 Mingming Xin
Mingming Xin Guanghui Yang
Guanghui Yang- State Key Laboratory for Agrobiotechnology, Key Laboratory of Crop Heterosis Utilization (MOE), Beijing Key Laboratory of Crop Genetic Improvement, China Agricultural University, Beijing, China
In angiosperms, the endosperm nurtures the embryo and provides nutrients for seed germination. To identify the expression pattern of small interfering RNA in the developing maize endosperm, we have performed high-throughput small RNA transcriptome sequencing of kernels at 0, 3, and 5 days after pollination (DAP) and endosperms at 7, 10, and 15 DAP using B73 and Mo17 reciprocal crosses in previous study. Here, we further explored these small RNA-seq data to investigate the potential roles of miRNAs in regulating the gene expression process. In total, 57 conserved miRNAs and 18 novel miRNAs were observed highly expressed in maize endosperm. Temporal expression profiling indicated that these miRNAs exhibited dynamic and partitioned expression patterns at different developmental stages between maize reciprocal crosses, and quantitative RT-PCR results further confirmed our observation. In addition, we found a subset of distinct tandem miRNAs are generated from a single stem-loop structure in maize that might be conserved in monocots. Furthermore, a SNP variation of Zma-miR408-5p at 11th base position was characterized between B73 and Mo17 which might lead to completely different functions in repressing targets. More interestingly, Zma-miR408-5p exhibited B73-biased expression pattern in the B73 and Mo17 reciprocal hybrid endosperms at 7, 10, and 15 DAP according to the reads abundance with SNPs and CAPS experiment. Together, this study suggests that miRNA plays a crucial role in regulating endosperm development, and exhibited distinct expression patterns in developing endosperm between maize reciprocal crosses.
Introduction
In flowering plants, the triploid endosperm is derived from the fertilization of the central cell (2n) and the sperm cell (1n) (Yadegari and Drews, 2004). Although it is a terminal tissue, it plays a crucial role not in only accumulating nutrients but also in supporting the development of the diploid embryo. In maize, the endosperm initiates a phase of syncytial development upon fertilization, where the nucleus undergoes mitosis in the absence of cytokinesis. This process is followed by a period of cellularization at approximately 3 days after pollination (DAP), during which existing endosperm nuclei are bound by cell walls (Berger, 2003; Olsen, 2004; Sabelli and Larkins, 2009). Subsequently, intensive cell division occurs. Following these proliferative processes, the differentiation, and specialization of endosperm cells begins at ~4–6 DAP and results in four primary endosperm compartments: the starchy endosperm, the basal endosperm transfer layer, the aleurone, and the embryo-surrounding region. At approximately 10 DAP, nutrient uptake and zein storage protein synthesis initiate, coincident with the rapid increase of auxin accumulation. Finally at ~15 DAP, the rate of fresh weight grain reaches a peak (Lur and Setter, 1993).
Temporal transcriptome profiling has revealed that gene expression patterns are dramatically changed during the development of maize endosperms (Li et al., 2014). These development-dependent genes involved in distinct functional categories, including amino acid biosynthesis, carbohydrate biosynthesis, and energy reserve metabolic processes etc (Li et al., 2014). Evidence shows that the disruption of gene expression in the maize endosperm is likely to result in a developmental defection and lead to the failure of seed maturation, possibly due to an inability to provide nutrients and signals for embryo growth. For example, the mutation of miniature1, encoding an endosperm-specific isoform of cell wall invertase, reduces kernel fresh weight by approximately 70% by negatively controlling sink strength (Cheng et al., 1996). In addition, the misexpression of the endosperm-specific gene Meg1 results in malformed transfer cells, which in turn adversely regulates nutrient uptake, sucrose partitioning, and finally seed biomass yield (Costa et al., 2012). These findings indicate precise that the regulation of gene expression in endosperm is required to coordinate proper seed development (Berger et al., 2006).
MiRNAs, which range from 20 to 24 nucleotides (nt) in length, negatively control gene expression through transcript cleavage or translation inhibition through near-perfect complementarity to their targets (Voinnet, 2009). With the advent of high-throughput sequencing technologies, diverse sets of miRNAs have been identified in different plant species on a genome-wide level, exhibiting tissue-specific or/and development-dependent expression patterns (Xue et al., 2009; Jiao et al., 2011; Sun et al., 2014), and our understanding of the functions of these regulatory molecules has also been greatly improved. Various miRNAs have been shown to regulate genes involved in cell differentiation, hormone signaling, stress responses, and organ development, including seeds (Mallory and Vaucheret, 2006). In wheat, 605 miRNAs were identified from developing grains at 5, 15, 25, and 30 DAP. Compared to miRNA abundance in 5-DAP kernels, 114, 147, and 211 miRNAs were found to be significantly up-regulated at the later stages, respectively, among which 86 were potentially associated with the control of grain-filling (Meng et al., 2013). In rice, Zhu et al. (2008) identified 59 miRNA families in two seed developmental stages of japonica species,—0–5 DAP and 6–10 DAPand found that their expression levels changed dynamically, with the highest expression ratio being 18.4:1 (6–10/0–5 DAP), which was observedfor miR408 (Zhu et al., 2008). Moreover, seven out of 36 miRNAs, including osa-miR167, osa-miR397, osa-miR398, osa-miR408, osa-miR528, osa-miR1866-3p, and osa-miRc11 were also preferentially expressed in rice seeds according to miRNA chip data in subspecies of japonica (Xue et al., 2009). In addition, a total of 112 indica miRNA families were discovered in developing grains, although a discrepancy in expression trends was observed in comparison to previous results (Xue et al., 2009; Lan et al., 2012). In maize, Gu et al. discovered 95 conserved miRNAs and 18 novel miRNAs expressed in 4–6- and 7–23-DAP endosperms, and nearly half of the conserved miRNAs (45/95) were shown to have higher expression levels in later stages than in earlier, including miR408 which is similar to the findings reported in rice (Zhu et al., 2008; Gu et al., 2013). Jiao et al. also identified 38 novel miRNAs from mixed maize endosperms harvested at 12, 16, 20, and 24 DAP (Jiao et al., 2011). Furthermore, gene ontology (GO) enrichment analysis demonstrated that the putative targets of these grain-abundant miRNAs could be involved in the regulation of carbohydrate and protein metabolism, transcription, and cellular transport functional groups, suggesting their crucial roles in gene regulatory networks in seed development (Xue et al., 2009; Lan et al., 2012; Gu et al., 2013; Meng et al., 2013).
However, the miRNA expression patterns in the reciprocal developing endosperm and how the gene expression of the filial genomes is regulated by miRNA have not been explored. More interestingly, the proportions of the parental miRNAs contribute to endosperm development remain unknown, although we have recently reported that a subset of siRNAs exhibited imprinted expression patterns in the developing maize endosperm (Xin et al., 2014). In the present study, we performed genome-wide analysis of endosperm-abundant miRNAs in B73 and Mo17 reciprocal kernels at 0, 3, and 5 DAP and in endosperms at 7, 10, 15 DAP, and investigated their dynamic and potentially biased expression patterns by using the small RNA-seq data published in our previous study (Xin et al., 2014). Our data revealed that all 18 newly identified miRNAs were enriched in the developing endosperms compared to the kernels and that they also showed partitioned expression levels in the reciprocal crosses. Additionally, we characterized a group of tandem miRNAs which were generated from a single precursor in maize. Finally, our data demonstrated that Zma-miR408-5p exhibited a parent-biased expression pattern based on the SNP information and Cleaved Amplified Polymorphic Sequence (CAPS) experiment, further suggesting that parental genomes might not contribute equally to controlling endosperm development in terms of miRNA-mediated regulation networks.
