 Elena M. Kramer
Elena M. Kramer- Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA, USA
One of the major goals of the modern study of evodevo is to understand the evolution of gene function across a range of contexts, including sub/neofunctionalization, co-option of genetic modules, and the evolution of morphological novelty. To these ends, comparative studies of gene expression can be useful for constructing hypotheses, but cannot provide direct evidence of functional evolution. Unfortunately, determining endogenous gene function in non-model species is often not an option. Faced with this dilemma, a common approach is to use heterologous expression (HE) in genetically tractable model species as a proxy for functional analyses. Such experiments have important limitations, however, and require caution in the interpretation of their results. How do we dissociate biochemical function from its original genomic context? In the end, what does HE actually tell us? Here, I argue that HE only sheds light on specific types of biochemical conservation, but can be useful when experiments are carefully interpreted.
As developmental biologists, it is important to remember that when we speak of “gene function,” we are conflating, by necessity, a complex array of different factors. At a fundamental level, we can think of gene function as representing two complementary components: the first being biochemical function and the second being developmental role (Figure 1). The former is determined by the coding sequence of the gene itself and encompasses everything from secondary/tertiary protein structure, to enzymatic capacity, to co-factor and/or DNA binding site affinity. These aspects of gene function may change as the sequence of your favorite gene (YFG) itself evolves. As if this weren’t complicated enough, the actual developmental role played by YFG is the product of all of these primary components interacting with a wide array of cis- and trans-acting phenomena, including the expression patterns of YFG in relation to its co-factors, the epigenetic state of target loci, the position of binding sites within the genome, post-translation regulation of all interacting proteins, etc. Obviously, these secondary components evolve as well, to varying degrees in a coordinated fashion with YFG. So when we talk about the evolution of gene function, we are really considering the evolution of the whole genomic context of YFG—its protein sequence, cis- and trans-regulation, interacting partners, and target gene repertoire.
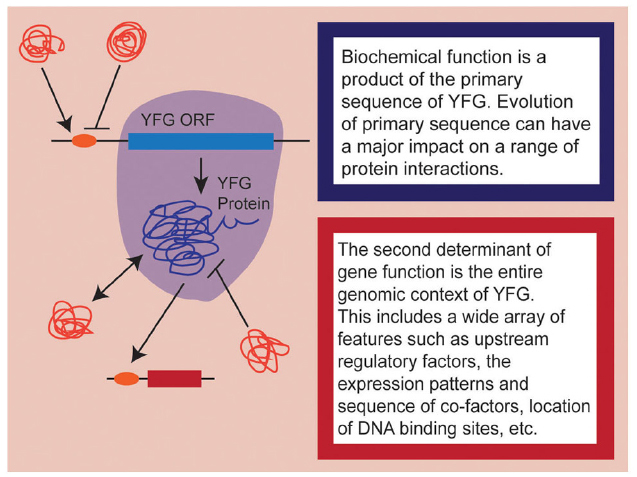
Figure 1. A schematic representation of the dual nature of gene function. Aspects influencing biochemical function are highlighted in shades of blue while aspects of the genomic context are highlighted in shades of red. Note that here, I am only considering heterologous expression of coding sequences, so upstream regulatory elements are considered to be part of the endogenous genomic context.
Heterologous expression (HE) takes the primary component of gene function—the sequence of the coding region itself—and plugs it into the second component—the genomic context—of a different species. We are essentially performing a site-directed mutagenesis experiment in which we ask whether the sequence differences between YFG and its endogenous homolog disrupt the functional roles normally played by the endogenous locus in its own genomic environment. Of course, HE can be conducted with varying degrees of rigor. The most rigorous approach is to drive expression with the endogenous promoter and ask whether YFG can rescue the phenotype of a null mutation in the endogenous locus. With surprising frequency, however, the heterologous locus is simply over-expressed in a wild type background (e.g., Lee et al., 2012; Perilleux et al., 2013; Lovisetto et al., 2015), such that the real question being asked is: Can this alien protein perturb development in the same manner as the endogenous protein when it is over-expressed? Such an approach creates new problems, including the nature of protein interactions, which are subject to reaction equilibria and therefore sensitive to the concentrations of the interacting factors.
