 Takayuki Fujiwara1*
Takayuki Fujiwara1* Yu Kanesaki2
Yu Kanesaki2 Shunsuke Hirooka1,3
Shunsuke Hirooka1,3 Atsuko Era1,3
Atsuko Era1,3 Nobuko Sumiya1,3
Nobuko Sumiya1,3 Hirofumi Yoshikawa3,4
Hirofumi Yoshikawa3,4 Kan Tanaka3,5
Kan Tanaka3,5 Shin-Ya Miyagishima1,3,6*
Shin-Ya Miyagishima1,3,6*- 1Department of Cell Genetics, National Institute of Genetics, Mishima, Japan
- 2NODAI Genome Research Center, Tokyo University of Agriculture, Tokyo, Japan
- 3Japan Science and Technology Agency, Core Research for Evolutional Science and Technology, Kawaguchi, Japan
- 4Department of Bioscience, Tokyo University of Agriculture, Tokyo, Japan
- 5Chemical Resources Laboratory, Tokyo Institute of Technology, Yokohama, Japan
- 6Department of Genetics, Graduate University for Advanced Studies, Mishima, Japan
The unicellular red alga Cyanidioschyzon merolae is a model organism for studying the basic biology of photosynthetic organisms. The C. merolae cell is composed of an extremely simple set of organelles. The genome is completely sequenced. Gene targeting and a heat-shock inducible gene expression system has been recently established. However, a conditional gene knockdown system has not been established, which is required for the examination of function of genes that are essential to cell viability and primary mutant defects. In the current study, we first evaluated the expression of a transgene from two chromosomal neutral loci located in the intergenic region between CMD184C and CMD185C, and a region upstream of the URA5.3 gene. There was no significant difference in expression between them and this result suggests that both may be used as neutral loci. We then designed an inducible and repressible gene expression by using promoters of nitrate-assimilation genes. The expression of nitrate-assimilation genes such as NR (nitrate reductase), NIR (nitrite reductase), and NRT (the nitrate/nitrite transporter) are reversibly regulated by their dependence on nitrogen sources. We constructed stable strains in which a cassette containing the NR, NIR, or NRT promoter and sfGFP gene was inserted in a region upstream of URA5.3 and examined the efficacy of the promoters. The NR, NIR, and NRT promoters were constitutively activated in the nitrate medium, whereas their activities were extremely low in presence of ammonium. The activation of each promoter was immediately inhibited within a period of 1 h by the addition of ammonium. Thus, a conditional knockdown system in C. merolae was successfully established. The activity varies among the promoters, and each is selectable according to the expression level of a target gene estimated by RNA-sequencing. This method is applicable to defects in genes of interest in photosynthetic organism.
Introduction
In studies on photosynthetic eukaryotes, molecular genetic techniques have been developed and used extensively for certain land plants and the unicellular green alga Chlamydomonas reinhardtii widely used as model systems. For the investigation of phenomena that are generally shared by algae and land plants, unicellular algae offer several experimental advantages. For example, a relatively homogeneous population is available in unicellular algae, in contrast to land plants, in which cells differentiate into heterogeneous populations. The generation time of unicellular algae is much shorter than that required for multicellular land plants. Thus far, C. reinhardtii has been the most extensively studied green alga, because it is genetically tractable. However, overexpression of a given gene of interest and expressing the genes of other organisms are very difficult because of the silencing of transgenes (Rosales-Mendoza et al., 2012; Liu and Benning, 2013). In certain other microalgae, methods for transformation have been reported, but as yet are still far from having come into practical use (Gong et al., 2011).
The unicellular red alga Cyanidioschyzon merolae inhabits sulfate-rich hot springs. The cell organization is very simple and the cell contains a single nucleus, mitochondrion and chloroplast along with a minimum set of membranous structures. The cell also has simple architectures of the endoplasmic reticulum and Golgi body, a single peroxisome and a small number of lysosomes (vacuoles; Kuroiwa et al., 1998; Misumi et al., 2005). The cell division and organelle division are tightly synchronized by the light/dark cycle (Suzuki et al., 1994). The nuclear, chloroplast, and mitochondrial genomes have been completely determined (i.e., without any gaps; Ohta et al., 1998, 2003; Matsuzaki et al., 2004; Nozaki et al., 2007). It possesses an extremely simple nuclear genome (16.5 Mbp; 4,775 protein-coding genes) among the photosynthetic eukaryotes (c.f. the green alga C. reinhardtii, 120 Mb; the land plant Arabidopsis thaliana, 157 Mb) with low genetic redundancy (Matsuzaki et al., 2004; Misumi et al., 2005; Nozaki et al., 2007). Therefore, this alga is suitable for various “omics” analyses. Various methods for genetic manipulation have been developed, such as the transient expression of proteins from plasmids (Ohnuma et al., 2008, 2009, 2014; Watanabe et al., 2011) and gene targeting by homologous recombination (Minoda et al., 2004; Imamura et al., 2010).
By virtue of a combination of these salient features and the experimental techniques of molecular genetics, C. merolae has become a promising model organism for the study of cell biology and metabolism in photosynthetic eukaryotes, such as chloroplast and mitochondrial division (Miyagishima et al., 2001, 2003; Nishida et al., 2003, 2007; Yoshida et al., 2009, 2010), vacuolar inheritance (Yagisawa et al., 2007, 2009; Fujiwara et al., 2010), the dynamics of other organelles (Miyagishima et al., 1998; Yagisawa et al., 2012, 2013; Fujiwara et al., 2013a; Imoto et al., 2013), nitrogen assimilation (Imamura et al., 2008, 2010) and circadian rhythms (Miyagishima et al., 2014). However, to study the mechanisms underlying the activities that are essential to cellular growth and survival, inactivation or modification of the mechanisms by gene manipulation would likely be lethal. Even when a certain gene manipulation does not lead to cell death, it is likely that prolonged cultivation of such genetically modified cells would lead to secondary indirect defects in the cells, which makes it extremely difficult to evaluate any primary defects that are directly caused by the gene manipulation.
To overcome this critical problem, analyses of primary defects, using conditional knockdown, an approach which is not effective under permissive environmental conditions but can be effective under certain tightly restricted conditions, are greatly desired. In contrast to C. reinhardtii, in which the expression of transgenes is often silenced, transgenes (either endogenous or exogenous genes) are stably expressed in C. merolae (Fujiwara et al., 2013b; Sumiya et al., 2014; Watanabe et al., 2014), which is an important advantage for constructing a gene-regulation system. To this end, an inducible gene expression system using the heat-shock promoter has recently been developed in C. merolae (Sumiya et al., 2014). This heat-shock system enables an examination of the effect of the expression of modified genes (e.g., dominant negative forms of the protein) on the cells, but it is difficult to evaluate the effect of repression of gene expression in this system. This is because culturing at such a high temperature (50°C) for a long period, which is required to maintain the expression of a target gene, damages cells and ultimately leads to cell lethality before the repression of the gene expression. Thus, an inducible/repressible gene expression system based on a less severe environmental condition is desired, as in the case in budding yeast, in which the regulatory systems for gene expression have been developed by using either carbon-source dependent promoters or a tetracycline-inducible/repressible system (Kaufmann and Knop, 2011).
To develop an inducible/repressible gene expression system, we focused on the promoters of the nitrate assimilation genes in C. merolae based on the following characteristics. A large number of eukaryotes and prokaryotes assimilate nitrogen from ammonium and nitrate (Ohmori et al., 1977; ter Schure et al., 2000). In many cases, ammonium is preferred over nitrate as an inorganic nitrogen source, because nitrate must be reduced to ammonium by expending energy for the purpose of its assimilation (Warner and Kleinhofs, 1992). When the balance between ammonium and nitrate changes in the environment, cell metabolism changes in order to adapt (Engelsberger and Schulze, 2012). Previous transcriptome analyses in C. merolae showed that transcription of certain key nitrogen assimilation genes is induced when the cells are transferred from the nitrogen (ammonium)-replete medium to the nitrogen-depleted medium (Imamura et al., 2008). The induced genes include NRT (the nitrate/nitrite transporter, CMG018C, the gene number is taken from http://merolae.biol.s.u-tokyo.ac.jp/), NR (nitrate reductase, CMG019C), NIR (nitrite reductase, CMG021C), AMT (the high affinity ammonium transporter, CMT526C), and GS (glutamine synthetase, CMI233C; Imamura et al., 2008).