Materials and Methods
Identification of Novel miRNAs and Target Prediction
After removing adaptors, reads shorter than 15 nucleotides were discarded, and the rest were parsed into FASTA format. Then, the reads were mapped to the reference genome of ZmB73_RefGen_v3.tar.gz using the miRDeep-P program with default algorithms. Next, flanking sequence of approximate 250 bp were extracted to predict secondary structures, and stem-loop structures with qualifying scores were further processed by the miRDeep core algorithm. Only small RNAs consistent with the requirements for plant miRNAs were retained for further analysis. Finally, to obtain high-confidence miRNAs in maize endosperm, we only kept endosperm abundant small RNAs with “T” at the beginning of the 5' end. Putative targets prediction was performed using the psRNATarget interface server with default settings.
Quantitative RT-PCR Validation for Mature miRNA
The SYBR® PrimeScript miRNA RT-PCR Kit (Code No. RR716) was used following the manufacturer's instructions (TaKaRa). Briefly, the reverse transcription was performed by using 1 μg of total RNA incubated with 10 μL of 2 × miRNA Reaction Buffer Mix with Universal Adaptor Primer containing Oligo-dT, 2 μL of 0.1% BSA, 2 μL of miRNA PrimeScript RT Enzyme Mix, and 5 μL of RNase Free dH2O in a 20-μL reaction mixture. The temperature program was adjusted to run for 60 min at 37°C, 5 s at 85°C, and then 4°C forever, the Universal Adaptor Primer contained the Uni-miR qRT-PCR primer binding site, the universal reverse primer for qRT-PCR of miRNA.
QRT-PCR of miRNA was conducted on a Bio-Rad CFX96TM Real-Time System. Each reaction included 2 μL of product from the diluted RT reactions, 1.0 μL of miRNA primer (10 μM), 12.5 μL of SYBR Premix Ex Taq II(2 ×), and 8.5 μL of RNase Free water. The reactions were incubated in a 96-well plate at 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 59°C for 30 s, and 72°C for 30 s. All reactions were run in three replicates for each sample. The value of 2−ΔΔct and standard deviation were then calculated from three replicates, and statistical analysis was performed by using t-test (p < 0.05). The Zma-actin gene was used as the endogenous control. All of the primers used in these analyses are listed in Table S1, the Uni-miR qRT-PCR primer binding to Universal Adaptor Primer for the real time PCR reaction was provided by the kit.
Cleaved Amplified Polymorphic Sequence (CAPS)
Experimental validation of the biased expression patterns of Zma-miR408-5p was performed using CAPS assays as previously described (Konieczny and Ausubel, 1993). RT-PCR was performed using the primers listed in Table S1 which is available online. Amplification products were digested with the restriction enzyme McluI to differentiate the transcripts derived from the B73 and Mo17 alleles.
Results
Global Comparison of Conserved miRNA Profiling Reveals their Complexity During Maize Kernel and Endosperm Development
To study the potential roles of miRNAs in developing maize kernels and endosperms, 12 siRNA libraries of 0-, 3-, and 5-DAP kernels and 7-, 10-, and 15-DAP endosperms from the B73 and Mo17 reciprocal crosses were sequenced (Xin et al., 2014)to observe key events in endosperm development such as cellularization, proliferation, differentiation, and nutrient accumulation (Lopes and Larkins, 1993; Sabelli and Larkins, 2009). The majority siRNAs of our dataset were 24 nt in length and accounted for approximately 68% of total distinct reads, whereas ~12.5% siRNA reads belong to the 21-nt family, this result is similar to those of previous studies (Jiao et al., 2011; Gu et al., 2013) (Figure 1A). Further investigation revealed that a large proportion of 24-nt siRNA reads begin with A at the 5′ end (46.0% average, 29.2 ~ 60.3%), whereas more 21-nt siRNAs begin with T (54.4% average, 40.2 ~ 66.6%) (Figures 1B,C).
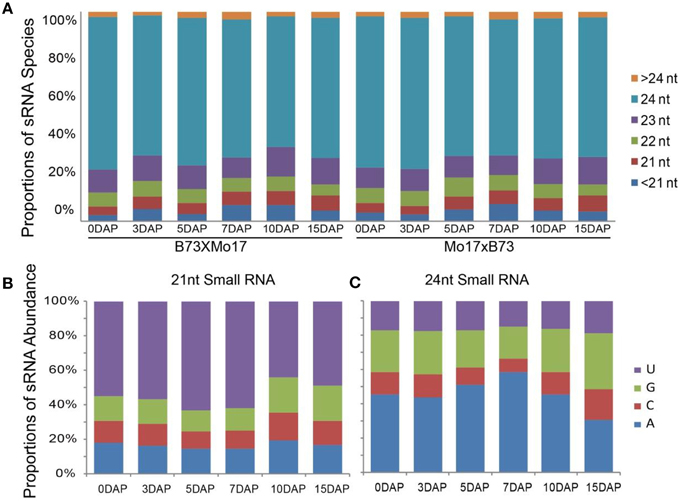
Figure 1. Distribution features of small RNA species in kernel and endosperm small RNA libraries. (A) Proportions of distinct small RNA spceies in 12 sequencing libraries. (B) Proportions of abundance for 21-nt small RNAs with A, C, G, and U at the beginning of the sequence, during kernel and endosperm developmental stages. (C) Proportions of abundance for 24-nt small RNAs with A, C, G, and U at the beginning of the sequenceduring kernel and endosperm developmental stages. DAP, days after pollination.
In previous study, we mainly focused on siRNA expression patterns, while here, we paid more attention to miRNA expression trends in the B73 and Mo17 reciprocal endosperms. After removing adaptors and low-quality sequences, the ~163.2 million filtered reads were aligned against mature maize miRNAs registered at miRBase (Release 21), idenfying 57 known miRNA members belonging to 25 families during maize kernel and endosperm developmental stages (Figure 2A). The expression levels of these miRNAs were normalized to the total reads of each library (RPM, Reads Per Million). Among them, Zma-miR168, Zma-miR166, Zma-miR156, Zma-miR528, Zma-miR827, and Zma-miR167 were the top six most abundantly expressed miRNAs in both reciprocal crosses, corresponding to more than 98% of the total number of known miRNAs (Table S2, Figure 2A). As predicted, a proportion of these conserved miRNAs exhibited temporal expression patterns in the kernels and endosperms, e.g., Zma-miR166 was predominantly expressed in 0-, 3-, and 5-DAP kernels, on average, and was ~8.7-fold higher than in 7, 10, 15 DAP endosperms. In contrast, the expression level of Zma-miR156 was significantly higher in the endosperm stages than in the kernel stages. In addition, Zma-miR528 and Zma-miR167 showed dramatically varied expression patterns during endosperm development. While the expression of Zma-miR528 was significantly down-regulated from 7- to 15-DAP endosperms, a rapid up-regulation of Zma-miR167 was observed at 10- and 15-DAP. Although the expression level of Zma-miR827 showed a sharp up-regulation at 15 DAP compared with 7 and 10 DAP, it was more highly expressed in maize kernels than in the endosperms. Unlikely, Zma-miR168 was consistently expressed with high levels during both kernels and endosperms, possibly because of its key role in regulating the production of miRNAs (Mallory and Vaucheret, 2006) (Figure 2A).
