 Keiichi Mochida
Keiichi Mochida- 1Cellulose Production Research Team, Biomass Engineering Research Division, RIKEN Center for Sustainable Resource Science, Yokohama, Japan
- 2Gene Discovery Research Group, RIKEN Center for Sustainable Resource Science, Yokohama, Japan
- 3Kihara Institute for Biological Research, Yokohama City University, Yokohama, Japan
- 4Group of Genome Diversity, Institute of Plant Science and Resources, Okayama University, Kurashiki, Japan
- 5Group of Environmental Response Systems, Institute of Plant Science and Resources, Okayama University, Kurashiki, Japan
Crops are exposed to various environmental stresses in the field throughout their life cycle. Modern plant science has provided remarkable insights into the molecular networks of plant stress responses in laboratory conditions, but the responses of different crops to environmental stresses in the field need to be elucidated. Recent advances in omics analytical techniques and information technology have enabled us to integrate data from a spectrum of physiological metrics of field crops. The interdisciplinary efforts of plant science and data science enable us to explore factors that affect crop productivity and identify stress tolerance-related genes and alleles. Here, we describe recent advances in technologies that are key components for data driven crop design, such as population genomics, chronological omics analyses, and computer-aided molecular network prediction. Integration of the outcomes from these technologies will accelerate our understanding of crop phenology under practical field situations and identify key characteristics to represent crop stress status. These elements would help us to genetically engineer “designed crops” to prevent yield shortfalls because of environmental fluctuations due to future climate change.
Introduction
Abiotic stress conditions can have a negative effect on the productivity of agricultural systems. According to a recent report from the Intergovernmental Panel on Climate Change (IPCC), humanity is facing an increased risk of agricultural production shortfalls (https://www.ipcc.ch/report/ar5/). Modern plant science has achieved remarkable advances in elucidating the molecular systems associated with abiotic stress responses in plants under artificially controlled conditions inside the laboratory. This is especially true for the model plant species Arabidopsis thaliana, where functional genomic analyses after the completion of sequencing its genome have identified key genes involved in the regulatory network of abiotic stress responses (Hirayama and Shinozaki, 2010; Nakashima et al., 2014). However, several critical problems remain regarding the practical application of this laboratory-derived knowledge to molecular science based breeding of crops adapted to adverse environments. The next challenge in generating practical stress-tolerant crops that can withstand future climate changes requires an understanding of the responses of crops to multiple abiotic stresses under field growth conditions. Large fluctuations in multiple abiotic stress conditions and large heterogeneity between stress levels for different plant genotypes and developmental stages are the chief causes of the complexity underlying variations in abiotic stress responses in crops under field conditions (Mittler and Blumwald, 2010).
The considerable recent advances in analytical technologies in omics-based research have provided crucial resources for investigating biological systems not only in model plants but also crop species (Mochida and Shinozaki, 2010). With large-scale transcriptome datasets, it will be feasible to perform correlated gene expression analyses to identify candidate genes involved in particular gene networks (Mochida et al., 2011; Obayashi et al., 2014). Metabolome analyses provide information on the accumulation patterns of metabolites in plants in various biological contexts, such as changes in environment, developmental stage, and genotype, and offer an efficient approach to revealing the metabolic systems underlying complex phenotypes (Tohge et al., 2011; Balmer et al., 2013; Fukushima and Kusano, 2013). Hormonomic analysis, which enables simultaneous profiling of phytohormones and their derivatives, also plays an important role in investigating phytohormone networks in different biological contexts (Kojima et al., 2009; Kanno et al., 2010). Integrated approaches using synergistic combinations of different omics systems, so called “trans-omics,” are increasingly an effective means of investigating plant cellular systems in response to abiotic stresses (Dinakar and Bartels, 2013; Deshmukh et al., 2014). Furthermore, in the last decade, rapid progress in next-generation sequencing (NGS) technologies has enabled access to genome-scale sequence information from a wide range of organisms, even those with large and complex genome structures such as wheat and barley (Mochida and Shinozaki, 2011, 2013). Whole genome resequencing is a feasible NGS application for exploring genome-scale polymorphisms in natural variations, and to identify the association between genetic polymorphisms and phenotypic variations including those induced by stress. Another NGS application, RNA-seq, is highly scalable and can be used to rapidly acquire comprehensive transcriptome data in any species. The effective use of genome-scale datasets from various types of omics analyses rely on computer-aided approaches that have become increasingly important in studies to determine the responses of plant cellular systems to environmental changes. A broad range of bioinformatics techniques are essential to access large-scale omics datasets and to efficiently discover biologically significant information and then use this to answer specific questions on stress responses in plants. Systems approaches with mathematical modeling have recently received much attention for understanding biological phenomena under both controlled laboratory conditions and fluctuating field conditions.
With the currently available methods and resources for studying plant stress responses, it is expected that interdisciplinary efforts involving plant science and data science will enable exploration of factors that affect crop productivity and will aid discovery of genes and alleles associated with quantitative traits of stress tolerance in crops. It is essential not only to examine a snapshot of the cellular network under multiple stress conditions at a particular moment but also to monitor throughout the life cycle, since changes in physiological status over time might influence the eventual phenotype. The identification and estimation of the effects of parameters, based on an understanding of the genetics and physiology of responses to environmental changes of crops throughout their life cycle, are required to design a crop with the required performance of stress tolerances in the field condition. In this mini review, we provide an overview of recent advances in technologies that are key components for data driven crop design, such as crop population genomics, chronological trans-omics analysis, and computer-aided molecular network prediction (Figure 1).
