 Berline Fopa Fomeju
Berline Fopa Fomeju Cyril Falentin
Cyril Falentin Gilles Lassalle
Gilles Lassalle Maria J. Manzanares-Dauleux
Maria J. Manzanares-Dauleux Régine Delourme
Régine Delourme- 1Institut National de la Recherche Agronomique, UMR1349 IGEPP, Le Rheu, France
- 2Agrocampus Ouest, UMR1349 IGEPP, Le Rheu, France
All crop species are current or ancient polyploids. Following whole genome duplication, structural and functional modifications result in differential gene content or regulation in the duplicated regions, which can play a fundamental role in the diversification of genes underlying complex traits. We have investigated this issue in Brassica napus, a species with a highly duplicated genome, with the aim of studying the structural and functional organization of duplicated regions involved in quantitative resistance to stem canker, a disease caused by the fungal pathogen Leptosphaeria maculans. Genome-wide association analysis on two oilseed rape panels confirmed that duplicated regions of ancestral blocks E, J, R, U, and W were involved in resistance to stem canker. The structural analysis of the duplicated genomic regions showed a higher gene density on the A genome than on the C genome and a better collinearity between homoeologous regions than paralogous regions, as overall in the whole B. napus genome. The three ancestral sub-genomes were involved in the resistance to stem canker and the fractionation profile of the duplicated regions corresponded to what was expected from results on the B. napus progenitors. About 60% of the genes identified in these duplicated regions were single-copy genes while less than 5% were retained in all the duplicated copies of a given ancestral block. Genes retained in several copies were mainly involved in response to stress, signaling, or transcription regulation. Genes with resistance-associated markers were mainly retained in more than two copies. These results suggested that some genes underlying quantitative resistance to stem canker might be duplicated genes. Genes with a hydrolase activity that were retained in one copy or R-like genes might also account for resistance in some regions. Further analyses need to be conducted to indicate to what extent duplicated genes contribute to the expression of the resistance phenotype.
Introduction
Polyploidization is a common mode of evolution in flowering plants which occurs through genome merging (allopolyploidy) or genome doubling (autopolyploidy) and results in an increased gene set (Doyle et al., 2008; Wang et al., 2012). The evolution of duplicated genes after whole-genome duplication (WGD) has been studied extensively. In various plant species, a large proportion of duplicated genes are lost after a polyploidization event, a phenomenon called fractionation or diploidization, while a smaller proportion remains in a duplicated state (Cheng et al., 2012; Jiang et al., 2013). Several hypotheses have been put forward to describe the fate of duplicated genes after WGD, including loss of function, neo-functionalization, sub-functionalization or functional redundancy (Freeling, 2008; Edger and Pires, 2009; Freeling, 2009; Wang et al., 2012).
The loss/retention of duplicated genes is not a random process and, from studies in various plant species, it appears to depend on gene functional category. Thus for example, in Arabidopsis thaliana, over-retained genes are involved in basic cellular machinery, nucleotide-sugar metabolism, signal transduction or regulatory functions, while the diploidized genes are involved in DNA repair, tRNA ligation or defense (Blanc and Wolfe, 2004). The loss/retention of duplicated genes might also depend on their parental origin. In this case, one of the parental genomes is more likely to retain genes and has a higher gene density than the other(s) genome(s). This phenomenon, referred to as biased fractionation, has been demonstrated in several species including Zea mays (Schnable et al., 2011), Triticum aestivum (Pont et al., 2013), Arabidopsis thaliana (Thomas et al., 2006), and Brassica rapa (Cheng et al., 2012; Tang et al., 2012). Furthermore, expression bias, as described by Wang et al. (2011), has been reported in allopolyploid species: when considering homoeologous gene pairs in allopolyploids, mRNA transcripts from the sub-genome that retains more genes tend to be more highly expressed and thus contribute more to the total transcriptome (Bardil et al., 2011; Schnable et al., 2011; Yoo et al., 2013; Woodhouse et al., 2014).
In several polyploid crop species such as wheat, soybean, cotton and strawberry, it has been shown that duplicated regions were involved in controlling complex agronomical traits (Axelsson et al., 2001; Rong et al., 2007; Liu et al., 2011; Combes et al., 2012; Lerceteau-Kohler et al., 2012). However, there is a lack of studies dissecting the genomic structure and function of these duplicated regions. A recent study in soybean showed that one major QTL (Quantitative Trait Loci) involved in the control of oil and protein seed content on chromosome 20 and its homoeologous region on chromosome 10, were divergent in gene content and gene density (Severin et al., 2011; Lestari et al., 2013). Both expression bias and expression equivalence were observed at the transcriptomic level in these homoeologous regions: out of the 89 homoeologous gene pairs expressed in both regions, 40% showed bias (higher transcript accumulation) toward the homoeologs on chromosome 10. To fully understand both the genetic control and the mechanisms underlying complex traits in polyploid plant species, it is worthwhile to take into account the evolutionary processes through the dissection and comparison of the genomic regions underlying homoeologous duplicated QTL.
Brassica napus is an outstanding model for investigating structural and/or functional comparisons of duplicated regions involved in the control of complex traits. Firstly, the B. napus genome is highly duplicated. Indeed, B. napus (2n = 4x = 38, genome AACC) is an allotetraploid species formed from the hybridization between Brassica rapa (2n = 2x = 20, A genome) and Brassica oleracea (2n = 2x = 18, C genome) (U, 1935). Brassica ancestors have undergone two duplication events (named α and β) and two triplication events of which the most recent is specific to the Brassica clade (Jenczewski et al., 2013). These WGD events, along with the merger of the two progenitor genomes, have resulted in a large number of duplicated regions in the B. napus genome. Secondly, the organization of duplications in the B. napus genome has been well characterized. The ancestral Brassicaceae genome was reconstructed in 24 genomic blocks (A–X) also called the ancestral karyotype blocks (AK blocks). Theses blocks have been identified in B. rapa (Schranz et al., 2006; Cheng et al., 2013), B. oleracea (Kaczmarek et al., 2009), and B. napus (Parkin, 2011; Delourme et al., 2013). Moreover, the structural organization of the hexaploid Brassica ancestor genome was determined from whole-genome sequence analyses of the B. rapa (Tang and Lyons, 2012; Cheng et al., 2013) and B. oleracea (Liu et al., 2014; Parkin et al., 2014) genomes. The ancestral Brassica genome contains three sub-genomes showing different levels of fractionation: the LF sub-genome corresponds to the less fractionated genome with the highest gene density, the MF1 sub-genome is the moderately fractionated sub-genome with moderate gene density and sub-genome MF2 is the most fractionated genome with the lowest gene density (Cheng et al., 2013; Liu et al., 2014; Parkin et al., 2014). The organization of these sub-genomes has not yet been elucidated in B. napus. However, the recently released genomic sequence of B. napus showed good collinearity between the A/C genomic sequences of B. napus and the A and C genomes of B. rapa and B. oleracea (Chalhoub et al., 2014). The organization of LF, MF1 and MF2 can thus be extrapolated to the B. napus genome. Thirdly, B. napus is closely related to the model plant species Arabidopsis thaliana [diverged ~20 million years ago (MYA)] thus allowing functional divergences within B. napus duplicated regions to be explored further based on the function of the A. thaliana orthologous genes. This relatedness between A. thaliana and B. napus has been successfully used in previous comparative genetic and genomic analyses to identify candidate genes underlying QTL or to find putative functions of targeted genes (Cai et al., 2012; Zou et al., 2012). In B. napus, homoeologous regions were shown to be involved in the control of important traits such as seed glucosinolate content (Howell et al., 2003), flowering time (Wang et al., 2009), yield-related components (Chen et al., 2007) and resistance to sclerotinia stem rot (Zhao et al., 2006; Wei et al., 2014). Comparative genomic analyses of homoeologous B. napus regions involved in polygenic traits would then give the opportunity to study the impact of genome duplications on the structural and functional organization of such regions in a highly duplicated genome.