In this study, we first confirmed that at least two chromosomal loci are sufficient to express a transgene as neutral loci and then developed a nitrogen-source-dependent inducible/repressible gene expression system by using the promoters of nitrate-assimilation genes.
Results
Estimation of an Ability to Express Transgene of Two Different Neutral Loci by Comparing sfGFP Expression
In order to evaluate the activities of promoters of nitrate-assimilation genes, we planned to express reporter protein [green fluorescent protein (GFP)] under the control of respective promoters from a C. merolae neutral chromosomal locus.
We have used the convergent intergenic region of CMD184C and CMD185C as a neutral locus because it is very short and therefore not likely to contain promoter activities that would potentially affect gene expression in its vicinity (Fujiwara et al., 2013b). However, it has not been determined whether the intergenic region is effective compared to other putative neutral loci. Recently, Watanabe et al. integrated a GFP gene in the vicinity of the chromosomal URA5.3 locus (CMK046C) and succeeded in expressing the GFP protein (Watanabe et al., 2014). Based on that report, we first compared the two chromosomal loci, namely the intergenic region between CMD184C and CMD185C and the upstream of URA5.3 locus, at the level of GFP transgene expression.
We previously prepared a strain in which the APCC promoter-superfolder GFP (sfGFP) and URA5.3 genes are integrated into the intergenic region between CMD184C and CMD185C of the C. merolae uracil-auxotrophic strain M4, which possesses a point mutation in the chromosomal URA5.3 locus (Sumiya et al., 2014; termed D-APCCp in this study; Figure 1A). For comparison, we prepared a strain in which the APCC promoter-sfGFP and URA5.3 selection genes are integrated into the chromosomal URA5.3 locus of M4. In this strain, termed U-APCCp, the APCC promoter-sfGFP is integrated into the region upstream of the URA5.3 locus (Figure 1B, the left panel). The occurrence of the recombination events in an upstream region of the URA5.3 locus in the U-APCCp strains were confirmed by PCR (Figure 1B, right panel).
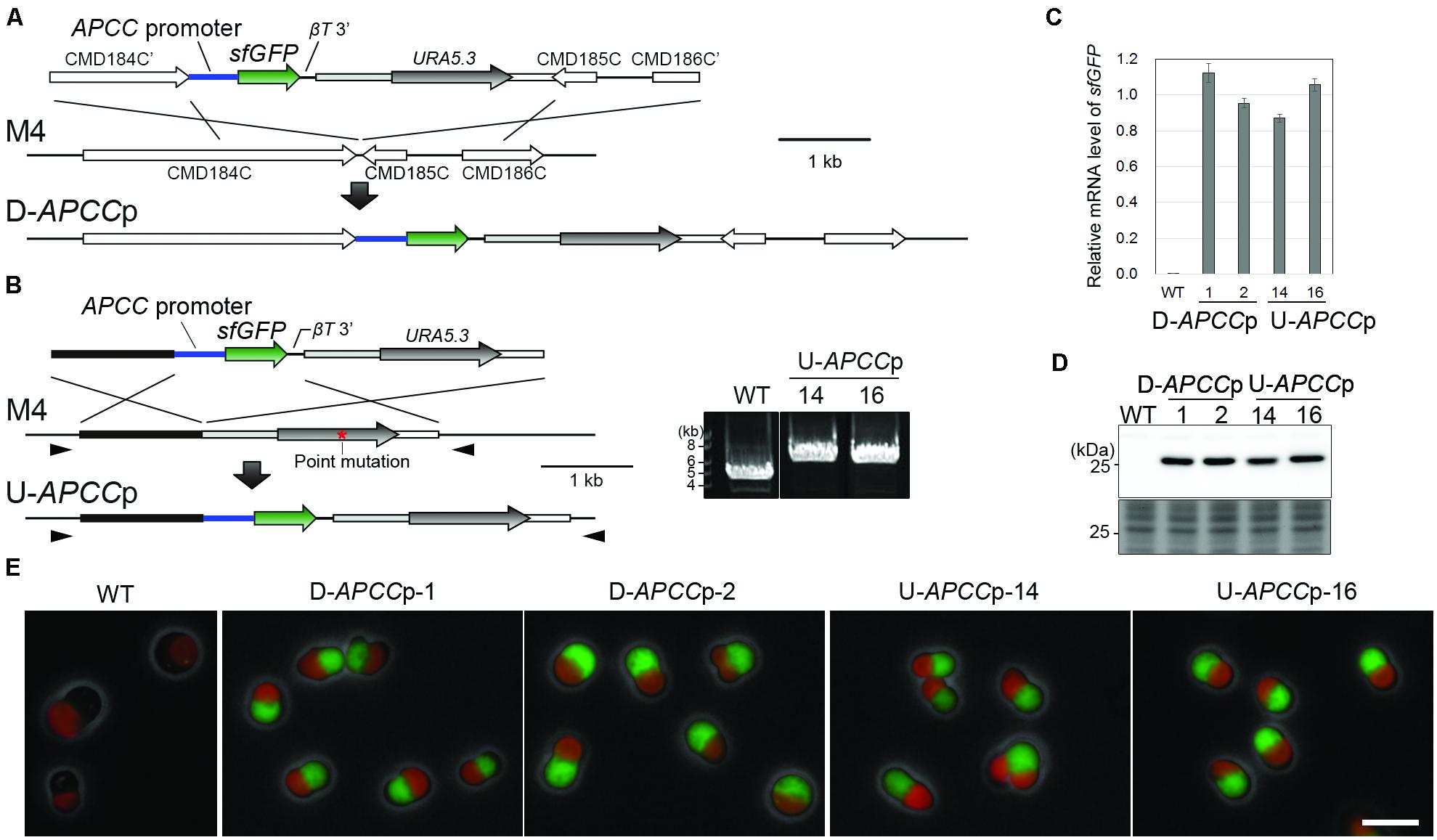
FIGURE 1. Construction and characterization of stable strains expressing sfGFP from two distinct chromosomal neutral loci. (A) Schematic diagram of the sfGFP gene insertion into the intergenic region between CMD184C and CMD185C by homologous recombination. The first line indicates the introduced DNA fragment and the second line indicates the genomic structure of the parental strain M4. For the efficient expression of sfGFP, the 600-bp upstream flanking sequence of APCC orf (APCC promoter) and the 200-bp downstream flanking sequence of the β-tubulin orf (βT 3′) were utilized as a promoter and a putative polyadenylation signal sequence, respectively. The third line indicates the expected genomic structure of the stable transformant. (B) Schematic diagram of the sfGFP gene insertion into the upstream region of the URA5.3 gene by homologous recombination. The same set of the promoter and the putative polyadenylation signal sequence as in (A) was flanked with the sfGFP orf. The red asterisk indicates the position of a point mutation in URA5.3 gene in the strain M4. The arrowheads indicate the positions of the PCR primers No. 14 and No. 15 (the exact positions and sequences are indicated in Supplementary Table S1). The PCR analysis of the independent U-APCCp strains confirmed the homologous recombination events (right panel). The WT strain was used as a negative control. The predicted size of the PCR product is 5.9 kb for U-APCCp and 4.2 kb for the WT, respectively. (C) Quantitative RT-PCR analysis of the D-APCCp and U-APCCp strains to estimate the sfGFP mRNA levels. The WT strain was used as a negative control. The sfGFP mRNA level in each strain was normalized with the data of DRP3/CmDnm1, and the average of the values in the D-APCCp and U-APCCp strains was defined as 1.0. The bar indicates the standard deviation (n = 3). (D) Immunoblot analysis of the total cell lysates with the anti-GFP antibody. An image of a gel stained with Coomassie Brilliant Blue (CBB) is shown as a loading control. (E) Fluorescent micrographs showing sfGFP fluorescent signals detected in the D-APCCp and U-APCCp strains. Green, GFP; red, autofluorescence of chlorophyll; gray, phase-contrast. Scale bar = 5 μm.
