 Qi Xie1
Qi Xie1 Jun Niu2
Jun Niu2 Xilin Xu3
Xilin Xu3 Lixin Xu1
Lixin Xu1 Yinbing Zhang1
Yinbing Zhang1 Bo Fan1
Bo Fan1 Xiaohong Liang1
Xiaohong Liang1 Lijuan Zhang4
Lijuan Zhang4 Shuxia Yin1*
Shuxia Yin1* Liebao Han1*
Liebao Han1*- 1Institute of Turfgrass Science, College of Forestry, Beijing Forestry University, Beijing, China
- 2Lab of Systematic Evolution and Biogeography of Woody Plants, College of Nature Conservation, Beijing Forestry University, Beijing, China
- 3Bioinformatics, College of Plant Protection, Hunan Agricultural University, Changsha, China
- 4Shenzhen Tourism College, Jinan University, Shenzhen, China
Japanese lawngrass (Zoysia japonica Steud.) is an important warm-season turfgrass that is able to survive in a range of soils, from infertile sands to clays, and to grow well under saline conditions. However, little is known about the molecular mechanisms involved in its resistance to salt stress. Here, we used high-throughput RNA sequencing (RNA-seq) to investigate the changes in gene expression of Zoysia grass at high NaCl concentrations. We first constructed two sequencing libraries, including control and NaCl-treated samples, and sequenced them using the Illumina HiSeq™ 2000 platform. Approximately 157.20 million paired-end reads with a total length of 68.68 Mb were obtained. Subsequently, 32,849 unigenes with an N50 length of 1781 bp were assembled using Trinity. Furthermore, three public databases, the Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-prot, and Clusters of Orthologous Groups (COGs), were used for gene function analysis and enrichment. The annotated genes included 57 Gene Ontology (GO) terms, 120 KEGG pathways, and 24 COGs. Compared with the control, 1455 genes were significantly different (false discovery rate ≤0.01, |log2Ratio |≥1) in the NaCl-treated samples. These genes were enriched in 10 KEGG pathways and 73 GO terms, and subjected to 25 COG categories. Using high-throughput next-generation sequencing, we built a database as a global transcript resource for Z. japonica Steud. roots. The results of this study will advance our understanding of the early salt response in Japanese lawngrass roots.
Background
Plant growth, development, and production depend largely on soil; however, more than 800 million hectares of land worldwide are subjected to salt stress. High-salinity soils prevent the reabsorption of water from plant roots, the first organ that responds to salt stress (Zhu, 2002; Bartels and Sunkar, 2005; Munns, 2005; Petricka et al., 2012); therefore plants can be poisoned by the excess uptake of salts. The level of sensitivity of the plant salt stress response plays a crucial role in determining resistance to high salt stress (Tracy et al., 2008; Schmidt et al., 2013; Wei et al., 2013). Given the current scarcity of water resources, recycled water, and water use have been the focus of many studies (Marinho et al., 2013). Improving the abiotic stress resistance of plants is an effective method for overcoming water shortages and for the cultivation of large areas of saline land.
A focus on early detection would assist the determination of plant stress responses (Schmidt et al., 2013). Model plants such as rice have evolved a regulatory network of early responses to chilling stress (Yun et al., 2010). As a vital signaling component in many biological processes (Mittler et al., 2004; Schippers et al., 2012), reactive oxygen species (ROS) can rapidly induce abiotic stress responses—H2O2 levels induced by salt stress rise within several minutes in rice (Hong et al., 2009). Understanding the physiological and molecular mechanisms underlying salt stress tolerance in halophytes is therefore of great importance. Several halophytes, including Thellungiella halophila, Mesembryanthemum crystallinum, Suaeda, and Populus, have been studied (Dyachenko et al., 2006; Sun et al., 2010; Fukao et al., 2011; Yoon et al., 2014); however, our understanding of the unique tolerance mechanisms of halophytes is limited. Thus, further study of various halophytes may provide new information regarding the ability of plants to resist salt damage.
Zoysia Willd (family Poaceae, subfamily Chloridoideae, tribe Zoysieae) are perennial grasses, consisting globally of about 10 recognized species (Tsuruta et al., 2011). Zoysia grasses are widely used as a warm-season turfgrass for home lawns, golf courses, athletic fields, and parks (Ge et al., 2006). The species belonging to this genus are the most salt and cold tolerant of the C4 grass species in the family Poaceae. They are indigenous to China, Japan, and Korea. As one of the three most important commercial species, Zoysia japonica Steud. also exhibits marked tolerance to abiotic stress. It is distributed naturally in mountainous areas, along riversides, and in coastal areas, and it shows moderate tolerance to weak shade. Further, it can survive in various soils, ranging from infertile sands to clays (Tsuruta et al., 2011).
Few genomic sequence resources are available for Zoysia grasses. Only 11 mRNA sequences (complete cDNAs) are available in the database of the National Center for Biotechnology Information (NCBI) for Z. japonica (as of April 2015). This grass has an advanced transformation system (Asano, 1989; Inokuma et al., 1998; Ge et al., 2006), but the lack of sequence resources has limited the exploitation of Z. japonica's genetic resources. To determine the molecular mechanism underlying the salinity tolerance of plants (Uddin et al., 2012), a large-scale analysis of differentially expressed genes (DEGs) is necessary.
Sequencing technology, which is used to analyze the transcriptome of a species without complete genome information, has undergone rapid progress in recent years with respect to the number of base calls and cost. Therefore, the technology is used in studies of many species and by researchers in many fields; for example, it has been used to study tissue regeneration in newts (Looso et al., 2013), glucosinolate metabolism in radish (Wang et al., 2013), and the leaf transcriptome of Camelina sativa (Liang et al., 2013).
In this study, we used the HiSeq™ 2000 platform to perform RNA sequencing (RNA-seq) of Zoysia grass roots. We compared the transcriptomes of plants grown under saline and normal conditions to identify differences in gene expression, and to identify the function of key transcripts and genes in the initiation of ROS-related signal transduction. Significant expression differences were found among genes involved in many metabolic pathways. Many novel genes were also identified and inferred to be expressed, specifically in the salt-treated plants. To the best of our knowledge, this is the first report of a transcriptome of Zoysia grass. We used H2O2 as a marker to identify DEGs between the control and salt-treated plants. This will improve our understanding of the mechanisms involved in short-term stress responses of Zoysia. These sequence data may also enhance our understanding of the molecular mechanisms in plants under salt stress, and provide a public dataset for use in future studies of Zoysia.
Materials and Methods
Plant Materials and Treatment
Z. japonica Steud. cv. Zenith was grown from seed in soil in a propagation tray. Three weeks after germination, individual seedlings were transplanted to pots (diameter: 15 cm, depth: 14.5 cm) filled with a mixture of topsoil and coarse river sand (1:1) in a greenhouse (25°C during the day/20°C at night, 16 h of light/8 h of dark, 800 μmol m−2s−1 photosynthetically active radiation, and 75% relative humidity). The plants were irrigated with water during the first 3 weeks to keep the soil moist. After that, they were transferred to pots and maintained with 1/2 Murashige and Skoog cultivation solution once a week and water irrigation twice a week. NaCl treatment (150 mM) was initiated 3 months after germination.
H2O2 Assay
To visualize H2O2 accumulation, samples (from control and salt-treated plants) were immediately placed in a 1 mg/ml 3,3′-diaminobenzidine (DAB)-HCl solution in 10 mM phosphate buffer (pH 3.8) at 25°C for 4 h in the dark. After staining, the root of each plant was boiled in 95% (v/v) ethanol for 30 min and rehydrated in 70% (v/v) ethanol for 48 h at 25°C. H2O2 was visualized as a reddish-brown coloration. Each experiment was repeated using 10 plants.
RNA Preparation and Library Preparation for Analysis
Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). The extracted RNA was treated with RNase-free DNase I (Takara Inc., Kyoto, Japan) for 45 min at 37°C to remove residual DNA. The quality of the RNA was evaluated using a NanoDrop 2000. The cDNA was prepared by pooling 10 μg of RNA each from the control and salt-treated samples.
Total RNA was extracted from normal and salt-treated plant roots. Poly A+ mRNA was obtained using a NEBNext Poly(A)mRNA Magnetic Isolation Module. Then, according to the instructions of the NEBNext mRNA Library Prep Master Mix Set for Illumina and NEBNext Multiplex Oligos for Illumina, a mixed cDNA library of salt-stressed (Case) seedlings and control (CK) plants was prepared.