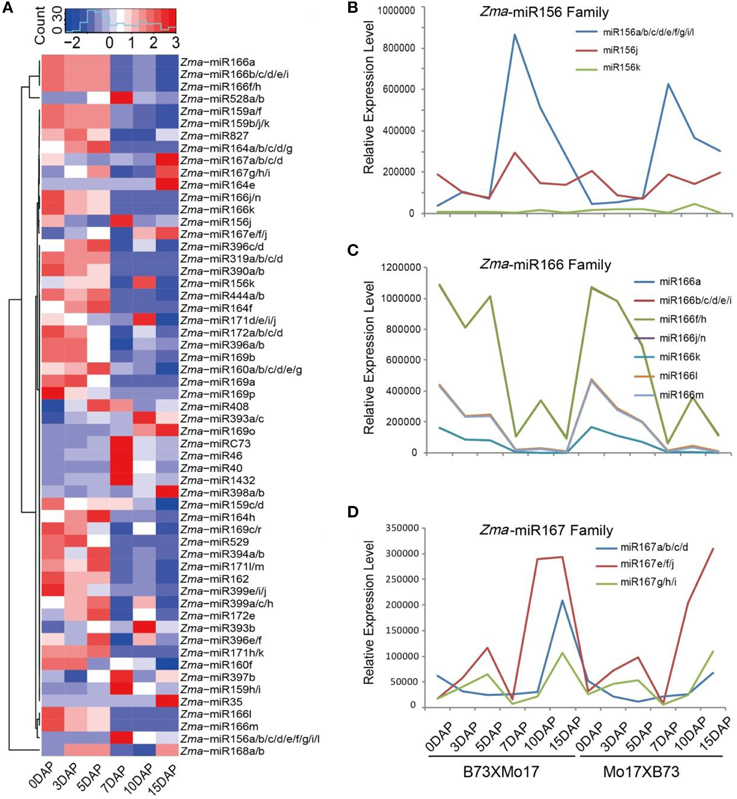
Figure 2. Conserved miRNA expression pattern during maize kernel and endosperm development in reciprocal crosses. (A) Hierarchical tandem of 57 conserved miRNAs with relatively high expression levels in at least one developmental stage. Diverse and tissue-specific miRNA expression patterns were exhibited, and a large proportion of miRNAs were highly expressed in kernels whereas only a few miRNAs were abundantly expressed during endosperm stages. (B) Dynamic and differential expression patterns of maize miR156 family members. The Zma-miR156a group has the highest expression level compared with other family members during all the developmental stages. (C) Kernel-abundant expression patterns of maize miR166 family members. Despite different expression levels, Zma-miR166 family members showed similar expression trends: they were highly expressed in 0-, 3-, and 5-DAP kernels but not in endosperms. (D) Endosperm-abundant expression patterns of maize miR167 family members. All Zma-miR167 members exhibited higher expression levels in endosperm stages than in the kernel stages. DAP, days after pollination.
Furthermore, we examined the expression patterns of different members in one miRNA family and observed a expression discrepancy among then. For example, the reads abundance and expression trends of Zma-miR156 family are mainly contributed by the member of Zma-miR156a, which peaks at 7 DAP and then decreases gradually at later stages, whereas Zma-miR156k and Zma-miR156j have low and moderate expression levels, respectively, and the latter is expressed during all kernel and endosperm stages with no obvious variation (Figure 2B). Unlike Zma-miR156, Zma-miR166 family members exhibited similar expression patterns, but their dynamic expression status was likely the results of the transient expression of Zma-miR166a/b/f in kernel stages (Figure 2C). In contrast, Zma-miR167 family members were all highly expressed in the endosperm stages, although they showed quantitative differences (Figure 2D).
Identification of Novel, Temporally Expressed miRNAs, and their Putative Targets during Maize Kernel and Endosperm Development
Upon double fertilization, endosperm undergoes a series of biological processes to form the nutritive tissue, which provides nutrients to support embryogenesis and seedling germination. To identify high-confidence miRNAs and investigate their expression profiles during developmental endosperms, we only focused on 21 nt small RNAs with T at the beginning of the 5′ end and reads number exceeding 100 rpm in 12 libraries. Finally, 18 novel putative miRNAs were identified in these six developmental stages based on the mirdeep-p program with the default algorithm (Yang and Li, 2011). Unexpectedly, all 18 miRNAs exhibit significantly increased expression levels in the endosperm compared to the kernels, corresponding to an average of 53.1-fold (2.4 ~ 410.0-fold) higher, although their expression levels among 7-, 10-, and 15-DAP endosperms varied (Table 1). Specifically, after combining the reads amount between two reciprocal crosses, four temporal gene-expression patterns were identified according to their expression trends in endosperms that we described as one-step-up, one-step-down (miRNA level transitions from low to high or high to low, respectively, in the consecutive 7-, 10-, 15-DAP stages), two-step-up-down or two step-down-up (miRNA level transitions from low to high and back down or from high to low and back up, respectively, in a series of endosperm developmental stages). The majority of newly identified miRNAs (14, 77.8%) exhibited a single transition point, of which 27.8% showed the one-step-up pattern, whereas 50% exhibited the one-step down pattern. In contrast, only 22.2% of miRNAs reached their expression peak or trough at 10 DAP (two-step-up-down or two step-down-up) (Figure 3A). Of these newly identified maize miRNAs, the most enriched is Zma-miR2001, whose abundance peaks at 7 DAP and then declines at 10 and 15 DAP; overall, its expression level in endosperms is significantly higher than in kernels by approximately 410.0-fold (Figure 3A). To validate the expression patterns of the newly identified miRNAs, quantitative RT-PCR were performed on mature miRNAs of Zma-miR2006 and Zma-miR2011, and their expression profiles were similar to the results obtained from RNA-seq data (Figure 3B).
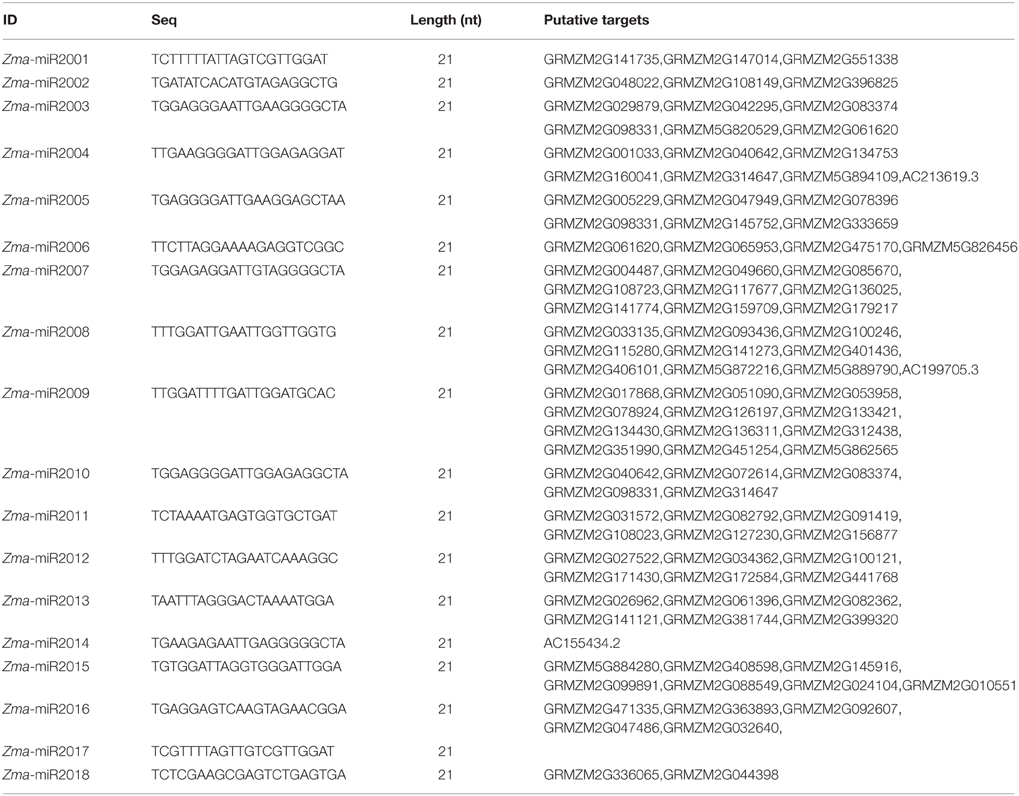
Table 1. New identified miRNAs and their putative targets.