In order to estimate the level of transgene expression at both neutral loci, we compared the mRNA and protein levels along with the fluorescent intensity of sfGFP between the D-APCCp and U-APCCp strains. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay and immunoblotting showed that almost the same levels of the sfGFP mRNA and sfGFP protein were expressed in the two strains (Figures 1C,D). By fluorescence microscopy, a similar level of GFP fluorescence was detected in D-APCCp and U-APCCp cells (Figure 1E). These results indicate that there is no significant difference in the ability to express a transgene between the two chromosomal loci and suggest that both may be used as neutral loci for the expression of transgenes.
The Activation and Strength of Activity of the NR, NIR, and NRT Promoters for the Expression of the sfGFP Transgene in the Nitrate Medium
Among NR, NIR, NRT, AMT and GS, the expression of which were induced by ammonium-depletion in a previous study (Imamura et al., 2008), we focused on the promoters of NR, NIR and NRT, because AMT and GS exhibited leaky transcription in ammonium-replete medium (Imamura et al., 2008). To test the effectiveness of the promoters in turning the expression of transgenes on or off, we integrated the NR, NIR, or NRT promoter (the 800-bp upstream flanking sequence of the respective genes) -sfGFP orf into the upstream region of the chromosomal URA5.3 locus in the same manner as in the U-APCCp strain (Figure 2A). The recombination events in the upstream region of URA5.3 locus was confirmed by PCR with primers No. 14 and No. 15 (Figure 2B). The resultant transformants were termed NRp, NIRp, and NRTp. The NRp-1, NIRp-12, and NRTp-1 strains were used for further investigation.
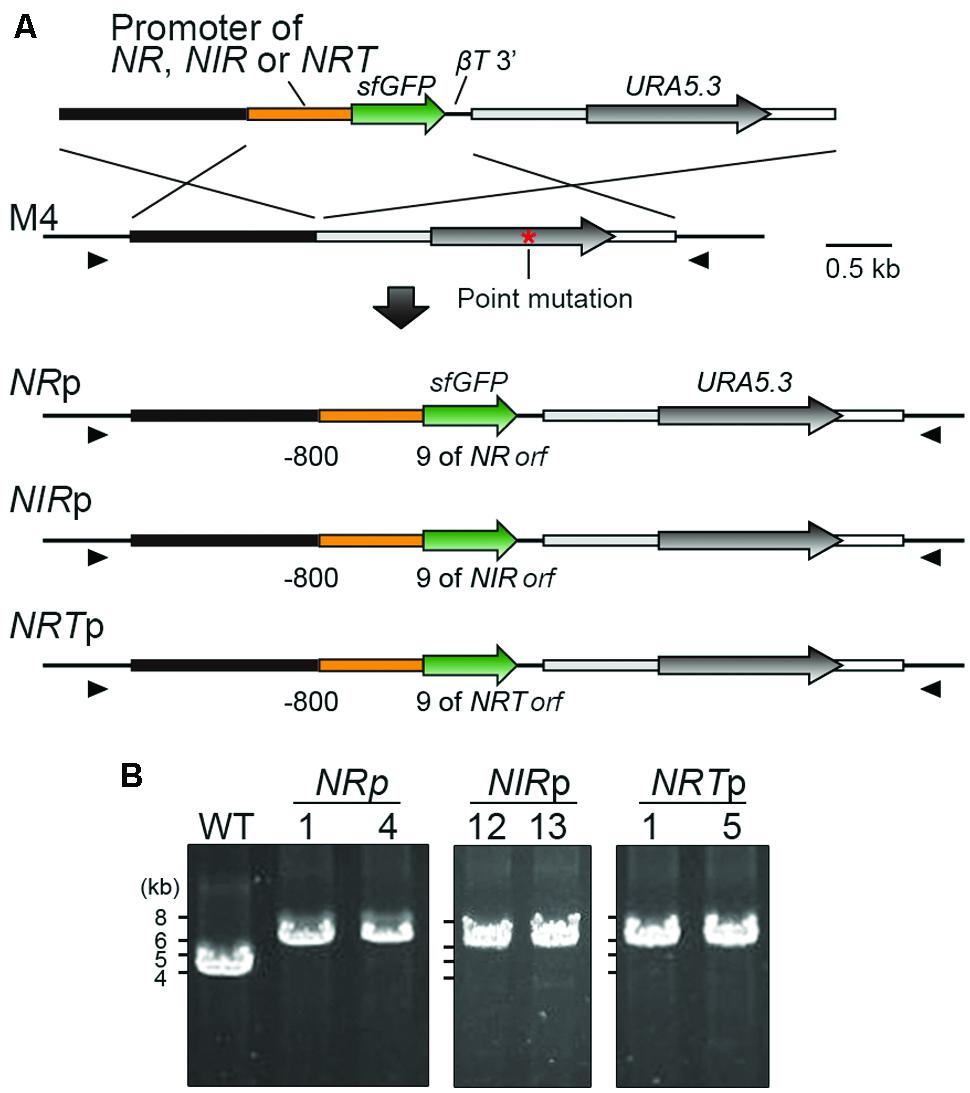
FIGURE 2. Construction of transformants expressing sfGFP by using the promoters of the nitrate assimilation genes. (A) Schematic diagram showing the targeted insertion of sfGFP flanked with the NR, NIR, or NRT promoter into the chromosomal region upstream of the URA5.3 gene. The third to fifth lines indicate the expected genomic structures in which a single copy of the transformed sequence is inserted by double-crossover homologous recombination in the respective cases. The red asterisk shows the position of the point mutation in the URA5.3 gene in strain M4. The arrowheads indicate the positions of the PCR primers used in (B). (B) PCR analysis of independent transformants confirmed the homologous recombination events. The WT strain was used as a negative control. The genomic sequence of the WT strain is identical to that of M4 except for the point mutation in the URA5.3 gene in M4. The predicted size of the PCR product is 6.1-kb for NRp, NIRp, and NRTp or 4.2 kb for the WT, respectively. The positions and sequences of the primers No. 14 and No. 15 are shown in Supplementary Table S1.
In order to activate the nitrate-assimilation gene promoters, NRp, NIRp, and NRTp cells in logarithmic culture were transferred from the ammonium medium to the nitrate medium.
Quantitative reverse transcription polymerase chain reaction revealed that the mRNA level of the sfGFP transgene in NRp, NIRp, and NRTp was extremely low before the medium exchange (0 h in Figure 3A; Based on the mean sfGFP mRNA levels in U-APCCp-14 and -16 that were cultivated in the ammonium medium and used as reference values).