Sequencing, De novo Assembly, and Annotation
The cDNA library was sequenced using the Illumina HiSeq™ 2000 platform. After cleaning the raw reads and discarding low-quality reads, we ran Trinity (Yoon et al., 2014) to assemble the clean reads into transcripts. The clean reads were used to construct a K-mer dictionary and each K-mer was used as an initial contig. The most frequent K-mer, which was in the dictionary with K-1 overlaps with the current contig end, was extended until neither direction could be extended further. The contigs that shared at least one K-1-mer were then read across the junction sites to build a pool. Each contig was used to construct a de Brujin graph with trimmed spurious edges and compacted linear paths. It then reconciled the graph with reads and pairs, and output one linear sequence for each splice form and/or paralogous transcript represented in the graph. The final selection of the most important section from the partial transcript was the unigene. To annotate sequences obtained by de novo assembly, we used BLASTX with a significance threshold of E≤10−5. The assembly unigenes were aligned against the plant protein datasets of the non-redundant protein (NR), Swiss-prot protein, TrEMBL, Gene Ontology (GO), Clusters of Orthologous Groups (COGs), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases.
Identification and Functional Annotation of DEGs
We calculated the expression levels as reads per kilobase exon model per million mapped reads (RPKM) for each root sample. DEGseq software was then used to determine significant DEGs defined as a fold change ≥2 and false discovery rate (FDR) < 0.01. Using BLAST, DEGs were aligned against the NR, GO, COGs, and KEGG databases.
Gene Comparison and Sequence Alignment within Relative Species
We previously analyzed published results on the salt response of model plants that are most homologous to Zoysia, including Setaria, barley, rice, and maize (Ueda et al., 2004, 2006; Qing et al., 2009; Puranik et al., 2011). We used Blastn with a significance threshold of E ≤ 10−10, sequence identified ≥30% and the length ≥30 aa. The DEGs were blasted against the salt-response genes that were identified in relative plants.
Identification of Zoysia Transcription Factors (TFs)
The TF database was downloaded from PlanTFDB (http://plntfdb.bio.uni-potsdam.de/v3.0/downloads.php). All unigenes were searched against those in the TF database by BLASTx (E < 10E−5).
Quantitative Reverse Transcription PCR (RT-qPCR) Verification
We subjected nine ERF salt stress-related unigenes (Comp132815_c0, Comp211710_c0, Comp196695_c0, Comp233109_c5, Comp223421_c0, Comp217707_c0, Comp206878_c0, Comp206878_c1, and Comp195676_c0) to RT-qPCR analysis. Root samples were collected from additional plants grown and tested under identical conditions. As per the Invitrogen protocol, total RNA was extracted from the root samples and then subjected to reverse transcription. The primers were designed using an online tool (http://www.idtdna.com/site). ZjActin (GenBank: GU290545.1) was used as a housekeeping gene. RT-qPCR was performed on a Bio-Rad RCR platform using a SYBR® Green Real-Time PCR Mix (TransStart Green qPCR SuperMix) to detect transcript abundance. Amplification was performed as follows: denaturation at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s, annealing at 60°C for 15 s, and extension at 72°C for 10 s. All amplifications were performed with three replicates. We calculated the relative expression levels of the selected unigenes using the 2−ΔΔCt method. The data from the reactions were analyzed using Bio-Rad software.
Phylogenetic Tree Construction
Each of nine ERFs had more than one homologous genes in the PlnTFDB database. We selected 21 homologous genes (Supplementary File 1) from that database to perform phylogenetic analysis. Multiple sequence alignments were performed using the Clustal X program with default parameters. Phylogenetic analysis was performed by aligning a maximum-likelihood phylogenetic tree using MEGA 6 software, which was supported by a bootstrap test with 1000 iterations.
Putative Molecular Markers
To investigate the distribution of simple sequence repeats (SSRs), the MIcroSAtellite identification tool (MISA version 1.0; http://pgrc.ipk-gatersleben.de/misa) was used to search the assembled transcripts. We scanned and counted the SSRs in all selected unigenes. The parameters were adjusted for the identification of perfect mono-, di-, tri-, tetra-, and penta-nucleotide motifs with a minimum of 12, 6, 5, 5, and 4 repeats, respectively.
Results and Discussion
Physiological Responses in the Roots of Salt-treated Plants
Most previous studies focused on long-term adaptations to salt stress in grasses (Wang and Jiang, 2007; Bian and Jiang, 2009; Du et al., 2009; Hu et al., 2012); few have investigated the initiation of the stress response that plays an important role in this type of stress signaling. Changes in the environment of plants increase the level of ROS (Hong et al., 2009), with the production of ROS being an early response to abiotic stress (Zhu, 2002; Mittler et al., 2004). H2O2 is the predominant salt-induced ROS (Yang et al., 2007); thus, it was used as a marker of the degree of salt stress in root samples.
As shown in Figure 1, the H2O2 concentration in roots increased shortly after salt treatment. Compared with the control plants, a yellow substance was visible on the roots. After 30 min of salt treatment, oxidative stress caused by H2O2 was visible throughout the roots. Thus, we selected representative samples at 0 and 30 min for transcriptomic sequencing.
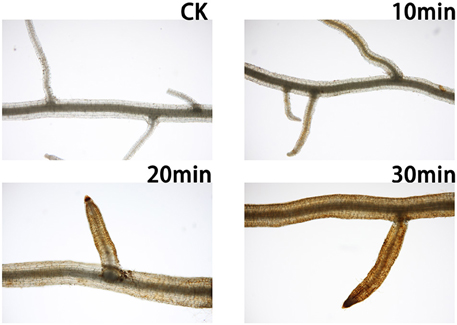
Figure 1. Sample roots were treated with DAB to detect H2O2 in the dark for 4 h. CK, Plants grown under normal conditions produced little H2O2; 10 min, in plants treated with salt for 10 min, the H2O2 level was slightly increased in lateral roots, while H2O2 was also detected in the main root; 20 min, in plants treated with salt for 20 min, H2O2 accumulation was detected in the entire root; 30 min, in plants treated with salt for 30 min, the roots were submerged in H2O2.
Illumina Sequencing and De novo Assembly of Zoysia Root Transcripts
RNA-seq technology is an indispensable tool for the whole-genome analysis of complex stress treatments. Compared with traditional large-scale sequencing, de novo whole-genome analysis is less costly and more efficient. It is suitable for those plants whose genomic sequences are unknown. We used RNA sequencing to analyze the transcriptome of Z. japonica Steud. roots.
To obtain a comprehensive transcriptome, two cDNA libraries denoted “CK” and “Case” prepared from three repeat RNA samples from normal and salt-treated roots were subjected to paired-end read sequencing using the Illumina platform. Paired-end read technology increases the depth and improves de novo assembly efficiency. Sequencing using the Illumina HiSeq™ 2000 platform resulted in the generation of 21.30 G raw reads. More than 80% had Phred-like quality scores at the Q30 level (error < 0.1%). After removing reads with adaptors, reads with unknown nucleotides, and low-quality reads, we obtained 105.47 million clean reads with an average GC content of 55.83% (Table 1A).
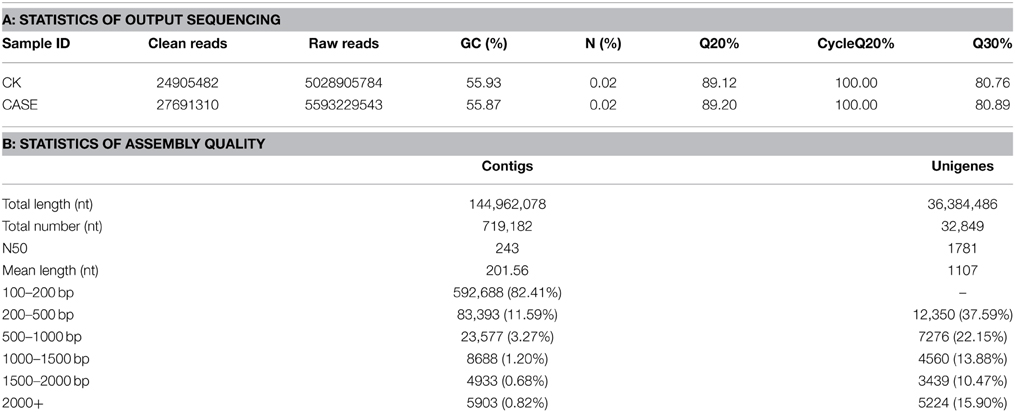
Table 1. Summary of RNA-seq and de novo assembly of Zoysia japonica Steud.