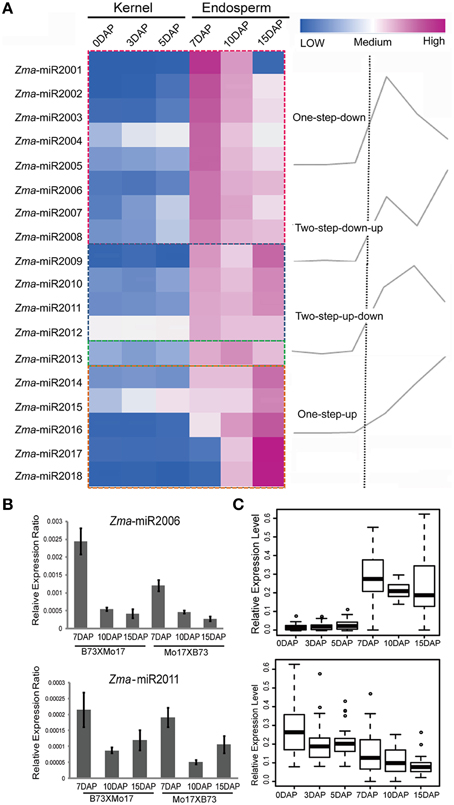
Figure 3. Developmentally dependent expression patterns of newly identified maize miRNAs negatively correlated with their targets expression. (A) 18 novel miRNAs were identified in 7-, 10-, and 15-DAP maize endosperms, which can be clustered into four groups according to their expression patterns, namely, one-step-down, one-step-up, two-step-down-up, and two-step-up-down. (B) Quantitative RT-PCR results of Zma-miR2006 and Zma-miR2011. Zma-miR2006 showed gradually decreased expression patterns in 7-, 10-, and 15-DAP endosperms in both reciprocal crosses, whereas Zma-miR2011 reached its lowest expression level in 10-DAP endosperm. (C) Boxplot of expression levels of novel miRNA and their putative targets in kernels and endosperms. MiRNA shows higher abundance in endosperm stages compared to kernel stages; in contrast, their targets exhibited lower expression levels in endosperm stages than in kernel stages, which is consistent with the negative correlation between miRNAs and their targets.
Subsequently, we predicted the putative targets of these abundantly endosperm-expressed miRNAs by running the psRNATarget program with default settings (http://plantgrn.noble.org/psRNATarget/?function=1). In total, 105 potential miRNA target genes were identified. To further confirm the correlation between miRNAs and target genes, we examined the expression patterns of these putative targets in our previous RNA-Seq data obtained in parallel using the same materials. Forty-seven out of 105 genes were found to be expressed (FPKM ≥ 1) in at least one of the developmental stages (Table S3), and interestingly, more than 80% of the target genes showed a higher expression level in the kernels than in endosperms. Further statistical analysis indicated that endosperm-abundant miRNAs have a significant negative correlation with their putative targets (r = −0.47, p = 8.15E-06) (Figure 3C).
Endosperm cell proliferation occurs at approximately 6–8 DAP (Sabelli and Larkins, 2009), suggesting that miRNAs with an expression pattern of one-step-down at 7 DAP are likely associated with cell cycle regulation. Accordingly, cyclin p4;1 (GRMZM2G029879) and cyclin-dependent kinase B2;1 (GRMZM2G141774) were found to be targeted by Zma-miR2003 and Zma-miR2007, respectively, which reached their peak expression levels in 7-DAP endosperm. Programmed cell death in the starch endosperm facilitates nutrient hydrolysis and uptake by the embryo at germination and begins at approximately 16 DAP in maize (Sabelli and Larkins, 2009). Consistently, senescence-related gene 1 (GRMZM2G363893) was potentially regulated by Zma-miR2016, which exhibited a one-step-up pattern in the 15-DAP endosperm. Interestingly, one of the putative targets of Zma-miR2010 is GRMZM2G083374, which encods a protein of unknown function but for which the mutation of its homolog in Arabidopsis caused a defective embryo. Therefore, although the exact biological functions of these miRNAs in the endosperm remain ambiguous, our results indicate that the dynamic activity and temporal expression patterns of miRNAs might play a role in the regulation of the maize endosperm or even embryo development.
MiRNAs Contribute Unequally Between Reciprocal Crosses during Maize Endosperm Development
Parental alleles are immediately activated in their filial genomes after fertilization, and their gene expression pattern, level, and timing are undoubtedly associated with plant development (Xin et al., 2013; Li et al., 2014). However, how miRNAs are expressed between reciprocal crosses during maize endosperm development remains undetermined. Our cytological analysis revealed that the endosperm in Mo17 × B73 undergoes faster development than that in B73 × Mo17 (Figure 4A), which led us to determine whether there are differences in the miRNA expression patterns between them. Not surprisingly, a proportion of miRNAs exhibited partitioned expression levels during kernel and endosperm developmental stages between B73 × Mo17 and Mo17 × B73, including both conserved and novel ones. For example, the expression levels of Zma-miR528 and Zma-miR397 were clearly higher in the Mo17 × B73 cross than in B73 × Mo17 in 5-DAP kernels and 7-DAP endosperms (Figure 4B). Similarly, Zma-miR408 also exhibited similar expression trends to Zma-miR528 and Zma-miR397, which might contribute to the phenotypic difference between these two reciprocal crosses (Figure 4B). It has been predicted that one target of miR528 and miR397 is the L-ascorbate oxidases (AO) gene, which would increase the accumulation of dehydroascorbate (DHA) and in turn restrict cell-division ability (Potters et al., 2000; Pignocchi et al., 2006). Therefore, a high abundance of miR397 and miR528 would directly repress the expression of AO and indirectly repress that of DHA, maintain a high rate of cell division, and thereby promote plant growth. Furthermore, the over-expression of miR408 promotes vegetative growth, but the knockdown of miR408 results in impaired growth in Arabidopsis; both of these trends are consistent with the rapid development of Mo17 × B73 kernels and endosperms (Zhang and Li, 2013).