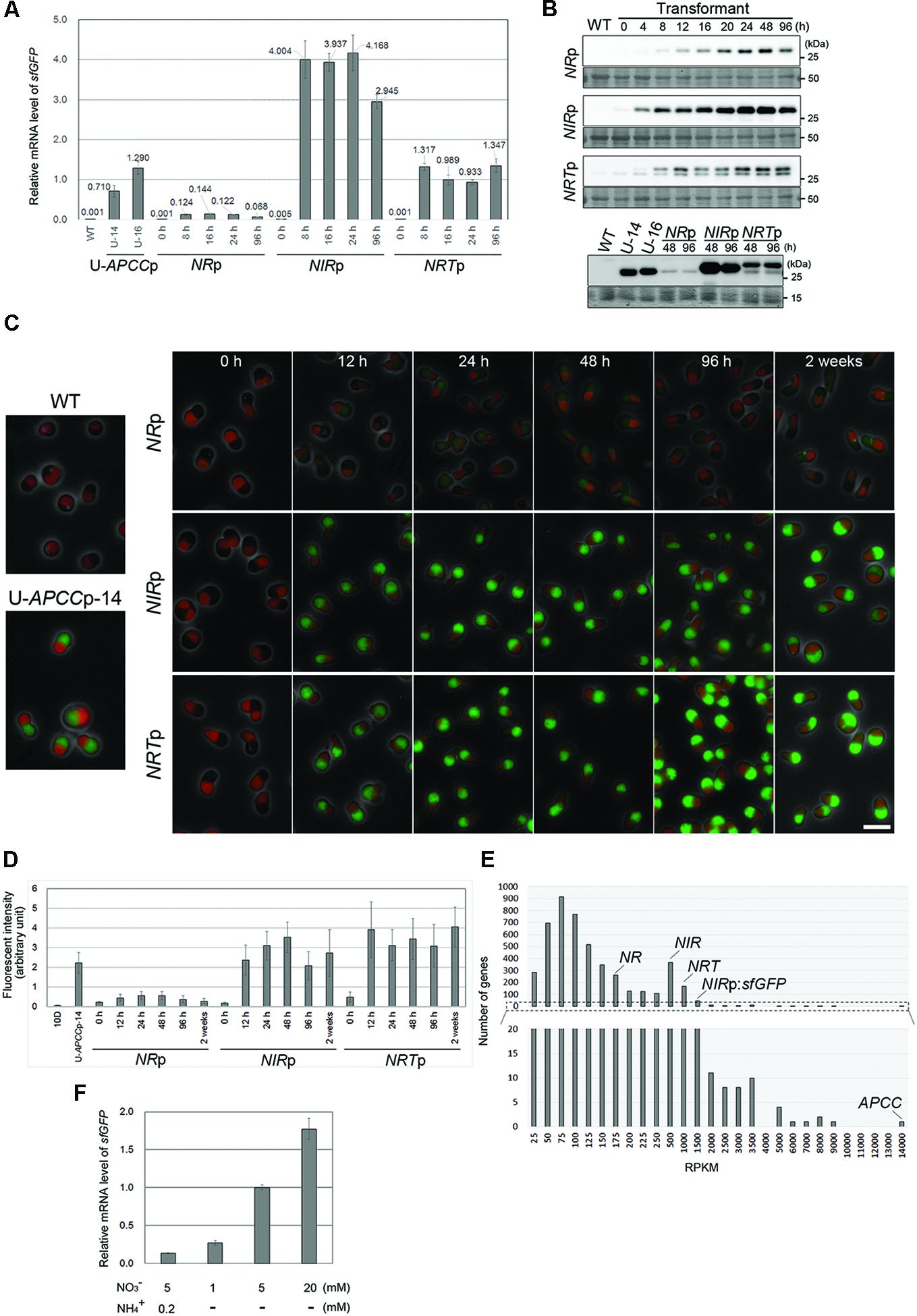
FIGURE 3. The induction of sfGFP expression by medium exchange in the NRp, NIRp, and NRTp strains. (A) Quantitative RT-PCR analyses showing the change in the sfGFP mRNA level in the NRp, NIRp, and NRTp strains before and after the exchange from the ammonium medium to the nitrate medium. The values were standardized by the average of the U-APCCp-14 and U-APCCp-16 which were cultured in the ammonium medium. The WT strain was used as a negative control. The time elapsed since the medium exchange is indicated below the graph and the time point 0 is just before the medium exchange. The bar indicates the standard deviation (n = 3). (B) Immunoblot analysis of the total cell lysates of the respective strains with the anti-GFP antibody. The upper panel shows the change in the sfGFP protein level just before (0 h) and after the medium exchange. The WT strain was used as a negative control. The lower panel compares the sfGFP protein level of the respective strains after the medium exchange. The sfGFP level in the U-APCCp strains (stably expressing GFP in both the ammonium and nitrate media: the results in the ammonium medium shown) is also shown as an index of the protein level. The WT strain was used as a negative control. An image of the PVDF membrane stained with Ponceau S is shown as a loading control. Two bands were detected in the NRTp strain with the anti-GFP antibody, most likely because there are two translational start sites. (C) Fluorescent micrographs showing the change in the level of the sfGFP fluorescent signal just before (0 h) and after the medium exchange. The green fluorescence of sfGFP was overlaid with the phase-contrast image and autofluorescence of the chloroplast. The WT and the U-APCC strain were used as a positive and a negative control, respectively. The exposure time to capture the sfGFP signal was 1 s for all images. Green, GFP; red, autofluorescence of chlorophyll; gray, phase-contrast. The scale bar = 5 μm. (D) The change in the intensity of sfGFP fluorescence in cells in (C). The bar indicates the standard deviation (n = 15). (E) RNA-seq analysis showing the relative mRNA levels of endogenous NR, NIR, and NRT in the transcriptome of the NIRp strain (logarithmic growth phase) cultured in the nitrate medium. The reads per kilo-base (RPKM) data indicates the relative mRNA abundance. The RPKM of endogenous NR, NIR, NRT, APCC, and sfGFP transcription of which is driven by the NIR promoter, was 158, 377, 787, 13384, and 1337, respectively. The lower histogram shows the magnification of the range from 0 to 20. (F) QRT-PCR analyses showing the sfGFP mRNA level in the NRTp strain 16 h after the medium exchange. The values were standardized by the data from culture in medium containing 5 mM nitrate without ammonium. The concentration of nitrate or ammonium is indicated below the graph. The bar indicates the standard deviation (n = 3).
After the medium exchange, the sfGFP mRNA level increased and reached a maximum in 8 h (Figure 3A). The maximum level of sfGFP mRNA varied with the promoter used and increased on the order of NRp, NRTp and NIRp, with this last one the highest.
Immunoblot analyses showed that the sfGFP protein was below the detectable level before the medium exchange (0 h). sfGFP accumulated and the level reached a maximum in a period from 24 to 48 h after the medium exchange in each strain (Figure 3B, the upper panel). Two bands were detected in the NRTp strain with the anti-GFP antibody, likely because there are two translational start sites. When the maximum level of sfGFP was compared, the level was the highest in NIRp, followed by NRTp, U-APCCp and NRp (Figure 3B, the lower panel), as was also the case for the mRNA level (Figure 3A).
Under fluorescence microscopy, sfGFP fluorescence was not detected before the medium exchange (0 h in Figure 3C). After the medium exchange, the fluorescent signal became evident and the signal in NRp cells was the weakest (Figures 3C,D), in accord with the immunoblotting results (Figure 3B). However, the sfGFP fluorescent signal level in NRTp was equivalent to or higher than that in NIRp (Figures 3C,D), in contrast to the result of immunoblotting (Figure 3B). The exact reason for this inconsistency is unclear at present, but it is likely that there are two sizes of sfGFP, and the larger one, which is specific to NRTp (Figure 3B), emits greater fluorescence. The sfGFP fluorescence was detectable for at least 2 weeks after the medium exchange (Figures 3C,D).
The above results indicate that the NR, NIR, and NRT promoters are inactive in the ammonium medium, with little evidence of leaky transcriptional activity, and are activated by the exchange of the ammonium medium for the nitrate medium. In addition, it has been shown that the transcriptional potency was the highest in the NIR promoter, followed by the NRT, APCC (a constitutive promoter) and NR promoters.
To further evaluate the strength of the transcriptional activity of the NR, NIR, and NRT promoters in the activated state (i.e., in the nitrate medium), we compared the mRNA levels of the NR, NIR, NRT, and sfGFP transgenes with other nucleus-encoded genes in NIRp cells grown in the nitrate medium by RNA sequence (RNA-seq) analysis. To compare mRNA level within a sample, the counts of the RNA-seq reads were normalized by the GC content bias correction method (Risso et al., 2011). The RNA-seq analysis detected transcripts of 4,740 genes among 4,774 C. merolae nucleus-encoded genes. It was ascertained that the APCC gene was very strongly transcribed in the cell in the nitrate medium (Figure 3E). The mRNA level was the highest in sfGFP, transcription of which was driven by the NIR promoter, followed by NRT, NIR and NR genes, whose ranking was the 59-th, 127-th, 352-th, and 1162-th highest among the nucleus-encoded genes, respectively (Figure 3E and Supplementary Data 1). It should be noted that strength of promoter activity and a corresponding mRNA level was not always consistent: the mRNA level of the sfGFP transgene was higher than that of the NIR, and the order of the promoter activities and the mRNA level of the NIR and NRT became inverse. It is likely that the orf sequence and the 3′ untranslated region are also involved in its mRNA turnover.
