplification. Each 25 μl PCR consisted of 10 μl of DNA digested/ligated library), 5 μl of 5x NEB MasterMix, 2 μl of 10 uM Forward and Reverse primers at 10 uM (Supplementary Table S1). Reactions were performed in a BioRad T100 thermocycler for eight cycles consisting of 95°C (30 s), 62°C (30 s), 68 °C (30 s). All eight PCR products were pooled and then purified first using a PureLink® PCR Purification Kit (Life Technologies) according to manufacturer’s instructions (resuspended in 30 μl). Excess adaptor was finally removed using Ampure XP magnetic beads (Beckman) by mixing 30 μl of the pooled PCRs with 22.5 μl of beads. Captured fragments were eluted in 30 μl of water. Next, 125 bp paired-end sequencing was performed in one Illumina HiSeq 2000 v4lane (Illumina Inc., San Diego, CA, USA) by QBI Centre for Brain Genomics.
Statistical Analysis
Analysis of Genetic/Epigenetic Variability using MSAP
The MSAP technique uses MspI or HpaII as isoschizomers; both can cleave the motif CCGG in the absence of methylation. MspI can also cleave hemi-methylated dsDNA (mC in one DNA strand only) or fully methylated DNA sequences where the internal cytosine is methylated CmCGG. However, it cannot digest hemi-methylated and fully methylated at the external cytosine site, viz: mCCGG and mCmCGG motifs (Walder et al., 1983; Reyna-Lopez et al., 1997). In contrast, HpaII is more sensitive to methylation but can cleave hemimethylated DNA at the external cytosine position (mCCGG; Mann and Smith, 1977; Reyna-Lopez et al., 1997). Direct comparison of MspI profiles with those generated by the more methylation-sensitive HpaII therefore does not provide a definitive contrast between genetic variation and that attributable to changes in methylation (Fulneček and Kovařík, 2014). For these reasons, simple comparisons were made between profiles generated from various tissues of plants grown in the two settings (micropropagation and field cuttings) under the reasonable assumption that consistent differences will arise from differential methylation (driven by RdDM) rather than by repeated chance mutations.
MSAP profiles were visualized using GeneMapper Software v4 (Applied Biosystems, Foster City, CA, USA). Two matrices containing allelic information were generated. First, a qualitative analysis was carried out in which epiloci were scored as “present” (1) or “absent” (0) to form a presence/absence binary matrix. In this case, the selection of MSAP fragments was limited to allelic sizes between 100 and 580 bp to reduce the potential impact of size homoplasy (Caballero et al., 2008). Profile polymorphisms between DNA samples from the same cassava variety but extracted from different tissues (young leaves, newly mature leaves, and primary root tips) were retained as inter-tissue methylation differences. Polymorphisms between DNA samples from in vitro culture plants and from field grown plants were considered as in vitro culture induced methylation differences. Second, a matrix containing peak heights of fragments with allelic size between 50 and 550 bp was created for quantitative analysis (Rodríguez López et al., 2012). In both cases, different levels of hierarchy were generated to group the samples. Samples were first grouped according to cassava variety. Then, samples were divided into field grown and in vitro grown. Finally, samples were separated into the three different tissues of origin (young leave, mature leave, and roots).
For the analysis of the MSAP qualitative data, GenAlex v6.4 software (Peakall and Smouse, 2006) was used to infer pairwise epigenetic PhiPT distances (estimation of genetic/epigenetic distances) between different cassava samples. Analysis of molecular variance (AMOVA) was then performed using the same software to test the significance of the estimated PhiPT between tissues (Michalakis and Excoffier, 1996). An allele frequency table was generated using GenAlex 6.4 to find in vitro/field specific qualitative markers for each cultivar and for all cultivars. Finally, the visualization of the patterns of tissue epigenetic variations in this study was done by constructing a Principal Coordinates Analysis (PCoA).
For each variety, the peak height intensities of the epiloci generated using MSAP were compared between field grown and in vitro tissue samples as well as compared between samples from different tissue origins within field and in vitro groups. Preceding comparative analysis the data was filtered by removing epicloci containing excessively low peak height intensities across the complete set of samples. From this reduced set of epiloci the peak height libraries were normalized using the model based weighted trimmed mean method derived in Robinson and Oshlack (2010). For each pair of tissue groups being investigated, the normalized peak heights were extracted and compared using the approach described in Robinson and Smyth (2007, 2008). This approach initially assumes the normalized peak heights are distributed as a negative binomial with a common dispersion calculated across the complete set of epiloci for the two groups. From this, individual epiloci dispersions were calculated using the empirical Bayes methods of Robinson and Smyth (2007). An exact statistical test was then conducted for each epiloci to determine differences in peak heights between the two groups (Robinson and Smyth, 2008). The p-values obtained from these tests were then appropriately adjusted for multiple comparisons using the false discovery rate (FDR) method of Benjamini and Hochberg (1995). All analyses were performed using the differential expression analysis R package edgeR (Robinson et al., 2010) available in the R statistical computing environment (R Development Core Team Foundation, 2015).
Analysis of Genetic/Epigenetic Variability using GBS Data
For the processing of Illumina HiSeq 2000 v4 data, the sequences from the unfiltered fastq Illumina output were separated into samples using the barcode sequence and trimmed to 64 bp using the software TASSEL (sourceforge.net/projects/tassel/). Only sequences with one of the exact used barcodes followed by the expected sequence of three nucleotides remaining from an MspI cut-site (5′-CGG-3′) were retained for analysis. Sequences present in the negative water control were also removed from the analysis. Finally only sequences present in three or more different samples were kept for analysis. A matrix of sequence abundance of was then generated for further analysis.
Using the differential expression analysis procedure outlined in Section “Analysis of Genetic/Epigenetic Variability Using MSAP,” the sequence abundances were compared between field grown and in vitro tissue samples and also compared between samples from different tissue origins within field and in vitro groups. Sequences presenting significantly different number of reads between all in vitro and all field grown samples for each variety were isolated. Finally, only those sequences that presented the same variation (increase or reduction of number of reads in all varieties when comparing in vitro against field grown samples) in at least four of the five studied varieties were considered micropropagation induced markers. Due to the extremely low probability of a mutational event leading to the generation of these markers happening in all plants from all varieties during culture we can consider that such markers are differentially methylated regions (DMRs) induced by the micropropagation procedure. Detected DMRs were then selected for blast analysis against the cassava (BLASTN, nucleotide query to cassava nucleotide database genome blast tool in Phytozome. Top hits indicating differential methylation of a genic region were sought by comparing exons, introns, and flanking sequences (5 kb upstream of the Transcription Start Site and 5 kb downstream of the Transcription Termination Site).
Results
Analysis of Genetic/Epigenetic Variability using MSAP
Estimation of Genetic/Epigenetic Differences Based on Qualitative Analysis
Methylation-sensitive amplified polymorphism profiles generated a total of 164 loci (13 unique to HpaII, 22 unique to MspI, and 129 common to both enzymes) for the 86 samples of five cassava cultivars used in this study. PCoA analysis created from a simple presence/absence similarity matrix of the combined MSAP profiles revealed clear separation between in vitro propagated cultivars and their field counterparts for all cultivars (Figure 1). Calculated genetic/epigenetic distances between field and in vitro samples were significant for all cultivars (Table 2). In general, calculated distances between in vitro and field samples where higher for samples restricted using MspI (Table 2). All pairwise PhiPT between in vitro-grown samples and those grown in the field tissues were significant for all varieties using both HpaII and MspI (Table 2). In general, genetic/epigenetic distances were higher between tissues recovered from plants grown in the field than between those taken from in vitro-grown plants (Table 3). The reduced divergence between tissues taken from in vitro-grown material was also evident from the PCoA analysis, with samples from different tissues of in vitro propagated plants occupying less eigen space than those of the same tissues obtained from field grown plants (Figure 1). However, the level of variability observed within tissue types did not differ significantly between in vitro-grown and field-grown material.