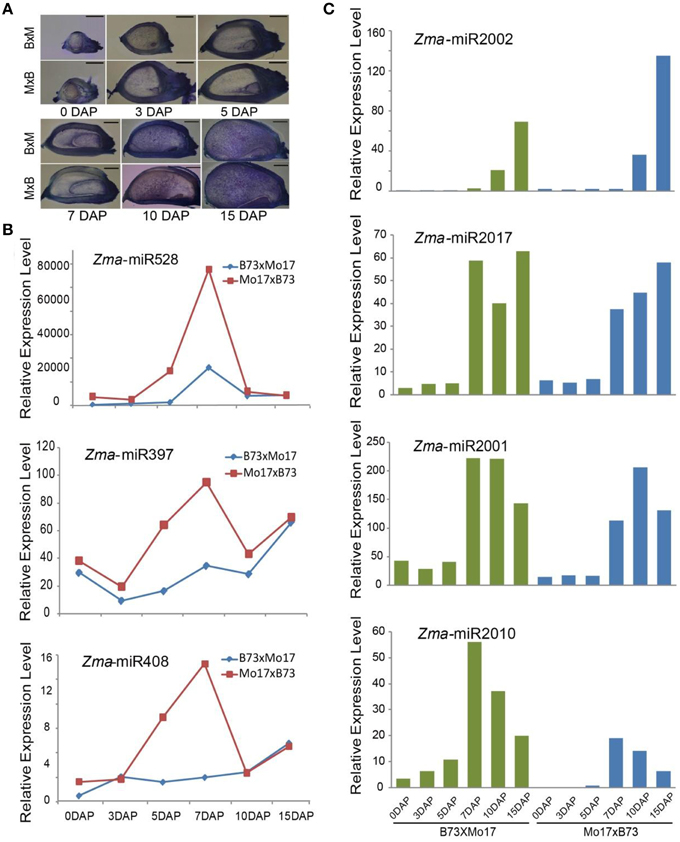
Figure 4. Phenotypic difference and temporal expression patterns of miRNAs in 0-, 3-, 5-DAP kernels and 7-, 10-, 15-DAP endosperms between B73 and Mo17 reciprocal crosses. (A) The kernel and endosperm of Mo17 × B73 grow faster than in B73 × Mo17 at all six stages. (B) Zma-miR528, Zma-miR397, and Zma-miR408 were more highly expressed in Mo17 × B73 compared to B73 × Mo17 in 5-DAP kernel and 7-DAP endosperm. (C) Newly identified Zma-miR2002, Zma-miR2017, Zma-miR2001, and Zma-miR2010 exhibited differential expression patterns between reciprocal crosses.
Similarly, although the expression level of Zma-miR2002 was predominantly expressed in the late stage of endosperms in both reciprocal crosses, it was approximately 2-fold higher in the Mo17 × B73 cross compared to B73 × Mo17 (Figure 4C). Moreover, Zma-miR2017 was barely expressed in all developing kernels, and its expression level exhibited a trough in the 10-DAP endosperm of the B73 × Mo17 cross but a gradual increase from 7 to 15 DAP in the Mo17 × B73 cross (Figure 4C). By contrast, we observed a peak expression level of Zma-miR2001 in the 10-DAP endosperm in the Mo17 × B73 cross but no significant change in the Mo17 × B73 cross (Figure 4C). In addition, several newly identified miRNAs were found to be expressed in kernels of one cross but not the other, including Zma-miR2010, Zma-miR2015, Zma-miR2016, and Zma-miR2018. Furthermore, the expression level of Zma-miR2010 exhibited a sharper decrease in B73 × Mo17 than in Mo17 × B73 (Figure 4C). Taken together, these results indicate that miRNAs might contribute to the phenotypic differences between reciprocal crosses in maize kernel and endosperm development.
Dynamic Expression of Tandem miRNAs Generated from a Common Hairpin Structure during Reciprocal Maize Endosperm Development
Recent analyses of massive amounts of data from high-throughput sequencing have identified subclasses of miRNA species derived from alternative biogenesis pathways except for the canonical miRNAs, including NAT-miRNA, miRtron, snoRNA-derived, and tRNA-derived miRNA (Miyoshi et al., 2010). In this study, we identified a group of miRNAs termed tandem miRNAs which were co-generated from a single hairpin locus. For example, in addition to Zma-miR319 and Zma-miR319*, their precursor also produces another pair of miRNA-like that were closer to the loop structure compared to Zma-miR319, these are defined as Zma-miR319.1 and Zma-miR319.1*, respectively (Figure 5A). Similarly, the precursor of Zma-miR169 also yields an accompanying miRNA (Zma-miR169.1), although its Zma-miR169.1* was not detected, possibly due to a relatively low expression level (Figure 5A). In addition, of the newly identified maize miRNAs, Zma-miR2001.1, and Zma-miR2013.1 were also detected during developing maize endosperm (Figure 5A). Interestingly, the precursors of five members of the miR159 family all exhibited this tandem miRNA expression pattern including Zma-miR159a, Zma-miR159b, Zma-miR159f, and Zma-miR159j/k (Figure S1A). Moreover, a previous study has reported that tandem miR159.1 and miR159.2 were co-produced in rice and its relatives (Lacombe et al., 2008), which raised an intriguing question of their evolution in plant species. Thus, we next examined the conservation of miR159 precursors among maize, rice, wheat, and barley, and surprisingly, that they all produced tandem miRNAs, although with several mismatches in miR159.1 among different species (Figure S1B). To summarize, tandem miRNA expression manner is evolutionarily conserved at least in monocots.
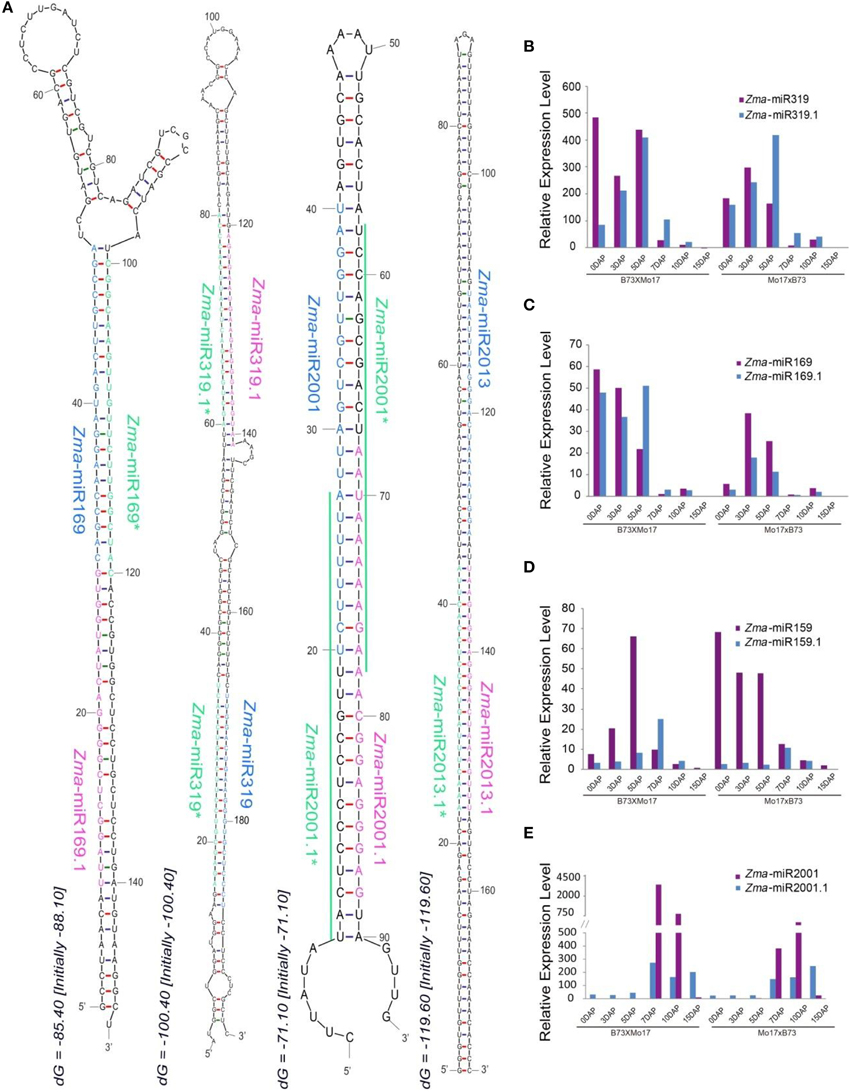
Figure 5. Biosynthesis and dynamic expression patterns of tandem miRNAs. (A) Stem-loop structures of tandem miRNAs. Zma-miR169&Zma-miR169.1, Zma-miR319 and Zma-miR319.1, Zma-miR2001 and Zma-miR2001.1, Zma-miR2013 and Zma-miR2013.1 were generated from a single precursor. (B) Varied expression patterns of Zma-miR319 and Zma-miR319.1 during kernel and endosperm developmental stages. (C) Dynamically changed expression patterns of Zma-miR169 and Zma-miR169.1 during kernel and endosperm developmental stages. (D) Temporal expression patterns of Zma-miR169 and Zma-miR169.1 during kernel and endosperm developmental stages. (E) Development-dependent expression patterns of Zma-miR2001 and Zma-miR2001.1 during kernel and endosperm developmental stages.