Given this perspective, we should consider the variety of ways that HE is typically used in the evodevo field. These include to bolster evidence of genetic orthology (e.g., Serrano et al., 2009), to assess homology of a genetic module or an organ (e.g., Halder et al., 1995; Whipple et al., 2004), and to broadly assess conservation of gene “function” between taxa (e.g., Alvarez-Buylla et al., 2010; Kachroo et al., 2015). The first of these uses should be rejected since similarity of function is absolutely not a criterion for genetic homology in general or orthology in particular (Theissen, 2002; Gabaldon and Koonin, 2013). It is even true that positive HE results can be misleading when it comes to assessing orthology. Perhaps the best understood instance of this phenomenon is the AGAMOUS (AG) lineage of floral organ identity genes in flowering plants. The functions of AG homologs were first described in the core eudicot model systems Arabidopsis and Antirrhinum (snapdragon; Coen and Meyerowitz, 1991). In Arabidopsis, the ag mutant phenotype results in homeotic transformation of fertile organs into sterile organs and a loss of determinacy in the floral meristem. The plena (ple) mutant in Antirrhinum has the identical phenotype and PLE is clearly homologous to AG. However, PLE and AG are not orthologous but, rather, are derived from a whole genome duplication that occurred at the base of the core eudicots (Davies et al., 1999; Kramer et al., 2004). The orthologs of PLE in Arabidopsis are a pair of recent duplicates called SHATTERPROOF1/2, which participate in fruit and ovule development (Liljegren et al., 2000), while the ortholog of AG in Antirrhinum is called FARINELLI (FAR), a gene that primarily contributes to stamen development (Davies et al., 1999; Causier et al., 2005). These distinct functions appear to be due to independent patterns of subfunctionalization that occurred along the lineages leading to the rosid Arabidopsis on the one hand and the asterid Antirrhinum on the other. Furthermore, while the paralogs AG and PLE are biochemically equivalent in Arabidopsis, the orthologs AG and FAR are not (Causier et al., 2005; Airoldi et al., 2010). This is most likely due to changes in selection as FAR became specialized to function in stamen identity. Therefore, while it may commonly be true that orthologs are more likely than not to have both functional similarity and biochemical conservation, we cannot take it for granted.
If you will permit me a digression, I would also like to strongly discourage the common use of the term “functional ortholog.” It is important to remember that function is generally considered not to be a criterion for homology, even among genes (Theissen, 2002). I actually agree with Mindell and Meyer (2001) on this point, that there should be some leeway for discussing the inheritance of genetic function, but we should recognize that it is widely held that functions of any kind cannot be homologous. What information are we trying to convey when we say “functional ortholog?” We want to say that we have a pair of genes that are genetic orthologs and also appear to play similar functional roles. This is an important piece of information; certainly, we often want to know if function is conserved among orthologs. However, this terminology seems to suggest that “functional” orthologs have an additional quality of greater orthology because they show conserved function. This is simply untrue. Orthology is a feature of genetic relationship, of inheritance and patterns of gene duplication. It does not increase or decrease based on functional similarity. It is much more informative to say that you have performed a rigorous phylogenetic and/or syntenic analysis and have determined that the genes in question are orthologs and, further, appear to share conserved functions. We must recognize that this statement can really only be made if you have conducted endogenous functional studies in the taxa being compared. If you have only performed HE, then the best you can say is that there is some degree of biochemical conservation.
The use of HE to assess homology of a genetic module or an organ is more complex and relates to the need to distinguish between process homology and morphological homology, which has been well-covered by many previous authors (Bolker and Raff, 1996; Abouheif, 1997; Abouheif et al., 1997). These authors recognized quite early during the molecular renaissance of our field that shared expression of genetic homologs, and even shared developmental control by homologous genetic modules, should not be used as the basis for assessment of morphological homology. Hodin (2000) succinctly addressed the issue while discussing the limited value of HE with Pax6 homologs: “A positive result tells you only that the biochemical properties of the protein have been conserved, not necessarily that its function within a certain morphological structure has also been conserved. The commonplace use of the same gene within an organism performing distinct functions in a multitude of tissue reveals why this experiment is generally uninformative with respect to evolutionary history (see also Abouheif et al., 1997).” Here, Hodin seeks to highlight the fact that conservation of biochemical interactions within a particular genetic module does not inform on the myriad of ways in which that module can be developmentally deployed. In this regard, I should note that HE can provide some relevant information if you are simply trying to assess homology of a genetic module, but I would argue that phylogeny-based homology assessment of the genes involved and tests of endogenous regulatory interactions are even more useful.