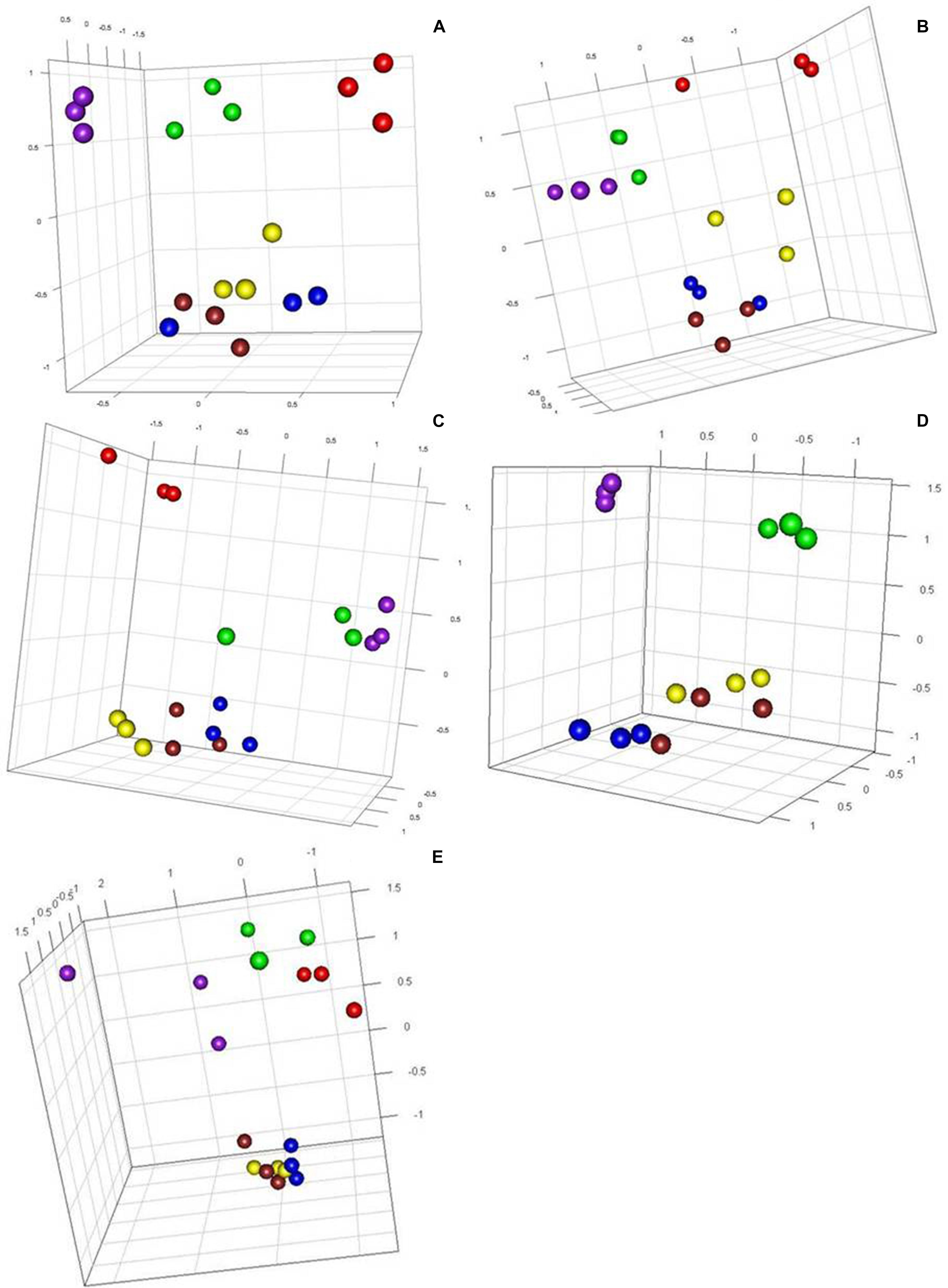
FIGURE 1. Principal component analysis showing tissue separation in five cassava varieties using generated epigenetic profiles. Effects of tissue culture and field environment on epigenetic variation. Principal coordinate diagrams based on the Euclidean analysis of methylation-sensitive amplified polymorphism (MSAP) distances obtained from three different tissues of Kiroba (A) and Mzungu (B), Kizimbani (C), Mfaransa (D) and Kibandameno (E) varieties (in vitro and field cuttings) using primer combination HpaII2.2/EcoRI5. Green, field young leaves; Red, field mature leaves; Purple, field roots; Yellow, in vitro young leaves; Blue, in vitro mature leaves; and Brown, in vitro roots.
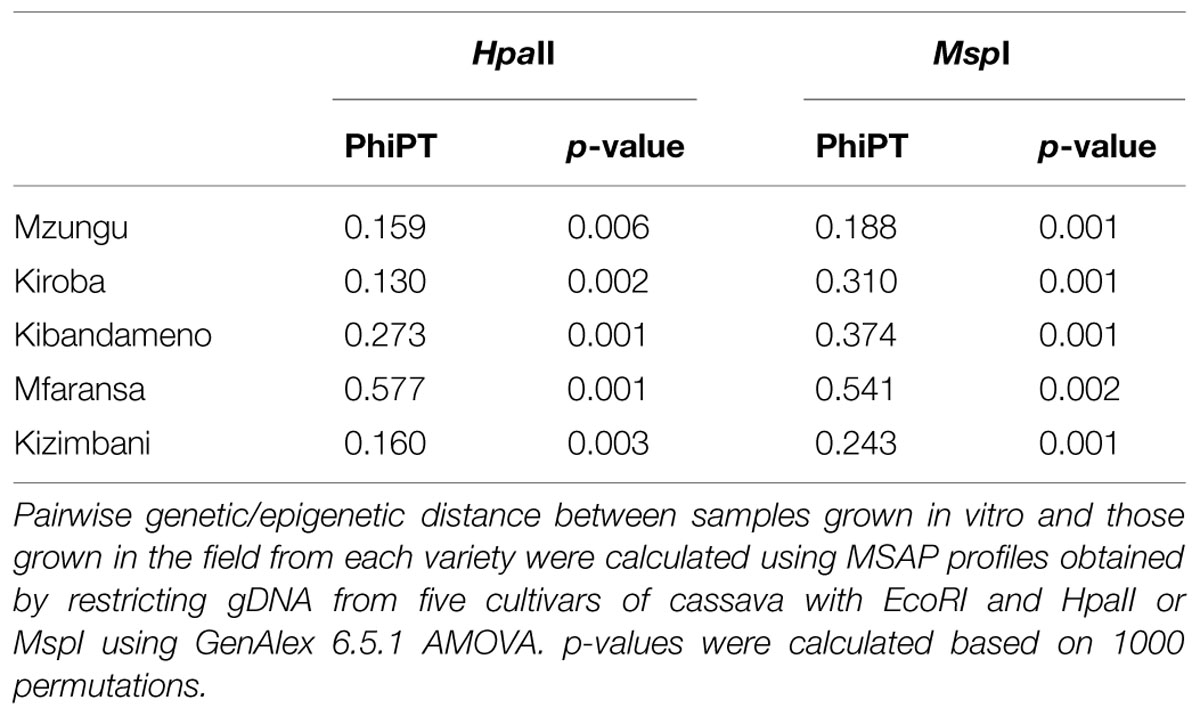
TABLE 2. Summary of calculated genetic/epigenetic distance (PhiPT) between in vitro- and field-grown samples.