Further investigation indicated that these tandem miRNAs also exhibited altered expression patterns during kernel and endosperm development, e.g., both Zma-miR319.1 and Zma-miR169.1 were predominantly expressed in 0, 3, and 5 DAP kernels, whereas Zma-miR159.1 and Zma-miR2001.1 were preferentially expressed in endosperms (Figures 5B–E). In addition, these tandem miRNAs showed differential expression trends compared with their “neighbors” during the kernel and endosperm developmental stages in both reciprocal crosses, although they were located in a single hairpin locus. For example, the expression level of Zma-miR319.1 gradually increased in 0-, 3-, and 5-DAP kernels, unlike the expression pattern of Zma-miR319. In contrast to the observation in the kernels, Zma-miR319.1 exhibited a higher expression level in developing endosperms than that of Zma-miR319 (Figure 5B). Surprisingly, although the expression level of Zma-miR159 was clearly higher in kernels than in endosperms, Zma-miR159.1 exhibited the opposite expression patterns: its average expression level was higher in endosperms compared to kernels (Figure 5D). Moreover, the expression abundance of Zma-miR2001 was significantly enriched in 7- and 10-DAP endosperms compared with Zma-miR2001.1 in both reciprocal crosses; however, the expression patterns were reversed in 15-DAP endosperms (Figure 5E).
Parentally Biased Expression of Zma-miR408-5p in Maize Reciprocal Crosses
A proportion of small interfering RNAs exhibited possible parent-biased expression patterns in the developing plant endosperm between B73 and Mo17 reciprocal crosses (Xin et al., 2014), leading us to investigate whether miRNAs also showed allele-biased expression patterns. We first examined the SNPs located in the endosperm-abundant miRNAs between B73 and Mo17 cultivars. However, no informative SNPs were identified in these small regulatory molecules between the two inbred lines, probably because of their high evolutionary conservation, except for Zma-miR408*. Further comparison found Zma-miR408* was more abundantly expressed than Zma-miR408 in both kernels and endosperms, consistent with previous reports (Jiao et al., 2011; Gu et al., 2013) and indicating Zma-miR408* probably also has a biological relevance rather than the useless, degraded sequences. Therefore, we designed the original Zma-miR408 and Zma-miR408* to be Zma-miR408-3p and Zma-miR408-5p, respectively, based on 3′- or 5′-arm derivation of the miRNA species. Although the well-known conservation of miRNA, a SNP (A/G) between B73 and Mo17 inbred lines in mature Zma-miR408-5p sequence was identified and confirmed by sequencing (Figure 6A). Based on the SNP information, we next examined whether this miRNA is subjected to allele-biased expression between the two reciprocal crosses. Interestingly, Zma-miR408-5p showed a significantly B73-allele biased expression pattern in 7-, 10-, and 15-DAP reciprocal endosperms (Figure 6B, χ2-test, p-value ≤ 0.01), which was further confirmed by CAPS experiments (Figure 6C). Moreover, the SNP of Zma-miR408-5p was located at the 11th base position from the 5′ end between B73 and Mo17, indicating that this polymorphism was likely to cause functional variation of Zma-miR408-5p. Thus, these two should target complete different genes based on the principle of complementary base pairing because the 10/11 base position is very important for miRNA-directed mRNA cleavage (Figure 6B). The target prediction of B73 and Mo17 Zma-miR408-5p by the psRNATarget server is also consistent with our speculation. The putative targets of B73 Zma-miR408-5p were GRMZM2G034896, GRMZM2G093526, and GRMZM2G177518, while Mo17 Zma-miR408-5p likely negatively modulates five other targets, including AC196575.3_FG007, GRMZM2G169005, GRMZM2G162065, AC198481.3_FG004, and GRMZM2G349554.
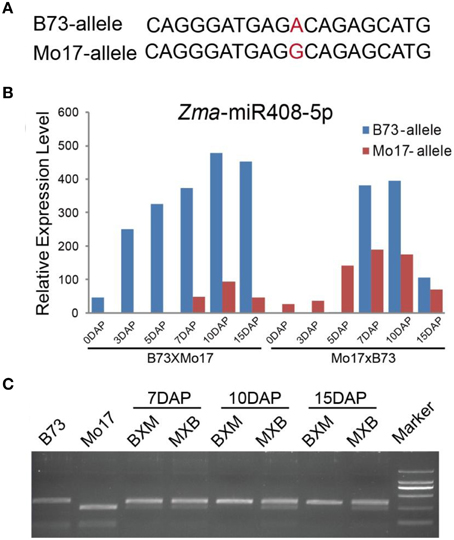
Figure 6. Biased expression pattern of Zma-miR408-5p in 7-, 10-, and 15-DAP endosperms in B73 and Mo17 reciprocal crosses. (A) SNP of Zma-miR408-5p between B73 and Mo17 confirmed by re-sequencing. (B) Zma-miR408-5p of B73 was more highly expressed compared to that of Mo17 during endosperm developmental stages in both reciprocal crosses based on the sequencing data. (C) Validation of biased expression pattern of Zma-miR408-5p using CAPS in 7, 10, and 15 DAP endosperms of reciprocal crosses.
Discussion
Complicated miRNA Interactions were Involved in the Regulation of Maize Endosperm Development
MiRNAs are key regulators of gene expression in plants, especially for transcription factors, which gives these small molecules a central role of plant phase transition, organ architecture, and response to stress. Furthermore, recent studies have revealed that different miRNAs are likely to be integrated into functional networks in cellular and physiological events during plant development. One of the most well understood examples is the antagonistic interplay of miR156 and miR172 node, that is, SPL9, the down-regulated target of miR156, is able to directly bind to the promoter of miR172b gene and correspondingly promote its expression, which coordinately regulated the transition from vegetative phase to reproductive phase. Specifically, increased abundance of miR156 prolongs the juvenile stage and delays the adult stage transition, while highly expressed miR172 leads to early flowering and premature features of vegetative growth. In addition, miR390 targets a non-protein coding gene and triggers the production of TAS3, and as a consequence, which in turn causes the degradation of AUXIN RESPONSE FACTOR3 (ARF3) and ARF4 mRNAs, and finally advance the process of precocious maturation.
Although these miRNA regulation networks were previously observed in Arabidopsis, these interplay nodes might also be applicable during maize kernel and endosperm development according to the miRNA expression profile determined here. For example, miR156 reaches its peak expression at 7 DAP, while miR172 exhibits its lowest expression level at the same developmental stage (Figure S2A). The biological significance underlying this observation is reasonable because the endosperm has completed cell differentiation and formed four compartments at 7 DAP (including basal endosperm transfer cell, aleurone, starch endosperm, and embryo surrounding region), and starch accumulation and storage protein are ready to begin, indicating the end of the “juvenile stage” and ready for “adult stage.” Correspondingly, the abundance of miR156 decreases at 10 and15 DAP while miR172 increases, consistent with the expression trends during leaf aging. Additionally, the miR390-TAS3 node might also play a role in the regulation of endosperm maturation process, as both were highly expressed in 0-, 3-, and 5-DAP kernels but barely expressed in 7-, 10-, and 15-DAP endosperms. This differential expression pattern would elevate the level of ARF3, a target of TAS3, and promote precocious entrance to the adult stage (Figure S2B). However, more work is needed to validate these miRNA functions in maize endosperms in the future.
