 Haibin Wang1,2
Haibin Wang1,2 Xiangyu Qi1Sumei Chen1Weimin Fang1Zhiyong Guan1Nianjun Teng1Yuan Liao1Jiafu Jiang1*Fadi Chen1,2*
Xiangyu Qi1Sumei Chen1Weimin Fang1Zhiyong Guan1Nianjun Teng1Yuan Liao1Jiafu Jiang1*Fadi Chen1,2*- 1College of Horticulture, Nanjing Agricultural University, Nanjing, China
- 2Jiangsu Province Engineering Lab for Modern Facility Agriculture Technology and Equipment, Nanjing, China
Polyploidy has been recognized as a widespread and common phenomenon among flowering plants. DNA-5′-CCGG site cytosine methylation (C-methylation) is one of the major and immediate epigenetic responses of the plant genome. Elucidating the ways in which altered C-methylation patterns, either at the whole genomic level or at specific sites can affect genome stability in polyploidy will require substantial additional investigation. Methylation sensitive amplification polymorphism profiling was used to evaluate variation in C-methylation among a set of 20 Chrysanthemum species and their close relatives of varying ploidy levels from diploid to decaploid. The range in relative C-methylation level was within 10%, and there was no significant difference neither between different ploidy levels nor between different species in the same ploidy level (U-values < 1.96). The transcript abundances of MET1 and DDM1 genes, which both involved in the regulation of C-methylation at CpG sites, were enhanced with increased ploidy level, but only MET1 was positively correlated with the nuclear DNA content. Considering the key role and efficiency of MET1 in maintaining CpG methylation, the limited variation observed with respect to C-methylation may reflect a balance between the increased activity of MET1 in the higher ploidy genomes and the larger number of CpG dinucleotide sites available for methylation.
Introduction
Polyploidy is widely recognized as a significant driver of higher plant evolution (Chen, 2007). The majority of extant angiosperm species have undergone at least one whole genome duplication (WGD) event to form either an auto- or an allopolyploid (Jiao et al., 2011). Polyploidization induces changes in both the genome sequence and the transcriptome. At least some of the latter are induced by alterations in cytosine (C-) methylation (DNA-5′-CCGG sites; Salmon et al., 2005; Parisod et al., 2010), a process which underlies much of the epigenetic variation in eukaryotic genomes. Actively transcribed sequences tend to be less heavily methylated than non-active ones, especially in their promoter region (Chan et al., 2005). The phenotypic consequences of altered C-methylation patterns can also give rise to evolutionary opportunities (Rangwala and Richards, 2004; Rapp and Wendel, 2005), particularly in the context of polyploidization events (Chen and Ni, 2006; Chen, 2007).
C-methylation in higher plant genomes is concentrated within CpG dinucleotides (Lister et al., 2008), and is strongly influenced by the activity of MET1, a gene encoding a DNA cytosine-5-methyltransferase, since the suppression of this gene results in a reduction in global C-methylation, particularly at CpG sites (Saze et al., 2003). A second important gene in this context is DDM1 (decrease in DNA methylation), which encodes a likely SNF2/SWI2 class chromatin remodeling protein (Jeddeloh et al., 1999). The functions of DDM1 and MET1 are both involved in the regulation of C-methylation at CpG sites (Wang et al., 2004).
The direct detection of C-methylation requires a modified form of sequencing (Martienssen and Colot, 2001; Riddle and Richards, 2002; Leitch and Leitch, 2008; Koh et al., 2010), but an indirect and more convenient means is provided by the MSAP (methylation sensitive amplification polymorphism) technique, which exploits the differential sensitivity shown by certain pairs of isoschizomeric restriction enzymes (REs) to C-methylation in their recognition site (DNA-5′-CCGG). MSAP based on the enzyme pair HpaII and MspI discriminates between hemi- (mCCGG) and fully methylated (CmCGG) sites (McClelland et al., 1994; Sha et al., 2005), and has been used to explore the variation in DNA methylation in a number of plant species (Zhao et al., 2007; Wang et al., 2009, 2014b), as well as to demonstrate C-methylation induced by polyploidization (Liu et al., 2001; Shaked et al., 2001; Qi et al., 2010).
The Asteraceae genus Chrysanthemum includes several polyploid species (Liu et al., 2012; Wang et al., 2013b, 2014c). Considerable variation at the ploidy level is present in this genus (from 2n = 2x = 18, to 2n = 36, 54, 72, up to 90; Liu et al., 2012). In previous studies, we investigated the genomic and epigenomic alterations during intergeneric hybridization in Chrysanthemum sp. using MSAP method. Surprisingly, in genus Chrysanthemum, the global DNA methylation concentration in the diploids (51.9–53.1%) was not much lower than that in the decaploid (55.4%; Wang et al., 2013a, 2014a,b). The aim of the present study was to characterize species-to-species variation for C-methylation in this genus, based on MSAP profiling. In addition, given the key role of MET1 and DDM1 in maintaining CpG methylation (Chen et al., 2010), an analysis of their transcript abundance was undertaken in Chrysanthemum species varying in ploidy level from diploid to decaploid.
Materials and Methods
Plant Material and DNA Extraction
The plants sampled for DNA analysis have been maintained by vegetative reproduction for at least 8 years under a constant environment (22°C during the day, a minimum at 15°C at night, a relative humidity of 70–75% and under natural light) at the Chrysanthemum Germplasm Resource Preserving Centre, Nanjing Agricultural University, China. The diploids selected were Chrysanthemum nankingense, C. dichrum, C. japonicum, C. boreale, C. lavandulifolium, and Tanacetum vulgare; the tetraploids were C. indicum, C. yoshinaganthum, C. okiense, C. japonicum var. wakasaense and C. chanetii; the hexaploids were C. vestitum, C. morifolium, C. japonense, and C. zawadskii; the octoploids were C. ornatum, Ajania shiwogiku, and A. × marginatum; and the decaploids were C. crassum and A. pacificum (Table 1). Their DNA was extracted from fully expanded fourth and fifth leaves harvested from three plants per species, using a modified CTAB method (Stewart and Via, 1993). DNA integrity was confirmed by running a 2% agarose gel. The concentration and purity of the DNA preparations were monitored using the Nano-Drop ND-1000 Spectrophotometer (Nano-Drop Technologies, Wilmington, DE, USA), and preparations were stored at -20°C for subsequent analysis.

TABLE 1. Taxa used as plant materials and their ploidy level.
MSAP Profiling
The interpretation of MSAP data is predominantly based on known RE activities at recognition sequences modified by methylation. Data concerning the methylation sensitivity of REs and corresponding literature can be found at the website of The Restriction Enzyme Database (REBASE1) (Fulneček and Kovařík, 2014). The MSAP profiling procedure was based on the protocol given in (Reyna-Lopez et al., 1997; Xiong et al., 1999). About 500 ng DNA per entry was double-digested at 37°C for 12 h in parallel with either 10 U EcoRI (New England Biolabs, China, EC 3.1.23.13) and 20 U HpaII (NEB, EC 3.1.23.24), or 10 U EcoRI, and 10 U MspI (NEB, EC 3.1.23.24). The products were ligated with 5 pmol EcoRI adaptor and 50 pmol HpaII-MspI adaptor (sequences given in Supplementary Table S1) in a reaction containing 4 U T4 DNA ligase held at 16°C for 4 h, after which the ligation reaction was heat-inactivated (65°C, 10 min). A 5 μL aliquot of the product was pre-amplified in the presence of 0.2 μM EcoRI and 0.2 μM HpaII-MspI non-selective primers (sequences given in Supplementary Table S1) in a 25 μL reaction containing 2.5 μL 10x PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTP, and 2 U Taq polymerase (Takara, Japan). The reactions were first denatured (94°C/3 min), then subjected to 24 cycles of 94°C/30 s, 56°C/60 s, 72°C/60 s, after which a final extension step (72°C/10 min) was given (Xiong et al., 1999). The pre-amplification product was diluted 1:29 in ddH2O to provide the template for the subsequent selective PCR, which included a fluorescently-labeled EcoRI primer and a non-labeled HpaII-MspI selective (three selective bases) primer. Primer sequences are given in Supplementary Table S1. The set of primer combinations (PCs) used were EcoRI selective primer #2 combined with HpaII/MspI selective primer #5 (abbreviated E2 + HM5), E2 + HM7, E3 + HM3, E4 + HM3, E4 + HM7, E4 + HM8, E6 + HM6, E6 + HM8, E7 + HM1, E7 + HM3, E8 + HM2, and E8 + HM8, and the remaining constituents of the reaction were identical to those used for the pre-amplification reaction. The reactions were first denatured (95°C/3 min), then subjected to 13 cycles of 94°C/30 s, 65°C/30 s (reduced by 0.7°C per cycle per cycles), 72°C/30 s, then to 25 cycles of 94°C/30 s, 55°C/30 s, 72°C/ 30 s, and finished by a final extension step of 72°C/7 min. The reaction products were denaturized by heating at 98°C for 3 min followed by cooling on ice and separated using an ABI3730xl (Applied Biosystems, Foster City, CA, USA) device, following the manufacturer’s instructions. Individual fragment sizes were estimated from the migration of the GeneScan LIZ500 size standards, as determined by GeneMapper® v3.7 software (Applied Biosystems). Fragments in the size range 120–480 bp were scored. Two replicate reactions per entry were run, and only reproducible fragments were retained. The statistical test used to interpret variation in MSAP fingerprint followed the suggestion made in (Wang et al., 2013a, 2014a,b):
Where, n1 represents the total sites for a given sample; n2 represents the total sites the mid-values; y1 represents the total DNA methylation sites, hemimethylation sites or fully methylation sites for a given sample, y2 represents the total DNA methylation sites, hemimethylation sites, or fully methylation sites of the mid-values. p1 is the percentage of total methylation sites, hemimethylation sites or fully methylation sites for a given sample; p2 is the percentage of total methylation sites, hemimethylation sites or fully methylation sites of the mid-values. The Pearson’s R2 coefficient between the ploidy level and relative C-methylation were evaluated using SPSS v19 software.