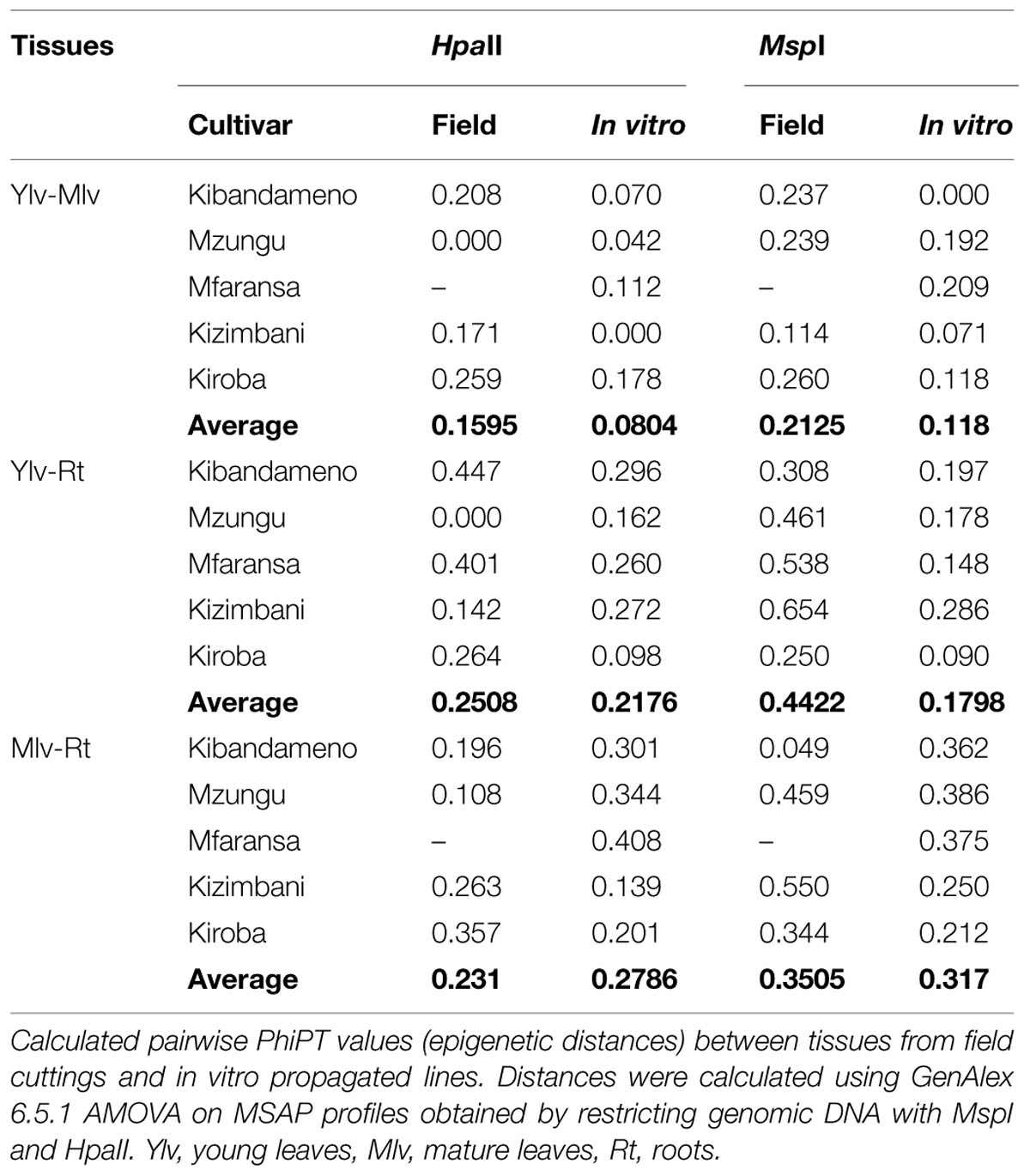
TABLE 3. Effect of in vitro culture on epigenetic differentiation between tissues.
We further analyzed the differences existing between each tissue derived from the field grown plants and all samples from plants grown in vitro. The aim here was to investigate which of the in vitro-grown tissues generated MSAP profiles were most similar to the field samples. Distance estimates were significant for all pairs, but it was consistently smaller between young leaves from field grown plants and bulked in vitro tissues (Table 4).
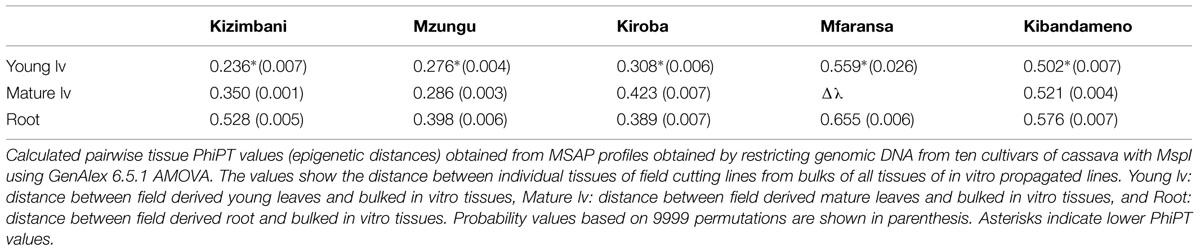
TABLE 4. Epigenetic distance between field cutting tissues and all tissues from in vitro conditions.
Estimation of Genetic/Epigenetic Differences Based on Quantitative Analysis
We selected 106 markers for quantitative analysis of MSAP profiles based on peak height data. In general, both enzymes, yielded more markers separating between tissues from field-grown plants than those taken from in vitro material (62 vs. 44 for MspI and 44 vs. 42 for HpaII; Table 5; For a list of all fragments and their levels of significance see Supplementary Table S2 for MspI and Supplementary Table S3 for HpaII). However the number and scale of these differences varied between cultivars.
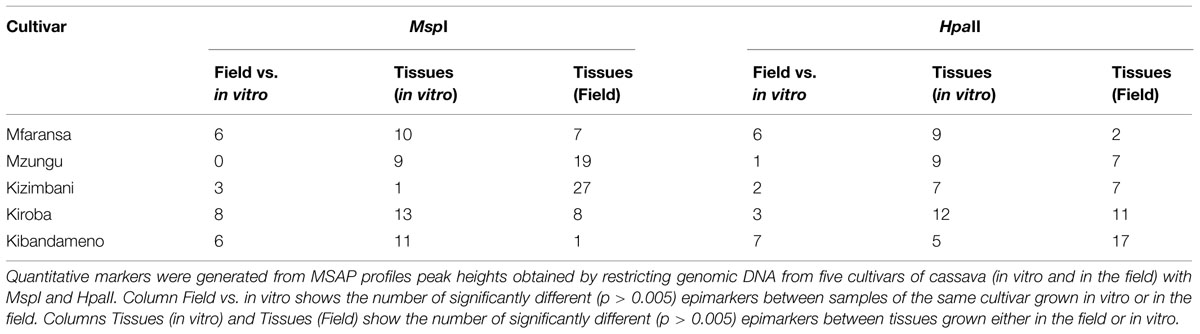
TABLE 5. Number of significantly different quantitative epimarkers across all cultivars.
A total of 14 and 15 markers for MspI and HpaI respectively were found to be significantly different (p < 0.005) between all in vitro- and field-derived material. Two of these markers generated using HpaII, epiloci 55 and 101 bp, were able to diagnose in vitro from field samples of three varieties, Kiroba, Kibandameno, and Kizimbani (Figure 2). The epilocus 55 bp generated using MspI was also significantly different between propagation systems for the same varieties but not epilocus 101 bp (Figure 2).