Process homology is especially relevant to cases of co-option of genetic modules to novel developmental functions. For instance, in butterflies Distal-less (Dll) orthologs have been recruited to promote the development of wing spots (Brunetti et al., 2001). The wing spot developmental program is very unlikely to be recapitulated by simply expressing the butterfly Dll in Drosophila because this developmental program is a product of what I defined as the second component of gene function, the endogenous genomic architecture of the butterfly. However, reciprocal HE of Dll orthologs between Drosophila and butterflies would be perfectly useful if your goal was to determine whether the evolution of the wing spot involved biochemical divergence in the butterfly Dll sequence. This type of co-option is just one extreme on a spectrum of evolutionary change that could also include morphological remodeling events such as the derivation of halteres from hindwings (Hersh et al., 2007), lodicules from petals (Whipple et al., 2007; Yoshida, 2012) or staminodia from stamens (Sharma and Kramer, 2013). Such evolutionary transitions may involve biochemical changes in upstream transcription factors but clearly also involve changes in target gene repertoires (e.g., Hersh et al., 2007). HE is much more likely to shed light on any biochemical changes rather than changes in target gene repertoires, which primarily depend on the positions of downstream binding sites dispersed throughout the genome.
The third common use of HE, to investigate conservation of “function,” is perfectly legitimate in many cases but less so in others. It is probably useful to start with a consideration of what can go wrong with HE. For instance, a lack of rescue or the failure to produce a phenotype may simply be due to the divergence between your species of interest and the reference model system. Even proteins that are likely to serve conserved functions can experience the process of developmental system drift (True and Haag, 2001) at the level of primary sequence. In other words, this is a site directed mutagenesis experiment in which the altered protein cannot function in the model system’s genomic context but may be perfectly functional in its original environment. On occasion, HE results in novel or dominant negative phenotypes (e.g., Lee et al., 2012; Katahata et al., 2014; Sun et al., 2014). These may be due to the disruptive effects of an alien protein being introduced to a system for which it is not adapted. If the heterologous protein can interact with some co-factors but not others, it may act as a dominant negative allele, especially when over-expressed. Perhaps most surprisingly though, even positive results can be misleading. Zarrinpar et al. (2003) tested the ability of SH3-domain protein homologs to rescue the function of one specific family member in yeast. They found that while endogenous paralogs were highly functionally specific and could not rescue, diverse metazoan homologs showed higher frequency of rescue. These results reflect the fact that members of the same genome, especially when co-expressed, will tend to co-evolve for a high degree of functional specificity. Homologs from divergent genomic contexts that have not experienced the same patterns of co-evolution may actually be quite promiscuous in a heterologous genome. Thus, we see that a range of results from HE can be uninformative or misleading, especially when you do not have functional data from the original organism.
So am I suggesting that HE is never useful for examining the evolution of gene function? Certainly not. In cases where biochemical divergence is specifically being assessed, this approach can be the best experiment to use, albeit with some caveats. Let’s consider a classic HE experiment, Ronshaugen et al. (2002), in which they tested the ability of Artemia Ubx to suppress limb development in Drosophila (Figure 2). Interestingly, the authors found that while the full length Artemia Ubx had little limb-suppressing capacity, a relatively minor C-terminal deletion allowed the Artemia protein to repress limbs in Drosophila. In light of this finding, the authors proposed a model in which the Ubx protein of a crustacean/insect ancestor experienced mutation in the C-terminus of the protein that uncovered a limb-repression function. This is certainly a plausible scenario that fits the presented data, but we should also recognize a weakness in that the experiment was only performed in the Drosophila genomic context where Ubx has a limb repressing function. If you could put the mutated Artemia Ubx back into Artemia, would it have the capacity to repress limbs or is that primarily a product of the Drosophila genome? As it turns out, further studies in Artemia have revealed a more complex situation that suggests that there may be multiple reasons why Ubx does not repress limbs in Artemia (Hsia et al., 2010). These findings underscore the fact that accurate interpretation of HE data really hinges on having as much information as possible in both taxa, including functional results whenever possible.