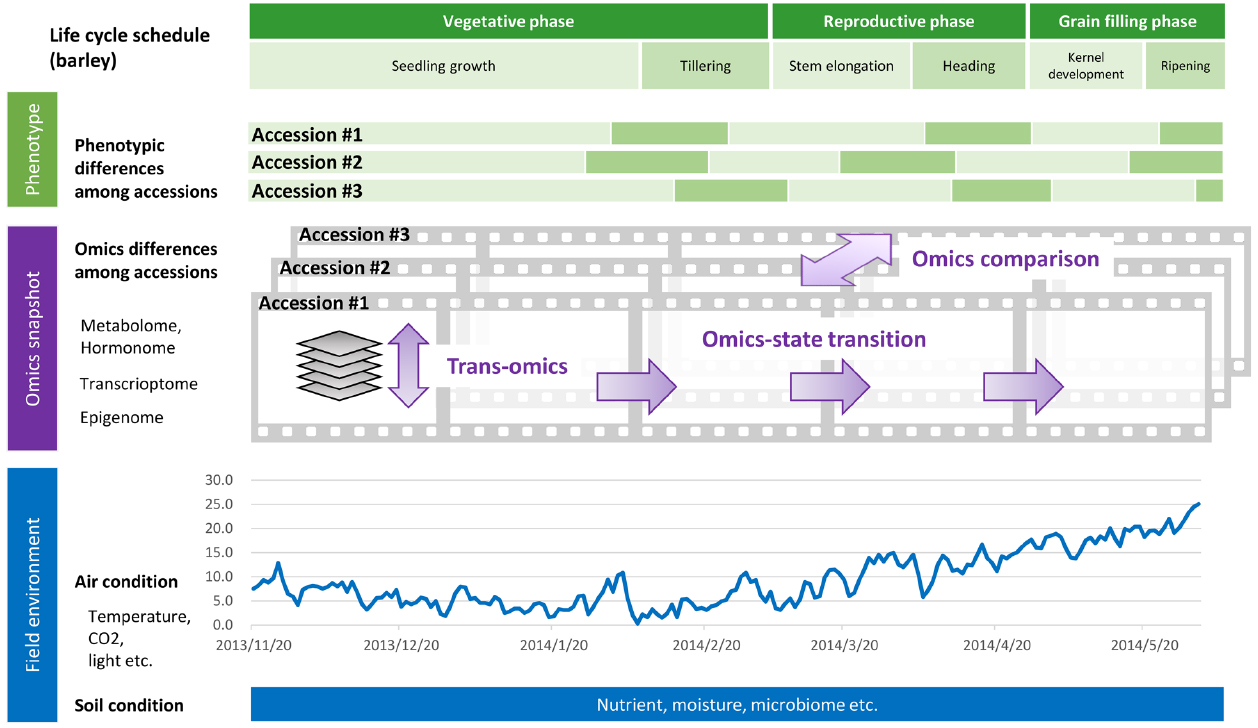
Figure 1. Phenology datasets during the life cycle of a crop grown under field conditions. Time series omics data such as transcriptome, epigenome, and metabolome/hormonome data can be acquired as sequential “snapshots” throughout the crop life cycle. Parameters regarding field environment including air and soil conditions can be acquired as “streamed” datasets.
Population Genomics in Crops
Genetic diversity in a crop population is a valuable resource for identifying alleles that can be exploited to improve crop productivity under a variety of adverse conditions (Huang and Han, 2014). Population-wide molecular phylogeographic analysis of a crop species can provide molecular evidence on its demographic history as a domesticated species (Saisho and Purugganan, 2007). Additionally, such analysis may identify relationships between biased geographic distributions and genetic differentiation, such as the particular genotype associated with a trait providing adaptation to a particular local environment. For example, in barley, a population-wide analysis of bio-geography and the degree of vernalization requirement showed a biased geographic distribution pattern of a quantitative growth habit trait (Saisho et al., 2011). As another example, a population-scale evolutionary analysis of HvAACT1, which encodes a citrate transporter involved in aluminum tolerance in barley and has a 1-kb insertion for Al-tolerance in the upstream region, only occurs in Al-tolerant cultivars in Japan, Korea, and China, suggesting adaptation to the acid soils of these areas (Fujii et al., 2012). These examples in barley demonstrate that population-scale exploration of the association between geographic distributions and genotypes could be an efficient strategy to identify alleles for locally adapted traits. The development of NGS has allowed high-throughput genotyping such as whole-genome re-sequencing, genotyping by sequencing (GBS), RNA-seq based genotyping, and exome sequencing, to rapidly generate genome-scale datasets on genetic polymorphism.
Whole genome re-sequencing analysis with information from a reference genome is a straightforward method to characterize genome-wide polymorphism patterns among accessions. Representative accessions, for example, elite lines in tomato, soybean, maize, and rice, have been investigated by whole genome re-sequencing, which has identified useful resources for further genetic studies in each crop (Lai et al., 2010; Arai-Kichise et al., 2011; Subbaiyan et al., 2012; Causse et al., 2013; Li et al., 2013). In some species with smaller genomes, the whole genome re-sequencing approach has been applied to population-wide analyses of genome-wide polymorphism patterns; this approach has been employed in poplar tree, tomato, common bean, and rice (Evans et al., 2014; Lin et al., 2014; Schmutz et al., 2014). Huang et al. (2012) carried out a whole-genome resequencing analysis in wild rice populations to generate a genome variation map, which also provided insights into the domestication history of domesticated rice. More recently, a core collection of 3000 rice accessions from 89 countries were re-sequenced and 18.9 million single nucleotide polymorphisms (SNPs) were found (Li et al., 2014a). A whole-genome resequencing dataset on a population wide scale can provide an important resource especially for understanding the demographic history of a domesticated species, and facilitate recognition of alleles associated with adaptive phenotypic variations, for example, tolerance of particular environments, by applying the resequencing dataset together with a dataset of the trait based on a genome-wide association study (GWAS).