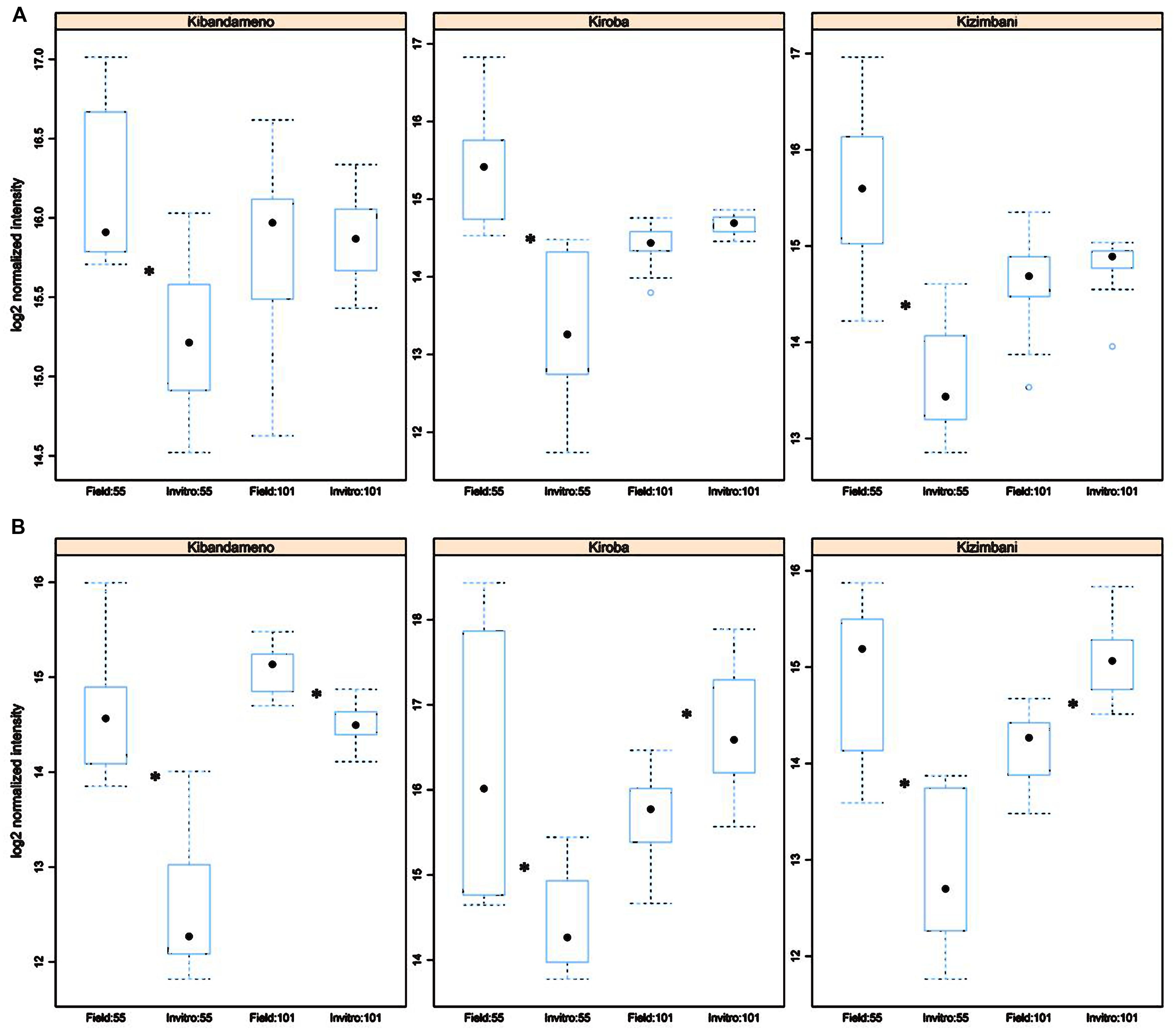
FIGURE 2. Differential peak intensity of informative epiloci (55 and 101 bp) between in vitro tissues and field tissues for three cassava cultivars (Kibandameno, Kiroba, Kizimbani). Peak intensities were obtained from MSAP profiles generated restricting genomic DNA from three different tissues (i.e., young leaf, mature leaf, and roots) in three plants grown either in vitro or in the field with MspI (A) and HpaII (B) and amplifying using primer combination HpaII2.2/EcoRI5. Box plots show the average normalized intensity scores for a 55 and a 101 bp MSAP fragment selected using. Asterisk indicates p < 0.005.
Analysis of Epigenetic Variability using GBS Data
In total, we generated 236,624,193 raw reads from the HiSeq 2000 v4 lane, of which 71,723,843 (32.3%) passed quality filter and contained the expected exact matches to sequences of the barcode adapter, MspI restriction product site and the EcoRI adapter, and which appeared in at least three biological samples but were absent from the negative (water) control. On average, 754,980 high quality reads were produced per DNA sample. Collectively, this included 357,271 unique sequence tags across all samples. The number of these markers that differed significantly in abundance between in vitro-grown and field-grown samples varied considerably between varieties: 3,298 for Kiroba; 25,683 for Kizimbani; 2,029 for Mfaransa; 34,098 for Mzungu and 17,702 for Kibandameno. Most of these sequences were more abundant in the field-grown samples (Table 6) and the overwhelming majority was variety-specific responses. We next sought to identify candidate generic epimarks that differentiate between propagation systems across all varieties. When the most stringent filter for differential abundance was applied (i.e., reads with an FDR lower than 0.05; the phase of differential abundance being conserved across all genotypes and tissues, and the absence of variety-specific SNPs) just 105 (0.03%) of unique differential sequences featured in the profiles of all varieties and showed a common pattern of phasing (Supplementary Table S4). There was also a marked difference in the phase of these marks, with just four tags being more abundant among in vitro-grown samples compared with 101 that were more numerous in the field-grown plants.
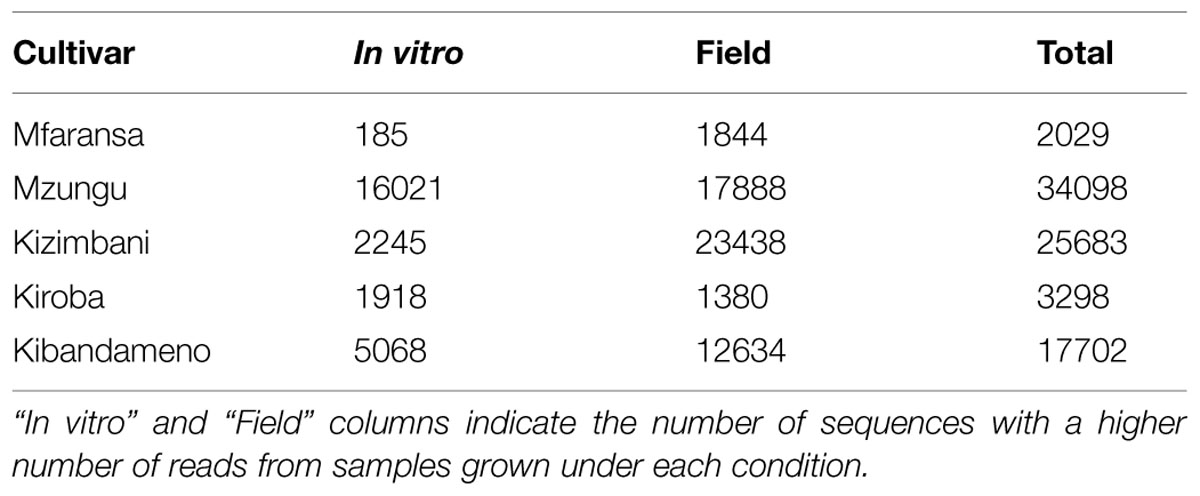
TABLE 6. Number of significantly different msGBS sequences between in vitro and field grown samples.
When compared against the cassava nucleotide database genome using the blast tool in Phytozome2, 102 differentially methylated sequences generated one or multiple hits against the cassava genome. Eighty nine top hits were associated to a gene (i.e., mapped within a window of 5 kb from the gene). BLAST results indicate that the homologs of these genes are involved in many processes, including cell differentiation, plant development, sugar metabolism, nucleic acid methylation, stress response, photosynthesis, signaling and transposon activation (Supplementary Table S5). Of the 89 differentially methylated genes 45 have been previously mentioned in the literature as having homologous genes that are: regulated by DNA methylation (14) or other epigenetic mechanisms (10), methylated or differentially methylated under different growing conditions (4), implicated in the regulation of DNA methylation (9) or other epigenetic mechanisms (1) and DNA binding proteins affected by methylation of their target sequences (3).