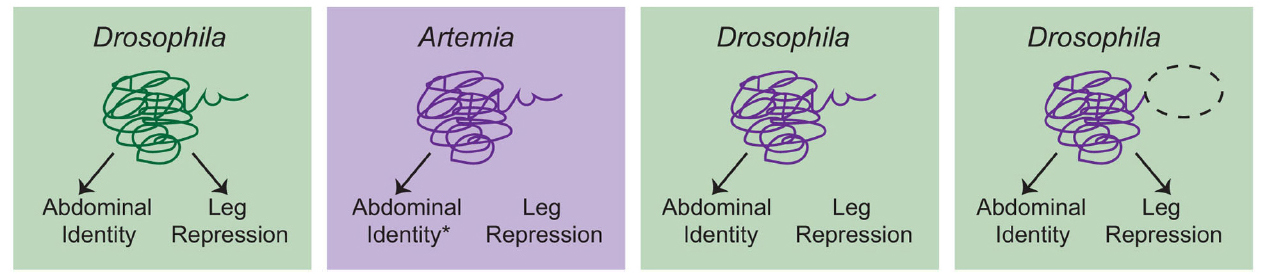
Figure 2. A schematic overview of the Ronshaugen et al. (2002) experiments. Drosophila Ubx promotes abdominal identity and acts to repress limb development in the abdomen. Artemia Ubx is assumed (*) to promote abdominal identity but not repress leg development. Consistent with this, when full-length Artemia Ubx was expressed in Drosophila, it was capable of promoting abdominal identity but could not repress leg development. Deletion of a C-terminal region of the Artemia Ubx (dashed oval) conferred leg repression capacity. This led to the conclusion that evolution of the C-terminal domain was a critical aspect of the evolution of leg repression function in the Ubx lineage in arthropods. However, there are several considerations that should be kept in mind. First, additional studies suggest that there may be multiple reasons why Artemia Ubx does not repress leg development (Hsia et al., 2010). Second, the ideal test would be to determine whether the truncated Artemia Ubx could repress legs if placed back into the endogenous Artemia context. Without this experiment, it remains possible that the observed function is simply a product of the Drosophila genomic context, in which Ubx normally represses leg development. Given everything we know now, the most conservative interpretation is that clearly Artemia Ubx is not biochemically equivalent to Drosophila Ubx. These biochemical differences may have been critical for the evolution of limb repressing functions, but studies in Artemia itself, as well as other arthropods would be necessary to confirm this hypothesis.
One especially elegant demonstration of how powerful HE can be when paired with functional studies in both the donor and recipient is work done on the control of flowering time in sugar beet (Beta vulgaris ssp. vulgaris, Pin et al., 2010). In flowering plants, homologs of the PEBP lineage defined by the Arabidopsis gene FLOWERING LOCUS T (FT) are broadly involved with promoting the transition from vegetative to reproductive development (reviewed Ballerini and Kramer, 2011). The FT protein has been identified as the classic Florigen factor that moves from leaves, where it is produced, to the apical meristem in order to change meristem identity. Consistent with this role, most FT homologs are only expressed at significant levels after the initiation of reproductive development. In cultivated sugar beet, however, a very recent gene duplication has given rise to two copies: BvFT1, which is primarily expressed during vegetative development, and BvFT2, which is expressed as expected during the reproductive stage (Pin et al., 2010). Using RNAi and overexpression in beet, Pin et al. (2010) clearly established that the BvFT1 paralog had acquired a dominant negative effect that represses flowering until vernalization (cold treatment) represses BvFT1 and allows expression of the floral promoting paralog BvFT2. This dramatic difference in function between the two paralogs can be recapitulated in Arabidopsis, where BvFT2 activates flowering while BvFT1 represses it. This demonstrates that there is a biochemical change in BvFT1 relative to the otherwise highly conserved function of FT proteins. The use of chimeric proteins and site-directed mutagenesis in the more tractable Arabidopsis system allowed the authors to identify the specific amino acid changes that are responsible for the neofunctionalization, and further demonstrate that these changes are associated with BvFT1 alleles that were selected during domestication. This kind of study relies heavily on HE but uses it in exactly the right way—by targeting an otherwise highly conserved genetic module, and in combination with detailed expression and functional studies in the original system, which allows the heterologous results to be accurately interpreted.