The Isolation of CnMET1 and CnDDM1 and Measurement of Transcript Abundance
Total RNA Isolation System (Takara) was used to isolate RNA from fully expanded C. nankingense leaves, following the manufacturer’s instructions. A 1 μL aliquot of the resulting RNA (containing about 600 ng) provided the template for the synthesis of the first cDNA strand by SuperScriptIII Reverse Transcriptase (Takara) with random hexamer primers. The subsequent PCR used two degenerate primer pairs (DP1/DP2 and DP3/DP4: sequences given in Supplementary Table S2) which targeted MET1, designed from an alignment of the MET polypeptides of Medicago truncatula (XP_003619753.1), Hieracium pilosella (ACX83570.1), Nicotiana tabacum (BAF36443.1), Prunus persica (AAM96952.1), Elaeis guineensis (ABW96888.1), and Arabidopsis thaliana (NP_199727.1). To obtain the full length cDNA prior to a RACE PCR, the sequences were first validated by amplification with a pair of gene-specific primers (SP-F/SP-R: sequences given in Supplementary Table S2; Supplementary Figure S1). For the 3′ RACE, the first cDNA strand was synthesized using an oligo (dT) primer incorporating the sequence of the adaptor primer, followed by a nested PCR using the gene-specific primer pair GSP3-1/3-2/3-3 and the adaptor primer (sequences given in Supplementary Table S2). For the 5′ RACE, the nested PCR used the 5′ RACE adaptor primer (Abridged Anchor Primer, AAP), the Abridged Universal Amplification Primer (AUAP) provided with the 5′ RACE System kit v2.0 (Takara) and the internal gene-specific primer pair (GSP5-1/5-2/5-3, sequences given in Supplementary Table S2). The gene’s open reading frame (ORF) was identified using www.ncbi.nlm.nih.gov/gorf/gorf.html and amplified using primers Full-F/Full-R (sequences given in Supplementary Table S2). A multiple sequence alignment of the predicted gene product with homologs present in M. truncatula, H. pilosella, N. tabacum, P. persica, E. guineensis, A. thaliana, Daucus carota (AAC39355.1), H. piloselloides (ACX83569.1), N. sylvestris (CAQ18900.1), Oryza sativa (BAG15930.1), Pisum sativum (AAC49931.1), Populus trichocarpa (XP_002305346.1), Ricinus communis (XP_002518029.1), Solanum lycopersicum (NP_001234748.1), and Zea mays (NP_001105186.1) was carried out using DNAMAN software v5.2.2 (Lynnon Biosoft, Canada), and a subsequent phylogenetic analysis was carried out using MEGA 5.0 software2. The same strategy was applied for the isolation of CnDDM1 (primer sequences given in Supplementary Table S2).
Transcription profiling of MET1 and DDM1 was based on RNA extracted from fully expanded fourth and the fifth leaves of C. nankingense (2x), C. indicum (4x), C. morifolium (6x), C. ornatum (8x), and C. crassum (10x) which are species that were intensively investigated in the previous studies. Prior to its reverse transcription, 30 ng RNA was treated with 10 U of RNase-free DNaseI (Takara) at 37°C for 30 min to remove any contaminating genomic DNA. The first cDNA strand was synthesized by SuperScriptIII Reverse Transcriptase (Takara), according to the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed using SYBR Premix Ex TaqTMII (Takara). The reactions were first denatured (95°C/2 min), then subjected to 40 cycles of 95°C/30 s, 55°C/30 s, 72°C/30 s. A previous study suggests that when making cross-species comparison of transcript abundance involving different ploidy levels, care needs to be taken in the selection of reference gene(s) (Wang et al., 2014d), especially in Chrysanthemum sp. (Wang et al., 2015). Here, EF1α (GenBank accession KF305681), TUB (KF305685), ACTIN (KF305683), and PP2A (KF305684) were used as candidate reference genes (Primer sequences were listed in Supplementary Table S2). The selection of a reference gene was based on geNORM (Vandesompele et al., 2002) and NormFinder (Andersen et al., 2004) Software. The former calculates a stability value (M) for each gene, with a lowest M-value being taken as an indicator of stable transcription. The latter provides a direct measure of the variation using an ANOVA-based model and ranks the candidate genes accordingly. The data were shown as mean ± SE (n = three biological replicates). Each qRT-PCR amplicon was cloned using a PMD19 TA cloning kit (Takara) and sequenced for verification.
Flow Cytometric Acquisition of Nuclear DNA Content
Flow cytometry was used to determine the relative nuclear DNA contents of C. nankingense, C. indicum, C. morifolium, C. ornatum, and C. crassum. A 200 mg sample of fresh leaf material (three biological replicates per species) was macerated in 2 mL 15 mM Tris-HCl, pH 7.5, 80 mM KCl, 20 mM NaCl, 20 mM Na2EDTA, 2% (v/v) β-mercaptoethanol, 0.05% (v/v) Triton X-100 (Liu et al., 2011). The homogenate was passed through a 50 μm nylon mesh and centrifuged (1,500 × g, 10 min, 4°C). Prior to flow cytometry, a 200 μL aliquot of 3 U μL-1 RNAase, 50 μg mL-1 presidiums iodide was added, and the reaction was kept in the dark at 4°C for 30 min. Flow cytometry was affected with a Coulter EpicsxL device (Beckman Coulter, Miami, FL, USA). Nuclear DNA content was estimated with respect to that of the reference species C. nankingense. Each measurement was based on the mean of three technical replicates and only those associated with a coefficient of variation <5% were accepted.
Results
MSAP Fingerprinting
The methylation status of each fragment (Figures 1A,B, Supplementary Data S1) and the proportion of methylated fragments present in the 20 species is presented in Table 2. A. pacificum (10x) had the highest proportion of methylated sites (59.2%), followed by C. ornatum (8x, 57.1%), A. shiwogiku (8x, 56.7%), A. × marginatum (8x, 56.6%) and C. japonense (6x, 55.6%). The lowest proportions were present in C. lavandulifolium (2x, 49.8%), T. vulgare (2x, 51.9%) and C. dichrum (2x, 52.2%). The range in relative C-methylation level was within 10%.