Discussion
Micropropagation via nodal cuttings relies on the regeneration of pre-existing meristems and so is widely considered to be analogous to field cuttings because they do not pass through a state of disorganized (dedifferentiated) tissue state (Dale and McPartlan, 1992). Nevertheless, ramets recovered from nodal micropropagation can still exhibit signs of increased morphological variability (somaclonal variation) when compared to those recovered from field cuttings (Debnath, 2005). Us-Camas et al. (2014) suggested that such observations might be explained by the stressful environment experienced by in vitro-grown plants (i.e., high relative humidity, low ventilation rate, high concentrations of sugars and plant growth regulators, and low light availability). Under these conditions, cultured plants cells are also forced to change their molecular make ups in order to generate different cell types. Cell division to generate tissues and organs require a precise coordination of genetic and epigenetic processes (Miguel and Marum, 2011; Smulders and de Klerk, 2011). For micropropagation systems that rely on dedifferentiation and de novo organogenesis, in vitro culture can often yield occasional regenerants that are phenotypically off-type (e.g., Lakshmanan and Taji, 2000; Rout et al., 2000; Da Silva et al., 2015). In contrast, those recovered from meristem micropropagation are widely reported to remain more faithful to the phenotype of the parental plant in range of species (e.g., Villordon and LaBonte, 1996) including cassava (e.g., Santana et al., 2009). There is nevertheless a large body of evidence indicating that changed growing conditions often induces moderations in global methylation patterns in culture (for review see Pastor et al., 2013) and this leads to the plausible expectation of epigenetic divergence between plants cloned by meristem micropropagation and field cuttings. Evidence supporting this assertion came from a study by Baranek et al. (2010), who used MSAP profiles to compare daughter plants recovered from field cuttings and micropropagated nodal segments of two grape vine varieties. The authors found consistent differences between the two systems in their clustering on resultant dendrogams. However, the work failed to further characterize the variation in terms of tissue type or to provide sequence identity for the differential epimarks. Characterizing such epigenetic differences may prove useful not only for the mere detection of putative somaclonal variants (Causevic et al., 2006) but for use in epiallele discovery, and as a tool for directed crop epigenetic improvement. In this study we combine MSAP and msGBS (Xia et al., 2014) to survey for C-methylation perturbations associated with the micropropation of elite clones of cassava (M. esculenta).
Analysis of Genetic/Epigenetic Variability using MSAP
Both quantitative (Figure 1) and qualitative analysis (Table 5) of MSAP generated profiles showed clear separation of all tissues in all five varieties studied. Higher levels of diversity and divergence were observed when using MspI than HpaII (Tables 2 and 4). Care should be exercised before tentatively assigning this variability as likely to have arisen through genetic or epigenetic causes. Moreover, polymorphic markers between propagation systems that appear in the profiles generated of both isoschizomer restriction enzymes (HpaII and MspI; Figure 2) could be caused by either a genetic or an epigenetic change. Conversely, a polymorphic marker detected by only one of the enzymes can only be epigenetic in nature (Pérez-Figueroa, 2013). Application of this reasoning implies that variation at the propagation system diagnostic 55 epilocus could be explained by both genetic and epigenetic changes whereas that of epilocus 101 was due to differential methylation arising from the tissue culture conditions (Figure 2). However, since the chance of a genetic mutation occurring at exactly the same location on more than one occasion is extremely low (Rodríguez López et al., 2010b) combined with the fact that these two markers polymorphic were found in three different cultivars implies that both markers probably have an epigenetic origin rather than one caused by genetic mutation.
The variability in MSAP profiles seen between DNA extracted from the same tissue type was both modest and consistent, regardless of the propagation system used to produce the plants (Figure 1). This finding suggests that these DNA methylation changes induced by micropropagation are not random, as would be expected for genetic somaclonal variation (Bairu et al., 2011) and so more likely to be associated with methylation events associated with cell and tissue differentiation. Circumstantial evidence in support of this inference can be taken from the PhiPT distance estimate, which showed that samples from in vitro nodal micropropagation ramets were always (epigenetically) closer to young leaves of their field counterparts (Table 4).
Genetic variation induced during in vitro nodal micropropagation cannot be ruled out in this study. However, our results suggest that the majority, if not all, the variability detected using MSAPs is epigenetic in nature. This is supported by the lack of higher levels of variation between micropropagated samples than in field grown samples and the fact that the observed variability seems to be conserved between different plants and between different varieties (Figure 1) and also by previous studies that show that micropropagated plants using this approach present high levels of genetic stability (Lata et al., 2010) but measurable levels of epigenetic variability (Baranek et al., 2010).
Our MSAP results suggest that (1) in vitro nodal micropropagation introduces de novo variability in the global methylation patterns; (2) micropropagation induced epigenetic variability does not seem to be random.
Analysis of Epigenetic Variability using GBS Data
Most studies of the epigenetic basis of somaclonal variation have used MSAPs to characterize culture-induced epigenetic variation. This technique is reliable and does not require previous knowledge of the studied organism. Conversely, it presents the disadvantage that the generated markers are anonymous. It is possible to isolate and sequence the differential markers (Massicotte et al., 2011), although the process can be cumbersome, expensive and time-consuming (Schrey et al., 2013), especially when many markers and samples are involved. The use of Next-generation sequencing can significantly reduce the cost of epiallele sequence characterization. The recent development of GBS (Elshire et al., 2011; Poland et al., 2012) and its methylation-sensitive version (ms-GBS; Xia et al., 2014) has allowed for a simple, time and cost effective system for the sequencing of multiple DMRs in non-model organisms.
Our study uncovered 105 unique sequences (Supplementary Table S4; 0.03% of those generated) with different levels of methylation between propagation systems. Although total sequence reads were similar between systems, the vast majority of differential tags (101/105) were more abundant among ramets recovered from field cuttings, suggesting again lower global levels of methylation in field grown plants. This is in contradiction with previous evidence suggesting that in vitro culture is related to low DNA methylation (Valledor et al., 2007). However, deciphering global hyper/hypomethylation from restriction products is not a reliable approach (Fulneček and Kovařík, 2014). What is more, other studies have shown that methylation levels during in vitro propagation are related to the donor tissue (Fang et al., 2009; Wang et al., 2012), to the length of the culture (Diaz-Sala et al., 1995; Rodríguez López et al., 2010a,b), and the media components (LoSchiavo et al., 1989; Arnholdt-Schmitt, 1993).
BLAST analysis against the cassava genome of the micropropagation induced DMRs generated in this study yielded significant hits for 102 sequences of which the 89 top hits were each associated to a gene (i.e., mapped within a window of 5 kb from the gene; Supplementary Table S5). BLAST results indicate that the homologs of these fragments are involved in many processes, including cell differentiation, plant development, sugar metabolism, nucleic acid methylation, stress response, photosynthesis, cell wall modifications, signaling and transposon activation (Supplementary Table S5). However, it is important to remember that the mere presence of differential methylation in or around a gene is not sufficient evidence to infer that expression of the gene is actually regulated by methylation. There are nevertheless enticing hints to suggest that these candidate methylation markers for propagation system may indeed also play a role in metabolic divergence between field cuttings and meristem micropropagated plants.
A series of studies have implicated DNA methylation in the regulation of genes controlling pathways in plant developmental progression or tissue differentiation (Messeguer et al., 1991), during embryogenesis, seed formation (Xiao et al., 2006), apical dominance regulation, flowering, and floral and leaf formation (Finnegan et al., 1996). Several differentially abundant loci identified in the present study showed high sequence homology to loci in cassava that have been previously implicated in cell differentiation and development: CWF19, XPMC2, EXO70, TAP42-like, Sterile alpha motif (SAM) domain-containing protein, AP3M, Enhancer of polycomb-like transcription factor protein, cassava protein containing a transcription factor UCC1, GFS9, GT-2, EMB71, and ARF2 (Supplementary Table S5). Even if it were shown that the changes in methylation among loci identified are causally linked to changes in gene expression, further work would still be required to establish whether such changes are sufficient to cause a biological meaningful change in cell metabolism and phenotype. Once again, however, there are some grounds to reason that at least some loci might.