Stem canker caused by the fungus Leptosphaeria maculans is one disease of oilseed rape causing serious losses in Europe, Australia and North America (West et al., 2001; Fitt et al., 2006). Both qualitative and quantitative resistances were identified in B. napus or in related species (Delourme et al., 2006; Hayward et al., 2012; Raman et al., 2012). Qualitative resistance is based on a gene-for-gene interaction, which is expressed from the seedling stage. More than 10 specific resistance genes (R genes) have been identified in B. napus and related Brassica species B. rapa, B. juncea, and B. nigra (Rlm1–11, RlmS, LepR1-4) (Yu et al., 2005, 2008, 2013; Delourme et al., 2006; Van de Wouw et al., 2009; Balesdent et al., 2013), some of which are organized in clusters on B. napus chromosomes (Delourme et al., 2004, 2006). Since this resistance is total, it exerts strong selection pressure on the fungal populations that rapidly adapt and varieties whose resistance is based solely on such major genes lose their effectiveness only three-four years after their release (Brun et al., 2000; Li et al., 2003; Rouxel et al., 2003). Quantitative adult-plant resistance, which is a partial, polygenic resistance mediated by QTL is considered as non-race specific (Delourme et al., 2006) and more durable but its effectiveness varies between cropping seasons due to environmental conditions. Qualitative resistance protects the plant from development of leaf lesions at the young plant stage and subsequently prevents the development of stem canker at the adult plant stage. Thus, effective detection of quantitative resistance in field conditions is only possible in the absence of effective R genes. The use of quantitative resistance alone or in combination with qualitative resistance is a way to maximize the effectiveness and durability of the resistance (Brun et al., 2010). These findings highlight the interest and need to strengthen quantitative resistance studies in order to help breeding and improve disease management. Genetic studies of quantitative resistance showed that many QTL in different genetic backgrounds controlled this resistance (Pilet et al., 1998, 2001; Kaur et al., 2009; Jestin et al., 2011, 2012, 2015; Raman et al., 2012). Recently, using linkage and genome-wide association (GWA) analyses, we showed that a large proportion of regions (at least 44%) involved in this quantitative resistance corresponded to duplicated homoeologous regions. These regions were located mainly in duplications of the five AK blocks E, J, R, U, and W (Fopa Fomeju et al., 2014). In the present study, we conducted a comparative genomic analysis of these duplicated homoeologous regions involved in the control of the quantitative resistance to stem canker in B. napus. We investigated the similarity/divergence of the duplicated regions in terms of gene content and putative functions of retained genes. We took advantage of the recently released B. napus genome sequence (Chalhoub et al., 2014) for the structural analysis of the duplicated regions. B. napus—A. thaliana relatedness was used to explore gene ontology (GO) categories and the function of genes located in the duplicated regions. According to the functions of the genes that were retained in all duplicated regions or were specific to some regions, we discussed the putative functions associated to the stem canker quantitative resistance.
Materials and Methods
Association Mapping Panels, Genotyping, and Phenotyping Data
A panel of 170 winter B. napus varieties was used (Table S1). The panel comprises modern oilseed rape (OSR) varieties with low seed erucic acid and glucosinolate content (00 quality) and old varieties with high seed erucic acid and high glucosinolate content (++ quality). The varieties were chosen to represent a wide range of winter oilseed rape diversity and derived from different breeding programs. The absence of any effective specific resistance genes conferring total resistance in our field trial conditions was also checked in order to only evaluate the level of quantitative resistance. This was deduced from cotyledon tests using isolates carrying known AvrLm genes (Balesdent et al., pers. comm.). Thus, we excluded OSR varieties known as carrying Rlm7, a highly effective gene that was recently introduced in OSR varieties in France. This panel was genotyped with single nucleotide polymorphism (SNP) markers using a Brassica 60K SNP BeadChip Array (Liu et al., 2013) and evaluated for resistance to stem canker in a field trial in 2013. One hundred and nine OSR varieties were common to a panel of 116 OSR varieties phenotyped for resistance to stem canker in a field trial in 2006 (Table S1) (Fopa Fomeju et al., 2014).
DNA was isolated from young leaves and DNA extracted using the DNeasy 96 Plant Kit (Qiagen, Courtaboeuf, France). DNA was quantified with the Quant-iT™ PicoGreen® Assay (Invitrogen, Carlsbad, USA), using the Appliskan multiplate reader (Thermo Scientific, Courtaboeuf, France). Concentrations were adjusted to a minimum of 50 ng/μL and were submitted to a provider, where the Infinium® assay was performed following the manufacturer's protocol (Illumina Inc., San Diego, USA). The automatic allele calling for each locus was accomplished using the Genome Studio software (Illumina Inc., San Diego, USA). The clusters were manually edited when necessary. Technical replicates and signal intensities were controlled and only the most reliable calls were retained.
Stem canker was evaluated at one location in a α-design (3 columns and 5 rows) with three replicates (INRA Le Rheu) in France in 2012/13. In each replicate, three control varieties (“Eurol” and “Goeland” as moderately susceptible and “Jet Neuf” as resistant) were replicated in each row × column block in order to check the homogeneity of the trial. Contaminated residues collected from the previous season were scattered throughout the plots 1 month after sowing in order to ensure homogeneous disease infection. Leaf lesions were assessed on each plot at the autumn using a 0–4 scale according to the number of plants with leaf lesions and the number of leaf lesions per plant. Stem canker severity was assessed 2–3 weeks before harvest (mid-June). Forty plants per plot were uprooted and stem canker was assessed on a 0–9 disease index (DI) (Aubertot et al., 2004). DI increases with crown canker severity, starting from zero for healthy plants to nine for completely lodged plants. The 2006 phenotypic data were obtained from a similar experimental design (Jestin et al., 2011).
Association Mapping
Genome wide association (GWA) analyses were performed on two panels (i) panel 1: the sub-panel of 109 OSR varieties phenotyped in 2006 and (ii) panel 2: the whole panel of 170 OSR varieties phenotyped in 2013. A total of 13,973 informative SNPs that were mapped on our “Darmor-bzh” × “Yudal” doubled haploid population was selected for association analyses. GWA was carried out as described in Fopa Fomeju et al. (2014) using the GAPIT package (Lipka et al., 2012). Markers with a major allele frequency (MajAF) greater than 0.95 and genotypes or markers with more than 10% of missing genotyping data were eliminated to prevent analyses bias. We used a mixed linear model taking into account the structure within the panel as a fixed effect, and the relatedness between genotypes as a random effect. The calculation of the P matrix, which took into account the panel structure, was based on Principle Component Analysis (PCA) and was performed with EIGENSTRAT software (Price et al., 2006). The calculation of the kinship, or K, matrix was based on an identity by state matrix for all SNP markers and was directly performed by GAPIT (Lipka et al., 2012). The proportion of false positive associations was controlled by a False Discovery Rate (FDR) test at 10%.
Delineation of the A. thaliana and B. napus Genomic Sequences to Investigate
The flanking sequences (100–300 bp, median size: 120 bp) of SNPs were used as queries for BLASTn alignments against A. thaliana nucleic sequences in TAIR10 (https://www.arabidopsis.org/) with an E-value threshold of 10−6. Best gene hits and the corresponding AK block in A. thaliana were anchored on the genetic B. napus reference map. Thus for each genetic region with resistance-associated markers in B. napus, the corresponding A. thaliana gene interval was deduced. We took advantage of our present GWA studies and of the one of Fopa Fomeju et al. (2014) to delineate these intervals. They included all the resistance-associated markers identified in all corresponding B. napus duplicated regions.
The genomic regions thus delineated were extracted from the A. thaliana genome (https://www.arabidopsis.org/) and aligned against the whole B. napus genome sequence (Chalhoub et al., 2014) to find the corresponding orthologous duplicated regions in B. napus. This reciprocal alignment was conducted to make sure that (i) we investigated all the corresponding duplicated regions in B. napus and (ii) we searched for the same ancestral interval on all the duplicates in the B. napus genome. The software MumMer v3.0 (Kurtz et al., 2004) was used to conduct a nucleotide versus nucleotide alignment (“NUCmer” command) between each A. thaliana region and the B. napus genome. The default alignment parameters were used (http://mummer.sourceforge.net/manual/#nucmer).
For a visual overview of the synteny and the collinearity between the identified B. napus duplicated regions thus identified, we used the software MAUVE v2.3.1 (Darling et al., 2010).
Structural Analysis of the Duplicated Regions with Resistance-associated Markers
We took advantage of the close relatedness between B. napus and A. thaliana to identify the duplicated genes in the B. napus regions. The annotated B. napus genes (Chalhoub et al., 2014) were translated and aligned against the bank of A. thaliana proteins (https://www.arabidopsis.org/). When two different translated B. napus genes showed the best matches to the same A. thaliana protein then we considered that the two B. napus genes were likely to be homoeologous (if one gene was on the A genome and the other on the C genome) or paralogous (if the two genes were on the same genome).
We compared the duplicated regions in terms of interval length, number of genes and gene density. For each block, we also estimated the level of retention of A. thaliana genes in the duplicated regions. The results were examined in relation to the putative organization of the ancestral sub-genomes LF, MF1, and MF2. The organization of these sub-genomes was reported in B. napus from findings in B. rapa (Cheng et al., 2013) and B. oleracea (Liu et al., 2014; Parkin et al., 2014) genomes. We then looked at the distribution of the retained gene copies in the different duplicated regions in B. napus. Indeed, for a given block, the A. thaliana genes can be present in one of the duplications in B. napus or in up to six duplicated regions. Furthermore, in a given B. napus region, genes can be present in one copy (single-copy) or in two or more copies (multi-copies).