Genotyping by sequencing or RNA-seq based genotyping are more affordable approaches than whole genome sequencing for genome-wide and population wide genotyping. GBS is a popular method that provides a rapid and robust approach for identifying sequences with a low level of representation in multiplex samples (Elshire et al., 2011; Poland et al., 2012). A number of genome-wide polymorphism datasets have been obtained from GBS analysis, for example, 2815 accessions of the USA national maize inbred seed bank using 681,257 SNPs (Romay et al., 2013), 971 worldwide accessions of sorghum with ∼265,000 SNPs (Morris et al., 2013), and 304 short-season soybean lines with >47,000 SNPs (Sonah et al., 2015); these have also been applied to GWAS analysis (so called GBS-GWAS analysis).
High-throughput genome-scale genotyping is a key technology to finding adaptive genes that might be of promise for improving crop productivity in particular environments. Careful analysis of associations between genome-wide patterns of polymorphism and phenotypic variations in adaptive traits holds great promise for elucidating crop species domestication histories at both the ecological and evolutionary levels (Huang and Han, 2014). Furthermore, such analyses enable the estimation of the genetic effects of candidate allelic combinations and quantification of heritability, which are critical parameters to production of reliable allelic combinations in the designed crop varieties.
Omics-Based Elucidation of Crop Phenology
Crops in the field are exposed to multiple environmental stimuli. Crop life cycle changes are often triggered by environmental signals, for example, temperature- and photoperiod-related cues for flowering, and timely initiation of these developmental changes is critical to final productivity. Therefore, understanding the physiological responses of crops to seasonal and short-term fluctuations in the environment is vital to estimation of their potential impact on the crop life cycle and eventual yield. For this purpose, omics-based long-term chronological profiling of crops under field conditions is an efficient strategy for characterizing phenological responses in gene regulatory networks. Such analyses provide insights into the regulation of gene functions in response to environmental fluctuations and are an aid for the identification of genes that are key mediators between environmental signals and crop productivity (Gibson, 2008).
Time-series transcriptome analysis during plant life cycles has become an efficient approach to infer phenological responses under variable environmental conditions. Richards et al. (2012) performed a time-series transcriptome analysis in A. thaliana shoots in the field from seedling to reproductive stages and found enrichment of several co-expressed gene clusters that were induced by abiotic and biotic stresses. Several studies have investigated the dynamics of genome-scale gene expression patterns using transcriptome analysis of a life cycle sample series from cultivated rice plants grown under field conditions (Sato et al., 2011; Nagano et al., 2012; Matsuzaki et al., 2015). It was shown that mathematical modeling and prediction of genome-wide transcriptional changes under field conditions could be successfully carried out based on life cycle transcriptome datasets and meteorological datasets (Nagano et al., 2012). Similarly, the transcriptome in a single clone of a grapevine cultivar was recorded over three consecutive years in 11 vineyards and it was demonstrated that the additive effects of temperature and water availability particularly influenced grape quality (Dal Santo et al., 2013).
Gene expression in response to developmental and environmental signals are often regulated by epigenetic mechanisms through small RNAs, histone modifications and DNA methylation (Chinnusamy and Zhu, 2009; Kinoshita and Seki, 2014). Recent studies in plants have shown that epigenetic mechanisms are involved in some important biological processes such as genomic imprinting, defense responses to pathogens, acclimation to abiotic stresses, and vernalization responses (Ikeda, 2012; Kim et al., 2012; Woods et al., 2014; Liu et al., 2015). Furthermore, some of these epigenetic modifications are inherited through mitotic and meiotic cell divisions. The meiotically heritable epigenetic modifications are termed “epialleles” and can cause heritable phenotypic variation (Kalisz and Purugganan, 2004; Weigel and Colot, 2012). In epigenetic regulation of plant stress tolerance, non-heritable epigenetic modifications are involved in acclimation as a short-term stress resistance response. Mitotically and meiotically heritable epigenetic modifications function as a “stress memory” within and across generations, respectively (Chinnusamy and Zhu, 2009). A recent study of epigenetic recombinant inbred lines (epiRILs) of A. thaliana showed that variations in DNA methylation cause heritable variation of ecologically important plant traits, such as root allocation, drought tolerance and nutrient plasticity (Zhang et al., 2013). Plant epigenome data are therefore vital to the understanding of epigenetic and genetic regulation of phenotypic diversity (Schmitz et al., 2013). It is now recognized that epigenetic diversity in populations and epigenetic changes in response to environmental fluctuation are also considerable factors in adaptation and evolution and could be a resource for improvement of crop stress tolerance.
Phenome analyses provide datasets on a variety of phenotypes using mutants and/or natural variants. With large-scale loss-of-function or gain-of-function mutants, phenome analyses using artificially induced mutants have played an essential role in discovering genes involved in phenotypic changes and for determining their biological functions (Kuromori et al., 2006, 2009). Recent advances in technologies such as sensors, imaging, and internet communication have begun to provide various tools for high-throughput plant phenotyping under field conditions (Klukas et al., 2014; Li et al., 2014b; Fahlgren et al., 2015; Grosskinsky et al., 2015). Remote phenotyping of crops in the field is emerging as a feasible application for drones with multiple sensors, not only for trait analysis in genetics but also for precision agriculture (Liebisch et al., 2015). Hand-held devices that aid phenotyping can be an efficient tool to carry out high-throughput phenotypic data acquisition (Vankudavath et al., 2012). Integration of imaging and sensing technologies have provided tools for non-invasive approaches to monitor biometrics of growing crops (Busemeyer et al., 2013; Li et al., 2014b; Kjaer and Ottosen, 2015). High-throughput plant phenotyping approaches have been synergistically applied to genetics to accelerate gene discovery in crops. For example in rice, a high-throughput rice phenotyping facility (HRPF) makes it possible to monitor 15 traits during the rice growth period; and these data can be applied to GWAS (Yang et al., 2014). In addition to conventional phenotyping, quantitative molecular profiles from various high-throughput analytical techniques such as metabolomics could be used as a comprehensive dataset of molecular phenotypes.