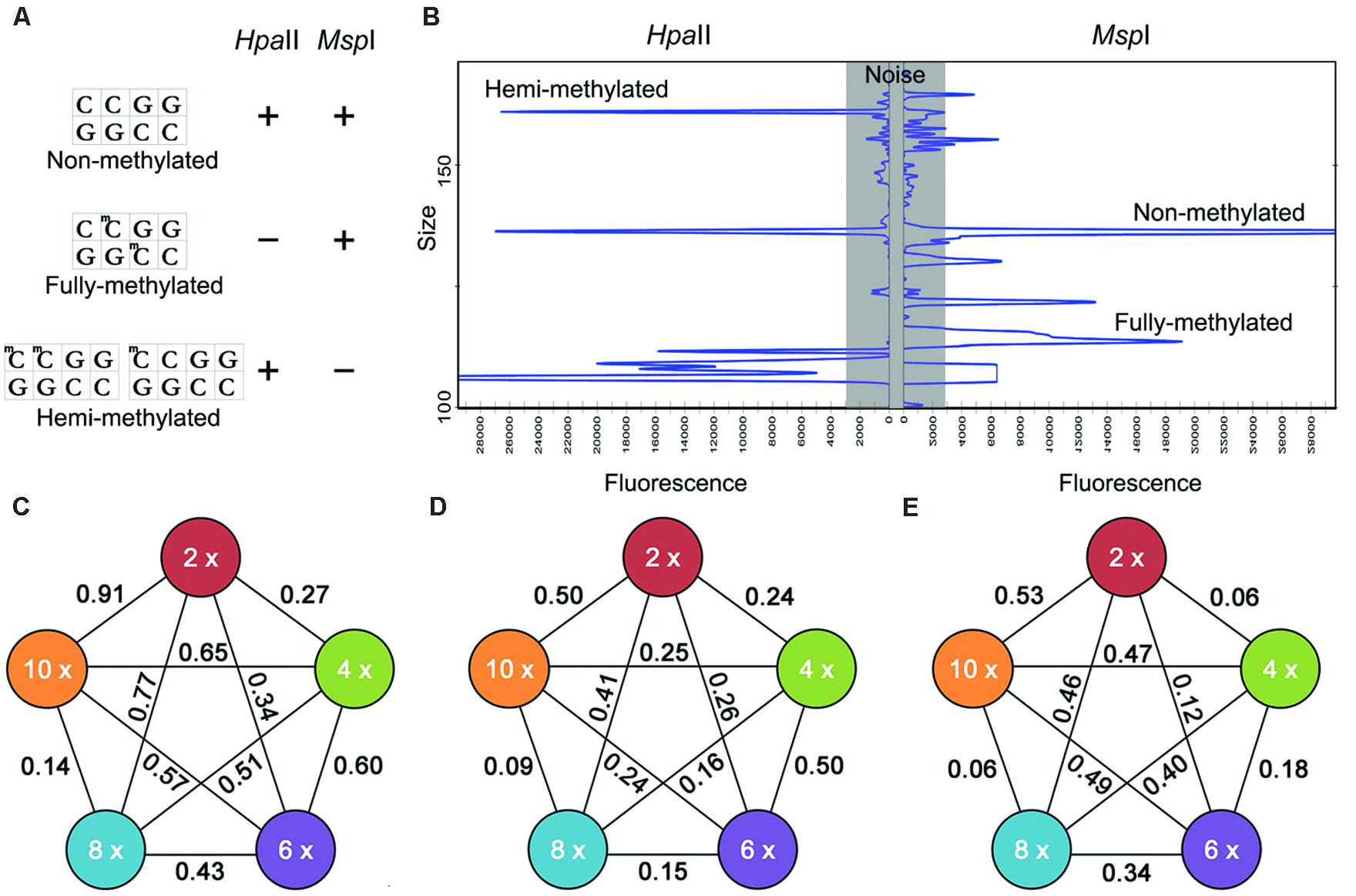
FIGURE 1. Representative fragments in methylation sensitive amplification polymorphism (MSAP) profiles (A,B) and the relationship between U-values and ploidy level (C–E). (A) Hpa II and Msp I sensitivities to 5-CCGG methylation status (“+”: enzyme cuts; “-”: enzyme does not cut): three types of fragment generated. Type I: non-methylated, appearing in both the H and M lanes; Type II: fully methylated, present in the M but not the H lanes; Type III: hemi-methylated, present in the H but not the M lanes; (B) Examples of MSAP electrophoresis patterns in ABI3730xl; The relationship between U-values and ploidy level (C-E). U-values associated with (C) total methylation, (D) full methylation, (E) hemi-methylation. A higher U-value implies a larger difference between different samples, but only U-values>1.96 were statistically significant.
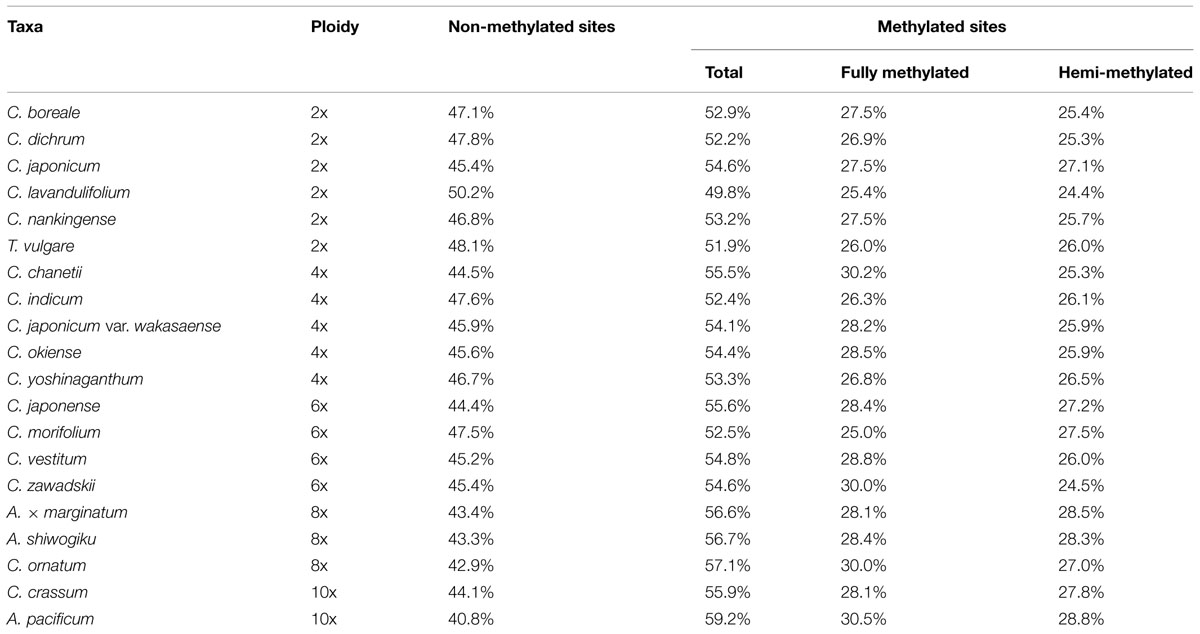
TABLE 2. Variation in C-methylation status among the 20 species.
Variation in Cytosine Methylation within a Given Ploidy Level
Based on the MSAP profiles, the numbers of non-methylated, hemi-methylated, and fully methylated CCGG sites derived from the MSAP profiles were used to calculate the relative methylation level of the six diploid species; this lay in the range 49.8–54.6% (U = 0.11–1.11, U0.05 = 1.96, a higher U-value implies a larger difference between different samples, but only U-values >1.96 were statistically significant), broken down into 25.4–27.5% (U = 0.05–0.68) fully methylated internal cytosines and 24.4–27.1% (U = 0.02–0.69) hemi-methylated external ones. None of the individual values departed statistically from the mid-value of the set of diploid species (Tables 2 and 3).