Another powerful application of HE is to use homologs from a series of diverging taxa to probe the conservation of specific biochemical properties, such as recognition of DNA binding sites. This is essentially a matter of letting evolution do the site-directed mutagenesis for you: as you move out to more deeply diverging taxa, there are more non-synonymous mutations, allowing you to ask whether the endogenous biochemical function is still retained. The land plant-specific transcription factor LEAFY (LFY) is ideal for this type of study because unlike most plant gene lineages, it has very few retained paralogs. Maizel et al. (2005), tested the ability of LFY homologs from across the land plants to rescue the lfy mutation in Arabidopsis, and then further complemented the phenotypic analysis with microarray studies of gene expression. They found that there was a gradual decreasing degree of phenotypic rescue as they moved out to more distantly related taxa. When paired with tests of protein/DNA interaction, their results suggest that “the declining ability to replace Arabidopsis LFY … is caused by a progressive failure to interact with the canonical LFY binding sites,” which, of course, are defined based on work done in Arabidopsis. The microarray analysis of the various transgenic lines demonstrated that in the weakest cases of rescue, one of the last target interactions to be lost was with the floral meristem identity gene APETALA1 (AP1). The authors quite correctly noted that this finding does not tell us anything about what the heterologous LFY homologs activate in their endogenous settings—AP1 homologs are not even present outside angiosperms. Rather, this reflects the extraordinarily high affinity of the LFY binding site present in the AP1 promoter, such that even deeply divergent homologs with many non-synonymous changes are still capable of recognizing it. This kind of study highlights evolutionary processes affecting both aspects of developmental gene function since it detects biochemical changes that have altered DNA affinity while also underscoring the fact that repertoires of target genes will simultaneously be evolving.
In summary, my argument is that HE can be very useful in specific cases where we want to investigate changes in the primary component of gene function, which is to say biochemical function. This includes enzymatic capacity as well as affinity for a range of interactions such as protein-DNA and protein–protein. It yields the best results when paired with functional studies in both the donor and recipient taxa so that potentially spurious phenotypes can be ruled out. I think it is also true that HE works best when you can target a genetic module that is otherwise very highly conserved, so that you can lessen the impact of drift and divergence in other components of the pathway (although this is hard to ever rule out completely!). HE does not inform upon homology in general or orthology in particular, nor does it give us much information on what developmental roles the gene may play in its original genomic context, so use it with care.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to acknowledge several other scientists with whom I have discussed this question over the years, particularly Vivian Irish, who set me straight on the subject to begin with, as well as Amy Litt, and Sarah Mathews.