A number of studies have shown that DNA methylation plays a central role in gene expression and plant development under stress (for extensive reviews see Chinnusamy and Zhu, 2009; Kinoshita and Seki, 2014). Not surprisingly perhaps, abiotic stresses like those encountered under in vitro culture conditions have been found to impose an effect on DNA methylation and have been correlated with subsequent organogenesis (Us-Camas et al., 2014). However, the comparative paucity of marks that appear at higher abundance in meristem culture (just 4 of the 105 generic marks; Supplementary Table S4) suggests that changes of this type lay in the minority. Explanation is therefore required for the far more commonly encountered appearance of marks among the field cutting samples only. Perhaps the most plausible hypothesis for this divergence lay in the more variable living environment experienced by field cutting plants when compared with the more homogeneous environment in culture. Field-grown plants are continuously exposed to pathogens throughout their lifetime and their DNA epigenetic patterns become altered by infection (Alvarez et al., 2010; De-La-Peña et al., 2012). It is therefore, not difficult to presume that in vitro grown plants will not experience the same pathogen or abiotic stress-induced DNA methylation marks as will those grown exposed to pathogens in the field. In our study, 13 of the 105 differentially methylated loci mapped to locations of the cassava genome associated to known stress response genes [i.e., Calcium-dependent lipid-binding (CaLB domain) family protein, Plastocyanin-like domain, Leucine-Rich Repeat Receptor-Like Protein Kinase (LRR-RK), Disease resistance protein (TIR-NBS-LRR class) family, ATBCB, ATHCHIB, ATTTM2, MLP-Like Protein 28, ATMGL, ATBZIP1, XTH1, Peroxidase superfamily protein, ATATG18F and HT1; Supplementary Table S5]. Plants have evolved two different strategies involving LRR proteins to perceive microbial pathogens. LRR-RKs are transmembrane host-encoded pattern-recognition receptors that directly recognize pathogens while NBS-LRR indirectly recognize pathogen effectors by sensing their effects on plant target proteins (Yu et al., 2013). The latest has been shown to be regulated by DNA methylation (Yu et al., 2013). Previous studies have shown that overexpression of NBS-LRRs induces a severe drop in fitness (Tao et al., 2000). Our results show several of the 105 candidate loci associated to both types of LRRs, so it would be tempting to speculate that the agronomic performance of micropropagated plants could be potentially affected if the observed changes on DNA methylation led to the overexpression of such genes. Calcium lipid-binding domain (CaLB domain) proteins are repressors of abiotic stress response in plants (de Silva et al., 2011) and have been shown to be regulated by environmental conditions through DNA methylation (Dubin et al., 2015). Curiously, our results showed one differentially abundant sequence matched to the xyloglucan endotransglucosylase/hydrolase1 (XTH1) gene. Previous studies have shown that XTHs have a function on cell wall modifications and that changes on their DNA methylation levels are associated to colonization of potato plants by beneficial bacterial endophytes (Da et al., 2012).
In all, 45 of the 89 differentially abundant sequences matched to genes that have been previously reported to be: regulated by DNA methylation (14) or other epigenetic mechanisms (10), methylated or differentially methylated under different growing conditions (4), associated to the regulation of DNA methylation (9) or other epigenetic mechanisms (1) and DNA binding proteins affected by methylation of their target sequences (3) (For references see Supplementary Table S5). Which (if any) of these genes is playing a role in a possible divergence in cell metabolism and phenotype between plants replicated by meristem-propagation and field cutting warrants further attention. Looking further ahead, identifying the developmentally important genes whose expression is sensitive to culture growth conditions may ultimately allow for the development of new culture regimes that yield regenerants with the lowest possible incidence of off-types. In the shorter term, however, the provision of methylation marks that consistently diverge in abundance between plants propagated by meristem culture and those recovered by field cuttings could have utility in the optimization of in vitro meristem propagation protocols and also in the diagnosis of the origin of clonal stocks.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was carried out as part of a biosafety capacity-building project for sub-Saharan Africa, being implemented by the ICGEB. This report is based on research funded by the Bill & Melinda Gates Foundation. The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the Bill & Melinda Gates Foundation nor the ICGEB.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00590
Footnotes
References
Agorio, A., and Vera, P. (2007). ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell 19, 3778–3790. doi: 10.1105/tpc.107.054494
Alvarez, M. E., Nota, F., and Cambiagno, D. A. (2010). Epigenetic control of plant immunity. Mol. Plant Pathol. 11, 563–576. doi: 10.1111/j.1364-3703.2010.00621.x
Arnholdt-Schmitt, B. (1993). Rapid changes in amplification and methylation pattern of genomic DNA in cultured carrot root explants (Daucus carota L.). Theor. Appl. Genet. 85, 793–800. doi: 10.1007/bf00225021
Bairu, M. W., Aremu, A. O., and Van Staden, J. (2011). Somaclonal variation in plants: causes and detection methods. Plant Growth Regul. 63, 147–173. doi: 10.1007/s10725-010-9554-x
Baranek, M., Krizan, B., Ondrusikova, E., and Pidra, M. (2010). DNA-methylation changes in grapevine somaclones following in vitro culture and thermotherapy. Plant Cell Tiss. Organ. Cult. 110, 11–22. doi: 10.1007/s11240-009-9656-1
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B 57, 289–300.
Caballero, A., Quesada, H., and Rolán-Alvarez, E. (2008). Impact of amplified fragment length polymorphism size homoplasy on the estimation of population genetic diversity and the detection of selective loci. Genetics 179, 539–554. doi: 10.1534/genetics.107.083246
Causevic, A., Gentil, M. V., Delaunay, A., El-Soud, W., Garcia, Z., Pannetier, C., et al. (2006). Relationship between DNA methylation and histone acetylation levels, cell redox and cell differentiation states in sugarbeet lines. Planta 224, 812–827. doi: 10.1007/s00425-006-0267-3
Chinnusamy, V., and Zhu, J. K. (2009). Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 12, 133–139. doi: 10.1016/j.pbi.2008.12.006
Da, K., Nowak, J., and Flinn, B. (2012). Potato cytosine methylation and gene expression changes induced by a beneficial bacterial endophyte, Burkholderia phytofirmans strain PsJN. Plant Physiol. Biochem. 50, 24–34. doi: 10.1016/j.plaphy.2011.09.013
Da Silva, J. A. T., Lema-Ruminska, J., Tymoszuk, A., and Kulpa, D. (2015). Regeneration from chrysanthemum flowers: a review. Acta Physiol. Plant 37:36. doi: 10.1007/s11738-015-1773-3
Dale, P. J., and McPartlan, H. C. (1992). Field performance of transgenic potato plants compared with controls regenerated from tuber disks and shoot cuttings. Theor. Appl. Genet. 84, 585–591.
de Silva, K., Laska, B., Brown, C., Sederoff, H. W., and Khodakovskaya, M. (2011). Arabidopsis thaliana calcium-dependent lipid-binding protein (AtCLB): a novel repressor of abiotic stress response. J. Exp. Bot. 62, 2679–2689. doi: 10.1093/jxb/erq468
Debnath, S. C. (2005). Morphological development of lingonberry as affected by in vitro and ex vitro propagation methods and source propagule. HortScience 40, 760–763.