In silico Functional Analysis of the Duplicated Regions with Resistance-associated Markers
We deduced Gene Ontology (GO) categories from the A. thaliana best blast hits in order to functionally characterize and compare the duplicated B. napus regions between each other and with A. thaliana orthologous regions. The GO terms were obtained from the most recent release of predicted gene locus in the TAIR database (https://www.arabidopsis.org/).
For GO term enrichment analysis, we used the online analysis tool “Singular Enrichment Analysis” (SEA) from AgriGO (http://bioinfo.cau.edu.cn/agriGO/analysis.php). A Fisher test with the Yekutielli multi-test correction method (significance threshold = 0.05) was used. The GO type chosen was the Plant GO slim. For comparison of GO terms of the genes present in 5–6 vs. 1–2 duplicated regions of our studied five blocks, we used a contingency Chi-2 test (α = 0.001).
Finally, nucleotide binding site—leucine rich repeat (NBS-LRR) genes identified in the B. napus genome (Chalhoub et al., 2014), genes predicted in the B. napus genome as being putative resistance gene analogs in A. thaliana, and known major stem canker resistance genes were searched for in our targeted regions. This last category included the major LepR3/Rlm2 gene which was cloned and identified as a receptor-like protein (Larkan et al., 2013, 2015) and other Rlm genes previously located in B. napus maps (Delourme et al., 2006). For the latter ones, the previously identified flanking markers were mapped on our SNP map in order to deduce their putative position.
Results
Phenotypic Analysis of the B. napus Panel
The results obtained confirmed that we have mainly assessed quantitative resistance in our panels. Indeed, all oilseed rape varieties in the experiment showed typical leaf lesions in the autumn, indicating their lack of effective specific total resistance conferred by major genes that prevent leaf lesion development. As expected, the varieties showed variability in stem canker resistance with DI ranging from 0.3 (resistant) to 8.4 (susceptible). Analysis of variance showed significant (P < 0.001) phenotypic variation among lines and replicates. The proportion of total phenotypic variation which resulted from the genotypic variation within the oilseed rape collection was estimated at 0.94. Analysis of variance on the three control varieties did not show a significant genotype x replicate interaction (α = 0.05). The mean disease index of each variety (Table S1) was then used for GWA.
GWA and Identification of Duplicated Homoeologous Regions Associated to Stem Canker Resistance
Association analyses were conducted on the two OSR panels with 13,973 informative SNP markers that were well distributed on our reference genetic map with an average density of 1 SNP every 0.15 centimorgans (cM). The average physical distance between two SNPs on the B. napus reference sequence was estimated at 60 kb, based on the physical position of the used SNP (Rousseau-Gueutin, pers. comm.). Linkage disequilibrium (LD) evaluated in the two panels extended up to 1.22 cM in the panel of 109 OSR and up to 1.37 cM in the panel of 170 OSR. GWA analyses revealed 686 and 1019 markers significantly (p-value < 0.05) associated with resistance in the 109 and 170 OSR panels, phenotyped in 2006 and 2013, respectively (Table S2). A set of 52 associated SNP markers was common to both panels. After false discovery rate (FDR) correction, no significant associated markers were detected in the 109 OSR panel, and one associated marker located on linkage group (LG) A08 in block U was observed in the 170 OSR panel. Out of the 686 and 1019 resistance-associated markers, 505 (73%) and 467 (45%) were clearly anchored on one AK block (Table S3). The remaining markers were located in regions including several nested blocks or were putatively located on two different blocks.
We compared results obtained in the present two GWA analyses with those published by Fopa Fomeju et al. (2014), and we found 28 and 32 common regions containing resistance-associated markers between the three or two analyses, respectively (Table S3). Of these 60 regions, 27 (45%) corresponded to duplications of the AK blocks that were already identified by Fopa Fomeju et al. (2014) i.e., the blocks E (six regions), J (six regions), R (four regions), U (six regions), and W (four regions). In addition in the present study, resistance-associated markers were identified on the six duplications of the block F. In further analyses we focused on the AK blocks E, J, R, U, and W, as these were consistent between our different studies.
Structural Organization of Homoeologous Duplicated B. napus Regions
The A. thaliana gene intervals corresponding to the duplicated B. napus regions that carry resistance associated markers were delineated (Table 1) and their genomic sequence was extracted. The alignment of the sequence of all the studied AK block intervals to the B. napus sequence (Figure S1) revealed the expected number (six duplicated regions) and the expected location (according to the genetic map) of the duplications in the B. napus genome. The dotplots indicated that there was a good collinearity between A. thaliana and B. napus nucleotide sequences. Few rearrangements, mainly inversions, were identified between A. thaliana and B. napus duplicates. One small translocation appeared to be located on U block between chromosomes C3 and C8. Multiple alignments of the duplicated B. napus regions indicated that there was globally a better collinearity between homoeologous regions than between paralogous regions (Figure S2).

Table 1. Interval definition of the five AK genomic blocks investigated in this study.
For each block, although the size of the duplicated regions was larger on the C genome than on the A genome, the gene density was higher in the A than in the C regions (Table 2). Both on the A and C B. napus genomes, the regions showing the highest number of conserved genes with A. thaliana corresponded to regions located on the ancestral less fractionated sub-genome (LF sub-genome) (Table 2). The only exception was for the duplication of AK block J on C4, which was not assigned to the same sub-genome in previous studies by Liu et al. (2014) and Parkin et al. (2014). Likewise, for the duplicated regions of blocks E, J, R, and U on the A genome and of block U on the C genome, the regions with an intermediate number and the lowest number of conserved genes with A. thaliana corresponded to the MF1 and MF2 sub-genomes, respectively. For the duplicated regions of block W on the A genome and blocks E, J, R, and W on the C genome, the differences between the number of conserved genes with A. thaliana was not significant enough to distinguish between the MF1 and MF2 sub-genomes in these regions. Except for the block E, there were more tested SNPs in the regions located on the A than on the C genome. There was no bias in the distribution of tested markers across the three sub-genomes. The proportion of resistance-associated markers identified in the duplicated regions was similar on the A or C genomes, and on the LF, MF1, and MF2 sub-genomes, except for a higher proportion observed on MF2 on A genome.
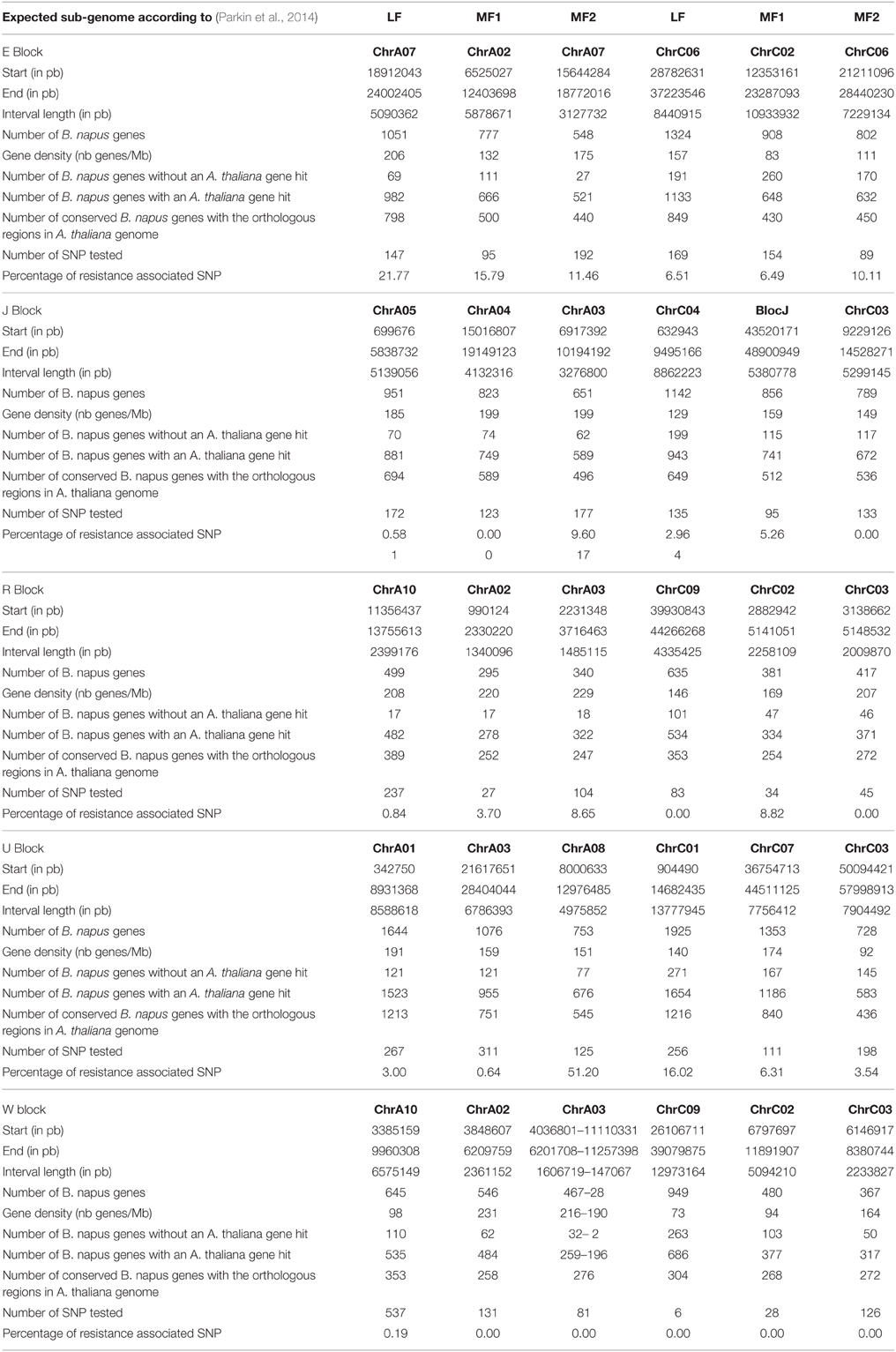
Table 2. Structural organization of the duplicated regions of AK blocks E, J, R, U, and W in B. napus genome.