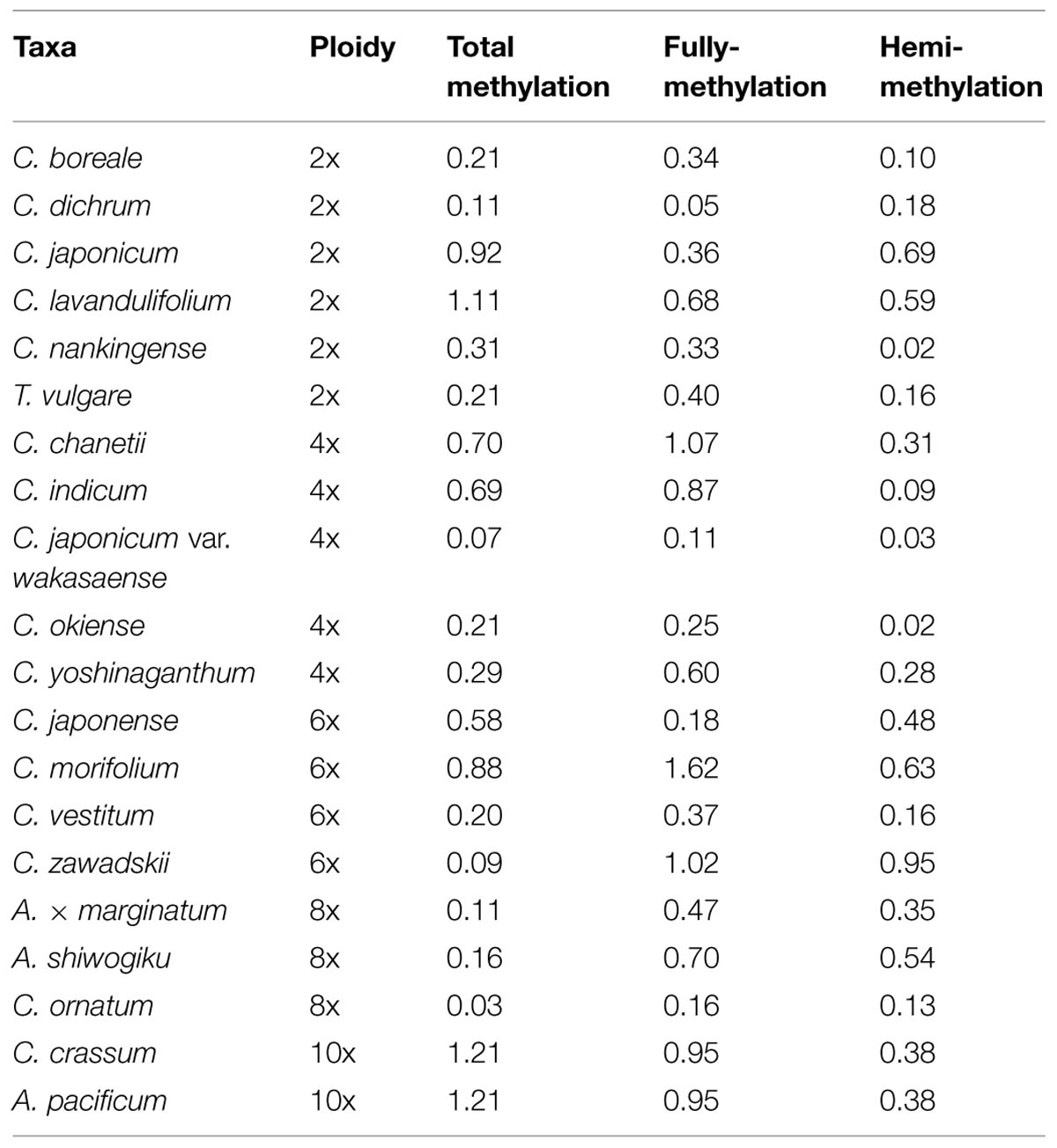
TABLE 3. U-values associated with species within a ploidy level.
For the five tetraploid entries, the proportion of methylated fragments lay between 52.4 and 55.5%. C. indicum had the lowest proportion of both total methylated and fully methylated sites, while the lowest proportion of hemi-methylated sites was present in C. chanetii. The U-values ranged from 0.07 to 0.70 (total methylation), 0.11 to 1.07 (full methylation), and 0.02 to 0.31 (hemi-methylation), and none of the individual values differed significantly from one another (Tables 2 and 3).
Among the higher ploidy entries (6–10x), the maximum U-values were 1.21 (total methylation), 1.62 (full methylation), and 0.95 (hemi-methylation); these values did not differ significantly from one another (Tables 2 and 3). In each species, the hemi-methylated sites were less variable than the fully methylated ones, and the U-values associated with the fully methylated sites were greater than those of the hemi-methylated ones in 16 of the 20 species, however, none of these differences were statistically significant (Table 3).
Variation in Cytosine Methylation Status Across Ploidy Levels
Correlation analysis yielded statistically correlations (R2 = 0.27–0.65) between the ploidy level and relative C-methylation. A highest correlation coefficient was obtained in total methylation, while the lowest R2 was obtained in fully methylation. However, with respect to total methylation level, the U-values ranged from 0.14 to 0.91, with the lowest values being associated with the 8 and 10x entries, and the highest with the 2 and 10x entries (Figure 1C). With respect to the fully methylated sites, the U-values ranged from 0.09 to 0.50, with the lowest associated with the 8 and 10x species and the highest distributed among the 2, 4, 6, and 10x ones (Figure 1D). For the hemi-methylated sites, the range in U-value was 0.06–0.53, with the lowest recorded in the 8 and 10x species and the highest in the 2, 4, and 10x species (Figure 1E). Overall therefore there was no significant difference between ploidy level, either total, full or hemi-methylation levels.
Isolation of CnMET1 and CnDDM1 Gene and Analysis of its Transcription
The full length CnMET1 (Genbank accession KF305682) cDNA was a 5,124 nt sequence, comprising a 4,803 nt ORF, a 92 nt 5′-UTR and a 229 nt 3′-UTR. The sequence showed significant homology to other plant MET genes. At the peptide level, the mean level of identity was 71.8%, reaching >80% in the most conserved regions (data not shown). A phylogenetic analysis showed that the most closely related sequences to CnMET1 were the homologs from H. pilosella and H. piloselloides (both Asteraceae species); in the conserved regions of the gene, the level of sequence identity was >90% (Figure 2). The CnDDM1 (Genbank KJ560359) sequence encodes a 752 residue product, sharing substantial homology with other plant DDM genes (data not shown). Its most closely related sequences were its homologs from Brassica rapa and A. thaliana (Figure 2) and the CnMET1/AtMET1 and CnDDM1/AtMET1 motifs lie parallel to one another and in the most conserved regions. The MET1 cytosine-C5 specific DNA methylase domain and the DDM1 N-terminal domain had an almost identical three-dimensional structure (Data not shown).