De-La-Peña, C., Rangel-Cano, A., and Alvarez-Venegas, R. (2012). Regulation of disease-responsive genes mediated by epigenetic factors: interaction of Arabidopsis–Pseudomonas. Mol. Plant Pathol. 13, 388–398. doi: 10.1111/j.1364-3703.2011.00757.x
Diaz-Sala, C., Rey, M., Boronat, A., Besford, R., and Rodriguez, R. (1995). Variations in the DNA methylation and polypeptide patterns of adult hazel (Corylus avellana L.) associated with sequential in vitro subcultures. Plant Cell Rep. 15, 218–221. doi: 10.1007/bf00193723
Dubin, M. J., Zhang, P., Meng, D., Remigereau, M.-S., Osborne, E. J., Casale, F. P., et al. (2015). DNA methylation variation in Arabidopsis has a genetic basis, and shows evidence of local adaptation. Elife 4:e05255. doi: 10.7554/eLife.05255
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 4:e19379. doi: 10.1371/journal.pone.0019379
Fang, J.-Y., Wetten, A., Adu-Gyamfi, R., Wilkinson, M., and Rodríguez-López, C. (2009). Use of secondary somatic embryos promotes genetic fidelity in cryopreservation of cocoa (Theobroma cacao L.). Agric. Food Sci. 18, 152–159. doi: 10.2137/145960609789267579
Finnegan, E. J., Peacock, W. J., and Dennis, E. S. (1996). Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. U.S.A. 93, 8449–8454. doi: 10.1073/pnas.93.16.8449
Fulneček, J., and Kovařík, A. (2014). How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet. 15:2. doi: 10.1186/1471-2156-15-2
Jaligot, E., Rival, A., Beule, T., Dussert, S., and Verdeil, J. L. (2000). Somaclonal variation in oil palm (ElaisguineensisJacq.): the DNA methylation hypothesis. Plant Cell Rep. 19, 684–690. doi: 10.1007/s002999900177
Kahn, R. (2012). “Tissue culture applications for plant quarantine,” in Practical Tissue Culture Applications, eds K. Maramorosch and H. Hirumi (New York, NY: Academic Press), 185–201.
Kinoshita, T., and Seki, M. (2014). Epigenetic memory for stress response and adaptation in plants. Plant Cell Physiol. 55, 1859–1863. doi: 10.1093/pcp/pcu125
Kinoshita, Y., Saze, H., Kinoshita, T., Miura, A., Soppe, W. J., Koornneef, M., et al. (2007). Control of FWA gene silencing in Arabidopsis thaliana SINE-related repeats. Plant J. 49, 38–45. doi: 10.1111/j.1365-313X.2006.02936.x
Konan, N., Schöpke, C., Carcamo, R., Beachy, R., and Fauquet, C. (1997). An efficient mass propagation system for cassava (Manihot esculenta Crantz) based on nodal explants and axillary bud-derived meristems. Plant Cell Rep. 16, 444–449. doi: 10.1007/BF01092763
Lakshmanan, P., and Taji, A. (2000). Somatic embryogenesis in leguminous plants. Plant Biol. 2, 136–148. doi: 10.1055/s-2000-9159
Lata, H., Chandra, S., Techen, N., Khan, I. A., and Elsohly, M. A. (2010). Assessment of the genetic stability of micropropagated plants of Cannabis sativa by ISSR markers. Planta Med. 76, 97–100. doi: 10.1055/s-0029-1185945
LoSchiavo, F., Pitto, L., Giuliano, G., Torti, G., Nuti-Ronchi, V., Marazziti, D., et al. (1989). DNA methylation of embryogenic carrot cell cultures and its variations as caused by mutation, differentiation, hormones and hypomethylating drugs. Theor. Appl. Genet. 77, 325–331. doi: 10.1007/BF00305823
Mann, M. B., and Smith, H. O. (1977). Specificity of Hpa II and Hae III DNA methylases. Nucleic Acids Res. 4, 4211–4221. doi: 10.1093/nar/4.12.4211
Massicotte, R., Whitelaw, E., and Angers, B. (2011). DNA methylation: a source of random variation in natural populations. Epigenetics 6, 421–427. doi: 10.4161/epi.6.4.14532
Matthes, M., Singh, R., Cheah, S.-C., and Karp, A. (2001). Variation in oil palm (Elaeis guineensis Jacq.) tissue culture-derived regenerants revealed by AFLPs with methylation-sensitive enzymes. Theor. Appl. Genet. 102, 971–979. doi: 10.1007/s001220000491
Matzke, M., Kanno, T., Huettel, B., Daxinger, L., and Matzke, A. J. (2007). Targets of RNA-directed DNA methylation. Curr. Opin. Plant Biol. 10, 512–519. doi: 10.1016/j.pbi.2007.06.007
Messeguer, T., Ganal, M. W., Steffens, J. C., and Tanksley, S. D. (1991). Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear DNA. Plant Mol. Biol. 16, 753–770. doi: 10.1007/BF00015069
Meyer, L. R., Zweig, A. S., Hinrichs, A. S., Karolchik, D., Kuhn, R. M., Wong, M., et al. (2013). The UCSC genome browser database: extensions and updates 2013. Nucleic Acids Res. 41, D64–D69. doi: 10.1093/nar/gks1048
Michalakis, Y., and Excoffier, L. (1996). A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142, 1061–1064.
Miguel, C., and Marum, L. (2011). An epigenetic view of plant cells cultured in vitro: somaclonal variation and beyond. J. Exp. Bot. 62, 3713–3725. doi: 10.1093/jxb/err155
Morcillo, F., Gagneur, C., Richaud, A. H., Singh, F., R, Cheah, S. C., Rival, A., et al. (2006). Somaclonal variation in micropropagated oil palm. Characterization of two novel genes with enhanced expression in epigenetically abnormal cell lines and in response to auxin. Tree Physiol. 26, 585–594. doi: 10.1093/treephys/26.5.585
Morgan, H. D., Santos, F., Green, K., Dean, W., and Reik, W. (2005). Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14, R47–R58. doi: 10.1093/hmg/ddi114
Nic-Can, G. I., López-Torres, A., Barredo-Pool, F., Wrobel, K., Loyola-Vargas, V. M., Rojas-Herrera, R., et al. (2013). New Insights into Somatic Embryogenesis: LEAFY COTYLEDON1, BABY BOOM1 and WUSCHEL-RELATED HOMEOBOX4 Are Epigenetically Regulated in Coffea canephora. PLoS ONE 8:e72160. doi: 10.1371/journal.pone.0072160
Pastor, V., Luna, E., Mauch-Mani, B., Ton, J., and Flors, V. (2013). Primed plants do not forget. Environ. Exp. Bot. 94, 46–56. doi: 10.1016/j.envexpbot.2012.02.013
Peakall, R., and Smouse, P. E. (2006). GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295. doi: 10.1111/j.1471-8286.2005.01155.x
Peraza-Echeverria, S., Herrera-Valencia, V. A., and Kay, A.-J. (2001). Detection of DNA methylation changes in micropropagated banana plants using methylation-sensitive amplification polymorphism (MSAP). Plant Sci. 161, 359–367. doi: 10.1016/S0168-9452(01)00421-6
Pérez-Figueroa, A. (2013). msap: a tool for the statistical analysis of methylation-sensitive amplified polymorphism data. Mol. Ecol. Resour. 13, 522–527. doi: 10.1111/1755-0998.12064
Poland, J. A., Brown, P. J., Sorrells, M. E., and Jannink, J.-L. (2012). Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253. doi: 10.1371/journal.pone.0032253
Quiroz-Figueroa, F., Rojas-Herrera, R., Galaz-Avalos, R., and Loyola-Vargas, V. (2006). Embryo production through somatic embryogenesis can be used to study cell differentiation in plants. Plant Cell Tiss. Organ. Cult. 86, 285–301. doi: 10.1007/s11240-006-9139-6
R Development Core Team Foundation. (2015). R: A language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0.
Reyna-Lopez, G. E., Simpson, J., and Ruiz-Herrera, J. (1997). Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet. 253, 703–710. doi: 10.1007/s004380050374
Robert, M. L., Herrera, J. L., Chan, J. L., and Contreras, F. (1992). “Micropropagation of Agave spp,” in Biotechnology in Agriculture and Forestry, ed. J. P. Y. Bajaj (Berlin: Springer-Verlag), 306–329.