The analysis of the conservation of A. thaliana orthologous genes in the duplicated B. napus regions (Table 3) showed that (i) the A. thaliana genes were retained in one to six duplicated regions of a block and (ii) in a given B. napus region, one A. thaliana orthologous gene was present in 0, 1 or several copies. Thus, duplicated genes can be retained in two ways: one gene copy can be retained on all the duplicated regions of an AK block and/or several copies of the gene can be located in at least one of the duplicated regions of the AK block. For all the studied blocks, most of the A. thaliana orthologs (~60%) were located in one or two duplicated regions of a given block. In contrast, only 4% of the genes were present in all the duplicated regions of a given block and 6% on five of the six duplicated regions (Table 3). In most cases, when gene copies were located in two duplicated regions, one copy was on the A genome and the other was on the C genome (data not shown). Overall, for all AK blocks, a large majority of genes (85%) were maintained in one copy per duplicated region. In addition, the proportion of genes in multiple copies is lower for genes that are located in one region compared with genes that are retained in several duplicated regions. For example, 7% of the genes retained in only one duplicate of block J were found in multiple copies within this duplicated region whereas 36% of the genes that were retained on the six duplicates of block J were in multiple copies on at least one of the six duplicated regions.
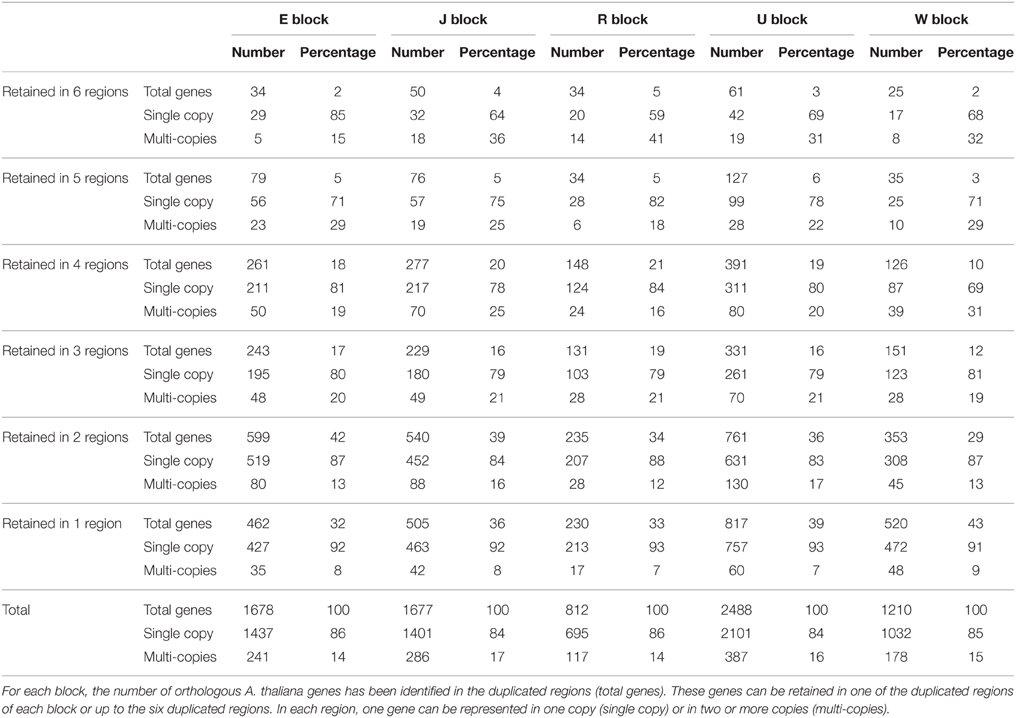
Table 3. Distribution of genes in the studied duplicated regions in B. napus genome.
Gene Ontology Term Analyses
We investigated gene categories in the duplicated regions involved in resistance to stem canker. B. napus genes with an ontology deduced from A. thaliana orthologs were used to perform GO slim enrichment analyses. No significant enrichment was identified when we compared GO terms of genes located in the duplicated B. napus regions with the GO terms of genes located in the corresponding A. thaliana orthologous regions. We then compared GO terms of genes located on five and six duplicated regions versus those located on one or two regions in B. napus. Among the genes located in five or six duplicated regions (Table S4), there were significantly more genes involved in “developmental processes,” “cell organization & biogenesis,” “response to stress,” “response to abiotic or biotic stimulus,” “transcription DNA-dependent,” “transport,” and “signal transduction” compared with genes located in one duplicated region (Figure 1). Furthermore, among the genes retained in five or six duplicated regions, more genes had molecular functions corresponding to “transcription factor activity,” “protein binding,” and “transferase activity.” In contrast, there were more genes with a molecular function corresponding to “hydrolase activity” within genes located on one or two duplicated regions (Figure 1).
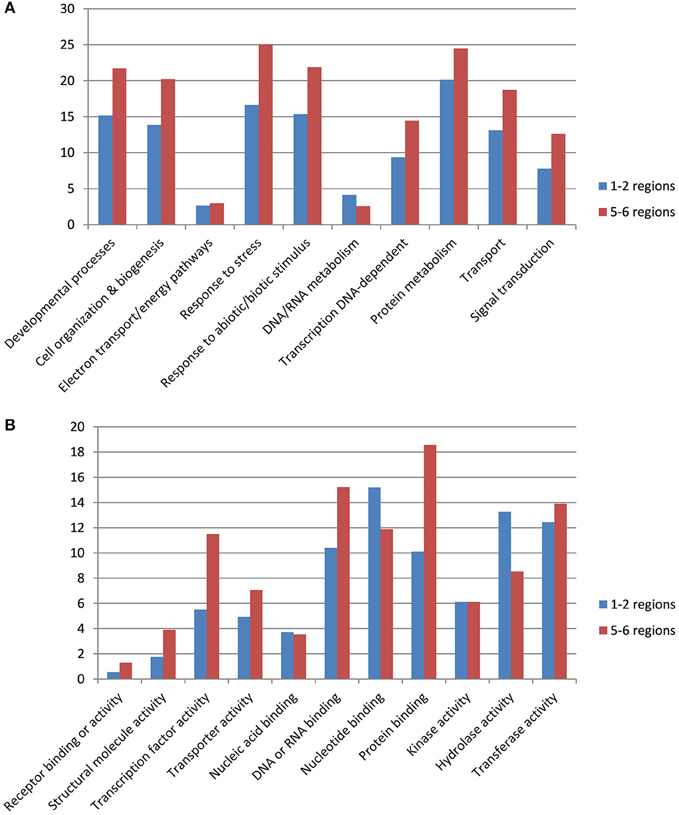
Figure 1. Gene ontology terms associated to genes retained in 1–2 or 5–6 duplicated regions. Gene ontology terms for biological process (A) and molecular activity (B).