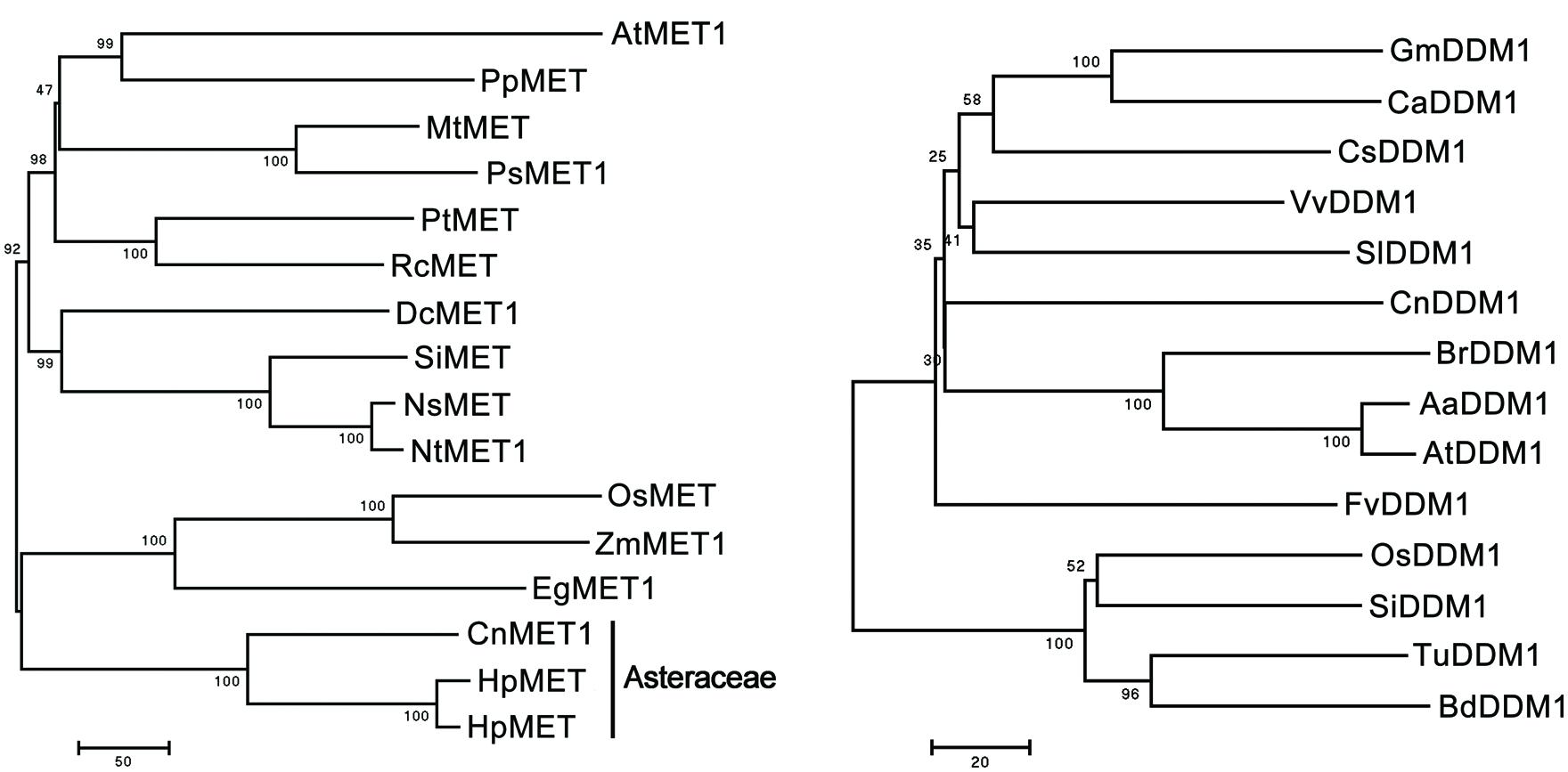
FIGURE 2. Phylogeny of MET and DDM1 proteins. Arabidopsis thaliana AtMET1 (NP_199727.1), Prunus persica PpMET (AAM96952.1), Medicago truncatula MtMET (XP_003619753.1), Pisum sativum PsMET (AAC49931.1), Populus trichocarpa PtMET (XP_002305346.1), Ricinus communis RcMET (XP_002518029.1), Daucus carota DcMET1 (AAC39355.1), Solanum lycopersicum SlMET (NP_001234748.1), Nicotiana sylvestris NsMET (CAQ18900.1), N. tabacum NtMET1 (BAF36443.1), Oryza sativa OsMET (BAG15930.1), Zea mays ZmMET1 (NP_001105186.1), Elaeis guineensis EgMET1 (ABW96888.1), Hieracium pilosella HpMET (ACX83570.1), H. piloselloides HpMET (ACX83569.1), Vitis vinifera VvDDM1 (XP_002267239.2), Glycine max GmDDM1(XP_003516571.1), S. lycopersicum SlDDM1 (XP_004232396.1), Crocus sativus CsDDM1 (XP_004149166.1), B. rapa BrDDM1 (BAG30707.1), Cicer arietinum CaDDM1 (XP_004512037.1), Fragaria vesca FvDDM1 (XP_004289144.1), O. sativa OsDDM1 (BAF34942.1), Setaria italica SiDDM1 (XP_004981943.1), Triticum urartu TuDDM1 (EMS59856.1), Brachypodium distachyon BdDDM1 (XP_003560489.1), Arabidopsis arenosa AaDDM1 (AAP92713.1), A. thaliana AtDDM1 (NP_201476.1).
Relative to the nuclear DNA content of C. nankingense, that of C. indicum (4x) was 1.94-fold greater, that of C. morifolium (6x) 2.91-fold greater, that of C. ornatum (8x) 3.25-fold greater and that of C. crassum (10x) 4.25 greater (Figure 3A). The relationship between relative nuclear DNA content and ploidy level was not completely linear. This finding is not surprising given the accumulated data across angiosperms which indicate genome down-sizing (due to equilibration or/and deletion of DNA) following hybridization or/and polyploidization.
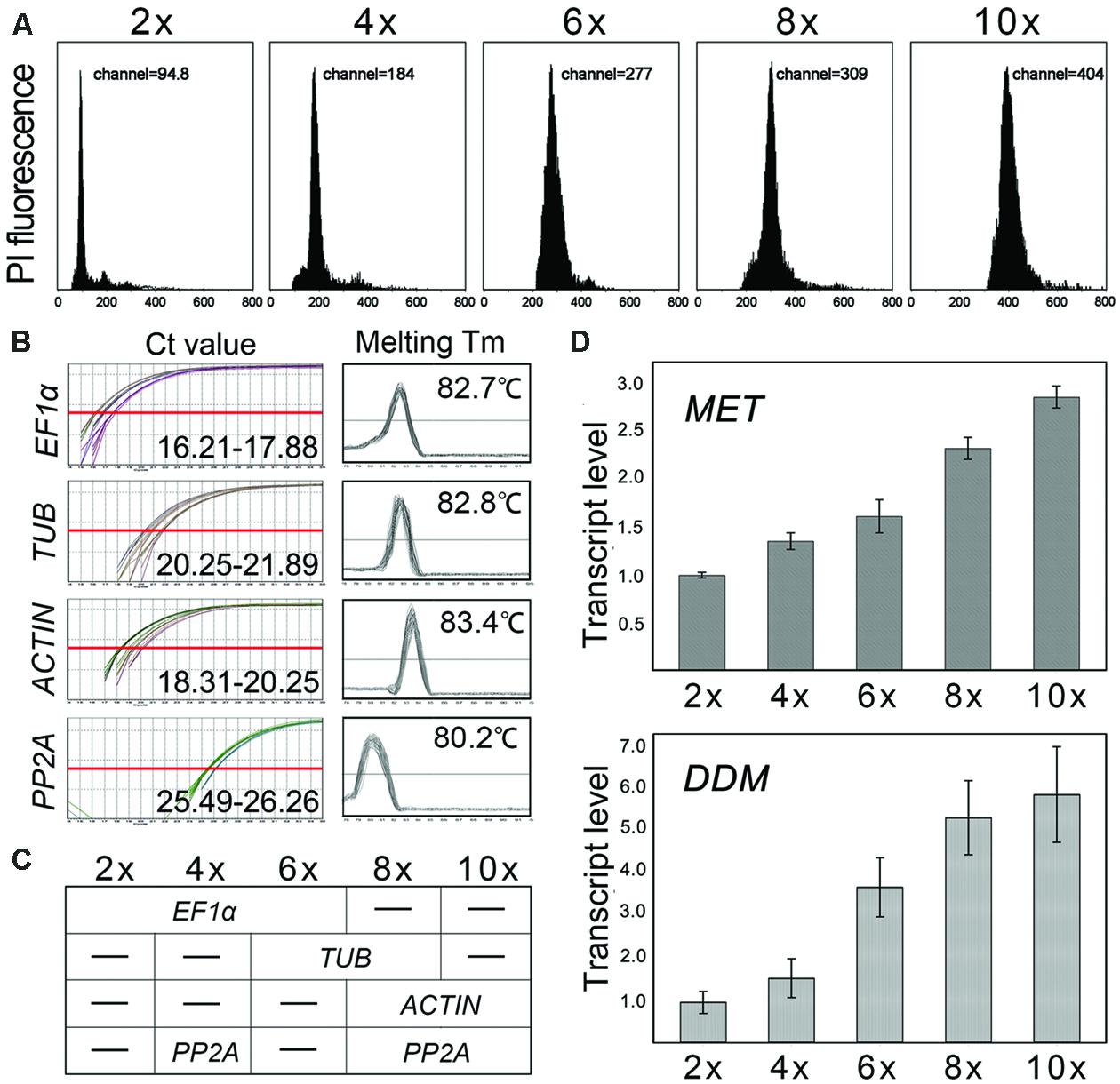
FIGURE 3. Nuclear DNA content obtained by flow cytometry and MET1/DDM1 transcription analysis. (A) Nuclear DNA content of Chrysanthemum nankingense (2x), C. indicum (4x), C. morifolium (6x), Ajania shiwogiku (8x) and C. crassum (10x); (B) The Ct value and melting temperature of each reference genes; the X axis represents the PCR cycle number. The red line represents the threshold fluorescence at which the Ct was determined. None of the reference genes were uniformly transcribed (Ct) in five ploidy levels and species. Melting curve analyses showed that each primer pair amplified a single PCR product. (C) Model of reference gene(s) used in five ploidy levels and species; (D) Transcript abundance was correlated with ploidy level for both MET1 and DDM1. PP2A was selected as inter-run calibrators (IRCs) for EF1α, TUB, and ACTIN, and the coefficient of variation (transcript abundance normalized using EF1α4x-6x-TUB6x-8x-ACTIN8x-10x vs. using PP2A4x-8x-10x) < 5%.