Metabolome analysis provides a comprehensive molecular snapshot based on metabolites synthesized in biological reactions. It can be affected by various factors, such as genetic and epigenetic factors, developmental stages and organs, environmental stimuli and diseases. Therefore, it could be thought that the metabolome can represent chemical phenotypes reflecting the physiological state in an organism (Mochida et al., 2009; Sakurai et al., 2013). The combinatorial use of high-throughput metabolome profiling and GWAS has become an efficient strategy to reveal the genetic architecture of biochemical properties in plants (Adamski and Suhre, 2013; Wen et al., 2014; Matsuda et al., 2015). Metabolome profiling at different plant developmental stages can provide stage-dependent information on the physiological state of the plant in response to the environment during the lifecycle (Onda et al., 2015). Therefore, chronological metabolome analysis throughout the plant life cycle under field conditions will also be a vital strategy to describe the physiological state and to extract state factors associated with traits in crops.
Computer Aided Understanding of Biological Phenomena in Plants
As described above, recent advances in omics analytical technologies have produced a wealth of genome-scale datasets even from crops growing in field conditions. One of the important issues in bioinformatics is how to deal with such large and heterogeneous datasets, and to establish heuristic procedures to accelerate gene discovery (Mochida and Shinozaki, 2011). Information resources such as databases and computational tools are extremely important for effectively handling genome-scale datasets. Additionally, data storage for omics datasets must ensure persistence and retrieval functionalities for shared use (Mochida and Shinozaki, 2011).
To gain a mechanistic understanding of biological systems, mathematical modeling and simulation approaches have been applied to the study of plant cellular metabolism, growth, developmental processes, and responses to the environment (Aikawa et al., 2010; De Vos et al., 2012; Katsuragi et al., 2013; Satake et al., 2013; Miyazaki et al., 2014). Mathematical modeling approaches are also used to understand a wide range of biological functions in growth, survival, and reproduction in plants, for example, in the circadian regulation of plant carbohydrate metabolism (Webb and Satake, 2015), phloem sucrose transport associated with rice grain yield (Seki et al., 2015), and silicon uptake in rice roots (Sakurai et al., 2015).
Machine learning is a field of computer science for the design of computational algorithms that automatically improve with experience. In the last two decades, this research field has dramatically advanced with the emergence of artificial intelligence and data science, and has been applied in various fields in science, technology and commerce (Jordan and Mitchell, 2015). Machine learning is also used in applications for the analysis of genome-scale datasets and other large-scale omics datasets in life science (Libbrecht and Noble, 2015). Learning methods in machine learning are usually classified into two primary categories of supervised and unsupervised learning. The supervised machine learning aims to produce an algorithm to predict output on unknown input via a training process using a dataset of known pairs of input and output. The unsupervised machine learning methods are used to extract structures and identify their features in a given dataset without examples for training. Computational modeling using machine learning has been performed recently with the aim of predicting gene networks based on large-scale transcriptome datasets in plants. For example, in Arabidopsis, supervised machine learning was used to build a network model of responses to stress conditions, to explore genes related to stress responses, and to predict molecular interactions (Ma et al., 2014; Nourani et al., 2015). Machine learning provides a data-driven approach to extract latent rules or patterns from a comprehensively collected dataset without any biased view on the biological phenomena of interest (Figure 2).
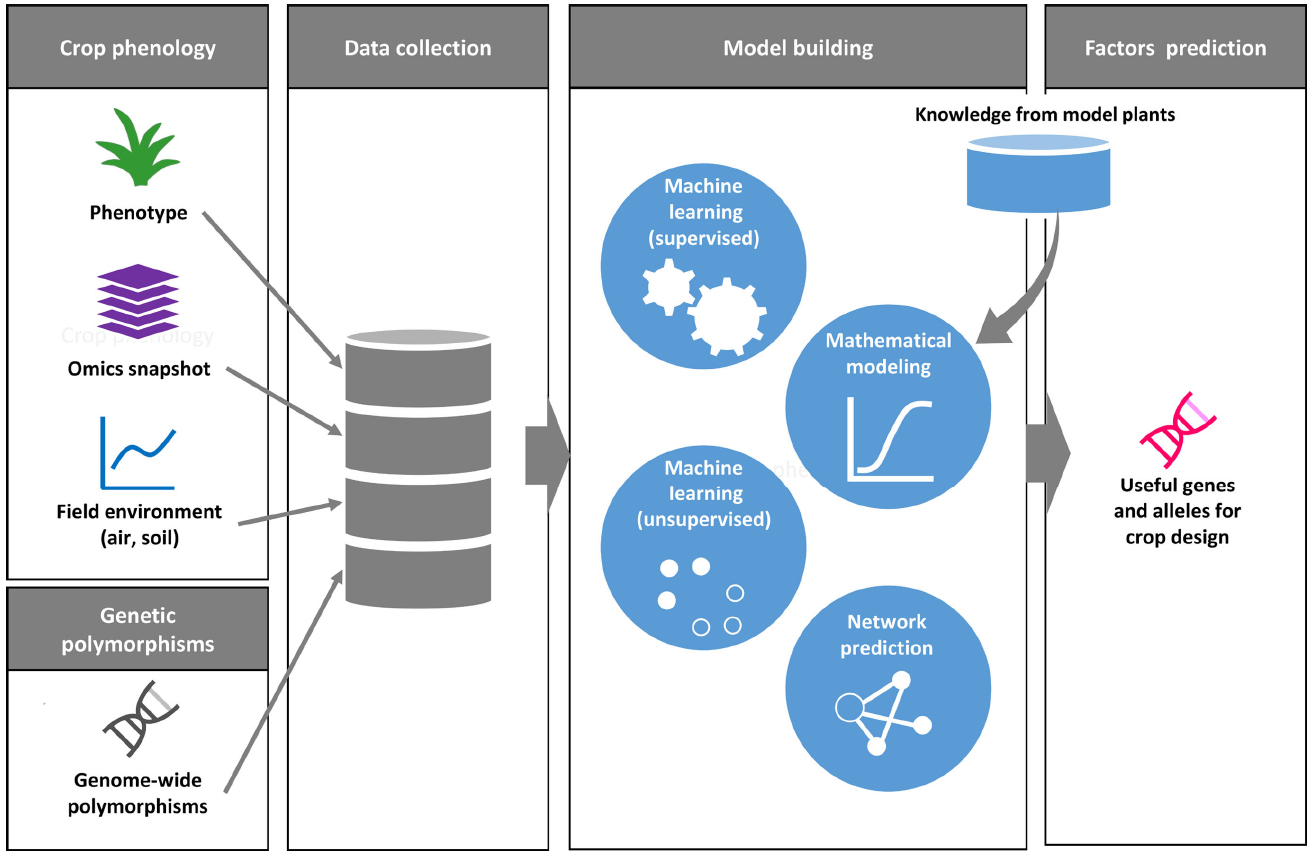
Figure 2. A conceptual framework for phenology data driven crop design. Phenology datasets include plant phenotypes, omics snapshots with environmental data. A genome-wide polymorphism dataset is useful to find genetic association between accessions with traits as well as with physiological state. For model building, the collected data are applied to various types of computer-aided methods. Digitized datasets can also be assessed against data on well-characterized gene functions in model plants. Machine learning-based data clustering and network prediction may help us to identify candidate genes and alleles for crop improvement.