We further examined the effect of nitrate concentration and co-existence of ammonium and nitrate in the medium on promoter activity. QRT-PCR showed that the level of sfGFP mRNA increased depending on concentration of nitrate (Figure 3F), and that addition of 0.2 mM ammonium to the medium with 5 mM nitrate decreased the mRNA level (Figure 3F).
Repression of the NR, NIR, and NRT Promoters in the Expression of the sfGFP Transgene by the Addition of Ammonium
The aim of this study was to develop a system that would be able to turn off, as well as turn on, the expression of genes of interest. To this end, we tested whether the expression of sfGFP was suppressed by adding ammonium to the culture of NRp, NIRp, and NRTp. After the cultivation for 16 h in the nitrate medium to express sfGFP constitutively, ammonium was supplied to the medium. qRT-PCR analysis showed that the mRNA level of sfGFP in each strain decreased to the basal level within 1 h after the addition of ammonium (Figure 4A). In contrast to the mRNA results, immunoblotting analysis and fluorescence microscopy showed that the level of the sfGFP protein gradually decreased and became hardly detectable at 96 h after the addition of ammonium (Figures 4B–D). These results showed that the transcription of a transgene driven by the NIR, NR, or NRT promoter effectively abrogated by the addition of ammonium.
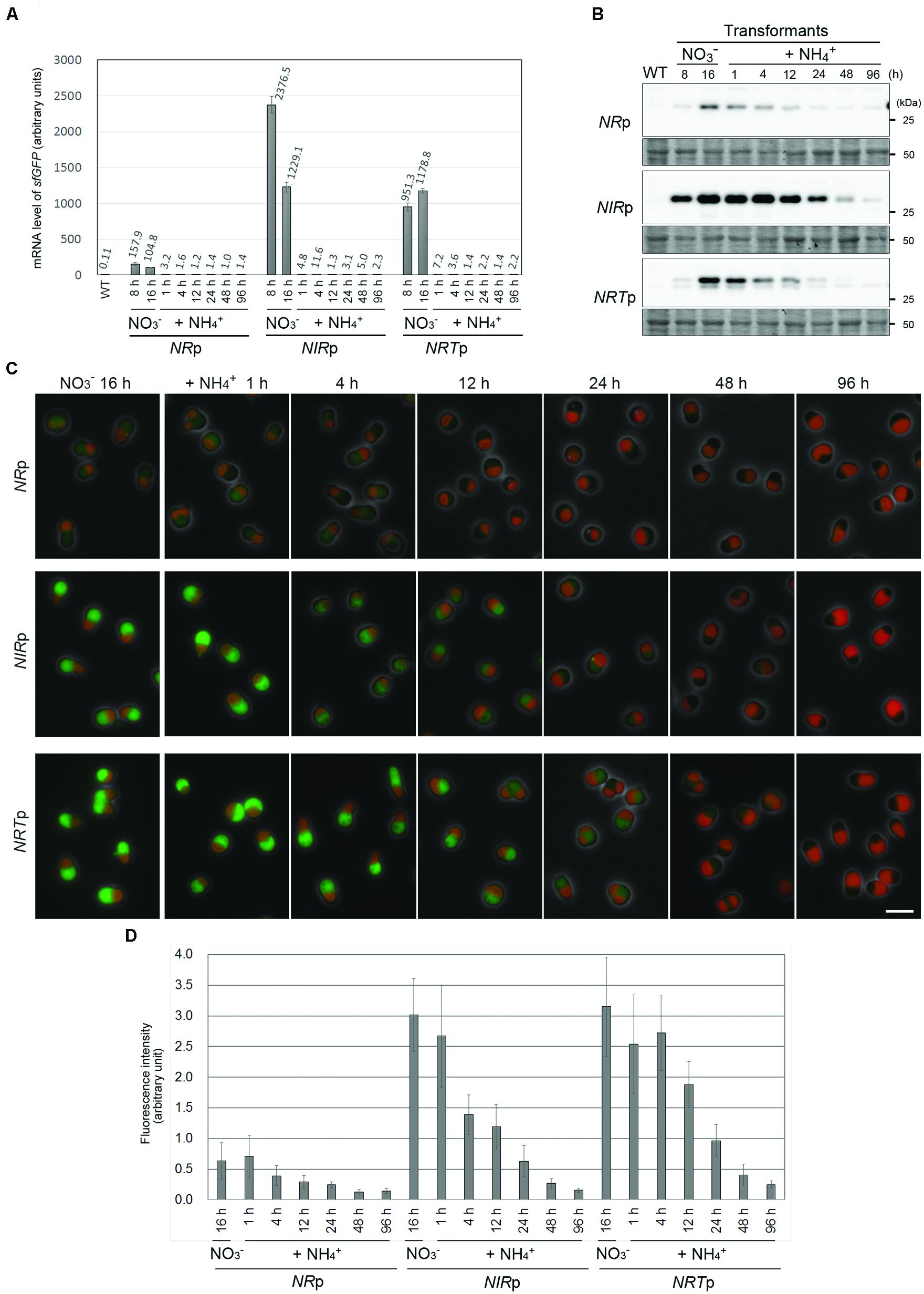
FIGURE 4. The suppression of sfGFP expression by the addition of ammonium in the NRp, NIRp, and NRTp strains. (A) Quantitative RT-PCR analyses showing the change in the sfGFP mRNA level in the NRp, NIRp, and NRTp strains before and after the addition of ammonium. The values in the respective stains were normalized with the data of DRP3/CmDnm1. The WT strain was used as a negative control. After cultivation of the cells in the nitrate medium for 16 h, ammonium was added to the medium and then the cells were cultured for 96 h. The bar indicates the standard deviation (n = 3). (B) Immunoblot analysis of the total cell lysates of the respective strains with the anti-GFP antibody showing the change in the sfGFP protein level before and after the addition of ammonium. An image of the PVDF membrane stained with Ponceau S is shown as a loading control. (C) Fluorescent micrographs showing the change in the level of the sfGFP fluorescent signal before and after the addition of ammonium. The exposure time to capture the sfGFP signal was 1 sec for all images. Green, GFP; red, autofluorescence of chlorophyll; gray, phase-contrast. The scale bar = 5 μm. (D) The change in the intensity of sfGFP fluorescence in cells in (C). The bar indicates the standard deviation (n = 15).
Discussion
In this study, we first evaluated the effectiveness of the two chromosomal loci of C. merolae, the intergenic region between CMD184C and CMD185C, and the upstream region of URA5.3 as neutral loci for the expression of transgenes. The results showed that these loci have the same ability to express a transgene. Thus far, only the URA5.3 selection gene marker has been available for the transformation of C. merolae. We are developing another marker. When the additional selection marker becomes available, multiple transgenes will be inserted into the two neutral chromosomal loci.
In addition, we have succeeded in developing an inducible and repressive transgene expression system in C. merolae by replacing nitrate (“on”) with ammonium (“off”) as the nitrogen source in the medium. The NR, NIR, and NRT promoters were constitutively activated in the nitrate medium, whereas the basal levels of the NR, NIR, and NRT promoter activity were extremely low in the presence of ammonium. This inducible system will be useful, as the previously developed heat-shock promoter system in C. merolae, for the expression of dominant-negative forms of proteins and the evaluation of the resultant primary defects in cells (Sumiya et al., 2014).
In terms of the repressive system, it will be useful for conditional gene knockdown, which has not been developed to date in photosynthetic eukaryotes. In this case, the URA5.3 gene and NR, NIR, or NRT promoter will be inserted into the upstream region of a gene of interest (i.e., the endogenous promoter will be replaced with that of a nitrate-assimilation gene) in cells cultured in the nitrate medium. The formation of colonies after transformation will be feasible, because the sfGFP will have been continuously expressed at that time for a period of at least for 2 weeks, which is sufficient for colony formation (Figure 3C). Then, the expression of the gene of interest will be immediately turned off by the addition of ammonium to the medium. In this study, the level of sfGFP immediately decreased after the inactivation of the promoters by the addition of ammonium, but it took ∼96 h for sfGFP to reach the undetectable level (Figure 4B). However, because GFP is a very stable protein (Chalfie et al., 1994), it probably will take a much shorter period of time for a protein of interest to be degraded after the inactivation of the promoters in most cases.