Cross species comparison of gene expression can be taken with the right reference genes (Wang et al., 2014d). Across all templates, EF-1a was the most abundantly transcribed gene, accompanied by the lowest Ct (16.21–17.88), followed by ACTIN (18.31–20.25) and TUB (20.25–21.89), while PP2A (25.49–26.26) was the least abundantly transcribed gene (Figure 3B). Melting curve analyses showed that each primer pair amplified a single PCR product (Figure 3B). However, none of the reference genes were uniformly transcribed in five ploidy levels and species. Therefore, it was necessary to evaluate the reference genes for normalization in the tested samples. Here, two algorithms, geNorm and NormFinder, were used to determine which of the reference genes would be most suitable in each group. The results shown EF1α/TUBULIN/ACTIN/PP2A were predicted to deliver the most reliable level of normalization for 2x vs. 4x vs. 6x/6x vs. 8x/8x vs. 10x/4x vs. 8x vs. 10x ploidy (Supplementary Figure S2; Supplementary Data S2), as the model shown in Figure 3C.
MET1 was transcribed in all five species, and all the amplicon sequences recovered after qRT-PCR shared the same sequence (data not shown). However, there were substantial inter-specific differences in transcript abundance, in general increasing with the ploidy level. Thus, MET1 transcript abundance in C. crassum (10x) was 1.23-fold that in C. ornatum (8x), 1.77-fold that in C. morifolium (6x), 2.11-fold that in C. indicum (4x) and 2.87-fold that in the C. nankingense (2x; Figure 3D). Similarly, for DDM1, the abundance of transcript in C. crassum, C. ornatum, C. morifolium, and C. indicum was, respectively, 6.19, 5.62, 3.88, and 1.60-fold that in C. nankingense (Figure 3D). The results showed that transcript abundance of two genes was increased with genome size, but only MET1 positively correlated with the nuclear DNA content (r = 0.765, P = 0.001), while DDM1 transcript abundance was not correlated (P > 0.005).
Discussion
The Asteraceae represent a relatively young family which has diversified substantially over the past 40 million years. At least three WGD events have occurred during the evolution of the family (Barker et al., 2008; Malinska et al., 2010), and many of its currently extant species are polyploid (Martin et al., 2011). The frequency of polyploidy in the Chrysanthemum genus suggests that these species still retain the potential to evolve rapidly (Yang et al., 2006; Liu et al., 2012). MSAP is based on the AFLP technology and can be used effectively to detect polymorphism in DNA methylation patterns within and among different species with no requirement for prior genome information other than the approximate genome size. We therefore consider MSAP to be a suitable technique to evaluate epigenetic changes at the level of DNA methylation in present studies (Wang et al., 2013a, 2014a,b).
The MSAP profiles of the various Chrysanthemum species and their close relatives have demonstrated numerous C-methylation polymorphisms. C-methylation in coding and promoter sequence can have a profound effect on a gene’s expression, so some of these epigenetic polymorphisms may have an impact on the phenotype of plant (Shaked et al., 2001; Adams and Wendel, 2005). C-methylation levels and patterns were variable between species sharing the same ploidy level, although the extent of this variation was not that great. Although the extent of C-methylation suggested there were little correlations with ploidy level (R2 = 0.27–0.65), the range in relative C-methylation level was within 10% and there was no significant difference between ploidy level, either total, full, or hemi-methylation levels (Table 2, Figure 1), for example, in the 10x species C. crassum, 55.9% of the MSAP fragments were methylated, while in the 8x species C. ornatum the proportion was 57.1%, in the 6x species C. morifolium 52.5%, in the 4x species C. indicum 52.4%, and in the 2x species C. japonicum 54.6%. A similar result has been reported in a number of other genera, which the characteristics of DNA methylation in ployploid may be not consistent with ploidy level (Fortune et al., 2007).
The MET sequence is highly conserved across the spectrum from plants to mammals. Its preferred target in plant genomes is the CpG dinucleotide (Lister et al., 2008). In A. thaliana at least, the MET1 enzyme acts to maintain the global level of C-methylation, since when suppressed via antisense technology, the global level of C-methylation, particularly at CpG sites, is strongly reduced, despite the presence of several other methyltransferase genes (Robertson and Jones, 2000; Steward et al., 2000; Kankel et al., 2003; Takeda and Paszkowski, 2006). Here, the sequence of the Chrysanthemum MET1 homolog is very similar to that of other MET genes, as would be expected if MET1 is a functional DNA methyltransferase (Figure 2). In contrast to the patterns of C-methylation in the Chrysanthemum polyploids, MET1 transcript abundance was positively correlated with genome size. MET1’s primary function is to control C-methylation, a form of DNA modification which is necessary to maintain the integrity and stability of the genome. The simplest and most probable explanation for the increased transcript abundance of MET1 in higher ploidy genomes, in which the level of C-methylation is not significantly different from that in low ploidy genomes, is that the quantity of DNA in a high ploidy genome (and therefore the number of CpG dinucleotides) is much higher. Notably, variability of MET1 transcript abundance was still in a lesser extent (max 2.87-fold). This might be due to MET1-mediated methylation was target-specific and has a high level of methylation efficiency on CG targets (Miki and Shimamoto, 2008; Meyer, 2011).
CpG-methylated sites are targeted by MET1 via its methyl-CpG binding domain, while DDM1 proteins interact with methyl-CpG binding domain proteins and affect their sub-nuclear localization (Zemach et al., 2005). The A. thaliana DDM1 mutation induces a reduction in DNA methylation level, and is stably inherited (Kakutani et al., 1999). In the present study, the transcript abundances of MET1 and DDM1 genes were all enhanced with increased ploidy level, however, only MET1 positively correlated with the nuclear DNA content, while DDM1 transcript abundance was not correlated. This can be explained by the difference in the DNA methylation efficiency between DDM1 and MET1. Because MET1 product is DNA methyltransferase, while DDM1 product functions as a modifier of DNA methyltransferases (Kakutani et al., 1995).
Conclusion
Inter-specific variability in C-methylation (DNA-5′-CCGG sites) within the same ploidy level in the Chrysanthemum genus is quite limited, and the nuclear DNA of higher ploidy species is also not necessarily more highly methylated than that of the low ploidy genomes. The transcript abundance of both MET1 and DDM1 was enhanced with increased ploidy level but only MET1 was positively correlated with the nuclear DNA content. Since the higher ploidy genomes harbor a larger number of CpG sites, the enhance of MET1 transcript abundance would likely have made no significant impact on the overall proportion of methylated sites.
Author contributions
Conceived and designed the experiments: HW, FC, NT, SC. Performed the experiments: HW, FC, ZG. Analyzed the data: HW, XQ. Contributed reagents/materials/analysis tools: WF, ZG. Wrote the paper: HW, SC. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (31272203, 31272196, 31372092, 31370699, 31372100), the Program for New Century Excellent Talents in University of Chinese Ministry of Education (NCET-12-0890), the Fund for Independent Innovation of Agricultural Sciences in Jiangsu Province [CX(12)2020], the Program for Hi-Tech Research, Jiangsu, China (BE2012350), and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00668
Figure S1 | The strategy for isolating CnMET1.
Figure S2 | Average expression stability (M) values of the reference genes, as calculated by geNorm, where a low M reflects stability and a high M instability, asterisk is the selected reference gene. (A) Reference gene selection in 2x vs. 4x/ 4x vs. 6x/6x vs. 8x/8x vs. 10x, EF1α, TUB, and ACTIN were selected; (B) As EF1α was predicted to deliver the most reliable level of normalization for 2x vs. 4x vs. 6x, PP2A has the lowest M-value in 4x vs. 8x vs. 10x ploidy, PP2A was selected as inter-run calibrators (IRCs) for normalizing EF1α, TUB, and ACTIN.
Data S1 | Binary allele calls for MSAP data.