Conclusion and Future Perspectives
Cross-disciplinary research, including computer science, functional genomics, and crop phenology, will provide a unique opportunity to establish technologies for data-driven crop design to prevent crop yield shortfall under changing future environments. It is expected that a synergy of life science and data science will allow us to perceive novel and latent values underlying the observed dataset by unbiased data-driven analyses. Unbiased illustration of physiological state dynamics of crops growing under field conditions could be an efficient strategy to figure out features of genetic factors but also “state factors” that determine eventual agronomical traits. Another issue for the data-driven approach is how we fill the gap between findings from model plants studied in laboratories and those from crops under field conditions to generalize our knowledge on plant systems including those in response to environmental changes. Complementary use of hypothesis-driven and data-driven approaches should be a practical way for further understanding of physiological responses to field environments with cross-referencing to knowledge from model plants that has been accumulated in laboratories. Therefore, platforms for computing and linking life science data will also play more significant roles in research on data-driven crop breeding.
Author Contributions
KM, DS, and TH conceived research and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by Joint Research Program implemented at the Institute of Plant Science and Resources, Okayama University in Japan (Grant No. 2641 to KM and DS), and by Grant-in-Aid for Scientific Research (B) (Grant No. 15KT0038 to KM, DS, and HT) of the Japan Society for the Promotion of Science (JSPS). This work was partially supported by Grants-in-Aid for Young Scientists (A) (Grant No. 26712003 to KM), and by Grant-in-Aid for Scientific Research on Innovative Areas (Grant No. 23119524 to KM and Grant No. 25119716 to DS) of JSPS. This work was partially supported by funds to KM from the Advanced Low Carbon Technology Research and Development Program (ALCA, J2013403C) of the Japan Science and Technology Agency (JST).
References
Adamski, J., and Suhre, K. (2013). Metabolomics platforms for genome wide association studies—linking the genome to the metabolome. Curr. Opin. Biotechnol. 24, 39–47. doi: 10.1016/j.copbio.2012.10.003
Aikawa, S., Kobayashi, M. J., Satake, A., Shimizu, K. K., and Kudoh, H. (2010). Robust control of the seasonal expression of the Arabidopsis FLC gene in a fluctuating environment. Proc. Natl. Acad. Sci. U.S.A. 107, 11632–11637. doi: 10.1073/pnas.0914293107
Arai-Kichise, Y., Shiwa, Y., Nagasaki, H., Ebana, K., Yoshikawa, H., Yano, M., et al. (2011). Discovery of genome-wide DNA polymorphisms in a landrace cultivar of Japonica rice by whole-genome sequencing. Plant Cell Physiol. 52, 274–282. doi: 10.1093/pcp/pcr003
Balmer, D., Flors, V., Glauser, G., and Mauch-Mani, B. (2013). Metabolomics of cereals under biotic stress: current knowledge and techniques. Front. Plant Sci. 4:82. doi: 10.3389/fpls.2013.00082
Busemeyer, L., Mentrup, D., Moller, K., Wunder, E., Alheit, K., Hahn, V., et al. (2013). BreedVision—a multi-sensor platform for non-destructive field-based phenotyping in plant breeding. Sensors (Basel) 13, 2830–2847. doi: 10.3390/s130302830
Causse, M., Desplat, N., Pascual, L., Le Paslier, M. C., Sauvage, C., Bauchet, G., et al. (2013). Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genomics 14:791. doi: 10.1186/1471-2164-14-791
Chinnusamy, V., and Zhu, J. K. (2009). Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 12, 133–139. doi: 10.1016/j.pbi.2008.12.006
Dal Santo, S., Tornielli, G. B., Zenoni, S., Fasoli, M., Farina, L., Anesi, A., et al. (2013). The plasticity of the grapevine berry transcriptome. Genome Biol. 14, r54. doi: 10.1186/gb-2013-14-6-r54
Deshmukh, R., Sonah, H., Patil, G., Chen, W., Prince, S., Mutava, R., et al. (2014). Integrating omic approaches for abiotic stress tolerance in soybean. Front. Plant Sci. 5:244. doi: 10.3389/fpls.2014.00244
De Vos, D., Dzhurakhalov, A., Draelants, D., Bogaerts, I., Kalve, S., Prinsen, E., et al. (2012). Towards mechanistic models of plant organ growth. J. Exp. Bot. 63, 3325–3337. doi: 10.1093/jxb/ers037
Dinakar, C., and Bartels, D. (2013). Desiccation tolerance in resurrection plants: new insights from transcriptome, proteome and metabolome analysis. Front. Plant Sci. 4:482. doi: 10.3389/fpls.2013.00482