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Robinson, M. D., and Oshlack, A. (2010). A scaling normalization method for differentia expression analysis of RNA-seq data. Genome Biol. 11:R25. doi: 10.1186/gb-2010-11-3-r25
Robinson, M. D., and Smyth, G. K. (2007). Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23, 2881–2887. doi: 10.1093/bioinformatics/btm453
Robinson, M. D., and Smyth, G. K. (2008). Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332. doi: 10.1093/biostatistics/kxm030
Rodríguez López, C. M., Morán, P., Lago, F., Espiñeira, M., Beckmann, M., and Consuegra, S. (2012). Detection and quantification of tissue of origin in salmon and veal products using methylation sensitive AFLPs. Food Chem. 131, 1493–1498. doi: 10.1016/j.foodchem.2011.09.120
Rodríguez López, C. M., Wetten, A. C., and Wilkinson, M. J. (2010a). Progressive erosion of genetic and epigenetic variation in callus-derived cocoa (Theobroma cacao) plants. New Phytologist. 186, 856–868. doi: 10.1111/j.1469-8137.2010.03242.x
Rodríguez López, C. M., Sicilia Bravo, H., Wetten, A. C., and Wilkinson, M. J. (2010b). Detection of somaclonal variation during cocoa somatic embryogenesis characterised using cleaved amplified polymorphic sequence and the new freeware Artbio. Mol. Breed. 25, 501–516. doi: 10.1007/s11032-009-9348-x
Rodríguez López, C. M., and Wilkinson, M. J. (2015). Epi-fingerprinting and epi-interventions for improved crop production and food quality. Front. Plant Sci. 6:397. doi: 10.3389/fpls.2015.00397
Rout, G. R., Samantaray, S., and Das, P. (2000). Biotechnology of the banana: a review of progress. Plant Biol. 2, 512–524. doi: 10.1055/s-2000-7470
Ruiz-García, L., Cervera, M. T., and Martínez-Zapater, J. M. (2005). DNA methylation increases throughout Arabidopsis development. Planta 222, 301–306. doi: 10.1007/s00425-005-1524-6
Santana, M. A., Romay, G., Matehus, J., Vicente-Villardon., J. L., and Demey, J. R. (2009). A simple and low-cost strategy for micropropagation of cassava (Manihot esculenta Crantz). Afr. J. Biotechnol. 8, 3789–3797.
Schrey, A. W., Alvarez, M., Foust, C. M., Kilvitis, H. J., Lee, J. D., Liebl, A. L., et al. (2013). Ecological epigenetics: beyond MS-AFLP. Integr. Comp. Biol. 53, 340–350. doi: 10.1093/icb/ict012
Smulders, M., and de Klerk, G. (2011). Epigenetics in plant tissue culture. Plant Growth Regul. 63, 137–146. doi: 10.1007/s10725-010-9531-4
Su, Z., Han, L., and Zhao, Z. (2011). Conservation and divergence of DNA methylation in eukaryotes: new insights from single base-resolution DNA methylomes. Epigenetics 6, 134–140. doi: 10.4161/epi.6.2.13875
Tao, Y., Yuan, F., Leister, R. T., Ausubel, F. M., and Katagiri, F. (2000). Mutational analysis of the Arabidopsis nucleotide binding site-leucine-rich repeat resistance gene RPS2. Plant Cell 12, 2541–2554.
Tricker, P. J., Gibbings, G., Rodríguez López, C. M., Hadley, P., and Wilkinson, M. J. (2012). Low relative humidity triggers RNA-directed de novo DNA methylation and suppression of genes controlling stomatal development. J. Exp. Bot. 63, 3799–3813. doi: 10.1093/jxb/ers076
Tricker, P., Rodríguez López, C. M., Gibbings, G., Hadley, P., and Wilkinson, M. J. (2013). Transgenerational, dynamic methylation of stomata genes in response to low relative humidity. Int. J. Mol. Sci. 14, 6674–6689. doi: 10.3390/ijms14046674
Us-Camas, R., Rivera-Solís, G., Duarte-Ake, F., and de-La-Peña, C. (2014). In vitro culture: an epigenetic challenge for plants. Plant Cell Tiss. Organ. Cult. 118, 187–201. doi: 10.1007/s11240-014-0482-8
Valledor, L., Hasbún, R., Meijón, M., Rodríguez, J., Santamaría, E., Viejo, M., et al. (2007). Involvement of DNA methylation in tree development and micropropagation. Plant Cell Tiss. Org. 91, 75–86. doi: 10.1007/s11240-007-9262-z
Varga, A., Thoma, L. H., and Bruinsma, J. (1988). Effects of auxins and cytokinins on epigenetic instability of callus-propagated Kalanchoe blossfeldiana Pollen. Plant Cell Tiss. Organ. Cult. 15, 223–231. doi: 10.1007/BF00033646
Verdel, A., Vavasseur, A., Le Gorrec, M., and Touat-Todeschini, L. (2009). Common themes in siRNA-mediated epigenetic silencing pathways. Int. J. Dev. Biol. 53, 245–257. doi: 10.1387/ijdb.082691av
Villordon, A. Q., and LaBonte, D. R. (1996). Genetic variation among sweetpotatoes propagated through nodal and adventitious sprouts. J. Am. Soc. Hortic. Sci. 121, 170–174.
Walder, R. Y., Langtimm, C. J., Chatterjee, R., and Walder, J. A. (1983). Cloning of the MspI modification enzyme. The site of modification and its effects on cleavage by MspI and HpaII. J. Biol. Chem. 258, 1235–1241.
Wang, Q. M., Wang, Y. Z., Sun, L. L., Gao, F. Z., Sun, W., He, J., et al. (2012). Direct and indirect organogenesis of Clivia miniata and assessment of DNA methylation changes in various regenerated plantlets. Plant Cell Rep. 31, 1283–1296. doi: 10.1007/s00299-012-1248-6
Xia, Z., Zou, M., Zhang, S., Feng, B., and Wang, W. (2014). AFSM sequencing approach: a simple and rapid method for genome-wide SNP and methylation site discovery and genetic mapping. Sci. Rep. 4:7300. doi: 10.1038/srep07300
Xiao, W., Custard, K. D., Brown, R. C., Lemmon, B. E., Harada, J. J., Goldberg, R. B., et al. (2006). DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 18, 805–814. doi: 10.1105/tpc.105.038836
Yu, A., Lepère, G., Jay, F., Wang, J., Bapaume, L., Wang, Y., et al. (2013). Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. U.S.A. 110, 2389–2394. doi: 10.1073/pnas.1211757110
Keywords: methylation sensitive GBS, genotyping by sequencing, micropropagation, cassava, somaclonal variation, DNA methylation, epigenetics, methylation-sensitive amplified polymorphisms
Citation: Kitimu SR, Taylor J, March TJ, Tairo F, Wilkinson MJ and Rodríguez López CM (2015) Meristem micropropagation of cassava (Manihot esculenta) evokes genome-wide changes in DNA methylation. Front. Plant Sci. 6:590. doi: 10.3389/fpls.2015.00590
Received: 01 May 2015; Accepted: 16 July 2015;
Published: 13 August 2015.
Edited by:
Raúl Alvarez-Venegas, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoCopyright © 2015 Kitimu, Taylor, March, Tairo, Wilkinson and Rodríguez López. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos M. Rodríguez López, Plant Research Centre, School of Agriculture Food and Wine, Faculty of Sciences, University of Adelaide, Waite Campus, Adelaide, SA 5064, Australia,Y2FybG9zLnJvZHJpZ3VlemxvcGV6QGFkZWxhaWRlLmVkdS5hdQ==
†Shedrack R. Kitimu, Sokoine University of Agriculture, P.O BOX 3000, Morogoro, Tanzania