Data S2 | Expression stability of the reference genes, as calculated by NormFinder.
Footnotes
References
Abd E-Twab, M. H., and Kondo, K. (2007). Rapid genome reshuffling induced by allopolyploidization in F1 hybrid in Chrysanthemum remotipinnum (formerly Ajania remotipinna) and Chrysanthemum chanetii (formerly Dendranthema chanetii). Chromosome Bot. 2, 1–9. doi: 10.3199/iscb.2.1
Abd El-Twab, M. H., and Kondo, K. (2006). Fluorescence in situ hydridization and genomic in situ hybridization to identify the parental genomes in the intergeneric hybrid between Chrysanthemum japonicum and Nipponanthemum nipponicum. Chromosome Bot. 1, 7–11. doi: 10.3199/iscb.1.7
Abd El-Twab, M. H., and Kondo, K. (2012). Genome mutation revealed by artificial hybridization between Chrysanthemum yoshinaganthum and Chrysanthemum vestitum assessed by FISH and GISH. J. Bot. 2012, 480310. doi: 10.1155/2012/480310
Adams, K. L., and Wendel, J. F. (2005). Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8, 135–141. doi: 10.1016/j.pbi.2005.01.001
Andersen, C. L., Jensen, J. L., and Ørntoft, T. F. (2004). Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250. doi: 10.1158/0008-5472.CAN-04-0496
Bao, D., Ji-Shuang, Y., and Xue, Y. (2012). Study on cytology of some species in Chrysanthemum. North. Hortic. 7, 049.
Barker, M. S., Kane, N. C., Matvienko, M., Kozik, A., Michelmore, R. W., Knapp, S. J., et al. (2008). Multiple paleopolyploidizations during the evolution of the Compositae reveal parallel patterns of duplicate gene retention after millions of years. Mol. Biol. Evol. 25, 2445–2455. doi: 10.1093/molbev/msn187
Chan, S. W., Henderson, I. R., and Jacobsen, S. E. (2005). Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 6, 351–360. doi: 10.1038/nrg1601
Chen, F. D., Zhao, H. B., Li, C., Chen, S. M., and Fang, W. M. (2008). Advances in cytology and molecular cytogenetics of the genus Dendranthema. J. Nanjing Agr. Univ. 31, 118–126.
Chen, P. Y., Cokus, S. J., and Pellegrini, M. (2010). BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics 11:203. doi: 10.1186/1471-2105-11-203
Chen, Z. J. (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58, 377–406. doi: 10.1146/annurev.arplant.58.032806.103835
Chen, Z. J., and Ni, Z. (2006). Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays 28, 240–252. doi: 10.1002/bies.20374
Fortune, P. M., Schierenbeck, K. A., Ainouche, A. K., Jacquemin, J., Wendel, J. F., and Ainouche, M. L. (2007). Evolutionary dynamics of Waxy and the origin of hexaploid Spartina species (Poaceae). Mol. Phylogenet. Evol. 43, 1040–1055. doi: 10.1016/j.ympev.2006.11.018
Fulneček, J., and Kovařík, A. (2014). How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet. 15:2. doi: 10.1186/1471-2156-15-2
Jeddeloh, J. A., Stokes, T. L., and Richards, E. J. (1999). Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet. 22, 94–97. doi: 10.1038/8803
Jiao, Y., Wickett, N. J., Ayyampalayam, S., Chanderbali, A. S., Landherr, L., Ralph, P. E., et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100. doi: 10.1038/nature09916
Kakutani, T., Jeddeloh, J. A., and Richards, E. J. (1995). Characterization of an Arabidopsis thaliana DNA hypomethylation mutant. Nucleic Acids Res. 23, 130–137. doi: 10.1093/nar/23.1.130
Kakutani, T., Munakata, K., Richards, E. J., and Hirochika, H. (1999). Meiotically and mitotically stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabidopsis thaliana. Genetics 151, 831–838.
Kankel, M. W., Ramsey, D. E., Stokes, T. L., Flowers, S. K., Haag, J. R., Jeddeloh, J. A., et al. (2003). Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163, 1109–1122.
Koh, J., Soltis, P. S., and Soltis, D. E. (2010). Homeolog loss and expression changes in natural populations of the recently and repeatedly formed allotetraploid Tragopogon mirus (Asteraceae). BMC Genomics 11:97. doi: 10.1186/1471-2164-11-97
Leitch, A. R., and Leitch, I. J. (2008). Genomic plasticity and the diversity of polyploid plants. Science 320, 481–483. doi: 10.1126/science.1153585
Lister, R., O’Malley, R. C., Tonti-Filippini, J., Gregory, B. D., Berry, C. C., Millar, A. H., et al. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536. doi: 10.1016/j.cell.2008.03.029
Liu, B., Brubaker, C. L., Mergeai, G., Cronn, R. C., and Wendel, J. F. (2001). Polyploid formation in cotton is not accompanied by rapid genomic changes. Genome 44, 321–330.
Liu, P. L., Wan, Q., Guo, Y. P., Yang, J., and Rao, G. Y. (2012). Phylogeny of the genus Chrysanthemum L.: evidence from single-copy nuclear gene and chloroplast DNA sequences. PLoS ONE 7:e48970. doi: 10.1371/journal.pone.0048970
Liu, S. Y., Chen, S. M., Chen, Y., Guan, Z. Y., Yin, D. M., and Chen, F. D. (2011). In vitro induced tetraploid of Dendranthema nankingense (Nakai) Tzvel. shows an improved level of abiotic stress tolerance. Sci. Hortic. 127, 411–419. doi: 10.1016/j.scienta.2010.10.012
Malinska, H., Tate, J. A., Matyasek, R., Leitch, A. R., Soltis, D. E., Soltis, P. S., et al. (2010). Similar patterns of rDNA evolution in synthetic and recently formed natural populations of Tragopogon (Asteraceae) allotetraploids. BMC Evol. Biol. 10:291. doi: 10.1186/1471-2148-10-291
Martienssen, R. A., and Colot, V. (2001). DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293, 1070–1074. doi: 10.1126/science.293.5532.1070
Martin, C., Cervera, M. T., and Gonzalez-Benito, M. E. (2011). Genetic stability analysis of chrysanthemum (Chrysanthemum x morifolium Ramat) after different stages of an encapsulation-dehydration cryopreservation protocol. J. Plant Physiol. 168, 158–166. doi: 10.1016/j.jplph.2010.06.025
McClelland, M., Nelson, M., and Raschke, E. (1994). Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res. 22, 3640–3659. doi: 10.1093/nar/22.17.3640
Meyer, P. (2011). DNA methylation systems and targets in plants. FEBS Lett. 585, 2008–2015. doi: 10.1016/j.febslet.2010.08.017
Miki, D., and Shimamoto, K. (2008). De novo DNA methylation induced by siRNA targeted to endogenous transcribed sequences is gene-specific and OsMet1-independent in rice. Plant J. 56, 539–549. doi: 10.1111/j.1365-313X.2008.03624.x
Parisod, C., Holderegger, R., and Brochmann, C. (2010). Evolutionary consequences of autopolyploidy. New Phytol. 186, 5–17. doi: 10.1111/j.1469-8137.2009.03142.x
Qi, B., Zhong, X., Zhu, B., Zhao, N., Xu, L., Zhang, H., et al. (2010). Generality and characteristics of genetic and epigenetic changes in newly synthesized allotetraploid wheat lines. J. Genet. Genome 37, 737–748. doi: 10.1016/S1673-8527(09)60091-6
Rangwala, S. H., and Richards, E. J. (2004). The value-added genome: building and maintaining genomic cytosine methylation landscapes. Curr. Opin. Genet. Dev. 14, 686–691. doi: 10.1016/j.gde.2004.09.009
Rapp, R. A., and Wendel, J. F. (2005). Epigenetics and plant evolution. New Phytol. 168, 81–91. doi: 10.1111/j.1469-8137.2005.01491.x
Reyna-Lopez, G., Simpson, J., and Ruiz-Herrera, J. (1997). Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet. 253, 703–710. doi: 10.1007/s004380050374
Riddle, N. C., and Richards, E. J. (2002). The control of natural variation in cytosine methylation in Arabidopsis. Genetics 162, 355–363.