For the conditional knockdown assays, it is preferable that the expression level of a gene of interest, the transcription of which is driven by a promoter of a nitrate-assimilation gene, is close to the expression level of the original endogenous promoter. In this regard, we have shown that the transcriptional activity level of the nitrate-assimilation genes varies and one may select the promoter according to the experimental design. To choose an appropriate promoter, the RNA-seq analysis performed in current study (Figure 3E and Supplementary Data 1). Even though the mRNA level does not always indicate strength of corresponding promoter, the data of the RNA-seq is useful for estimating and comparing the strength of the promoter of a target gene as well as the promoters of NR, NIR, and NRT. Based on the data of the RNA-seq (Figure 3E), the NR promoter will be applied to many genes exhibiting weak expressions whereas the NIR and NRT promoter will be applicable to highly expressional genes when conditional knockdown of a gene of interest is designed. In addition, it is possible to adjust promoter activity by changing nitrate and ammonium concentration (Figure 3F).
It is anticipated that the current results will extend the range of genetic analyses investigating gene function essential for cell survival, and will facilitate our understanding of the fundamental mechanisms of cell proliferation, organelle division and inheritance, environmental adaptation and related subjects in photosynthetic organisms.
Material and Methods
Cell Culture
The wild type C. merolae 10D (NIES-3377) along with the other strains were grown in 20 mL of MA2 medium (Ohnuma et al., 2008) in a tissue culture flask 25 (TPP Techno Plastic Products AG, Switzerland) with shaking at 130 rpm under continuous white light (50 μmol/m2s) at 40°C. The cultures of all of the strains were diluted to an OD750 of 0.2 in 60 mL of 2x Allen’s medium (Allen, 1959), incubated under continuous light (80 μmol/m2s) with aeration (400 mL ambient air/min) for 2 days and then collected for the experiments. 2x Allen’s medium, which contains (NH4)2SO4 as the sole nitrogen source, is termed the ammonium medium in this study. In order to induce the expression of sfGFP in the NRp, NRTp and NIRp strains, the medium was exchanged to a nitrate medium (NaNO3 5 mM, Na2SO4 20 mM, KH2PO4 4 mM, MgSO4 2 mM, CaCl2 1 mM, twofold concentration of P4 metal, H2SO4 0.03% v/v to adjust to pH 2.3) containing NaNO3 instead of (NH4)2SO4 as the sole nitrogen source (Imamura et al., 2008, with a minor modification). In order to suppress sfGFP transcription, 1 M (NH4)2SO4 solution was added to the nitrate medium at a final concentration of 20 mM 16 h after the medium exchange.
Plasmid Construction and the Preparation of the Stable Transformants of C. merolae
The primers used in this study are listed in Supplementary Table S1. The U-APCCp strains were prepared as follows. The URA5.3 (CMK046C) selection marker gene (URA5.3 orf flanked with 2,3-kb upstream and 1.9-kb downstream sequences) was amplified by PCR with primers No. 1 and No. 2 and C. merolae 10D genomic DNA as a template, and then cloned into the pGEM-Teasy vector (Promega) to prepare the pURA vector. To produce the pU-APCCp vector, the pURA vector was amplified by PCR with primers No. 3 and No. 4. The cassette containing APCC (CMO250C) promoter (the 600-bp upstream sequence flanking APCC orf), the sfGFP orf and the 200-bp downstream sequence flanking β-tubulin (CMN263C) orf was amplified by PCR with primers No. 5 and No. 6 and genomic DNA of the C. merolae D-APCCp stain (Sumiya et al., 2014) as a template, and then cloned into an amplified pURA vector using the In-Fusion HD Cloning Kit (Clontech). The resultant pU-APCCp vector was used as a template to amplify the assembled set of fragments (the ∼1.4-kb upstream sequence of URA5.3 orf [-2300 to -898], APCC promoter, sfGFP orf, 200-bp downstream sequence of β-tubulin orf and URA5.3 selection gene [-897 to +471]) with primers No. 1 and No. 2.
The NRp, NIRp, and NRTp strains were prepared as follows. To prepare the pU-APCCp vector without the APCC promoter, the vector was amplified by PCR with primers No. 3 and No. 7. Then, the NR (CMG019C) promoter (the upstream flanking sequence of NR orf [-800 to 9 bp]), NIR (CMG021C) promoter (the upstream flanking sequence of NIR orf [-800 to 9 bp]) or NRT (CMG018C) promoter (the upstream flanking sequence of NRT orf [-800 to 9 bp]) was amplified by PCR with C. merolae 10D genomic DNA as a template and the primer set No. 8 and 9, No. 10 and 11, or No. 12 and 13, respectively. The respective promoters were cloned into the amplified pU-APCCp without the APCC promoter by using the In-Fusion HD Cloning Kit. The resultant pNRp::sfGFP, pNIRp::sfGFP, or pNRTp::sfGFP plasmid was used as a template to amplify the assembled set of fragments (the ∼1.4-kb upstream flanking region of URA5.3 orf, the promoter of NR, NIR or NRT, sfGFP orf, 200-bp downstream sequence of β-tubulin orf and the URA5.3 selection gene with primers No. 1 and No. 2.
The respectively amplified DNA fragments for homologous recombination were transformed into C. merolae strain M4 (Minoda et al., 2004), a derivative of C. merolae 10D, which has a mutation in the URA5.3 gene. Transformation was performed as described previously (Ohnuma et al., 2008; Imamura et al., 2010). A 4-μg aliquot of the PCR fragment was used for transformation and cells were incubated in MA2 medium containing uracil (0.5 mg/mL) for 1 day. After exchange to uracil-free MA2 medium, the cells were inoculated into starch on solidified MA2 under continuous white light at 42°C in ambient air supplemented with 5% CO2 until colonies were formed (for ∼2 weeks). The colonies were then transferred to starch on solidified MA2 medium containing uracil (0.5 mg/ml). Solidified MA2 medium was prepared according to Fujiwara et al. (2013b). The occurrence of the recombination events in the chromosomal URA5.3 region in the U-APCCp, NRp, NIRp, and NRTp strains were confirmed by PCR with primers No. 14 and No. 15.
Quantitative RT-PCR Analyses
Total RNA was extracted from a frozen cell pellet according to Fujiwara et al., 2009. cDNA was synthesized from 1 μg of the total RNA with an oligo(dT)15 primer using Prime Script Reverse Transcriptase (TAKARA Bio). Real-time PCR was performed using a StepOne Real-Time PCR System (Life technologies) in a 20-μl reaction mixture containing 4-μl template DNA, primers (0.25 nM each; Supplementary Table S1) and 10-μl Power SYBR Green Master Mix (Life technologies). Standard curves were constructed using serially diluted solutions of cDNA mixture prepared from the NRp, NIRp, and NRTp strains as well as the relevant sets of primers. The sfGFP mRNA level in the respective strains was normalized with the data of DRP3/CmDnm1 (CME019C).
Immunoblot Analysis
Fifteen μg of protein prepared from whole cell lysate were separated by SDS-PAGE, then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was probed with an anti-GFP antibody (Abcam, ab6556) at a final concentration of 0.2 μg/ml, followed by goat anti-rabbit HRP-conjugated IgG (Vector Laboratories) at a dilution of 1:20,000. The chemiluminescent signals were detected with ECL Prime Western Blotting Detection Reagent (GE Healthcare) and a VersaDoc Imaging System (BIO-RAD).
Fluorescence Microscopy
Images were captured using a fluorescence microscope (BX51; Olympus) with a digital CCD camera system DP71 (Olympus). The filter sets U-MNIBA3 and U-MWIG3 (Olympus) were used for the GFP fluorescence and auto-fluorescence of the chloroplast, respectively. The fluorescence intensity of sfGFP was quantified by ImageJ1.