The Trinity method can generate full-length transcripts without reference genomes (Grabherr et al., 2011; Haas et al., 2013). Using this method for de novo assembly, all high-quality clean reads were assembled into 719,182 contigs (Supplementary File 2) with an average length of 201.56 bp. Contigs of 100–500 bp were most frequent, accounting for 94% of the total. Subsequently, the contigs were clustered into 32,849 unigenes of which the mean length was 1107 bp and the N50 value was 1781 bp (Table 1B). There were 20,499 unigenes of ≥500 bp, and 5224 unigenes of ≥2000 bp. Most unigenes were in the range 200–500 bp (37.59%). Among these genes, the longest and shortest were 15,772 and 201 bp, respectively. The unigene lengths facilitated annotation and classification. The random distribution of the unigenes is presented in Figure 2.
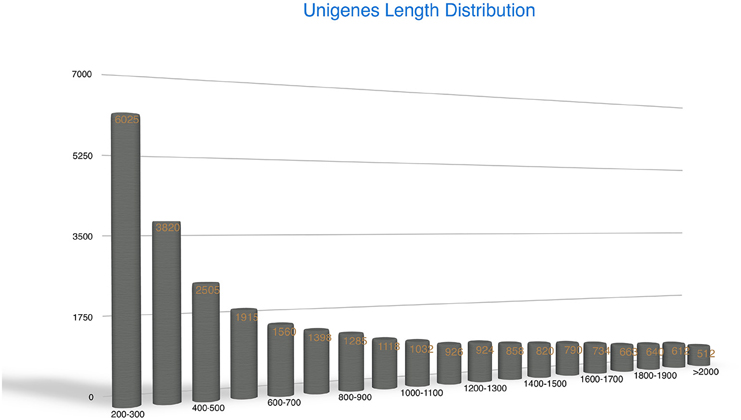
Figure 2. Random distribution of the assembled unigenes. The x-axis indicates the length of the unigenes. The y-axis indicates the number of unigenes.
Functional Annotation and Classification of Assembled Unigenes
The assembled sequences were first searched against the NR protein database, and analysis indicated that 55% of the sequences ranged from 1.0E−5 to 1.0E−50, while 18,006 of sequences with an E < 10E−50 displayed strong homology (Figure 3). The distribution of identity is shown in Figure 3B. The majority pattern was 60–80% similarity (12,111) and 40–60% similarity (9812). The closest species was Setaria itallica, with 14,950 genes (45%) matched. The next closest species was Sorghum bicolor, which showed 17% homology with Zoysia. This implies that the transcripts were assembled and annotated correctly (Dang et al., 2013; Wang et al., 2013). Figure 3C shows homologies with plants in the family Poaceae. Recent research (Ahn et al., 2015) supports our data showing that the most homologous model species to Zoysia is Setaria itallica, followed by Sorghum bicolor and Zea mays. Due to the differences in tissue between cultivar and treatment, our results were not completely consistent with that research.
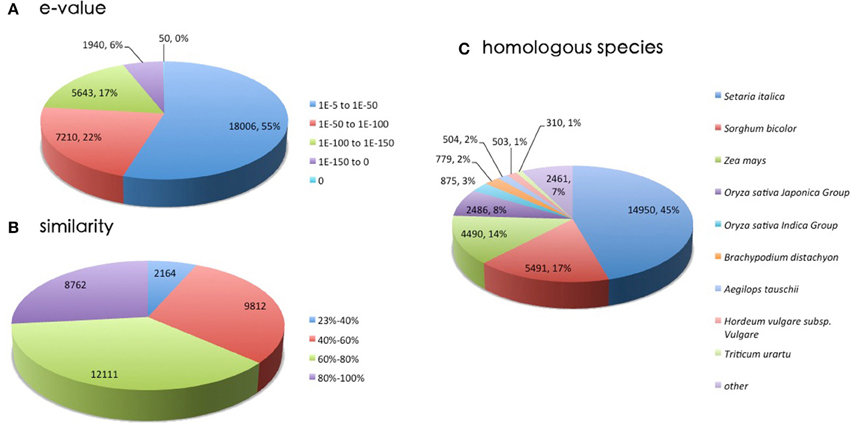
Figure 3. A homology search was conducted by BLASTx against the NR database. (A) E-value distribution of BLAST hits for matched unigene sequences. (B) Similarity distribution of top BLAST hits for each unigene. (C) Species distribution of the top BLAST hits.
GO annotation can provide a standardized vocabulary for assigning the functions of uncharacterized sequences. Based on sequence homology, 21,188 unigenes were categorized into 57 GO terms (Supplementary File 3) (Harris et al., 2004). The most frequently identified unigenes classified in these categories were “response to stimulus” (9918), “biological regulation” (8558) and “response to stress” (7371). A total of 13,480 unigenes were assigned to the COG classification (Supplementary File 3). There were 24 COG classes. The largest group was “general function prediction only” (2325, 17.24%), followed by “translation, ribosomal structure, and biogenesis” (1645, 12.20%), and “Posttranslational modification, protein turnover, chaperones” (1433, 10.63%). The smallest groups were “nuclear structures” (1, 0.0074%), and “cell motility” (25, 0.18%).
Identification of DEGs
We used the RPKM method (RPKM > 0.1) to calculate the expression levels of unigenes in the control and salt-treated samples. Using DESeq software (FDR < 0.01, FC > 2) to analyze the DEGs between salt- and control-treated root samples, a total of 1648 differentially expressed unigenes (Figure 4) were obtained. Among those genes, 948 were upregulated and 700 were downregulated (Supplementary File 4).
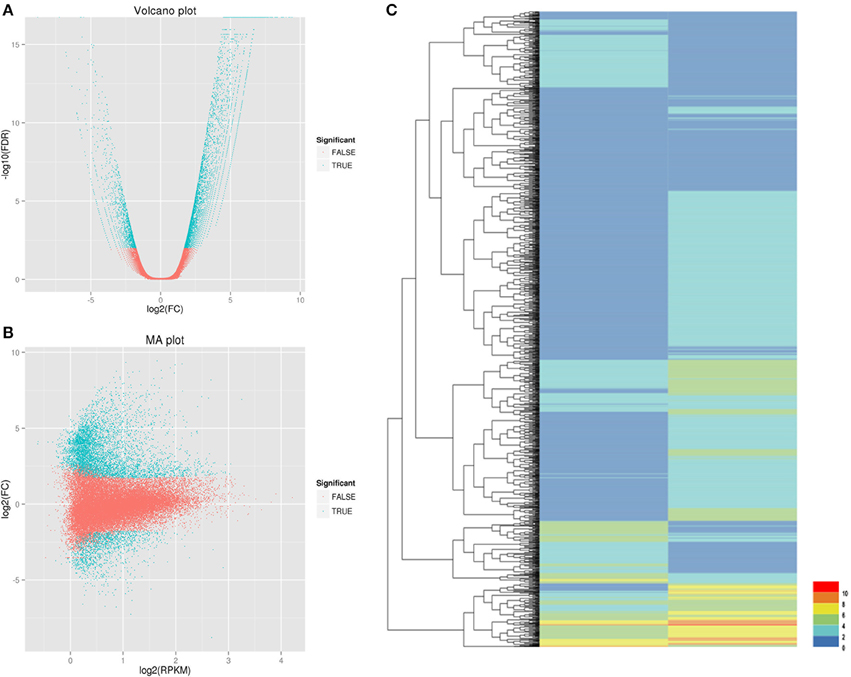
Figure 4. Identification of DEGs between the CK (control) and Case (salt-stressed) samples. (A) Volcano plot: The x-axis is the log of the fold change between the two conditions; the y-axis is the negative logarithm of the FDR. (B) MA plot: The x-axis is the average expression (log scale) level between the two conditions, indicating the basal expression level. The y-axis is the fold change (log scale), which indicates the difference between the two. (C) Each column represents a different sample. Each line represents a different gene. Each color represents a different gene expression level.
GO Classification
To categorize unigenes functionally, we assigned GO terms (P < 0.01). In total, 1,455 unigenes were enriched in 73 GO terms (Figure 5, Supplementary File 5). Among the unigenes, 798 were upregulated, and 647 were downregulated. After analyzing the statistically enriched GO functions related to DEGs, the majority of GO terms were assigned to biological processes distributed in 67 subcategories (91.78%), followed by cellular components and metabolic processes, with three subcategories each (4.11%).