References
Abouheif, E. (1997). Developmental genetics and homology: a hierarchical approach. Tren. Ecol. Evol. 12, 405–408. doi: 10.1016/S0169-5347(97)01125-7
Abouheif, E., Akam, M., Dickinson, W. J., Holland, P. W. H., Meyer, A., Patel, N. H., et al. (1997). Homology and developmental genes. Tren. Gen. 13, 432–433. doi: 10.1016/S0168-9525(97)01271-7
Airoldi, C. A., Bergonzi, S., and Davies, B. (2010). Single amino acid change alters the ability to specify male or female organ identity. Proc. Natl. Acad. Sci. U.S.A. 107, 18898–18902. doi: 10.1073/pnas.1009050107
Alvarez-Buylla, E. R., Ambrose, B. A., Flores-Sandoval, E., Vergara-Silva, F., Englund, M., Garay-Arroyo, A., et al. (2010). B-function expression in the flower center underlies the homeotic phenotype of Lacandonia schismatica (Triuridaceae). Plant Cell 22, 3543–3559. doi: 10.1105/tpc.109.069153
Ballerini, E. S., and Kramer, E. M. (2011). In the light of evolution: a reevaluation of conservation in the CO-FT regulon and its role in photoperiodic regulation of flowering time. Front. Plant Sci. 2:81. doi: 10.3389/fpls.2011.00081
Bolker, J. A., and Raff, R. A. (1996). Developmental genetics and traditional homology. Bioessays 18, 489–494. doi: 10.1002/bies.950180611
Brunetti, C. R., Selegue, J. E., Monteiro, A., French, V., Brakefield, P. M., and Carroll, S. B. (2001). The generation and diversification of butterfly eyespot color patterns. Curr. Biol. 11, 1578–1585. doi: 10.1016/S0960-9822(01)00502-4
Causier, B., Castillo, R., Zhou, J. L., Ingram, R., Xue, Y. B., Schwarz-Sommer, Z., et al. (2005). Evolution in action: following function in duplicated floral homeotic genes. Curr. Biol. 15, 1508–1512. doi: 10.1016/j.cub.2005.07.063
Coen, E. S., and Meyerowitz, E. M. (1991). The war of the whorls: genetic interactions controlling flower development. Nature 353, 31–37. doi: 10.1038/353031a0
Davies, B., Motte, P., Keck, E., Saedler, H., Sommer, H., and Schwarz-Sommer, Z. (1999). PLENA and FARINELLI: redundancy and regulatory interactions between two Antirrhinum MADS-box factors controlling flower development. EMBO J. 18, 4023–4034. doi: 10.1093/emboj/18.14.4023
Gabaldon, T., and Koonin, E. V. (2013). Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 14, 360–366. doi: 10.1038/nrg3456
Halder, G., Callaerts, P., and Gehring, W. J. (1995). Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 267, 1788–1792.
Hersh, B. M., Nelson, C. E., Stoll, S. J., Norton, J. E., Albert, T. J., and Carroll, S. B. (2007). The UBX-regulated network in the haltere imaginal disc of D. melanogaster. Dev. Biol. 302, 717–727. doi: 10.1016/j.ydbio.2006.11.011
Hodin, J. (2000). Plasticity and constraints in development and evolution. J. Exp. Zool. (Mol. Dev. Evol.) 288, 1–20. doi: 10.1002/(SICI)1097-010X(20000415)288:1<1::AID-JEZ1>3.0.CO;2-7
Hsia, C. C., Pare, A. C., Hannon, M., Ronshaugen, M., and Mcginnis, W. (2010). Silencing of an abdominal Hox gene during early development is correlated with limb development in a crustacean trunk. Evol. Dev. 12, 131–143. doi: 10.1111/j.1525-142X.2010.00399.x
Kachroo, A. H., Laurent, J. M., Yellman, C. M., Meyer, A. G., Wilke, C. O., and Marcotte, E. M. (2015). Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348, 921–925. doi: 10.1126/science.aaa0769
Katahata, S. I., Futamura, N., Igasaki, T., and Shinohara, K. (2014). Functional analysis of SOC1-like and AGL6-like MADS-box genes of the gymnosperm Cryptomeria japonica. Tree Genet. Genomes 10, 317–327. doi: 10.1007/s11295-013-0686-9
Kramer, E. M., Jaramillo, M. A., and Di Stilio, V. S. (2004). Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS-box genes in angiosperms. Genetics 166, 1011–1023. doi: 10.1534/genetics.166.2.1011
Lee, J. H., Park, S. H., and Ahn, J. H. (2012). Functional conservation and diversification between rice OsMADS22/OsMADS55 and Arabidopsis SVP proteins. Plant Sci. 185, 97–104. doi: 10.1016/j.plantsci.2011.09.003