RNA-seq Analysis
Total RNA was extracted from the NIRp cells after cultivation for 24 h in the nitrate medium using Trizol reagent (Invitrogen) and RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. mRNA was purified from 10 μg of total RNA of the NIRp cells using Dynabeads Oligo(dT)25 (Life technologies). Sequencing libraries were prepared by NEBNext mRNA library prep kit for Illumina (NEB) with following modifications. The oligo-dT primer was used for reverse transcription. After second strand synthesis, double stranded cDNA were fragmented to an average length of 300 bp using a Covaris S2 sonication system (Covaris, Woburn, CA, USA). One hundred cycles of paired-end sequencing were carried out using HiSeq2500 system according to the manufacturer’s specifications (Illumina). After the sequencing reactions were complete, the Illumina analysis pipeline (CASAVA 1.8.0) was used to process the raw sequencing data. The RNA-Seq reads were mapped to the C. merolae coding sequences2 using Bowtie2 with default parameters (Langmead and Salzberg, 2012). The Bowtie2 outputs were processed to obtain tag counts. Since it has been shown that the GC content can have a substantial impact on the read abundances in a RNA-Seq data set (Zheng et al., 2011), counts were full-quantile normalized within sample by the GC content bias correction methods implemented in the EDASeq R package (Risso et al., 2011). These normalized counts were used to calculate the expression level of each gene (in RPKM units) according to (Mortazavi et al., 2008).
Author Contributions
Conceived and designed the experiments: TF, KT; performed the experiments: TF; analyzed the data: TF; contributed reagents/materials/analysis tools: AE, NS, KT; RNA-seq: YK, SH, HY; wrote the paper: TF, S-YM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Grant-in-Aid for JSPS Fellows (3172 to TF) and by Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (no. 25251039 to S-YM and no. 24248061 to KT) and by Core Research for Evolutional Science and Technology (CREST) of Program of Japan Science and Technology Agency (JST; to S-YM, KT, and YH) and was partly supported by MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2013–2017 (S1311017). We also thank A. Yamashita and K. Hashimoto for technical assistance, and members of the Miyagishima lab. for technical advice. Pacific Edit reviewed the manuscript prior to submission.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00657
Footnotes
- ^ http://rsbweb.nih.gov/ij/download.html
- ^ http://merolae.biol.s.u-tokyo.ac.jp/download/cds_nt.fasta
References
Allen, M. B. (1959). Studies with Cyanidium caldarium, an anomalously pigmented chlorophyte. Arch. Microbiol. 32, 270–277. doi: 10.1007/bf00409348
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., and Prasher, D. C. (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. doi: 10.1126/science.8303295
Engelsberger, W. R., and Schulze, W. X. (2012). Nitrate and ammonium lead to distinct global dynamic phosphorylation patterns when resupplied to nitrogen-starved Arabidopsis seedlings. Plant J. 69, 978–995. doi: 10.1111/j.1365-313X.2011.04848.x
Fujiwara, T., Kuroiwa, H., Yagisawa, F., Ohnuma, M., Yoshida, Y., Yoshida, M., et al. (2010). The coiled-coil protein VIG1 is essential for tethering vacuoles to mitochondria during vacuole inheritance of Cyanidioschyzon merolae. Plant Cell 22, 772–781. doi: 10.1105/tpc.109.070227
Fujiwara, T., Misumi, O., Tashiro, K., Yoshida, Y., Nishida, K., Yagisawa, F., et al. (2009). Periodic gene expression patterns during the highly synchronized cell nucleus and organelle division cycles in the unicellular red alga Cyanidioschyzon merolae. DNA Res. 16, 59–72. doi: 10.1093/dnares/dsn032
Fujiwara, T., Tanaka, K., Kuroiwa, T., and Hirano, T. (2013a). Spatiotemporal dynamics of condensins I and II: evolutionary insights from the primitive red alga Cyanidioschyzon merolae. Mol. Biol. Cell 16, 2515–2527. doi: 10.1091/mbc.E13-04-0208
Fujiwara, T., Ohnuma, M., Yoshida, M., Kuroiwa, T., and Hirano, T. (2013b). Gene targeting in the red alga Cyanidioschyzon merolae: single- and multi-copy insertion using authentic and chimeric selection markers. PLoS ONE 8:e73608. doi: 10.1371/journal.pone.0073608
Gong, Y., Hu, H., Gao, Y., Xu, X., and Gao, H. (2011). Microalgae as platforms for production of recombinant proteins and valuable compounds: progress and prospects. J. Ind. Microbiol. Biotechnol. 38, 1879–1890. doi: 10.1007/s10295-011-1032-6
Imamura, S., Kanesaki, Y., Ohnuma, M., Inouye, T., Sekine, Y., Fujiwara, T., et al. (2008). R2R3-type MYB transcription factor, CmMYB1, is a central nitrogen assimilation regulator in Cyanidioschyzon merolae. Proc. Natl. Acad. Sci. U.S.A. 106, 12548–12553. doi: 10.1073/pnas.0902790106
Imamura, S., Terashita, M., Ohnuma, M., Maruyama, S., Minoda, A., Weber, A., et al. (2010). Nitrate assimilatory genes and their transcriptional regulation in a unicellular red alga Cyanidioschyzon merolae: genetic evidence for nitrite reduction by a sulfite reductase-like enzyme. Plant Cell Physiol. 51, 707–717. doi: 10.1093/pcp/pcq043
Imoto, Y., Kuroiwa, H., Yoshida, Y., Ohnuma, M., Fujiwara, T., Yoshida, M., et al. (2013). Single-membrane-bounded peroxisome division revealed by isolation of dynamin-based machinery. Proc. Natl. Acad. Sci. U.S.A. 110, 9583–9588. doi: 10.1073/pnas.1303483110
Kaufmann, A., and Knop, M. (2011). “Genomic promoter replacement cassettes to alter gene expression in the yeast Saccharomyces cerevisiae,” in Methods in Molecular Biology, Vol. 765, ed. J. A. Williams (New York, NY: Springer Publishing), 275–294.