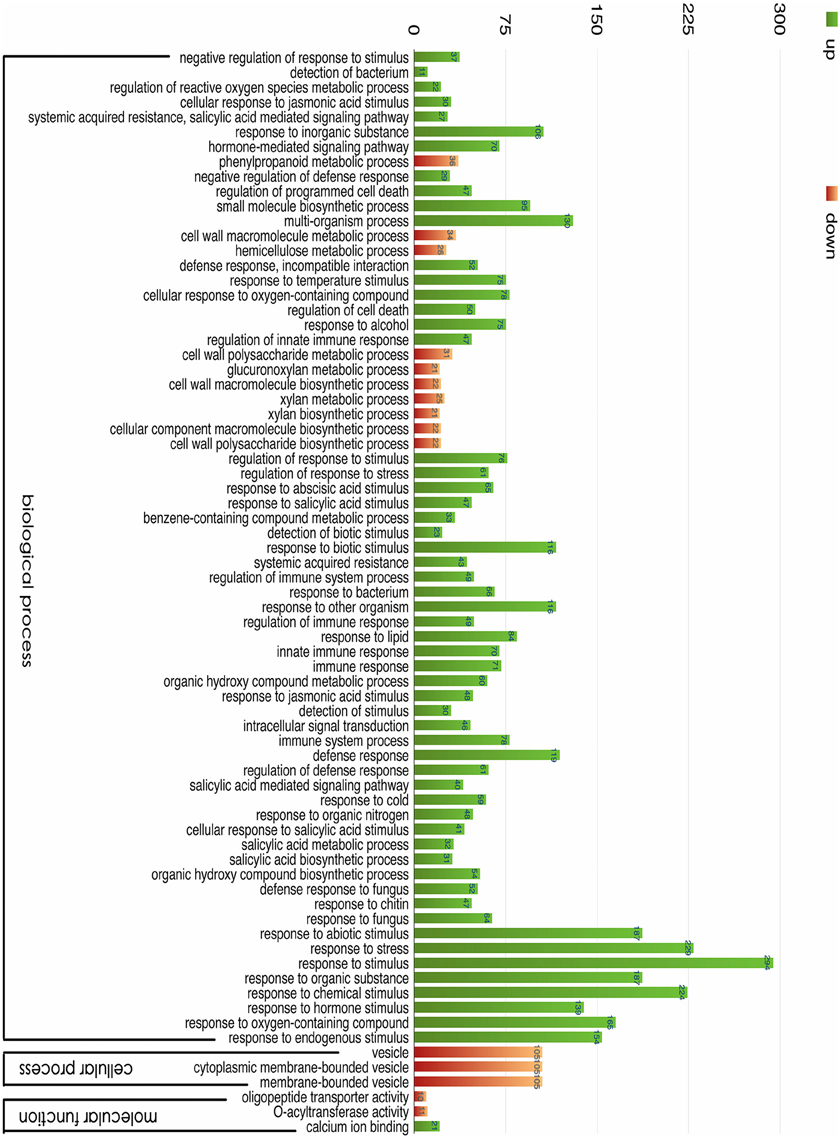
Figure 5. GO classification of the unigenes. The results are summarized for three main categories: biological process, cellular component, and molecular function. In total, 1445 DEGs with BLAST matches to known proteins were assigned to GO categories.
The most over-represented GO terms in response to salt stress were “response to stimulus” (294 unigenes), “response to stress” (229 unigenes), “response to chemical stimulus” (224 unigenes), “response to abiotic stimulus” (187 unigenes), and “response to an organic substance” (187 unigenes). Many genes in these GO terms responded to short-term salt stress, suggesting that they play important roles in the early salt response. We also found nine significantly enriched hormone-related GO terms, including ABA-related (“response to abscisic acid stimulus”), JA-related (“response to jasmonic acid stimulus” and “cellular response to jasmonic acid stimulus”), and SA-related (“response to salicylic acid stimulus,” “cellular response to salicylic acid stimulus,” “salicylic acid-mediated signaling pathway,” “salicylic acid metabolic process,” “salicylic acid biosynthetic process,” “systemic acquired resistance,” and “salicylic acid-mediated signaling pathway”) terms. These hormones were upregulated by salinity, and they induced genes involved in alleviating salt stress (Wang et al., 2001). A GO terms analysis may help in understanding the salt tolerance mechanism involved when plants suffer a sudden increase in soil salinity.
Calcium Ion (Ca2+) Signaling
Ca2+ acts as a secondary messenger in salt stress responses (Xuan et al., 2013). The cytosolic free Ca2+ concentration ([Ca2+]cyt) changes within seconds in plants subjected to salt stress (Knight et al., 1997), leading to downstream signaling. RSA1, a nuclear Ca2+-binding protein, can sense changes in the free Ca2+ level elicited by salt stress in the nucleus and transduce this signal by activating the SOS1 promoter (Guan et al., 2013). P-type Ca2+ channels can sense changes in the [Ca2+]cyt and transduce the signal downstream by activating specific targets (Huda et al., 2013). Among the DEGs, we identified 12 unigenes belonging to a major Ca2+ sensor family; of these, 10 were upregulated and two were downregulated (Table 2A).
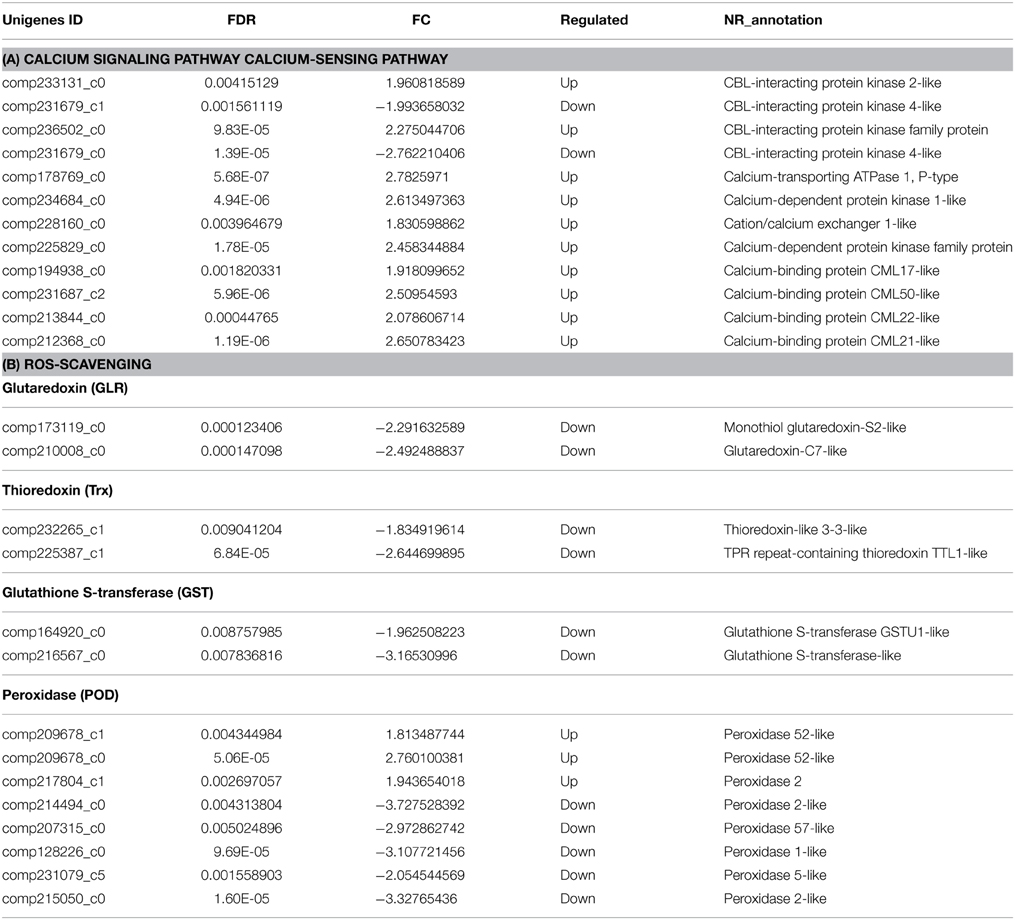
Table 2. List of some early salt stress response genes.
ROS Scavenging
Salt stress can cause the rapid accumulation of ROS, including superoxide, H2O2, and hydroxyl radicals. ROS can perturb cellular redox homeostasis leading to DNA damage and changes in protein or membrane function. Plants possess ROS scavenging systems to control ROS levels and cope with oxidative stress. Z. japonica has a similar ROS scavenging system to that of other plant species. Cold treatment significantly increases antioxidant enzyme levels (Xuan et al., 2013), which may provide protection against oxidative damage in this species (Xu et al., 2013a). Among our DEGs, we identified 14 unigenes belonging to the ascorbate-glutathione cycle, GPX pathway, and Prx/Trx pathway. Among these unigenes, three were upregulated while 11 were downregulated (Table 2B).
Comparison of Salt-responsive Genes in Zoysia and Relative Species
To further characterize the identified genes, we used BLAST to compare 255 DEGs (Table 3, Supplementary File 6) with previously identified salt-responsive genes in four relative of the model plants. Genes in barley and rice had similar DEGs, and more than 60% of these DEGs were homologous sequences with Zoysia. Only 27 of 296 genes in maize were matched with those in Zoysia. These results suggest that barley and Zoysia use a similar mechanism to respond to salt stress, whereas maize has a different system for this response.