Multiple Pathways Exist to Generate miRNAs in Plants
In flowering plants, the classical mature miRNAs are typically derived from pri-miRNAs transcribed by RNA polymerase II, which is then processed into miRNA precursor and form a stem-loop structure (Kim, 2005). However, a recent study identified two special classes of miRNAs, namely, mirtron and NAT-miRNA, which are originating from splicing-dependent introns of the host genes and the overlapping transcript antisense to the target gene, respectively(Lu et al., 2008; Zhu et al., 2008). In addition, in several cases, tandem stem-loop structures have been reported to be produced within a common precursor in rice, maize, Medicago truncatula, and soybean (Guddeti et al., 2005; Chuck et al., 2007; Wang et al., 2007; Zhang et al., 2008). However, in this study, we confirmed that tandem but distinct miRNAs can be generated from a single transcribed locus in maize and verified that this pathway of producing miRNA could be conserved in monocot plants. This seems to be a more efficient way to produce miRNAs in terms of energy consumption, similar to the production of siRNAs. In previous study, it has been reported that miR159.1 and miR159.2 were simultaneously generated from a single precursor in rice (Lacombe et al., 2008), however, we found this phenomena was not a unique case and expression of tandem miRNAs could represent a new mechanism to produce multiple miRNAs in monocot plants. More interestingly, although co-transcribed from a common precursor, tandem miRNAs are not expressed completely synchronously; instead, they sometimes exhibit different, even antagonistic expression trends during maize kernel and endosperm development (Figure 5), indicating the production of these tandem miRNAs might be controlled at multi-levels, e.g., transcription of the pri-miRNA during any of the biogenesis steps, or at turnover of the mature miRNA. However, more efforts are needed to validate our prediction of these potential miRNAs, especially the confirmation of their biosynthesis in dcl1 maize mutants.
A Subset of miRNAs Probably Exhibited Allele-biased Expression Patterns in Maize Endosperm
Allele-biased expression occurs to small interfering RNAs in the endosperm of Arabidopsis, rice, and maize and their expression patterns are subjected to dynamic changes during endosperm development (Mosher et al., 2009; Rodrigues et al., 2013; Xin et al., 2013). Further analysis of these siRNAs with allele-biased expression in Arabidopsis indicates that they likely influence the expression of AGAMOUS-LIKE genes and result in small seeds, leading us to examine whether miRNAs exhibit similar expression patterns (Lu et al., 2012).
Unfortunately, because of the high conservation of sequence, SNP information is unavailable for allele-biased expression analysis of most miRNAs except for Zma-miR408-5p, which contains an A/G transition at the 11th position from the 5′ end of the sequence. Based on the reads count in deep sequencing data, the expression of Zma-miR408-5p is B73 allele-biased. In addition, CAPS validation of the miRNA408 precursor further confirmed our hypothesis. More interestingly, the SNP variation in Zma-miR408-5p between B73 and Mo17 would cause unexpected target changes. In this scenario, a subset of genes (e.g., targets for Mo17 Zma-miR408-5p), which will not be down-regulated in inbred lines (e.g., B73), would be unavoidably repressed after hybridization, and vice versa. This regulation pattern maybe fit dominant model to partially explain heterosis in F1, and their distinct expression profile during kernel and endosperm development in the reciprocal crosses might be associated with phenotypic difference between each other. However, we cannot excluded the possibility that Zma-miR408* of B73 is a star miRNA that might be more abundantly expressed or just more stable compared to that of Mo17 without further biological validation. In conclusion, our study proved that not only protein coding gene and small interfering RNA but also a proportion of miRNAs might exhibit allele-biased expression pattern, but their biosynthesis and biological relevance need further investigation.
Author Contributions
ZN, XW, QS conceived the project. MX and GY collected the plant materials. MX, YY, HP, and ZH analyzed data. MX, XW, and NZ wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Key Scientific Program (2012CB910900), the National Natural Science Foundation of China (31471479, 30925023 and 31230054), Chinese Universities Scientific Fund (15054038), and National Science Foundation (1261830).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00744
Table S1. The primer pairs for miRNA qRT-PCR.
Table S2. The expression level of conserved miRNAs in 0-, 3-, 5-DAP kernels and 7-, 10-, 15-DAP endosperms of B73 and Mo17reciprocal crosses (RPM: reads per million).
Table S3. The expression pattern of putative targets of 47 miRNA in 0-, 3-, 5-DAP kernels and 7-, 10-, and 15-DAP endosperm in B73 and Mo17 reciprocal crosses.
Figure S1. The stem-loop structures of precursors of miR159 and their tandem miRNAs in maize (A) and the sequences of miR159 in maize, wheat, barley, and rice (B).
Figure S2. The expression pattern of miR156 and miR172 (A) as well as miR390 and TAS3 (B) in 0-, 3-, 5-DAP kernels and 7-, 10-, and 15-DAP endosperm in maize. RPM, reads per million.
Abbreviations
DAP, days after pollination; CAPS, Cleaved Amplified Polymorphic Sequence; nt, nucleotide; ARF, AUXIN RESPONSE FACTOR3; AO, L-ascorbate oxidases; DHA, dehydroascorbate; GO, gene ontology; RPM, reads per million.