Structural and Functional Characterization of Genes with Resistance-associated Markers
From the present GWA studies and the one of Fopa Fomeju et al. (2014), we identified 294 resistance-associated markers in the duplicated regions corresponding to the five studied AK blocks. Of these, 148 markers were anchored in B. napus genes. They corresponded to 129 genes, of which 124 had a blast hit with A. thaliana and their putative function was thus investigated (Table S5). These 124 B. napus genes represented a large proportion of the resistance associated markers on the blocks W (65%) and E (51%) and about 30% of resistance-associated markers on blocks J, R, and U. Seven resistance-associated genes were retained in only one duplicated region and the 117 others were retained in two or more duplicated regions. Among these 117 genes, 18 (15%) were present in multiple copies in at least one of the duplicated regions. The 124 resistance-associated genes were mainly related to the biological processes “response to stress,” “cell organization & biogenesis,” and “protein metabolism” (Figure 2). The three main molecular functions found were “hydrolase activity,” “nucleotide binding,” and “transferase activity.” These GO categories described genes located on five or six duplicated regions, except for “hydrolase activity” which was preferentially identified for genes located in one duplicated region.
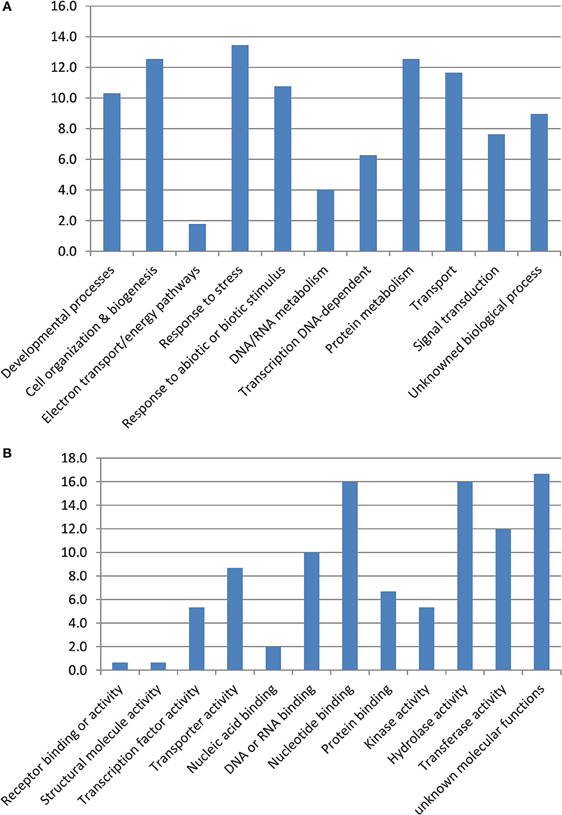
Figure 2. Gene ontology terms associated to SNP/genes associated to resistance to stem canker. Gene ontology terms for biological process (A) and molecular activity (B).
Investigation of R-like Genes
Out of the 425 NBS-LRR genes identified in the B. napus genome (Chalhoub et al., 2014), 135 genes were located in our studied B. napus duplicated regions. In addition, we found that 89 B. napus genes had a significant hit to NBS-LRR genes or resistance gene analogs (RGA) in A. thaliana. These were mostly distributed on duplicated regions corresponding to the blocks E, R, and U. Out of the 224 resistance-like genes (NBS-LRR and RGA), 21 were tested in the association study and four were significantly associated with resistance to stem canker (Table S2). Two were located on block E on LG A7 (LF sub-genome) and C2 and the two others on block U on LG C7. According to genetic linkage maps, the Rlm3-Rlm4-Rlm7-Rlm9 cluster might be in the neighborhood of block E-LF sub-genome on chromosome A7 and Rlm1 was assigned to the studied region of block E-MF2 sub-genome on chromosome A7. The approximate mapping position of Rlm1 spread in a region of 1300 kb according to bridge SNP between linkage map and genomic sequence (Table S2). In this region, 22 resistance-associated markers have been tested and 12 were significantly associated to resistance to stem canker. LepR3/Rlm2 sequence aligned to the B. napus gene GSBRNA2G00135758001 which was located outside our investigated region on the block R on chromosome A10 and was not tested in association mapping.
Discussion
In this study, we analyzed the structural and functional organization of duplicated regions involved in quantitative resistance of B. napus to stem canker. We took advantage of the recently released B. napus genome sequence (Chalhoub et al., 2014) for structural analyses and of the B. napus—A. thaliana relatedness for exploring GO categories and the putative function of genes located in the duplicated regions.
Duplicated Regions of At Least Five AK Blocks Are Involved in Quantitative Resistance to Stem Canker
Our present GWA analyses confirmed that duplicated regions of AK blocks E, J, R, U, and W are involved in quantitative resistance to stem canker as shown in our previous GWA study (Fopa Fomeju et al., 2014). Results from FDR corrected data indicated that there might be false positives among the resistance-associated markers. However, the duplicated regions of the blocks E, J, R, U, and W were identified with three datasets, suggesting that these regions are likely to be “true” quantitative resistance regions. Moreover, at least 15 of these duplicated resistance associated regions (on LG A01, A02, A03, A05, A08, C01, C02, C03, C04, C06, and C07) were located within or in the vicinity of QTL that were previously identified in biparental segregating populations and in a connected multiparental population (Pilet et al., 2001; Jestin et al., 2012, 2015). New potential duplicated regions were highlighted (such as duplications of block F) indicating that, as suggested in (Fopa Fomeju et al., 2014), more than 44% of duplicated genomic regions are involved in the control of this complex trait. This proportion is quite high compared to that observed in other polyploid plant species such as strawberry (Lerceteau-Kohler et al., 2012) or cotton (Rong et al., 2007) for other traits. This could be tightly linked to either the good structural conservation between the B. napus A and C genomes (Cheung et al., 2009; Chalhoub et al., 2014), the highly duplicated nature of the Brassicaceae genomes resulting from several WGD events, or the fact that loci involved in defense responses are often over-retained in plants after WGD (Blanc and Wolfe, 2004; Moghe et al., 2014).
Structural Organization of the Homoeologous Duplicated Regions Involved in Stem Canker Resistance
All the expected duplicated regions of our targeted blocks were found on the B. napus genomic sequence. This reflects the good coverage of the B. napus sequence, which was estimated at 79% (Chalhoub et al., 2014). Alignment of B. napus duplicated regions revealed better collinearity between homoeologous regions than between paralogous regions, homoeologous duplicated regions having more genes in common than paralogous duplicated regions, as generally observed in the genomes of B. napus and its progenitors (Cheung et al., 2009). The size of the extracted duplicated regions varied depending on their location on either the A or C genome. For a given block, the studied regions on the C genome were larger than their homoeologous regions on the A genome. It has been shown that the C genome is larger due to insertions of transposable elements; almost 40% of the C vs. 26% of the A genome corresponds to transposable elements in B. napus (Chalhoub et al., 2014). Similar results were observed for the A and C genomes of B. rapa and B. oleracea, respectively (Cheung et al., 2009; Cheng et al., 2012; Liu et al., 2014).
By analyzing gene distribution in our studied regions, we observed that the B. napus duplicated regions with the highest number of retained genes compared with A. thaliana were orthologous to regions located on the less fractionated sub-genome (LF) of B. rapa (Cheng et al., 2012) and B. oleracea (Liu et al., 2014; Parkin et al., 2014). This agrees with the hypothesis that the fractionation of the sub-genomes took place before the B. rapa—B. oleracea divergence (Tang et al., 2012) and suggests that, probably due to the recentness of B. napus, no significant new fractionation levels were observed in this allotetraploid. However, the occurrence of homoeologous exchanges, which may shape fractionation levels by modifying gene number, on the sub-genomes have been reported (Chalhoub et al., 2014), suggesting that fractionation might be ongoing in the B. napus genome. The proportions of resistance associated markers on the LF, MF1 and MF2 subgenomes were similar, which suggests that all the subgenomes were involved in the control of the resistance trait. This remains to be further studied since the number of associated markers identified does not provide information on the number of underlying causative genes, nor on their contribution to the phenotype expression.
When considering gene distribution across the duplicated regions, we found that a large majority of genes (60%) were only present in one or two duplicated regions of the AK blocks and that these were mostly present in a single copy per region (on average, 85% of the genes). In general, genes present in two duplicated regions had one copy on the A genome and the other copy on the C genome. In a recent study, De Smet et al. (2013) estimated that 66% of the B. rapa genes were single-copy genes. This is consistent with our findings suggesting that these single-copy genes have not resulted from the allotetraploidization event but rather from the long-term evolution of diploid ancestors.