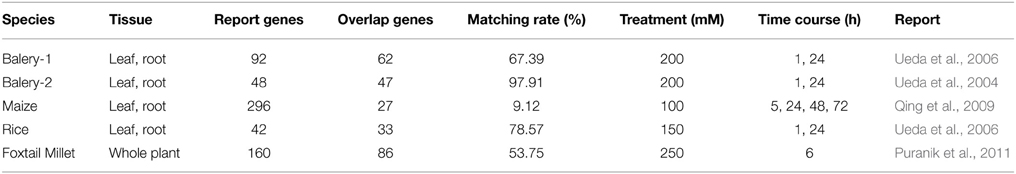
Table 3. Comparison of salt response genes identified in this study with those in C4 model-plant.
Major Regulators of the Salt-stress Response Transcriptome
Over the past decade, genes related to abiotic stress have been discovered and functionally characterized. Among these genes, TFs are master regulators that control gene clusters (Zhang et al., 2011). A single TF can regulate the expression of downstream genes by binding specifically to a cis-acting element in the promoter of a target gene. Members of the AP2/ERF, MYB, WRKY, and NAC families were shown to regulate salt tolerance. Enhanced expression of DREB2A can improve salt-stress tolerance in rice. Three rice NAC TFs (SNAC1, SNAC2, and NAC5) act as positive regulators of the salt stress response (Schmidt et al., 2013). A total of 3751 unigenes (Table 4) were identified as potential TFs with an average length of 1224 bp. The length distribution of the TFs is shown in Table 3. More than 43.05% of the TFs were longer than 1000 bp; the shortest TF was 201 bp, while the longest was 11,427 bp. In total, 80 TF families were identified (Supplementary File 7). The largest group was the AP2/EREBP family (187 unigenes), followed by the bHLH family (158 unigenes) and the WRKY family (137 unigenes). There were nine TF families containing more than 100 unigenes.
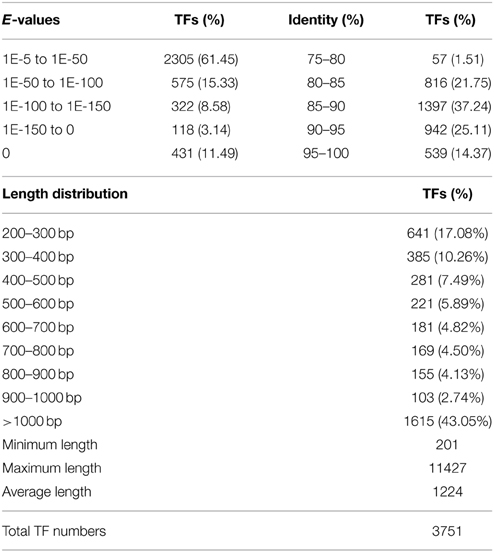
Table 4. Unigene against TF database.
We selected the TF family that was most closely related to abiotic stress from the DEGs (Supplementary File 8).
The AP2/EREBP Family
Increasing evidence indicates that ERF proteins are involved in the salt response. They adjust the expression of downstream genes and fine-tune crosstalk between signaling pathways (Agarwal et al., 2006; Nakano et al., 2006; Fukao et al., 2011). Many studies have shown that ERFs enhance salt tolerance (Gilmour et al., 2000; Park et al., 2001; Jung et al., 2007). Among the unigenes we identified were 187 ERF genes, including 34 DEGs. About 33 DEGs were upregulated and only 1 DEG (comp223372_c0) was downregulated. In both libraries, comp232359_c0 had the highest expression level, but the expression of this gene was slightly increased in the case libraries. The largest change was found for comp234321_c1, which was upregulated more than four-fold (log2 fold change).
The bZip Family
Members of the bZIP family of TFs respond to abiotic stress. They have important roles in the ABA response and the regulation of oxidative and pathogen defense responses (Yun et al., 2010). In total, 115 genes were identified in our database, with seven DEGs. Comp219239_c0 had a higher expression level, and its expression differed slightly between the two sample libraries.
The NAC Family
The NAC domain, which was identified based on consensus sequences from the Petunia NAM, Arabidopsis ATAF1/2, and CUC2 proteins, was plant-specific and contained the highly conserved NAC DNA-binding domain and variable C-terminal domains. The NAC family is considered to be important in plant development, senescence, auxin responses, and biotic stress responses (Aida et al., 1997; Kim et al., 2004; Olsen et al., 2005; Lu et al., 2007; Hao et al., 2011; Nakashima et al., 2012; Sun et al., 2012). After salt treatment, the expression of 131 NACs differed, and only 11 unigenes were considered to be DEGs. The transcript of comp231035_c0 was present at a high level in both libraries, and was upregulated almost two-fold under salt stress.
The WRKY Family
As one of the best-characterized TF families, the WRKY TF family has been suggested to play an important role in plant stress responses. The WRKY protein family contains a conserved amino acid sequence motif, WRKYGOK, at the N-terminus and a novel zinc-finger-like motif at the C-terminus. Numerous studies have demonstrated that WRKY family regulates various plant processes from development to biotic and abiotic stress responses (e.g., wounding, drought, and salinity). Eight WRKYs in wheat respond to low temperature, salt stress, and heat treatment (Wu et al., 2008). Enhancing the expression of WRKY25 and WRKY33 can increase the salt tolerance of Arabidopsis thaliana (Jiang and Deyholos, 2009; Li et al., 2011). The overexpression GmWRKY54 increases salt and drought tolerance in soybean. H2O2 treatment significantly induces the expression of AtWRKY30, AtWRKY75, AtWRKY48, AtWRKY39, AtWRKY6, AtWRKY53, and AtWRKY22 (Vanderauwera et al., 2005; Miao and Zentgraf, 2007; Zhou et al., 2011). In our study, we identified 137 WRKYs, including 20 DEGs. Of these, seven WRKYs (comp231105_c0, comp224918_c0, comp232021_c2, comp230256_c0, comp216034_c0, comp218505_c0, and comp190635_c0) had high transcription levels in both libraries.
The MYB Family
MYB proteins are the largest class of TFs in plants. Nearly 9% of the TFs in Arabidopsis are MYBs, and more than 1600 TFs have been identified (Riechmann et al., 2000). MYB proteins have DNA-binding domains, which contain one to four imperfect repeats in plants. Many studies have shown that MYBs are involved in abiotic stress responses. AtMYB41 is induced by drought, during which it modulates cell expansion and cuticle deposition (Cominelli et al., 2008). The overexpression of MdoMYB121 in tomato and apple confers improved tolerance to drought (Cao et al., 2013). TaMYB73 has been shown to enhance salt resistance by regulating the expression of related genes (He et al., 2012). A total of 129 genes were identified; among them, only 14 showed significant differences in expression.
The bHLH Family
TFs belonging to the bHLH family are important in plant development, circadian regulation, and stress responses. Here, we found 158 bHLH genes; 19 genes from this family were DEGs.
Putative Molecular Markers
Molecular markers are important for molecular breeding (Liu et al., 2012; Chen et al., 2013, 2014; Dai et al., 2013; Xu et al., 2013b; Li et al., 2014). SSRs are 1-6-bp iterations of DNA sequences that occur only in noncoding regions. The occurrence of SSRs in transcribed sequences has been established. The roles of SSRs in plants such as rice (Cho Yg et al., 2000), bread wheat (Gupta et al., 2003), and sugarcane (Cordeiro et al., 2001) have been reported.
In our study, 32,849 unigenes were used to detect SSRs, and a total of 4842 SSRs were identified in 3951 unigenes using MISA (Supplementary File 9). Among them, 702 unigenes contained at least 2 SSRs. The largest fraction was mono-nucleotide SSRs (2305), followed by tri-nucleotide SSRs (1694), and di-nucleotide SSRs (778). This phenomenon corresponds to natural selection. Molecular markers are a useful resource for determining functional genetic variation. Our results will facilitate the prediction of molecular markers for Zoysia.
Analysis of Genes of Interest
RT-qPCR
We identified nine ERFs with significant changes; their lengths varied from 260 to 873 bp (Figure 6). PCR amplification showed that all RT-qPCR primers (Supplementary File 10) used produced only single fragments of the expected lengths (100–250 bp; 100% success rate). All positive clones from the validation studies were subjected to Sanger sequencing, and the results confirmed those obtained using the Illumina method. Our qPCR results for the nine unigenes are in agreement with those from the DESeq analysis of our RNA-seq data.