References
Berger, F. (2003). Endosperm: the crossroad of seed development. Curr. Opin. Plant Biol. 6, 42–50. doi: 10.1016/S1369526602000043
Berger, F., Grini, P. E., and Schnittger, A. (2006). Endosperm: an integrator of seed growth and development. Curr. Opin. Plant Biol. 9, 664–670. doi: 10.1016/j.pbi.2006.09.015
Cheng, W. H., Taliercio, E. W., and Chourey, P. S. (1996). The Miniature1 seed locus of maize encodes a cell wall lnvertase required for normal development of endosperm and maternal cells in the pedicel. Plant Cell 8, 971–983. doi: 10.1105/tpc.8.6.971
Chuck, G., Cigan, A. M., Saeteurn, K., and Hake, S. (2007). The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 39, 544–549. doi: 10.1038/ng2001
Costa, L. M., Yuan, J., Rouster, J., Paul, W., Dickinson, H., and Gutierrez-Marcos, J. F. (2012). Maternal control of nutrient allocation in plant seeds by genomic imprinting. Curr. Biol. 22, 160–165. doi: 10.1016/j.cub.2011.11.059
Gu, Y., Liu, Y., Zhang, J., Liu, H., Hu, Y., Du, H., et al. (2013). Identification and characterization of microRNAs in the developing maize endosperm. Genomics 102, 472–478. doi: 10.1016/j.ygeno.2013.08.007
Guddeti, S., Zhang, D. C., Li, A. L., Leseberg, C. H., Kang, H., Li, X. G., et al. (2005). Molecular evolution of the rice miR395 gene family. Cell Res. 15, 631–638. doi: 10.1038/sj.cr.7290333
Jiao, Y., Song, W., Zhang, M., and Lai, J. (2011). Identification of novel maize miRNAs by measuring the precision of precursor processing. BMC Plant Biol. 11:141. doi: 10.1186/1471-2229-11-141
Kim, V. N. (2005). MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 6, 376–385. doi: 10.1038/nrm1644
Konieczny, A., and Ausubel, F. M. (1993). A procedure for mapping Arabidopsis mutations using codominant ecotype-specific Pcr-based markers. Plant J. 4, 403–410. doi: 10.1046/j.1365-313X.1993.04020403.x
Lacombe, S., Nagasaki, H., Santi, C., Duval, D., Piégu, B., Bangratz, M., et al. (2008). Identification of precursor transcripts for 6 novel miRNAs expands the diversity on the genomic organisation and expression of miRNA genes in rice. BMC Plant Biol. 8:123. doi: 10.1186/1471-2229-8-123
Lan, Y., Su, N., Shen, Y., Zhang, R., Wu, F., Cheng, Z., et al. (2012). Identification of novel MiRNAs and MiRNA expression profiling during grain development in indica rice. BMC Genomics 13:264. doi: 10.1186/1471-2164-13-264
Li, G., Wang, D., Yang, R., Logan, K., Chen, H., Zhang, S., et al. (2014). Temporal patterns of gene expression in developing maize endosperm identified through transcriptome sequencing. Proc. Natl. Acad. Sci. U.S.A. 111, 7582–7587. doi: 10.1073/pnas.1406383111
Lopes, M. A., and Larkins, B. A. (1993). Endosperm origin, development, and function. Plant Cell 5, 1383–1399. doi: 10.1105/tpc.5.10.1383
Lu, C., Jeong, D. H., Kulkarni, K., Pillay, M., Nobuta, K., German, R., et al. (2008). Genome-wide analysis for discovery of rice microRNAs reveals natural antisense microRNAs (nat-miRNAs). Proc. Natl. Acad. Sci. U.S.A. 105, 4951–4956. doi: 10.1073/pnas.0708743105
Lu, J., Zhang, C. Q., Baulcombe, D. C., and Chen, Z. J. (2012). Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl. Acad. Sci. U.S.A. 109, 5529–5534. doi: 10.1073/pnas.1203094109
Lur, H. S., and Setter, T. L. (1993). Role of auxin in maize endosperm development (timing of nuclear DNA endoreduplication, zein expression, and cytokinin). Plant Physiol. 103, 273–280. doi: 10.1186/1471-2229-14-142
Mallory, A. C., and Vaucheret, H. (2006). Functions of microRNAs and related small RNAs in plants. Nat. Genet. 38(Suppl.), S31–S36. doi: 10.1038/ng0706-850b
Meng, F., Liu, H., Wang, K., Liu, L., Wang, S., Zhao, Y., et al. (2013). Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol. 13:140. doi: 10.1186/1471-2229-13-140
Miyoshi, K., Miyoshi, T., and Siomi, H. (2010). Many ways to generate microRNA-like small RNAs: non-canonical pathways for microRNA production. Mol. Genet. Genomics 284, 95–103. doi: 10.1007/s00438-010-0556-1
Mosher, R. A., Melnyk, C. W., Kelly, K. A., Dunn, R. M., Studholme, D. J., and Baulcombe, D. C. (2009). Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature 460, 283–286. doi: 10.1038/nature08084
Olsen, O. A. (2004). Nuclear endosperm development in cereals and Arabidopsis thaliana. Plant Cell 16, S214–S227. doi: 10.1105/tpc.017111
Pignocchi, C., Kiddle, G., Hernandez, I., Foster, S. J., Asensi, A., Taybi, T., et al. (2006). Ascorbate oxidase-dependent changes in the redox state of the apoplast modulate gene transcript accumulation leading to modified hormone signaling and orchestration of defense processes in tobacco. Plant Physiol. 141, 423–435. doi: 10.1104/pp.106.078469
Potters, G., Horemans, N., Caubergs, R. J., and Asard, H. (2000). Ascorbate and dehydroascorbate influence cell cycle progression in a tobacco cell suspension. Plant Physiol. 124, 17–20. doi: 10.1104/pp.124.1.17
Rodrigues, J. A., Ruan, R., Nishimura, T., Sharma, M. K., Sharma, R., Ronald, P. C., et al. (2013). Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc. Natl. Acad. Sci. U.S.A. 110, 7934–7939. doi: 10.1073/pnas.1306164110
Sabelli, P. A., and Larkins, B. A. (2009). The development of endosperm in grasses. Plant Physiol. 149, 14–26. doi: 10.1104/pp.108.129437
Sun, F., Guo, G., Du, J., Guo, W., Peng, H., Ni, Z., et al. (2014). Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L.). BMC Plant Biol. 14:142. doi: 10.1186/1471-2229-14-142
Voinnet, O. (2009). Origin, biogenesis, and activity of plant microRNAs. Cell 136, 669–687. doi: 10.1016/j.cell.2009.01.046
Wang, S., Zhu, Q. H., Guo, X., Gui, Y., Bao, J., Helliwell, C., et al. (2007). Molecular evolution and selection of a gene encoding two tandem microRNAs in rice. FEBS Lett. 581, 4789–4793. doi: 10.1016/j.febslet.2007.09.002
Xin, M., Yang, R., Li, G., Chen, H., Laurie, J., Ma, C., et al. (2013). Dynamic expression of imprinted genes associates with maternally controlled nutrient allocation during maize endosperm development. Plant Cell 25, 3212–3227. doi: 10.1105/tpc.113.115592
Xin, M., Yang, R., Yao, Y., Ma, C., Peng, H., Sun, Q., et al. (2014). Dynamic parent-of-origin effects on small interfering RNA expression in the developing maize endosperm. BMC Plant Biol. 14:192. doi: 10.1186/s12870-014-0192-8
Xue, L. J., Zhang, J. J., and Xue, H. W. (2009). Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res. 37, 916–930. doi: 10.1093/nar/gkn998
Yadegari, R., and Drews, G. N. (2004). Female gametophyte development. Plant Cell 16(Suppl.), S133–S141. doi: 10.1105/tpc.018192
Yang, X., and Li, L. (2011). miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27, 2614–2615. doi: 10.1093/bioinformatics/btr430
Zhang, B., Pan, X., and Stellwag, E. J. (2008). Identification of soybean microRNAs and their targets. Planta 229, 161–182. doi: 10.1007/s00425-008-0818-x
Zhang, H., and Li, L. (2013). SQUAMOSA promoter binding protein-like7 regulated microRNA408 is required for vegetative development in Arabidopsis. Plant J. 74, 98–109. doi: 10.1111/tpj.12107
Keywords: maize endosperm, miRNA profiling, partitioned expression, reciprocal cross, tandem miRNA
Citation: Xin M, Yang G, Yao Y, Peng H, Hu Z, Sun Q, Wang X and Ni Z (2015) Temporal small RNA transcriptome profiling unraveled partitioned miRNA expression in developing maize endosperms between reciprocal crosses. Front. Plant Sci. 6:744. doi: 10.3389/fpls.2015.00744
Received: 03 June 2015; Accepted: 31 August 2015;
Published: 15 September 2015.
Edited by:
Stewart Gillmor, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Paula Casati, National Scientific and Technical Research Council, ArgentinaAlma Armenta-Medina, Laboratorio Nacioanl de Genomica para la Biodiversidad, Mexico
Copyright © 2015 Xin, Yang, Yao, Peng, Hu, Sun, Wang and Ni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingming Xin and Zhongfu Ni, Department of Plant genetics and breeding, China Agricultural University, No. 2 Yuanmingyuan Xi Road, Haidian District, Beijing 100193, China,bWluZ21pbmd4aW5AY2F1LmVkdS5jbg==;bml6ZkBjYXUuZWR1LmNu