Functional Analysis of the Genes Located in the Duplicated Regions Involved in Resistance to Stem Canker
Genes located on five or six of the duplicated regions of a given block represented about 10% of the total genes identified. The GO terms of these duplicated genes were related to response to stress, signaling and transcription regulation as identified in other plant species (Blanc and Wolfe, 2004; Paterson et al., 2006). Highly duplicated genes, i.e., genes present in several copies in a given region, were preferentially associated to transport, signal transduction and kinase activity functions. Similar results were reported in other plant species (De Smet et al., 2013; Han et al., 2014). Actually, genes involved in adaptive traits (such as resistance or tolerance to biotic and abiotic stresses) are preferentially over-retained after a WGD event allowing faster evolution and conferring an adaptive advantage to polyploid plants (Pont et al., 2013; Murat et al., 2014; Wu et al., 2014). This can lead to copy number or sequence variation between accessions in the homoeologous and paralogous regions and to phenotype differentiation as shown for flowering-related genes (Schiessl et al., 2014). Twenty-two of the genes retained in five or six duplicated regions were associated to resistance and were related to response to stress and transferase activity. However, at this point, we could not conclude on the involvement of duplicated genes in stem canker resistance due to the fact that only one of the duplicated copies was tested for 77% of the duplicated genes.
Genes maintained in only one duplicated region were enriched in “hydrolase activity” functions. An analysis conducted by Han et al. (2014) in 29 angiosperm genomes indicated that genes in single copy have binding and catalytic activities (including hydrolase activity). Previously, Schranz and Mitchell-Olds (2006) highlighted that genes involved in hydrolase activity were retained as single-copy genes after the alpha duplication in the Brassicaceae lineage. All these results suggest that the bias in retention of genes in single copy is common across different species. Thus our results may reflect a global pattern across the whole genome and may not be specific to the studied regions. Twenty-four associated markers were located in genes present in only one duplicated region and had GO terms related to “hydrolase activity.” Thus, we cannot exclude that genes encoding proteins with hydrolase activity are involved in response to stem canker in some regions. Indeed, the involvement of hydrolase activity has been highlighted in the response to tobacco virus infection (Guo et al., 1998). The HE-1 hydrolase protein may play a role in protection from oxidative damage associated with defense responses. It may also play a role in generating signals for activation of certain defense responses. The relationship between hydrolase activity and plant response to pathogens has been also demonstrated in A. thaliana (Kang et al., 2008) and potato (Reddy et al., 2000).
R and R-like genes could also account for resistance in some regions. Indeed, four resistance-associated markers located in NBS-LRR or RGA genes were found on blocks E and U. In addition, the major gene Rlm1 conferring blackleg resistance in B. napus was located in the neighborhood of the resistance-associated markers on the block E, chromosome A7, sub-genome MF2. The effect of this region on resistance could be due either to a residual effect of Rlm1 gene or to linked genes, as previously suggested for Rlm2 gene that was shown to co-localize with a resistance QTL to stem canker (Pilet et al., 2001).
Conclusion and Future Prospects
We identified a set of genes that were retained in duplicated regions and associated with quantitative resistance to stem canker and many were involved in stress responses, signaling and transcriptional regulation. It was shown in various other species that such genes were over-retained after WGD, indicating a common evolutionary pattern across species. We also found that almost 60% of the genes located in the studied duplicated regions were single- or two-copy genes, as expected in the whole B. napus genome, and that some of them e.g., genes with a hydrolase activity were associated with resistance. Thus, we cannot exclude that the genes underlying quantitative resistance to stem canker are not duplicated genes. Results also suggested that R-like genes might account for B. napus quantitative resistance to stem canker in some genomic regions. In future studies, the B. napus genes that are present in our studied regions but had no hit with A. thaliana genes, should be investigated to complete the results. The three B. napus sub-genomes (LF, MF1, and MF2) were found to contribute to quantitative stem canker resistance. In wheat, it was suggested that the most fractionated genome (B sub-genome) i.e., the sub-genome which is more sensitive to genome fractionation and more plastic, controls adaptive traits such as reaction to biotic and abiotic stresses (Pont et al., 2013). This has to be further analyzed in oilseed rape by investigating whether all duplicated copies are expressed and equally contribute to phenotype expression and if they have undergone sub-functionalization, which appears to be a major force in maintaining genes in a duplicated state.
Author Contributions
BF performed GWA studies, structural and functional sequence analyses. CF carried out genetic mapping and BLAST analyses for the construction of the integrated map used in this study and the positioning of duplicated blocks on this map. GL contributed to the structural and BLAST analyses. RD and MM helped analyse the results and coordinated the project. BF, RD, and MM wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
BF was a PhD student funded by INRA (Institut National de la Recherche Agronomique, Département de Biologie et Amélioration des Plantes) and the Région Bretagne. We gratefully acknowledge Nathalie Nesi, Cécile Baron and Laure Berton for providing the genotyping data obtained on the 170 OSR panel with the Brassica 60K SNP BeadChip Array in the RAPSODYN project. We acknowledge the Genetic Resource Center (BrACySol, UMR IGEPP Ploudaniel, France) for providing the seeds of some B. napus varieties. We thank the team of the INRA Experimental Unit at Le Rheu for performing the disease evaluation trials.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00772
References
Axelsson, T., Shavorskaya, O., and Lagercrantz, U. (2001). Multiple flowering time QTLs within several Brassica species could be the result of duplicated copies of one ancestral gene. Genome 44, 856–864. doi: 10.1139/gen-44-5-856
Aubertot, J. N., Schott, J. J., Penaud, A., Brun, H., and Doré, T. (2004). Methods for sampling and assessment in relation to the spatial pattern of phoma stem canker (Leptosphaeria maculans) in oilseed rape. Eur. J. Plant Pathol. 110, 183–192. doi: 10.1023/B:EJPP.0000015359.61910.3b
Balesdent, M. H., Fudal, I., Ollivier, B., Bally, P., Grandaubert, J., Eber, F., et al. (2013). The dispensable chromosome of Leptosphaeria maculans shelters an effector gene conferring avirulence towards Brassica rapa. New Phytol. 198, 887–898. doi: 10.1111/nph.12178
Bardil, A., de Almeida, J. D., Combes, M. C., Lashermes, P., and Bertrand, B. (2011). Genomic expression dominance in the natural allopolyploid Coffea arabica is massively affected by growth temperature. New Phytol. 192, 760–774. doi: 10.1111/j.1469-8137.2011.03833.x
Blanc, G., and Wolfe, K. H. (2004). Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16, 1679–1691. doi: 10.1105/tpc.021410
Brun, H., Chèvre, A. M., Fitt, B. D. L., Powers, S., Besnard, A. L., Ermel, M., et al. (2010). Quantitative resistance increases the durability of qualitative resistance to Leptosphaeria maculans in Brassica napus. New Phytol. 185, 285–299. doi: 10.1111/j.1469-8137.2009.03049.x
Brun, H., Levivier, S., Somda, I., Ruer, D., Renard, M., and Chèvre, A. M. (2000). A field method for evaluating the potential durability of new resistance sources: application to the Leptosphaeria maculans/Brassica napus pathosystem. Phytopathology 90, 961–966. doi: 10.1094/PHYTO.2000.90.9.961
Cai, G., Yang, Q., Yang, Q., Zhao, Z., Chen, H., Wu, J., et al. (2012). Identification of candidate genes of QTLs for seed weight in Brassica napus through comparative mapping among Arabidopsis and Brassica species. BMC Genet. 13:105. doi: 10.1186/1471-2156-13-105
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H., Wang, X., et al. (2014). Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chen, W., Zhang, Y., Liu, X., Chen, B., Tu, J., and Tingdong, F. (2007). Detection of QTL for six yield-related traits in oilseed rape (Brassica napus) using DH and immortalized F-2 populations. Theor. Appl. Genet. 115, 849–858. doi: 10.1007/s00122-007-0613-2
Cheng, F., Mandáková, T., Wu, J., Xie, Q., Lysak, M. A., and Wang, X. (2013). Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. Plant Cell 25, 1541–1554. doi: 10.1105/tpc.113.110486
Cheng, F., Wu, J., Fang, L., Sun, S., Liu, B., Lin, K., et al. (2012). Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS ONE 7:e36442. doi: 10.1371/journal.pone.0036442
Cheung, F., Trick, M., Drou, N., Lim, Y. P., Park, J. Y., Kwon, S. J., et al. (2009). Comparative analysis between homoeologous genome segments of Brassica napus and its progenitor species reveals extensive sequence-level divergence. Plant Cell 21, 1912–1928. doi: 10.1105/tpc.108.060376
Combes, M. C., Cenci, A., Baraille, H., Bertrand, B., and Lashermes, P. (2012). Homeologous gene expression in response to growing temperature in a recent allopolyploid (Coffea arabica L.). J. Hered. 103, 36–46. doi: 10.1093/jhered/esr120
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
De Smet, R., Adams, K. L., Vandepoele, K., Van Montagu, M. C. E., Maere, S., and Van de Peer, Y. (2013). Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc. Natl. Acad. Sci. U.S.A. 110, 2898–2903. doi: 10.1073/pnas.1300127110
Delourme, R., Pilet-Nayel, M. L., Archipiano, M., Horvais, R., Tanguy, X., Rouxel, T., et al. (2004). A cluster of major specific resistance genes to Leptosphaeria maculans in Brassica napus. Phytopathology 94, 578–583. doi: 10.1094/PHYTO.2004.94.6.578
Delourme, R., Chèvre, A. M., Brun, H., Rouxel, T., Balesdent, M. H., Dias, J. S., et al. (2006). Major gene and polygenic resistance to Leptosphaeria maculans in oilseed rape (Brassica napus). Eur. J. Plant Pathol. 114, 41–52. doi: 10.1007/s10658-005-2108-9
Delourme, R., Falentin, C., Fopa Fomeju, B. F., Boillot, M., Lassalle, G., André, I., et al. (2013). High-density SNP-based genetic map development and linkage disequilibroum assessment in Brassica napus L. BMC Genomics 14:120. doi: 10.1186/1471-2164-14-120
Doyle, J. J., Flagel, L. E., Paterson, A. H., Rapp, R. A., Soltis, D. E., Soltis, P. S., et al. (2008). Evolutionary genetics of genome merger and doubling in plants. Ann. Rev. Genet. 42, 443–461. doi: 10.1146/annurev.genet.42.110807.091524
Edger, P. P., and Pires, J. C. (2009). Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromos. Res. 17, 699–717. doi: 10.1007/s10577-009-9055-9
Fitt, B. D. L., Brun, H., Barnetti, M. J., and Rimmer, S. R. (2006). World-wide importance of phoma stem canker (Leptosphaeria maculans and L. biglobosa) on oilseed rape (Brassica napus). Eur. J. Plant Pathol. 192, 763–774. doi: 10.1007/s10658-005-2233-5
Fopa Fomeju, B., Falentin, C., Lassalle, G., Manzanares-Dauleux, M. J., and Delourme, R. (2014). Homoeologous duplicated regions are involved in quantitative resistance of Brassica napus to stem canker. BMC Genomics 15:498. doi: 10.1186/1471-2164-15-498
Freeling, M. (2008). The evolutionary position of subfunctionalization, downgraded. Genome Dynam. 4, 25–40. doi: 10.1159/000126004
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Ann. Rev. Plant Biol. 60, 433–453. doi: 10.1146/annurev.arplant.043008.092122
Guo, A., Durner, J., and Klessig, D. F. (1998). Characterization of a tobacco epoxide hydrolase gene induced during the resistance response to TMV. Plant J. 15, 647–656.