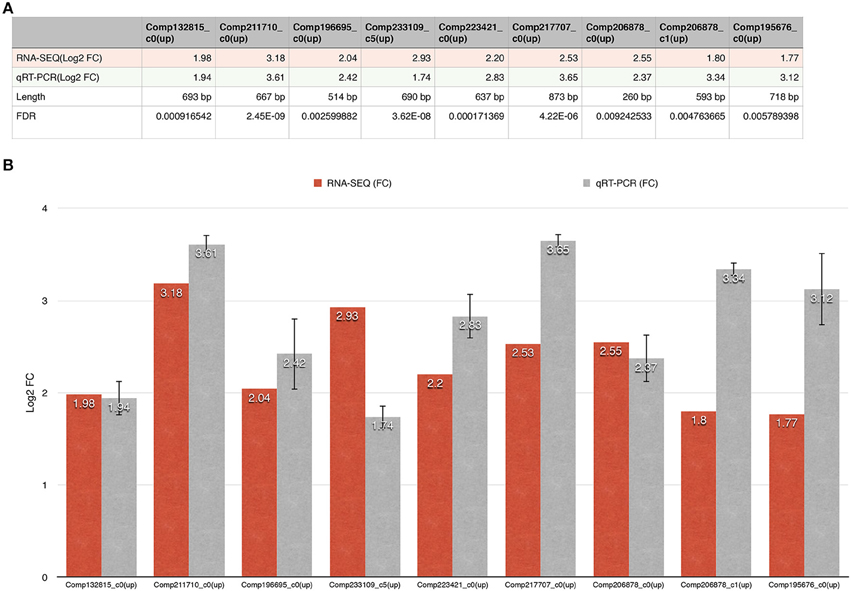
Figure 6. The fold changes in the DEGs as determined using RNA-seq and RT-qPCR are shown. (A) The first line represents the unigene ID; the second line represents the fold change in the DEG; the third line represents the RT-qPCR result; the fourth line represents the length of the unigene; and the fifth line represents the FDR of the unigene. (B) The x-axis represents the unigene ID while the y-axis represents the fold change in expression of the unigenes.
Phylogenetic Analysis
To further predict and distinguish the function of the nine unigenes, phylogenetic analysis was performed using 21 known plant AP2/EREBP protein sequences that were homologous to these unigenes. The analysis revealed the presence of five distinct clusters (Supplementary File 11), suggesting that these five groups might differ functionally in some respect. Comp206878_c1, Orza_sativa_subsp._indica_OsIBC009072, and Zea_mays_GRMZM2G129674_P01 belong to class I, whereas comp196695_c0, Zea_mays_GRMZM2G175525_P01, and Sorghum_bicolor_5041828 belong to the class II. In the phylogenetic tree, comp132815_c0, comp223421_c0, and comp211710_c0 were not closely related to the known AP2/EREBP genes, and comp233109_c5, comp217707_c0, comp195676_c0, and comp206878_c0 had a low bootstrap value. If these seven unigenes are novel ERFs or novel genes, the full lengths of these unigenes can be used further to inform our phylogenetic tree.
Conclusions
Here, we presented the first comprehensive transcriptome data of Z. japonica Steud. roots. In total, 32,849 unigenes were identified. The large number of transcripts identified will serve as a global resource for future studies. TFs play a crucial role in the early response to salt stress; thus, we identified candidate TFs related to salt stress, which will be the subject of future studies. In addition, a total of 4842 SSRs were identified. This information will facilitate future studies of plant biology and molecular breeding.
Author Contributions
HL conceived and designed the experiments and contributed the reagents. XQ and NJ wrote the manuscript. XQ, ZY, and FB performed the experiments. XX conducted the TF analysis. YS conducted the DEG analysis. ZL and SX conducted the H2O2 assay. XL and LX provided the materials and analytic tools.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Biomarker Technologies for providing assistance with the Illumina sequencing. We wish to thank Luo Yingfeng (Beijing Institute of Genomics, Chinese Academy of Sciences) for GO analysis and removal of redundant sequences. We also thank Drs. Lihuang Zhu and Li, and the members of Dr. Zhu's lab at the State Key Laboratory of Plant Genomics and National Plant Gene Research Center (Beijing, China), for their technical assistance and suggestions. This study was supported by the National High Technology Research and Development Program of China (863 Program) (No. 2013AA102607).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00610
Supplementary File 1. Detail of the 21 known AP2/EREBP genes.
Supplementary File 2. Length frequency distribution of contigs obtained by de novo assembly.
Supplementary File 3. COG and GO classification of zoysiagrass transcriptome.
Supplementary File 4. Summary and functional annotation of the identified DEGs.
Supplementary File 5. Gene ontology classification of DEGs.
Supplementary File 6. Detail of salt related genes that from homologous species.
Supplementary File 7. Transcript distribution across TF families.
Supplementary File 8. Major TFs identified in the DEG database.
Supplementary File 9. SSRs derived from the identified unigenes.
Supplementary File 10. Primers used for experimental validation.
Supplementary File 11. Phylogenetic tree of nine ERFs.
Availability of Supporting Data
The RNA sequence dataset supporting the results in this article is available from the NCBI Sequence Read Archive. The accession number is SRX838917.
References
Agarwal, P. K., Agarwal, P., Reddy, M. K., and Sopory, S. K. (2006). Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep. 25, 1263–1274. doi: 10.1007/s00299-006-0204-8
Ahn, J. H., Kim, J. S., Kim, S., Soh, H. Y., Shin, H., Jang, H., et al. (2015). De novo transcriptome analysis to identify anthocyanin biosynthesis genes responsible for tissue-specific pigmentation in zoysiagrass (Zoysia japonica Steud.). PLoS ONE 10:e0124497. doi: 10.1371/journal.pone.0124497
Aida, M., Ishida, T., Fukaki, H., Fujisawa, H., and Tasaka, M. (1997). Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. Plant Cell 9, 841–857. doi: 10.1105/tpc.9.6.841
Asano, Y. (1989). Somatic embryogenesis and protoplast culture in Japanese lawngrass (Zoysia japonica). Plant Cell Rep. 8, 141–143. doi: 10.1007/BF00716826
Bartels, D., and Sunkar, R. (2005). Drought and salt tolerance in plants. CRC. Crit. Rev. Plant Sci. 24, 23–58. doi: 10.1080/07352680590910410
Bian, S., and Jiang, Y. (2009). Reactive oxygen species, antioxidant enzyme activities and gene expression patterns in leaves and roots of Kentucky bluegrass in response to drought stress and recovery. Sci. Hortic. 120, 264–270. doi: 10.1016/j.scienta.2008.10.014
Cao, Z. H., Zhang, S. Z., Wang, R. K., Zhang, R. F., and Hao, Y. J. (2013). Genome wide analysis of the apple MYB transcription factor family allows the identification of MdoMYB121 gene confering abiotic stress tolerance in plants. PLoS ONE 8:e69955. doi: 10.1371/journal.pone.0069955
Chen, J., Hou, K., Qin, P., Liu, H., Yi, B., Yang, W., et al. (2014). RNA-Seq for gene identification and transcript profiling of three Stevia rebaudiana genotypes. BMC Genomics 15:571. doi: 10.1186/1471-2164-15-571
Chen, S., Huang, X., Yan, X., Liang, Y., Wang, Y., Li, X., et al. (2013). Transcriptome analysis in sheepgrass (Leymus chinensis): a dominant perennial grass of the Eurasian Steppe. PLoS ONE 8:e67974. doi: 10.1371/journal.pone.0067974
Cho Yg, I. T., Temnykh, S., Chen, X., Lipovich, L., and Et, A. L. (2000). Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor. Appl. Genet. 100, 713–722. doi: 10.1007/s001220051343
Cominelli, E., Sala, T., Calvi, D., Gusmaroli, G., and Tonelli, C. (2008). Over-expression of the Arabidopsis AtMYB41 gene alters cell expansion and leaf surface permeability. Plant J. 53, 53–64. doi: 10.1111/j.1365-313X.2007.03310.x
Cordeiro, G. M., Casu, R., McIntyre, C. L., Manners, J. M., and Henry, R. J. (2001). Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci. 160, 1115–1123. doi: 10.1016/S0168-9452(01)00365-X
Dai, H., Han, G., Yan, Y., Zhang, F., Liu, Z., Li, X., et al. (2013). Transcript assembly and quantification by RNA-Seq reveals differentially expressed genes between soft-endocarp and hard-endocarp hawthorns. PLoS ONE 8:e72910. doi: 10.1371/journal.pone.0072910
Dang, Z. H., Zheng, L. L., Wang, J., Gao, Z., Wu, S. B., Qi, Z., et al. (2013). Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna. BMC Genomics 14:29. doi: 10.1186/1471-2164-14-29
Du, H. M., Wang, Z. L., and Huang, B. R. (2009). Differential responses of warm-season and cool-season turfgrass species to heat stress associated with antioxidant enzyme activity. J. Am. Soc. Hortic. Sci. 134, 417–422. Available online at: http://journal.ashspublications.org/content/134/4/417.abstract
Dyachenko, O. V., Zakharchenko, N. S., Shevchuk, T. V., Bohnert, H. J., Cushman, J. C., and Buryanov, Y. I. (2006). Effect of hypermethylation of CCWGG sequences in DNA of Mesembryanthemum crystallinum plants on their adaptation to salt stress. Biochemistry (Mosc.) 71, 461–465. doi: 10.1134/S000629790604016X
Fukao, T., Yeung, E., and Bailey-Serres, J. (2011). The submergence tolerance regulator SUB1A mediates crosstalk between submergence and drought tolerance in rice. Plant Cell 23, 412–427. doi: 10.1105/tpc.110.080325
Ge, Y., Norton, T., and Wang, Z. Y. (2006). Transgenic zoysiagrass (Zoysia japonica) plants obtained by Agrobacterium-mediated transformation. Plant Cell Rep. 25, 792–798. doi: 10.1007/s00299-006-0123-8
Gilmour, S. J., Sebolt, A. M., Salazar, M. P., Everard, J. D., and Thomashow, M. F. (2000). Overexpression of the Arabidopsis CBF3 transcriptional activator mimics multiple biochemical changes associated with cold acclimation. Plant Physiol. 124, 1854–1865. doi: 10.1104/pp.124.4.1854