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. doi: 10.1371/journal.pone.0019379
Evans, L. M., Slavov, G. T., Rodgers-Melnick, E., Martin, J., Ranjan, P., Muchero, W., et al. (2014). Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat. Genet. 46, 1089–1096. doi: 10.1038/ng.3075
Fahlgren, N., Gehan, M. A., and Baxter, I. (2015). Lights, camera, action: high-throughput plant phenotyping is ready for a close-up. Curr. Opin. Plant Biol. 24, 93–99. doi: 10.1016/j.pbi.2015.02.006
Fujii, M., Yokosho, K., Yamaji, N., Saisho, D., Yamane, M., Takahashi, H., et al. (2012). Acquisition of aluminium tolerance by modification of a single gene in barley. Nat. Commun. 3, 713. doi: 10.1038/ncomms1726
Fukushima, A., and Kusano, M. (2013). Recent progress in the development of metabolome databases for plant systems biology. Front. Plant Sci. 4:73. doi: 10.3389/fpls.2013.00073
Gibson, G. (2008). The environmental contribution to gene expression profiles. Nat. Rev. Genet. 9, 575–581. doi: 10.1038/nrg2383
Grosskinsky, D. K., Svensgaard, J., Christensen, S., and Roitsch, T. (2015). Plant phenomics and the need for physiological phenotyping across scales to narrow the genotype-to-phenotype knowledge gap. J. Exp. Bot. 66, 5429–5440. doi: 10.1093/jxb/erv345
Hirayama, T., and Shinozaki, K. (2010). Research on plant abiotic stress responses in the post-genome era: past, present and future. Plant J. 61, 1041–1052. doi: 10.1111/j.1365-313X.2010.04124.x
Huang, X., and Han, B. (2014). Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 65, 531–551. doi: 10.1146/annurev-arplant-050213-035715
Huang, X., Kurata, N., Wei, X., Wang, Z. X., Wang, A., Zhao, Q., et al. (2012). A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501. doi: 10.1038/nature11532
Ikeda, Y. (2012). Plant imprinted genes identified by genome-wide approaches and their regulatory mechanisms. Plant Cell Physiol. 53, 809–816. doi: 10.1093/pcp/pcs049
Jordan, M. I., and Mitchell, T. M. (2015). Machine learning: trends, perspectives, and prospects. Science 349, 255–260. doi: 10.1126/science.aaa8415
Kalisz, S., and Purugganan, M. D. (2004). Epialleles via DNA methylation: consequences for plant evolution. Trends Ecol. Evol. 19, 309–314. doi: 10.1016/j.tree.2004.03.034
Kanno, Y., Jikumaru, Y., Hanada, A., Nambara, E., Abrams, S. R., Kamiya, Y., et al. (2010). Comprehensive hormone profiling in developing Arabidopsis seeds: examination of the site of ABA biosynthesis, ABA transport and hormone interactions. Plant Cell Physiol. 51, 1988–2001. doi: 10.1093/pcp/pcq158
Katsuragi, T., Ono, N., Yasumoto, K., Altaf-Ul-Amin, M., Hirai, M. Y., Sriyudthsak, K., et al. (2013). SS-mPMG and SS-GA: tools for finding pathways and dynamic simulation of metabolic networks. Plant Cell Physiol. 54, 728–739. doi: 10.1093/pcp/pct052
Kim, J. M., To, T. K., and Seki, M. (2012). An epigenetic integrator: new insights into genome regulation, environmental stress responses and developmental controls by histone deacetylase 6. Plant Cell Physiol. 53, 794–800. doi: 10.1093/pcp/pcs004
Kinoshita, T., and Seki, M. (2014). Epigenetic memory for stress response and adaptation in plants. Plant Cell Physiol. 55, 1859–1863. doi: 10.1093/pcp/pcu125
Kjaer, K. H., and Ottosen, C. O. (2015). 3D Laser Triangulation for Plant Phenotyping in Challenging Environments. Sensors (Basel) 15, 13533–13547. doi: 10.3390/s150613533
Klukas, C., Chen, D., and Pape, J. M. (2014). Integrated analysis platform: an open-source information system for high-throughput plant phenotyping. Plant Physiol. 165, 506–518. doi: 10.1104/pp.113.233932
Kojima, M., Kamada-Nobusada, T., Komatsu, H., Takei, K., Kuroha, T., Mizutani, M., et al. (2009). Highly sensitive and high-throughput analysis of plant hormones using MS-probe modification and liquid chromatography-tandem mass spectrometry: an application for hormone profiling in Oryza sativa. Plant Cell Physiol. 50, 1201–1214. doi: 10.1093/pcp/pcp057
Kuromori, T., Takahashi, S., Kondou, Y., Shinozaki, K., and Matsui, M. (2009). Phenome analysis in plant species using loss-of-function and gain-of-function mutants. Plant Cell Physiol. 50, 1215–1231. doi: 10.1093/pcp/pcp078
Kuromori, T., Wada, T., Kamiya, A., Yuguchi, M., Yokouchi, T., Imura, Y., et al. (2006). A trial of phenome analysis using 4000 Ds-insertional mutants in gene-coding regions of Arabidopsis. Plant J. 47, 640–651. doi: 10.1111/j.1365-313X.2006.02808.x
Lai, J., Li, R., Xu, X., Jin, W., Xu, M., Zhao, H., et al. (2010). Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 42, 1027–1030. doi: 10.1038/ng.684