Robertson, K. D., and Jones, P. A. (2000). DNA methylation: past, present and future directions. Carcinogenesis 21, 461–467. doi: 10.1093/carcin/21.3.461
Salmon, A., Ainouche, M. L., and Wendel, J. F. (2005). Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 14, 1163–1175. doi: 10.1111/j.1365-294X.2005.02488.x
Saze, H., Scheid, O. M., and Paszkowski, J. (2003). Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 34, 65–69. doi: 10.1038/ng1138
Sha, A., Lin, X., Huang, J., and Zhang, D. (2005). Analysis of DNA methylation related to rice adult plant resistance to bacterial blight based on methylation-sensitive AFLP (MSAP) analysis. Mol. Genet. Genomics 273, 484–490. doi: 10.1007/s00438-005-1148-3
Shaked, H., Kashkush, K., Ozkan, H., Feldman, M., and Levy, A. A. (2001). Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13, 1749–1759.
Steward, N., Kusano, T., and Sano, H. (2000). Expression of ZmMET1, a gene encoding a DNA methyltransferase from maize, is associated not only with DNA replication in actively proliferating cells, but also with altered DNA methylation status in cold-stressed quiescent cells. Nucleic Acids Res. 28, 3250–3259. doi: 10.1093/nar/28.17.3250
Stewart, C. N. Jr., and Via, L. E. (1993). A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. Biotechniques 14, 748–750.
Takeda, S., and Paszkowski, J. (2006). DNA methylation and epigenetic inheritance during plant gametogenesis. Chromosoma 115, 27–35. doi: 10.1007/s00412-005-0031-7
Tang, F., Chen, F., Chen, S., Teng, N., and Fang, W. (2009). Intergeneric hybridization and relationship of genera within the tribe Anthemideae Cass.(I. Dendranthema crassum (kitam.) kitam.× Crossostephium chinense (L.) Makino). Euphytica 169, 133–140.
Tang, F., Wang, H., Chen, S., Chen, F., Liu, Z., and Fang, W. (2011). Intergeneric hybridization between Dendranthema nankingense and Tanacetum vulgare. Sci. Hortic. 132, 1–6.
Tang, F., Wang, H., Chen, S., Chen, F., Teng, N., and Liu, Z. (2012). First intergeneric hybrids within the tribe Anthemideae Cass. III. Chrysanthemum indicum L. Des Moul.× Opisthopappus taihangensis (Ling) Shih. Biochem. Syst. Ecol. 43, 87–92.
Uehara, A., Nakata, M., Kitajima, J., and Iwashina, T. (2012). Internal and external flavonoids from the leaves of Japanese Chrysanthemum species (Asteraceae). Biochem. Syst. Ecol. 41, 142–149. doi: 10.1016/j.bse.2011.12.020
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genom. Biol. 3, research0034. doi: 10.1186/gb-2002-3-7-research0034
Wang, C. G., Li, H., Xue, Z. Y., Chen, C. B., Gu, Y., Sun, D. L., et al. (2009). Marker-based analysis of genome structure and DNA methylation in a watermelon (Citrullus lanatus) ploidy series. Bot. Stud. 50, 389–402.
Wang, H., Chen, S., Jiang, J., Zhang, F., and Chen, F. (2015). Reference gene selection for cross-species and cross-ploidy level comparisons in Chrysanthemum spp. Sci. Rep. 5, 8094. doi: 10.1038/srep08094
Wang, H., Dong, B., Jiang, J., Fang, W., Guan, Z., Liao, Y., et al. (2014a). Characterization of in vitro haploid and doubled haploid Chrysanthemum morifolium plants via unfertilized ovule culture for phenotypical traits and DNA methylation pattern. Front. Plant Sci. 5:738. doi: 10.3389/fpls.2014.00738
Wang, H., Jiang, J., Chen, S., Qi, X., Fang, W., Guan, Z., et al. (2014b). Rapid genetic and epigenetic alterations under intergeneric genomic shock in newly synthesized Chrysanthemum morifolium×Leucanthemum paludosum hybrids (Asteraceae). Genome Biol. Evol. 6, 247–259. doi: 10.1093/gbe/evu008
Wang, H., Qi, X., Gao, R., Wang, J., Dong, B., Jiang, J., et al. (2014c). Microsatellite polymorphism among Chrysanthemum sp. polyploids: the influence of whole genome duplication. Sci. Rep. 4, 6730. doi: 10.1038/srep06730
Wang, H., Wang, J., Jiang, J., Chen, S., Guan, Z., Liao, Y., et al. (2014d). Reference genes for normalizing transcription in diploid and tetraploid Arabidopsis. Sci. Rep. 4, 6781. doi: 10.1038/srep06781
Wang, H., Jiang, J., Chen, S., Fang, W., Guan, Z., Liao, Y., et al. (2013a). Rapid genomic and transcriptomic alterations induced by wide hybridization: Chrysanthemum nankingense x Tanacetum vulgare and C. crassum x Crossostephium chinense (Asteraceae). BMC Genomics 14:902. doi: 10.1186/1471-2164-14-902
Wang, H., Jiang, J., Chen, S., Qi, X., Peng, H., Li, P., et al. (2013b). Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PLoS ONE 8:e62293. doi: 10.1371/journal.pone.0062293
Wang, J., Tian, L., Madlung, A., Lee, H.-S., Chen, M., Lee, J. J., et al. (2004). Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167, 1961–1973. doi: 10.1534/genetics.104.027896
Watanabe, K. (1981). Studies on the control of diploid-like meiosis in polyploid taxa of Chrysanthemum. II. Octoploid Ch. ornatum Hemsley. Cytologia 46, 499–513. doi: 10.1508/cytologia.46.459
Watanabe, K. (1983). Studies on the control of diploid-like meiosis in polyploid taxa of Chrysanthemum. Theor. Appl. Genet. 66, 9–14. doi: 10.1007/BF00281840
Xiong, L., Xu, C., Maroof, M. S., and Zhang, Q. (1999). Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol. Gen. Genet. 261, 439–446. doi: 10.1007/s004380050986
Yang, W., Glover, B. J., Rao, G. Y., and Yang, J. (2006). Molecular evidence for multiple polyploidization and lineage recombination in the Chrysanthemum indicum polyploid complex (Asteraceae). New Phytol. 171, 875–886. doi: 10.1111/j.1469-8137.2006.01779.x
Zemach, A., Li, Y., Wayburn, B., Ben-Meir, H., Kiss, V., Avivi, Y., et al. (2005). DDM1 binds Arabidopsis methyl-CpG binding domain proteins and affects their subnuclear localization. Plant Cell 17, 1549–1558. doi: 10.1105/tpc.105.031567
Zhao, H., Chen, S., Tang, F., Jiang, J., Li, C., Miao, H., et al. (2012). Morphological characteristics and chromosome behaviour in F1, F2 and BC1 progenies between Chrysanthemum×morifolium and Ajania pacifica. Russ. J. Genet. 48, 808–818.
Zhao, H., Liu, Z., Hu, X., Yin, J., Li, W., Rao, G., et al. (2009). Chrysanthemum genetic resources and related genera of Chrysanthemum collected in China. Genetic Resour. Crop Evol. 56, 937–946. doi: 10.1007/s10722-009-9412-8
Keywords: DNA methylation, MET1, DDM1, polyploid, Chrysanthemum
Citation: Wang H, Qi X, Chen S, Fang W, Guan Z, Teng N, Liao Y, Jiang J and Chen F (2015) Limited DNA methylation variation and the transcription of MET1 and DDM1 in the genus Chrysanthemum (Asteraceae): following the track of polyploidy. Front. Plant Sci. 6:668. doi: 10.3389/fpls.2015.00668
Received: 31 January 2015; Accepted: 13 August 2015;
Published: 27 August 2015.
Edited by:
Thomas Debener, Leibniz University Hannover, GermanyReviewed by:
Marcus Linde, Leibniz Universität Hannover, GermanyCai-Zhong Jiang, United States Department of Agriculture – Agricultural Research Service, USA
Copyright © 2015 Wang, Qi, Chen, Fang, Guan, Teng, Liao, Jiang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fadi Chen and Jiafu Jiang, College of Horticulture, Nanjing Agricultural University, Nanjing 210095, China,Y2hlbmZkQG5qYXUuZWR1LmNu;amlhbmdqaWFmdUBuamF1LmVkdS5jbg==