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guan, Q., Wu, J., Yue, X., Zhang, Y., and Zhu, J. (2013). A nuclear calcium-sensing pathway is critical for gene regulation and salt stress tolerance in Arabidopsis. PLoS Genet. 9:e1003755. doi: 10.1371/journal.pgen.1003755
Gupta, P. K., Rustgi, S., Sharma, S., Singh, R., Kumar, N., and Balyan, H. S. (2003). Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol. Genet. Genomics 270, 315–323. doi: 10.1007/s00438-003-0921-4
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hao, Y. J., Wei, W., Song, Q. X., Chen, H. W., Zhang, Y. Q., Wang, F., et al. (2011). Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 68, 302–313. doi: 10.1111/j.1365-313X.2011.04687.x
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., et al. (2004). The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261. doi: 10.1093/nar/gkh036
He, Y., Li, W., Lv, J., Jia, Y., Wang, M., and Xia, G. (2012). Ectopic expression of a wheat MYB transcription factor gene, TaMYB73, improves salinity stress tolerance in Arabidopsis thaliana. J. Exp. Bot. 63, 1511–1522. doi: 10.1093/jxb/err389
Hong, C.-Y., Chao, Y.-Y., Yang, M.-Y., Cheng, S.-Y., Cho, S.-C., and Kao, C. H. (2009). NaCl-induced expression of glutathione reductase in roots of rice (Oryza sativa L.) seedlings is mediated through hydrogen peroxide but not abscisic acid. Plant Soil 320, 103–115. doi: 10.1007/s11104-008-9874-z
Hu, L., Li, H., Pang, H., and Fu, J. (2012). Responses of antioxidant gene, protein and enzymes to salinity stress in two genotypes of perennial ryegrass (Lolium perenne) differing in salt tolerance. J. Plant Physiol. 169, 146–156. doi: 10.1016/j.jplph.2011.08.020
Huda, K. M., Banu, M. S., Tuteja, R., and Tuteja, N. (2013). Global calcium transducer P-type Ca(2)(+)-ATPases open new avenues for agriculture by regulating stress signalling. J. Exp. Bot. 64, 3099–3109. doi: 10.1093/jxb/ert182
Inokuma, C., Sugiura, K., Imaizumi, N., and Cho, C. (1998). Transgenic Japanese lawngrass (Zoysia japonica Steud.) plants regenerated from protoplasts. Plant Cell Rep. 17, 334–338. doi: 10.1007/s002990050403
Jiang, Y., and Deyholos, M. K. (2009). Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 69, 91–105. doi: 10.1007/s11103-008-9408-3
Jung, J., Won, S. Y., Suh, S. C., Kim, H., Wing, R., Jeong, Y., et al. (2007). The barley ERF-type transcription factor HvRAF confers enhanced pathogen resistance and salt tolerance in Arabidopsis. Planta 225, 575–588. doi: 10.1007/s00425-006-0373-2
Kim, S., Kang, J. Y., Cho, D. I., Park, J. H., and Kim, S. Y. (2004). ABF2, an ABRE-binding bZIP factor, is an essential component of glucose signaling and its overexpression affects multiple stress tolerance. Plant J. 40, 75–87. doi: 10.1111/j.1365-313X.2004.02192.x
Knight, H., Trewavas, A. J., and Knight, M. R. (1997). Calcium signalling in Arabidopsis thaliana responding to drought and salinity. Plant J. 12, 1067–1078. doi: 10.1046/j.1365-313X.1997.12051067.x
Li, M.-Y., Wang, F., Jiang, Q., Ma, J., and Xiong, A.-S. (2014). Identification of SSRs and differentially expressed genes in two cultivars of celery (Apium graveolens L.) by deep transcriptome sequencing. Hortic. Res. 1:10. doi: 10.1038/hortres.2014.10
Li, S., Fu, Q., Chen, L., Huang, W., and Yu, D. (2011). Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 233, 1237–1252. doi: 10.1007/s00425-011-1375-2
Liang, C., Liu, X., Yiu, S. M., and Lim, B. L. (2013). De novo assembly and characterization of Camelina sativa transcriptome by paired-end sequencing. BMC Genomics 14:146. doi: 10.1186/1471-2164-14-146
Liu, S., Yeh, C. T., Tang, H. M., Nettleton, D., and Schnable, P. S. (2012). Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 7:e36406. doi: 10.1371/journal.pone.0036406
Looso, M., Preussner, J., Sousounis, K., Bruckskotten, M., Michel, C. S., Lignelli, E., et al. (2013). A de novo assembly of the newt transcriptome combined with proteomic validation identifies new protein families expressed during tissue regeneration. Genome Biol. 14:R16. doi: 10.1186/gb-2013-14-2-r16
Lu, P. L., Chen, N. Z., An, R., Su, Z., Qi, B. S., Ren, F., et al. (2007). A novel drought-inducible gene, ATAF1, encodes a NAC family protein that negatively regulates the expression of stress-responsive genes in Arabidopsis. Plant Mol. Biol. 63, 289–305. doi: 10.1007/s11103-006-9089-8
Marinho, L. E., Tonetti, A. L., Stefanutti, R., and Coraucci Filho, B. (2013). Application of reclaimed wastewater in the irrigation of rosebushes. Water Air Soil Pollut. 224, 1669. doi: 10.1007/s11270-013-1669-z
Miao, Y., and Zentgraf, U. (2007). The antagonist function of Arabidopsis WRKY53 and ESR/ESP in leaf senescence is modulated by the jasmonic and salicylic acid equilibrium. Plant Cell 19, 819–830. doi: 10.1105/tpc.106.042705
Mittler, R., Vanderauwera, S., Gollery, M., and Van Breusegem, F. (2004). Reactive oxygen gene network of plants. Trends Plant Sci. 9, 490–498. doi: 10.1016/j.tplants.2004.08.009
Munns, R. (2005). Genes and salt tolerance: bringing them together. New Phytol. 167, 645–663. doi: 10.1111/j.1469-8137.2005.01487.x
Nakano, T., Suzuki, K., Fujimura, T., and Shinshi, H. (2006). Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 140, 411–432. doi: 10.1104/pp.105.073783
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 97–103. doi: 10.1016/j.bbagrm.2011.10.005
Olsen, A. N., Ernst, H. A., Leggio, L. L., and Skriver, K. (2005). NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 10, 79–87. doi: 10.1016/j.tplants.2004.12.010
Park, J. M., Park, C. J., Lee, S. B., Ham, B. K., Shin, R., and Paek, K. H. (2001). Overexpression of the tobacco Tsi1 gene encoding an EREBP/AP2-type transcription factor enhances resistance against pathogen attack and osmotic stress in tobacco. Plant Cell 13, 1035–1046. doi: 10.1105/tpc.13.5.1035
Petricka, J. J., Winter, C. M., and Benfey, P. N. (2012). Control of Arabidopsis root development. Annu. Rev. Plant Biol. 63, 563–590. doi: 10.1146/annurev-arplant-042811-105501
Puranik, S., Jha, S., Srivastava, P. S., Sreenivasulu, N., and Prasad, M. (2011). Comparative transcriptome analysis of contrasting foxtail millet cultivars in response to short-term salinity stress. J. Plant Physiol. 168, 280–287. doi: 10.1016/j.jplph.2010.07.005
Qing, D. J., Lu, H. F., Li, N., Dong, H. T., Dong, D. F., and Li, Y. Z. (2009). Comparative profiles of gene expression in leaves and roots of maize seedlings under conditions of salt stress and the removal of salt stress. Plant Cell Physiol. 50, 889–903. doi: 10.1093/pcp/pcp038
Riechmann, J. L., Heard, J., Martin, G., Reuber, L., Jiang, C., Keddie, J., et al. (2000). Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290, 2105–2110. doi: 10.1126/science.290.5499.2105
Schippers, J. H., Nguyen, H. M., Lu, D., Schmidt, R., and Mueller-Roeber, B. (2012). ROS homeostasis during development: an evolutionary conserved strategy. Cell. Mol. Life Sci. 69, 3245–3257. doi: 10.1007/s00018-012-1092-4
Schmidt, R., Mieulet, D., Hubberten, H. M., Obata, T., Hoefgen, R., Fernie, A. R., et al. (2013). Salt-responsive ERF1 regulates reactive oxygen species-dependent signaling during the initial response to salt stress in rice. Plant Cell 25, 2115–2131. doi: 10.1105/tpc.113.113068
Sun, L. J., Li, D. Y., Zhang, H. J., and Song, F. M. (2012). [Functions of NAC transcription factors in biotic and abiotic stress responses in plants]. Yi Chuan 34, 993–1002. doi: 10.3724/SP.J.1005.2012.00993
Sun, Q., Gao, F., Zhao, L., Li, K., and Zhang, J. (2010). Identification of a new 130 bp cis-acting element in the TsVP1 promoter involved in the salt stress response from Thellungiella halophila. BMC Plant Biol. 10:90. doi: 10.1186/1471-2229-10-90
Tracy, F. E., Gilliham, M., Dodd, A. N., Webb, A. A., and Tester, M. (2008). NaCl-induced changes in cytosolic free Ca2+ in Arabidopsis thaliana are heterogeneous and modified by external ionic composition. Plant Cell Environ. 31, 1063–1073. doi: 10.1111/j.1365-3040.2008.01817.x
Tsuruta, S.-I., Kobayashi, M., and Ebina, M. (2011). “Zoysia,” in Wild Crop Relatives: Genomic and Breeding Resources, ed C. Kole (Berlin; Heidelberg: Springer), 297–309.