Liljegren, S. J., Ditta, G. S., Eshed, Y., Savidge, B., Bowman, J. L., and Yanofsky, M. F. (2000). SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404, 766–770. doi: 10.1038/35008089
Lovisetto, A., Baldan, B., Pavanello, A., and Casadoro, G. (2015). Characterization of an AGAMOUS gene expressed throughout development of the fleshy fruit-like structure produced by Ginkgo biloba around its seeds. BMC Evol. Biol. 15:139. doi: 10.1186/s12862-015-0418-x
Maizel, A., Busch, M. A., Tanahashi, T., Perkovic, J., Kato, M., Hasebe, M., et al. (2005). The floral regulator LEAFY evolves by substitutions in the DNA binding domain. Science 308, 260–263. doi: 10.1126/science.1108229
Mindell, D. P., and Meyer, A. (2001). Homology evolving. Trends Ecol. Evol. 16, 434–440. doi: 10.1016/S0169-5347(01)02206-6
Perilleux, C., Pieltain, A., Jacquemin, G., Bouche, F., Detry, N., D’aloia, M., et al. (2013). A root chicory MADS box sequence and the Arabidopsis flowering repressor FLC share common features that suggest conserved function in vernalization and de-vernalization responses. Plant J. 75, 390–402. doi: 10.1111/tpj.12208
Pin, P. A., Benlloch, R., Bonnet, D., Wremerth-Weich, E., Kraft, T., Gielen, J. J. L., et al. (2010). An antagonistic pair of FT homologs mediates the control of flowering time in sugar beet. Science 330, 1397–1400. doi: 10.1126/science.1197004
Ronshaugen, M., Mcginnis, N., and Mcginnis, W. (2002). Hox protein mutation and macroevolution of the insect body plan. Nature 415, 914–917. doi: 10.1038/nature716
Serrano, G., Herrera-Palau, R., Romero, J. M., Serrano, A., Coupland, G., and Valverde, F. (2009). Chlamydomonas CONSTANS and the evolution of plant photoperiodic signaling. Curr. Biol. 19, 359–368. doi: 10.1016/j.cub.2009.01.044
Sharma, B., and Kramer, E. M. (2013). Sub- and neofunctionalization of APETALA3 paralogs have contributed to the evolution of novel floral organ identity in Aquilegia (columbine, Ranunculaceae). New Phytol. 197, 949–957. doi: 10.1111/nph.12078
Sun, W., Huang, W. J., Li, Z. N., Song, C., Liu, D., Liu, Y. L., et al. (2014). Functional and evolutionary analysis of the AP1/SEP/AGL6 superclade of MADS-box genes in the basal eudicot Epimedium sagittatum. Ann. Bot. 113, 653–668. doi: 10.1093/aob/mct301
True, J. R., and Haag, E. S. (2001). Developmental system drift and flexibility in evolutionary trajectories. Evol. Dev. 3, 109–119. doi: 10.1046/j.1525-142x.2001.003002109.x
Whipple, C. J., Ciceri, P., Padilla, C. M., Ambrose, B. A., Bandong, S. L., and Schmidt, R. J. (2004). Conservation of B-class floral homeotic gene function between maize and Arabidopsis. Development 131, 6083–6091. doi: 10.1242/dev.01523
Whipple, C. J., Zanis, M. J., Kellogg, E. A., and Schmidt, R. J. (2007). Conservation of B class gene expression in the second whorl of a basal grass and outgroups links the origin of lodicules and petals. Proc. Natl. Acad. Sci. U.S.A. 104, 1081–1086. doi: 10.1073/pnas.0606434104
Yoshida, H. (2012). Is the lodicule a petal: molecular evidence? Plant Sci. 184, 121–128. doi: 10.1016/j.plantsci.2011.12.016
Keywords: evo-devo, heterologous expression, functional evolution, biochemical evolution, developmental genetics
Citation: Kramer EM (2015) A stranger in a strange land: the utility and interpretation of heterologous expression. Front. Plant Sci. 6:734. doi: 10.3389/fpls.2015.00734
Received: 30 June 2015; Accepted: 29 August 2015;
Published: 15 September 2015.
Edited by:
Sylvain Marcellini, University of Concepcion, ChileCopyright © 2015 Kramer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena M. Kramer, Department of Organismic and Evolutionary Biology, Harvard University, 16 Divinity Avenue, Cambridge, MA 02138, USA,ZWtyYW1lckBvZWIuaGFydmFyZC5lZHU=