Kuroiwa, T., Kuroiwa, H., Sakai, A., Takahashi, H., Toda, K., and Itoh, R. (1998). The division apparatus of plastids and mitochondria. Int. Rev. Cytol. 181, 1–41. doi: 10.1016/S0074-7696(08)60415-5
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Liu, B., and Benning, C. (2013). Lipid metabolism in microalgae distinguishes itself. Curr. Opin. Biotechnol. 24, 300–309. doi: 10.1016/j.copbio.2012.08.008
Matsuzaki, M., Mitsumi, O., Shin-I, T., Maruyama, S., Takahara, M., Miyagishima, S. Y., et al. (2004). Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428, 653–657. doi: 10.1038/nature02398
Minoda, A., Sakagami, R., Yagisawa, F., Kuroiwa, T., and Tanaka, K. (2004). Improvement of culture conditions and evidence for nuclear transformation by homologous recombination in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol. 45, 667–671. doi: 10.1093/pcp/pch087
Misumi, O., Matsuzaki, M., Nozaki, H., Miyagishima, S. Y., Mori, T., Nishida, K., et al. (2005). Cyanidioschyzon merolae genome. A tool for facilitating comparable studies on organelle biogenesis in photosynthetic eukaryotes. Plant Physiol. 137, 567–585. doi: 10.1104/pp.104.053991
Miyagishima, S. Y., Fujiwara, T., Sumiya, N., Hirooka, S., Nakano, A., Kabeya, Y., et al. (2014). Translation-independent circadian control of the cell cycle in a unicellular photosynthetic eukaryote. Nat. Commun. 5, 3807. doi: 10.1038/ncomms4807
Miyagishima, S. Y., Itoh, R., Toda, K., Takahashi, H., Kuroiwa, H., and Kuroiwa, T. (1998). Visualization of the microbody division in Cyanidioschyzon merolae with the fluorochrome brilliant sulfoflavin. Protoplasma 201, 115–119. doi: 10.1007/BF01280718
Miyagishima, S. Y., Nishida, K., Mori, T., Matsuzaki, M., Higashiyama, T., Kuroiwa, H., et al. (2003). A plant-specific dynamin-related protein forms a ring at the chloroplast division site. Plant Cell 15, 655–665. doi: 10.1105/tpc.009373
Miyagishima, S. Y., Takahara, M., Mori, T., Kuroiwa, H., Higashiyama, T., and Kuroiwa, T. (2001). Plastid division is driven by a complex mechanism that involves differential transition of the bacterial and eukaryotic division rings. Plant Cell 13, 2257–2268. doi: 10.2307/3871506
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nishida, K., Takahara, M., Miyagishima, S. Y., Kuroiwa, H., Matsuzaki, M., and Kuroiwa, T. (2003). Dynamic recruitment of dynamin for final mitochondrial severance in a primitive red alga. Proc. Natl. Acad. Sci. U.S.A. 100, 2146–2151. doi: 10.1073/pnas.0436886100
Nishida, K., Yagisawa, F., Kuroiwa, H., Yoshida, Y., and Kuroiwa, T. (2007). WD40 protein Mda1 is purified with Dnm1 and forms a dividing ring for mitochondria before Dnm1 in Cyanidioschyzon merolae. Proc. Natl. Acad. Sci. U.S.A. 104, 4736–4741. doi: 10.1073/pnas.0609364104
Nozaki, H., Takano, H., Misumi, O., Terasawa, K., Matsuzaki, M., Maruyama, S., et al. (2007). A 100%-complete sequence reveals unusually simple genomic features in the hotspring red alga Cyanidioschyzon merolae. BMC Biol. 5:28. doi: 10.1186/1741-7007-5-28
Ohmori, M., Ohmori, K., and Strotmann, H. (1977). Inhibition of nitrate uptake by ammonia in a blue-green alga, Anabaena cylindrica. Arch. Microbiol. 114, 225–229. doi: 10.1007/BF00446866
Ohnuma, M., Misumi, O., Fujiwara, T., Watanabe, S., Tanaka, K., and Kuroiwa, T. (2009). Transient gene suppression in a red alga, Cyanidioschyzon merolae 10D. Protoplasma 236, 107–112. doi: 10.1007/s00709-009-0056-5
Ohnuma, M., Yokoyama, T., Inouye, T., Sekine, Y., Kuroiwa, T., and Tanaka, K. (2014). Optimization of polyethylene glycol (PEG)-mediated DNA introduction conditions for transient gene expression in the unicellular red alga Cyanidioschyzon merolae. J. Gen. Appl. Microbiol. 60, 156–159. doi: 10.2323/jgam.60.156
Ohnuma, M., Yokoyama, T., Inouye, T., Sekine, Y., and Tanaka, K. (2008). Polyethylene glycol (PEG)-mediated transient gene expression in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol. 49, 117–120. doi: 10.1093/pcp/pcm157
Ohta, N., Matsuzaki, M., Misumi, O., Miyagishima, S. Y., Nozaki, H., Tanaka, K., et al. (2003). Complete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae. DNA Res. 10, 67–77. doi: 10.1093/dnares/10.2.67
Ohta, N., Sato, N., and Kuroiwa, T. (1998). Structure and organization of the mitochondrial genome of the unicellular red alga Cyanidioschyzon merolae deduced from the complete nucleotide sequence. Nucleic Acids Res. 26, 5190–5198. doi: 10.1093/nar/26.22.5190
Risso, D., Schwartz, K., Sherlock, G., and Dudoit, S. (2011). GC-content normalization for RNA-Seq data. BMC Bioinformatics 12:480. doi: 10.1186/1471-2105-12-480
Rosales-Mendoza, S., Paz-Maldonado, L., and Soria-Guerra, R. (2012). Chlamydomonas reinhardtii as a viable platform for the production of recombinant proteins: current status and perspectives. Plant Cell Rep. 31, 479–494. doi: 10.1007/s00299-011-1186-8
Sumiya, N., Fujiwara, T., Kobayashi, Y., Misumi, O., and Miyagishima, S. Y. (2014). Development of a heat-shock inducible gene expression system in the red alga Cyanidioschyzon merolae. PLoS ONE 9:e111261. doi: 10.1371/journal.pone.0111261
Suzuki, K., Ehara, T., Osafune, T., Kuroiwa, H., Kawano, S., and Kuroiwa, T. (1994). Behavior of mitochondria, chloroplasts and their nuclei during the mitotic cycle in the ultramicroalga Cyanidioschyzon merolae. Eur. J. Cell Biol. 63, 280–288.
ter Schure, E. G., van Riel, N. A., and Verrips, C. T. (2000). The role of ammonia metabolism in nitrogen catabolite repression in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 24, 67–83. doi: 10.1016/S0168-6445(99)00030-3
Warner, R. L., and Kleinhofs, A. (1992). Genetics and molecular biology of nitrate metabolism in higher plants. Physiol. Plant 85, 245–252. doi: 10.1034/j.1399-3054.1992.850217.x
Watanabe, S., Ohnuma, M., Sato, J., Yoshikawa, H., and Tanaka, K. (2011). Utility of a GFP reporter system in the red alga Cyanidioschyzon merolae. J. Gen. Appl. Microbiol. 57, 69–72. doi: 10.2323/jgam.57.69
Watanabe, S., Sato, J., Imamura, S., Ohnuma, M., Ohoba, Y., Chibazakura, T., et al. (2014). Stable expression of a GFP-reporter gene in the red alga Cyanidioschyzon merolae. Biosci. Biotechnol. Biochem. 78, 175–177. doi: 10.1080/09168451.2014.877823
Yagisawa, F., Fujiwara, T., Kuroiwa, H., Nishida, K., Imot, Y., and Kuroiwa, T. (2012). Mitotic inheritance of endoplasmic reticulum in the primitive red alga Cyanidioschyzon merolae. Protoplasma 249, 1129–1135. doi: 10.1007/s00709-011-0359-1
Yagisawa, F., Fujiwara, T., Ohnuma, M., Kuroiwa, H., Nishida, K., Imoto, Y., et al. (2013). Golgi inheritance in the primitive red alga, Cyanidioschyzon merolae. Protoplasma 250, 943–948. doi: 10.1007/s00709-012-0467-6
Yagisawa, F., Nishida, K., Kuroiwa, H., Nagata, T., and Kuroiwa, T. (2007). Identification and mitotic partitioning strategies of vacuoles in the unicellular red alga Cyanidioschyzon merolae. Planta 226, 1017–1022. doi: 10.1007/s00425-007-0550-y
Yagisawa, F., Nishida, K., Yoshida, M., Ohnuma, M., Shimada, T., Fujiwara, T., et al. (2009). Identification of novel proteins in isolated polyphosphate vacuoles in the primitive red alga Cyanidioschyzon merolae. Plant J. 60, 882–893. doi: 10.1111/j.1365-313X.2009.04008.x
Yoshida, Y., Kuroiwa, H., Hirooka, S., Fujiwara, T., Ohnuma, M., Yoshida, M., et al. (2009). The bacterial ZapA-like protein ZED is required for mitochondrial division. Curr. Biol. 19, 1491–1497. doi: 10.1016/j.cub.2009.07.035
Yoshida, Y., Kuroiwa, H., Misumi, O., Yoshida, M., Ohnuma, M., Fujiwara, T., et al. (2010). Chloroplasts divide by contraction of a bundle of nanofilaments consisting of polyglucan. Science 329, 949–953. doi: 10.1126/science.1190791
Keywords: algae, plants, homologous recombination, conditional knockdown, gene inducible system
Citation: Fujiwara T, Kanesaki Y, Hirooka S, Era A, Sumiya N, Yoshikawa H, Tanaka K and Miyagishima S-Y (2015) A nitrogen source-dependent inducible and repressible gene expression system in the red alga Cyanidioschyzon merolae. Front. Plant Sci. 6:657. doi: 10.3389/fpls.2015.00657
Received: 25 May 2015; Accepted: 10 August 2015;
Published: 26 August 2015.
Edited by:
John Love, University of Exeter, UKCopyright © 2015 Fujiwara, Kanesaki, Hirooka, Era, Sumiya, Yoshikawa, Tanaka and Miyagishima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takayuki Fujiwara and Shin-Ya Miyagishima, Department of Cell Genetics, National Institute of Genetics, 1111 Yata, Mishima 411-8540, Shizuoka, Japan,dGtmdWppd2FyYUBuaWcuYWMuanA=;c21peWFnaXNAbmlnLmFjLmpw