Li, J. Y., Wang, J., and Zeigler, R. S. (2014a). The 3,000 rice genomes project: new opportunities and challenges for future rice research. Gigascience 3, 8. doi: 10.1186/2047-217X-3-8
Li, L., Zhang, Q., and Huang, D. (2014b). A review of imaging techniques for plant phenotyping. Sensors (Basel) 14, 20078–20111. doi: 10.3390/s141120078
Li, Y. H., Zhao, S. C., Ma, J. X., Li, D., Yan, L., Li, J., et al. (2013). Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genomics 14:579. doi: 10.1186/1471-2164-14-579
Libbrecht, M. W., and Noble, W. S. (2015). Machine learning applications in genetics and genomics. Nat. Rev. Genet. 16, 321–332. doi: 10.1038/nrg3920
Liebisch, F., Kirchgessner, N., Schneider, D., Walter, A., and Hund, A. (2015). Remote, aerial phenotyping of maize traits with a mobile multi-sensor approach. Plant Methods 11, 9. doi: 10.1186/s13007-015-0048-8
Lin, T., Zhu, G., Zhang, J., Xu, X., Yu, Q., Zheng, Z., et al. (2014). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226. doi: 10.1038/ng.3117
Liu, J., Feng, L., Li, J., and He, Z. (2015). Genetic and epigenetic control of plant heat responses. Front. Plant Sci. 6:267. doi: 10.3389/fpls.2015.00267
Ma, C., Xin, M., Feldmann, K. A., and Wang, X. (2014). Machine learning-based differential network analysis: a study of stress-responsive transcriptomes in Arabidopsis. Plant Cell 26, 520–537. doi: 10.1105/tpc.113.121913
Matsuda, F., Nakabayashi, R., Yang, Z., Okazaki, Y., Yonemaru, J., Ebana, K., et al. (2015). Metabolome-genome-wide association study dissects genetic architecture for generating natural variation in rice secondary metabolism. Plant J. 81, 13–23. doi: 10.1111/tpj.12681
Matsuzaki, J., Kawahara, Y., and Izawa, T. (2015). Punctual transcriptional regulation by the rice circadian clock under fluctuating field conditions. Plant Cell 27, 633–648. doi: 10.1105/tpc.114.135582
Mittler, R., and Blumwald, E. (2010). Genetic engineering for modern agriculture: challenges and perspectives. Annu. Rev. Plant Biol. 61, 443–462. doi: 10.1146/annurev-arplant-042809-112116
Miyazaki, Y., Maruyama, Y., Chiba, Y., Kobayashi, M. J., Joseph, B., Shimizu, K. K., et al. (2014). Nitrogen as a key regulator of flowering in Fagus crenata: understanding the physiological mechanism of masting by gene expression analysis. Ecol. Lett. 17, 1299–1309. doi: 10.1111/ele.12338
Mochida, K., Furuta, T., Ebana, K., Shinozaki, K., and Kikuchi, J. (2009). Correlation exploration of metabolic and genomic diversity in rice. BMC Genomics 10:568. doi: 10.1186/1471-2164-10-568
Mochida, K., and Shinozaki, K. (2010). Genomics and bioinformatics resources for crop improvement. Plant Cell Physiol. 51, 497–523. doi: 10.1093/pcp/pcq027
Mochida, K., and Shinozaki, K. (2011). Advances in omics and bioinformatics tools for systems analyses of plant functions. Plant Cell Physiol. 52, 2017–2038. doi: 10.1093/pcp/pcr153
Mochida, K., and Shinozaki, K. (2013). Unlocking Triticeae genomics to sustainably feed the future. Plant Cell Physiol. 54, 1931–1950. doi: 10.1093/pcp/pct163
Mochida, K., Uehara-Yamaguchi, Y., Yoshida, T., Sakurai, T., and Shinozaki, K. (2011). Global landscape of a co-expressed gene network in barley and its application to gene discovery in Triticeae crops. Plant Cell Physiol. 52, 785–803. doi: 10.1093/pcp/pcr035
Morris, G. P., Ramu, P., Deshpande, S. P., Hash, C. T., Shah, T., Upadhyaya, H. D., et al. (2013). Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. U.S.A. 110, 453–458. doi: 10.1073/pnas.1215985110
Nagano, A. J., Sato, Y., Mihara, M., Antonio, B. A., Motoyama, R., Itoh, H., et al. (2012). Deciphering and prediction of transcriptome dynamics under fluctuating field conditions. Cell 151, 1358–1369. doi: 10.1016/j.cell.2012.10.048
Nakashima, K., Yamaguchi-Shinozaki, K., and Shinozaki, K. (2014). The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 5:170. doi: 10.3389/fpls.2014.00170
Nourani, E., Khunjush, F., and Durmus, S. (2015). Computational approaches for prediction of pathogen–host protein–protein interactions. Front. Microbiol. 6:94. doi: 10.3389/fmicb.2015.00094
Obayashi, T., Okamura, Y., Ito, S., Tadaka, S., Aoki, Y., Shirota, M., et al. (2014). ATTED-II in 2014: evaluation of gene coexpression in agriculturally important plants. Plant Cell Physiol. 55, e6. doi: 10.1093/pcp/pct178
Onda, Y., Hashimoto, K., Yoshida, T., Sakurai, T., Sawada, Y., Hirai, M. Y., et al. (2015). Determination of growth stages and metabolic profiles in Brachypodium distachyon for comparison of developmental context with Triticeae crops. Proc. Biol. Sci. 282.