Uddin, K., Juraimi, A. S., Ismail, M. R., Hossain, A., Othman, R., and Abdul Rahim, A. (2012). Physiological and growth responses of six turfgrass species relative to salinity tolerance. ScientificWorldJournal 2012, 905468. doi: 10.1100/2012/905468
Ueda, A., Kathiresan, A., Bennett, J., and Takabe, T. (2006). Comparative transcriptome analyses of barley and rice under salt stress. Theor. Appl. Genet. 112, 1286–1294. doi: 10.1007/s00122-006-0231-4
Ueda, A., Kathiresan, A., Inada, M., Narita, Y., Nakamura, T., Shi, W. M., et al. (2004). Osmotic stress in barley regulates expression of a different set of genes than salt stress does. J. Exp. Bot. 55, 2213–2218. doi: 10.1093/jxb/erh242
Vanderauwera, S., Zimmermann, P., Rombauts, S., Vandenabeele, S., Langebartels, C., Gruissem, W., et al. (2005). Genome-wide analysis of hydrogen peroxide-regulated gene expression in Arabidopsis reveals a high light-induced transcriptional cluster involved in anthocyanin biosynthesis. Plant Physiol. 139, 806–821. doi: 10.1104/pp.105.065896
Wang, K., and Jiang, Y. (2007). Antioxidant responses of creeping bentgrass roots to waterlogging. Crop Sci. 47, 232. doi: 10.2135/cropsci2006.07.0498
Wang, Y., Mopper, S., and Hasenstein, K. H. (2001). Effects of salinity on endogenous ABA, IAA, JA, AND SA in Iris hexagona. J. Chem. Ecol. 27, 327–342. doi: 10.1023/A:1005632506230
Wang, Y., Pan, Y., Liu, Z., Zhu, X., Zhai, L., Xu, L., et al. (2013). De novo transcriptome sequencing of radish (Raphanus sativus L.) and analysis of major genes involved in glucosinolate metabolism. BMC Genomics 14:836. doi: 10.1186/1471-2164-14-836
Wei, S., Wang, L., Zhang, Y., and Huang, D. (2013). Identification of early response genes to salt stress in roots of melon (Cucumis melo L.) seedlings. Mol. Biol. Rep. 40, 2915–2926. doi: 10.1007/s11033-012-2307-3
Wu, H. L., Ni, Z. F., Yao, Y. Y., Guo, G. G., and Sun, Q. X. (2008). Cloning and expression profiles of 15 genes encoding WRKY transcription factor in wheat (Triticum aestivem L.). Prog. Nat. Sci. 18, 697–705. doi: 10.1016/j.pnsc.2007.12.006
Xu, C., Li, X., and Zhang, L. (2013a). The effect of calcium chloride on growth, photosynthesis, and antioxidant responses of Zoysia japonica under drought conditions. PLoS ONE 8:e68214. doi: 10.1371/journal.pone.0068214
Xu, J., Li, Y., Ma, X., Ding, J., Wang, K., Wang, S., et al. (2013b). Whole transcriptome analysis using next-generation sequencing of model species Setaria viridis to support C4 photosynthesis research. Plant Mol. Biol. 83, 77–87. doi: 10.1007/s11103-013-0025-4
Xuan, J., Song, Y., Zhang, H., Liu, J., Guo, Z., and Hua, Y. (2013). Comparative proteomic analysis of the stolon cold stress response between the C4 perennial grass species Zoysia japonica and Zoysia metrella. PLoS ONE 8:e75705. doi: 10.1371/journal.pone.0075705
Yang, Y., Xu, S., An, L., and Chen, N. (2007). NADPH oxidase-dependent hydrogen peroxide production, induced by salinity stress, may be involved in the regulation of total calcium in roots of wheat. J. Plant Physiol. 164, 1429–1435. doi: 10.1016/j.jplph.2006.08.009
Yoon, S. K., Park, E. J., Choi, Y. I., Bae, E. K., Kim, J. H., Park, S. Y., et al. (2014). Response to drought and salt stress in leaves of poplar (Populus alba x Populus glandulosa): expression profiling by oligonucleotide microarray analysis. Plant Physiol. Biochem. 84, 158–168. doi: 10.1016/j.plaphy.2014.09.008. Available online at: http://www.sciencedirect.com/science/article/pii/S0981942814002939
Yun, K. Y., Park, M. R., Mohanty, B., Herath, V., Xu, F., Mauleon, R., et al. (2010). Transcriptional regulatory network triggered by oxidative signals configures the early response mechanisms of japonica rice to chilling stress. BMC Plant Biol. 10:16. doi: 10.1186/1471-2229-10-16
Zhang, L., Li, Z., Quan, R., Li, G., Wang, R., and Huang, R. (2011). An AP2 domain-containing gene, ESE1, targeted by the ethylene signaling component EIN3 is important for the salt response in Arabidopsis. Plant Physiol. 157, 854–865. doi: 10.1104/pp.111.179028
Zhou, X., Jiang, Y. J., and Yu, D. (2011). WRKY22 transcription factor mediates dark-induced leaf senescence in Arabidopsis. Mol. Cells 31, 303–313. doi: 10.1007/s10059-011-0047-1
Keywords: Zoysia japonica Steud., RNA sequencing (RNA-Seq), salt-stress, transcription factor, simple sequence repeats (SSRs)
Citation: Xie Q, Niu J, Xu X, Xu L, Zhang Y, Fan B, Liang X, Zhang L, Yin S and Han L (2015) De novo assembly of the Japanese lawngrass (Zoysia japonica Steud.) root transcriptome and identification of candidate unigenes related to early responses under salt stress. Front. Plant Sci. 6:610. doi: 10.3389/fpls.2015.00610
Received: 05 April 2015; Accepted: 23 July 2015;
Published: 20 August 2015.
Edited by:
Keqiang Wu, National Taiwan University, TaiwanReviewed by:
Ing-Feng Chang, National Taiwan University, TaiwanZhulong Chan, Wuhan Botanical Garden, Chinese Academy of Sciences, China
Copyright © 2015 Xie, Niu, Xu, Xu, Zhang, Fan, Liang, Zhang, Yin and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuxia Yin and Liebao Han, Beijing Forestry University, No.35, East Qinghua Road, Haidian District, Beijing 100083, China,eWluc3gzNjlAMTYzLmNvbQ==;aGFubGllYmFvQDE2My5jb20=