Han, F., Peng, Y., Xu, L., and Xiao, P. (2014). Identification, characterization, and utilization of single copy genes in 29 angiosperm genomes. BMC Genomics 15:504. doi: 10.1186/1471-2164-15-504
Hayward, A., McLanders, J., Campbell, E., Edwards, D., and Batley, J. (2012). Genomic advances will herald new insights into the Brassica: Leptosphaeria maculans pathosystem. Plant Biol. 14, 1–10. doi: 10.1111/j.1438-8677.2011.00481.x
Howell, P. M., Sharpe, A. G., and Lydiate, D. J. (2003). Homoeologous loci control the accumulation of seed glucosinolates in oilseed rape (Brassica napus). Genome 46, 454–460. doi: 10.1139/g03-028
Jenczewski, E., Chèvre, A., and Alix, K. (2013). Chromosomal and gene expression changes in Brassica Allopolyploids, in Polyploid and Hybrid Genomics, eds J. Chen and J. Birchle (Iowa: John Wiley and Sons, Inc.), 171–186
Jestin, C., Lodé, M., Domin, C., Falentin, C., Horvais, R., Coedel, S., et al. (2011). Association mapping of quantitative resistance for Leptosphaeria maculans in oilseed rape (Brassica napus L.). Mol. Breed. 27, 190–201. doi: 10.1007/s11032-010-9429-x
Jestin, C., Vallée, P., Domin, C., Manzanares-Dauleux, M. J., and Delourme, R. (2012). Assessment of a new strategy for selective phenotyping applied to complex traits in Brassica napus. Open J. Genet., 2, 190–201. doi: 10.4236/ojgen.2012.24025
Jestin, C., Bardol, N., Lodé, M., Duffé, P., Domin, C., Vallée, P., et al. (2015). Connected populations for detecting quantitative resistance factors to Phoma stem canker in oilseed rape (Brassica napus L.). Mol. Breed. 35, 167 doi: 10.1007/s11032-015-0356-8
Jiang, W. K., Liu, Y. L., Xia, E. H., and Gao, L. (2013). Prevalent role of gene features in determining evolutionary fates of whole-genome duplication duplicated genes in flowering plants. Plant Physiol. 161, 1844–1861. doi: 10.1104/pp.112.200147
Kaczmarek, M., Koczyk, G., Ziolkowski, P. A., Babula-Skowronska, D., and Sadowski, J. (2009). Comparative analysis of the Brassica oleracea genetic map and the Arabidopsis thaliana genome. Genome 52, 620–633. doi: 10.1139/g09-035
Kang, L., Wang, Y. S., Uppalapati, S. R., Wang, K., Tang, Y., Vadapalli, V., et al. (2008). Overexpression of a fatty acid amide hydrolase compromises innate immunity in Arabidopsis. Plant J. 56, 336–349. doi: 10.1111/j.1365-313X.2008.03603.x
Kaur, S., Cogan, N., Ye, G., Baillie, R., Hand, M., Ling, A., et al. (2009). Genetic map construction and QTL mapping of resistance to blackleg (Leptosphaeria maculans) disease in Australian canola (Brassica napus L.) cultivars. Theor. Appl. Genet. 120, 71–83. doi: 10.1007/s00122-009-1160-9
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Larkan, N. J., Lydiate, D. J., Parkin, I. A., Nelson, M. N., Epp, D. J., Cowling, W. A., et al. (2013). The Brassica napus blackleg resistance gene LepR3 encodes a receptor-like protein triggered by the Leptosphaeria maculans effector AVRLM1. New Phytol. 197, 595–605. doi: 10.1111/nph.12043
Larkan, N. J., Ma, L., and Borhan, M. H. (2015). The Brassica napus receptor-like protein RLM2 is encoded by a second allele of the LepR3/Rlm2 blackleg resistance locus. Plant Biotech. J. 13, 983–992. doi: 10.1111/pbi.12341
Lerceteau-Köhler, E., Moing, A., Guérin, G., Renaud, C., Petit, A., Rothan, C., et al. (2012). Genetic dissection of fruit quality traits in the octoploid cultivated strawberry highlights the role of homoeo-QTL in their control. Theor.Appl. Genet. 124, 1059–1077. doi: 10.1007/s00122-011-1769-3
Lestari, P., Van, K., Lee, J., Kang, Y. J., and Lee, S.-H. (2013). Gene divergence of homeologous regions associated with a major seed protein content QTL in soybean. Front. Plant Sci. 4:176. doi: 10.3389/fpls.2013.00176
Li, H., Sivasithamparam, K., and Barbetti, M. J. (2003). Breakdown of a Brassica rapa subsp. sylvestris single dominant blackleg resistance gene in Brassica napus rapeseed by Leptosphaeria maculans field isolates in Australia. Plant Dis. 87:752. doi: 10.1094/PDIS.2003.87.6.752A
Lipka, A. E., Tian, F., Wang, Q., Peiffer, J., Li, M., Bradbury, P. J., et al. (2012). GAPIT: genome association and prediction integrated tool. Bioinformatics 28, 2397–2399. doi: 10.1093/bioinformatics/bts444
Liu, W., Kim, M. Y., Kang, Y. J., Van, K., Lee, Y. H., Srinives, P., et al. (2011). QTL identification of flowering time at three different latitudes reveals homeologous genomic regions that control flowering in soybean. Theor. Appl. Genet. 123, 545–553. doi: 10.1007/s00122-011-1606-8
Liu, S. Y., Liu, Y. M., Yang, X. H., Tong, C. B., Edwards, D., Parkin, I. A. P., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nature Comm. 5, 3930. doi: 10.1038/ncomms4930
Liu, L., Qu, C., Wittkop, B., Yi, B., Xiao, Y., He, Y., et al. (2013). A high-density SNP map for accurate mapping of seed fibre QTL in Brassica napus L. PLoS ONE 8:e83052. doi: 10.1371/journal.pone.0083052
Moghe, G. D., Hufnagel, D. E., Tang, H., Xiao, Y., Dworkin, I., Town, C. D., et al. (2014). Consequences of whole-genome triplication as revealed by comparative genomic analyses of the wild radish Raphanus raphanistrum and three other brassicaceae species. Plant Cell 26, 1925–1937. doi: 10.1105/tpc.114.124297
Murat, F., Zhang, R., Guizard, S., Flores, R., Armero, A., Pont, C., et al. (2014). Shared subgenome dominance following polyploidization explains grass genome evolutionary plasticity from a seven protochromosome ancestor with 16K protogenes. Genome Biol. Evol. 6, 12–33. doi: 10.1093/gbe/evt200
Parkin, I. (2011). Ghosts: comparative mapping in the brassicaceae in Plant Genetics and Genomics: Crops and Models, Vol. 9, eds R. Schmidt and I. Bancroft (New York, NY; Dordrecht; Heidelberg; London: Springer), 153–170.