Poland, J. A., Brown, P. J., Sorrells, M. E., and Jannink, J. L. (2012). Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253. doi: 10.1371/journal.pone.0032253
Richards, C. L., Rosas, U., Banta, J., Bhambhra, N., and Purugganan, M. D. (2012). Genome-wide patterns of Arabidopsis gene expression in nature. PLoS Genet. 8, 482–495. doi: 10.1371/journal.pgen.1002662
Romay, M. C., Millard, M. J., Glaubitz, J. C., Peiffer, J. A., Swarts, K. L., Casstevens, T. M., et al. (2013). Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 14, R55. doi: 10.1186/gb-2013-14-6-r55
Saisho, D., Ishii, M., Hori, K., and Sato, K. (2011). Natural variation of barley vernalization requirements: implication of quantitative variation of winter growth habit as an adaptive trait in East Asia. Plant Cell Physiol. 52, 775–784. doi: 10.1093/pcp/pcr046
Saisho, D., and Purugganan, M. D. (2007). Molecular phylogeography of domesticated barley traces expansion of agriculture in the Old World. Genetics 177, 1765–1776. doi: 10.1534/genetics.107.079491
Sakurai, G., Satake, A., Yamaji, N., Mitani-Ueno, N., Yokozawa, M., Feugier, F. G., et al. (2015). In silico simulation modeling reveals the importance of the Casparian strip for efficient silicon uptake in rice roots. Plant Cell Physiol. 56, 631–639. doi: 10.1093/pcp/pcv017
Sakurai, T., Yamada, Y., Sawada, Y., Matsuda, F., Akiyama, K., Shinozaki, K., et al. (2013). PRIMe Update: innovative content for plant metabolomics and integration of gene expression and metabolite accumulation. Plant Cell Physiol. 54, e5. doi: 10.1093/pcp/pcs184
Satake, A., Kawagoe, T., Saburi, Y., Chiba, Y., Sakurai, G., and Kudoh, H. (2013). Forecasting flowering phenology under climate warming by modelling the regulatory dynamics of flowering-time genes. Nat. Commun. 4, 2303. doi: 10.1038/ncomms3303
Sato, Y., Antonio, B., Namiki, N., Motoyama, R., Sugimoto, K., Takehisa, H., et al. (2011). Field transcriptome revealed critical developmental and physiological transitions involved in the expression of growth potential in japonica rice. BMC Plant Biol. 11:10. doi: 10.1186/1471-2229-11-10
Schmitz, R. J., Schultz, M. D., Urich, M. A., Nery, J. R., Pelizzola, M., Libiger, O., et al. (2013). Patterns of population epigenomic diversity. Nature 495, 193–198. doi: 10.1038/nature11968
Schmutz, J., Mcclean, P. E., Mamidi, S., Wu, G. A., Cannon, S. B., Grimwood, J., et al. (2014). A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 46, 707–713. doi: 10.1038/ng.3008
Seki, M., Feugier, F. G., Song, X. J., Ashikari, M., Nakamura, H., Ishiyama, K., et al. (2015). A mathematical model of phloem sucrose transport as a new tool for designing rice panicle structure for high grain yield. Plant Cell Physiol. 56, 605–619. doi: 10.1093/pcp/pcu191
Sonah, H., O’Donoughue, L., Cober, E., Rajcan, I., and Belzile, F. (2015). Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 13, 211–221. doi: 10.1111/pbi.12249
Subbaiyan, G. K., Waters, D. L., Katiyar, S. K., Sadananda, A. R., Vaddadi, S., and Henry, R. J. (2012). Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 10, 623–634. doi: 10.1111/j.1467-7652.2011.00676.x
Tohge, T., Mettler, T., Arrivault, S., Carroll, A. J., Stitt, M., and Fernie, A. R. (2011). From models to crop species: caveats and solutions for translational metabolomics. Front. Plant Sci. 2:61. doi: 10.3389/fpls.2011.00061
Vankudavath, R. N., Bodanapu, R., Sreelakshmi, Y., and Sharma, R. (2012). High-throughput phenotyping of plant populations using a personal digital assistant. Methods Mol. Biol. 918, 97–116. doi: 10.1007/978-1-61779-995-2_8
Webb, A. A., and Satake, A. (2015). Understanding circadian regulation of carbohydrate metabolism in Arabidopsis using mathematical models. Plant Cell Physiol. 56, 586–593. doi: 10.1093/pcp/pcv033
Weigel, D., and Colot, V. (2012). Epialleles in plant evolution. Genome Biol. 13, 249. doi: 10.1186/gb-2012-13-10-249
Wen, W., Li, D., Li, X., Gao, Y., Li, W., Li, H., et al. (2014). Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 5, 3438. doi: 10.1038/ncomms4438
Woods, D. P., Ream, T. S., and Amasino, R. M. (2014). Memory of the vernalized state in plants including the model grass Brachypodium distachyon. Front. Plant Sci. 5:99. doi: 10.3389/fpls.2014.00099
Yang, W., Guo, Z., Huang, C., Duan, L., Chen, G., Jiang, N., et al. (2014). Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat. Commun. 5, 5087. doi: 10.1038/ncomms6087
Keywords: population genomics, transcriptome, epigenome, crop phenology, machine learning
Citation: Mochida K, Saisho D and Hirayama T (2015) Crop improvement using life cycle datasets acquired under field conditions. Front. Plant Sci. 6:740. doi: 10.3389/fpls.2015.00740
Received: 05 June 2015; Accepted: 31 August 2015;
Published: 22 September 2015.
Edited by:
Girdhar K. Pandey, University of Delhi, IndiaReviewed by:
Bjoern Usadel, RWTH Aachen University, GermanyKiyosumi Hori, National Institute of Agrobiological Sciences, Japan
Copyright © 2015 Mochida, Saisho and Hirayama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keiichi Mochida, Cellulose Production Research Team, Biomass Engineering Research Division, RIKEN Center for Sustainable Resource Science, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan,a2VpaWNoaS5tb2NoaWRhQHJpa2VuLmpw