Parkin, I. A., Koh, C., Tang, H., Robinson, S. J., Kagale, S., Clarke, W. E., et al. (2014). Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15:R77. doi: 10.1186/gb-2014-15-6-r77
Paterson, A. H., Chapman, B. A., Kissinger, J. C., Bowers, J. E., Feltus, F. A., and Estill, J. C. (2006). Many gene and domain families have convergent fates following independent whole-genome duplication events in Arabidopsis, Oryza, Saccharomyces and Tetraodon. Trends Genet. 22, 597–602. doi: 10.1016/j.tig.2006.09.003
Pilet, M. L., Delourme, R., Foisset, N., and Renard, M. (1998). Identification of loci contributing to quantitative field resistance to blackleg disease, causal agent Leptosphaeria maculans (Desm.) Ces. et de Not., in Winter rapeseed (Brassica napus L.). Theor. Appl. Genet. 96, 23–30.
Pilet, M. L., Duplan, G., Archipiano, H., Barret, P., Baron, C., Horvais, R., et al. (2001). Stability of QTL for field resistance to blackleg across two genetic backgrounds in oilseed rape. Crop Sci. 41, 197–205. doi: 10.2135/cropsci2001.411197x
Pont, C., Murat, F., Guizard, S., Flores, R., Foucrier, S., Bidet, Y., et al. (2013). Wheat syntenome unveils new evidences of contrasted evolutionary plasticity between paleo- and neoduplicated subgenomes. Plant J. 76, 1030–1044. doi: 10.1111/tpj.12366
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Raman, R., Taylor, B., Marcroft, S., Stiller, J., Eckermann, P., Coombes, N., et al. (2012). Molecular mapping of qualitative and quantitative loci for resistance to Leptosphaeria maculans causing blackleg disease in canola (Brassica napus L.). Theor. Appl. Genet. 125, 405–418. doi: 10.1007/s00122-012-1842-6
Reddy, P. S., Kumar, T. C., Reddy, M. N., Sarada, C., and Reddanna, P. (2000). Differential formation of octadecadienoic acid and octadecatrienoic acid products in control and injured/infected potato tubers. Biochem. Biophys. Acta Mol. Cell Biol. Lipids 1483, 294–300. doi: 10.1016/s1388-1981(99)00191-2
Rong, J., Feltus, F. A., Waghmare, V. N., Pierce, G. J., Chee, P. W., Draye, X., et al. (2007). Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176, 2577–2588. doi: 10.1534/genetics.107.074518
Rouxel, T., Willner, E., Coudard, L., and Balesdent, M. H. (2003). Screening and identification of resistance to Leptosphaeria maculans (stem canker) in Brassica napus accessions. Euphytica 133, 219–231. doi: 10.1023/A:1025597622490
Schiessl, S., Samans, B., Hüttel, B., Reinhard, R., and Snowdon, R. J. (2014). Capturing sequence variation among flowering-time regulatory gene homologs in the allopolyploid crop species Brassica napus. Front. Plant Sci. 5:404. doi: 10.3389/fpls.2014.00404
Schnable, J. C., Springer, N. M., and Freeling, M. (2011). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U.S.A. 108, 4069–4074. doi: 10.1073/pnas.1101368108
Schranz, M. E., Lysak, M. A., and Mitchell-Olds, T. (2006). The ABC's of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci. 11, 535–542. doi: 10.1016/j.tplants.2006.09.002
Schranz, M. E., and Mitchell-Olds, T. (2006). Independent ancient polyploidy events in the sister families Brassicaceae and Cleomaceae. Plant Cell 18, 1152–1165. doi: 10.1105/tpc.106.041111
Severin, A. J., Cannon, S. B., Graham, M. M., Grant, D., and Shoemaker, R. C. (2011). Changes in twelve homoeologous genomic regions in soybean following three rounds of polyploidy. Plant Cell 23, 3129–3136. doi: 10.1105/tpc.111.089573
Tang, H., and Lyons, E. (2012). Unleashing the genome of Brassica rapa. Front. Plant Sci. 3:172. doi: 10.3389/fpls.2012.00172
Tang, H., Woodhouse, M. R., Cheng, F., Schnable, J. C., Pedersen, B. S., Conant, G., et al. (2012). Altered patterns of fractionation and exon deletions in Brassica rapa support a two-step model of paleohexaploidy. Genetics 190, 1563–1574. doi: 10.1534/genetics.111.137349
Thomas, B. C., Pedersen, B., and Freeling, M. (2006). Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 16, 934–946. doi: 10.1101/gr.4708406
U, N. (1935). Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 7, 389–452.
Van de Wouw, A. P., Marcroft, S. J., Barbetti, M. J., Li, H. U., Salisbury, P. A., Gout, L., et al. (2009). Dual control of avirulence in Leptosphaeria maculans towards a Brassica napus cultivar with ‘sylvestris-derived’ resistance suggests involvement of two resistance genes. Plant Pathol. 58, 305–313. doi: 10.1111/j.1365-3059.2008.01982.x
Wang, J., Long, Y., Wu, B. D., Liu, J., Jiang, C. C., Shi, L., et al. (2009). The evolution of Brassica napus FLOWERING LOCUST paralogues in the context of inverted chromosomal duplication blocks. BMC Evol. Biol. 9:271. doi: 10.1186/1471-2148-9-271
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–U1157. doi: 10.1038/ng.919
Wang, Y., Wang, X., and Paterson, A. H. (2012). Genome and gene duplications and gene expression divergence: a view from plants. Year Evol. Biol. 1256, 1–14. doi: 10.1111/j.1749-6632.2011.06384.x
Wei, D., Mei, J., Fu, Y., Disi, J. O., Li, J., and Wei, Q. (2014). Quantitative trait loci analyses for resistance to Sclerotinia sclerotiorum and flowering time in Brassica napus. Mol. Breed. 34, 1797–1804. doi: 10.1007/s11032-014-0139-7
West, J. S., Kharbanda, P. D., Barbetti, M. J., and Fitt, B. D. L. (2001). Epidemiology and management of Leptosphaeria maculans (phoma stem canker) on oilseed rape in Australia, Canada and Europe. Plant Pathol. 50, 10–27. doi: 10.1046/j.1365-3059.2001.00546.x
Woodhouse, M. R., Cheng, F., Pires, J. C., Lisch, D., Freeling, M., and Wang, X. (2014). Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids (vol 111, pg 5283, 2014). Proc. Natl. Acad. Sci. U.S.A. 111, 6527–6527. doi: 10.1073/pnas.1405833111
Wu, P., Shao, Z., Wu, X.Z., Wang, Q., Wang, B., Chen, J. Q., et al. (2014). Loss/retention and evolution of NBS-encoding genes upon whole genome triplication of Brassica rapa. Gene 540, 54–61. doi: 10.1016/j.gene.2014.01.082
Yoo, M. J., Szadkowski, E., and Wendel, J. F. (2013). Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110, 171–180. doi: 10.1038/hdy.2012.94
Yu, F., Lydiate, D. J., and Rimmer, S. R. (2005). Identification of two novel genes for blackleg resistance in Brassica napus. Theor. Appl. Genet. 110, 969–979. doi: 10.1007/s00122-004-1919-y
Yu, F., Lydiate, D. J., and Rimmer, S. R. (2008). Identification and mapping of a third blackleg resistance locus in Brassica napus derived from B. rapa subsp. sylvestris. Genome 51, 64–72. doi: 10.1139/g07-103
Yu, F., Gugel, R. K., Kutcher, H. R., Peng, G., and Rimmer, S. R. (2013). Identification and mapping of a novel blackleg resistance locus LepR4 in the progenies from Brassica napus x B. rapa subsp. sylvestris. Theor. Appl. Genet. 126, 307–315. doi: 10.1007/s00122-012-1919-2
Zhao, J. W., Udall, J. A., Quijada, P. A., Grau, C. R., Meng, J. L., and Osborn, T. C. (2006). Quantitative trait loci for resistance to Sclerotinia sclerotiorum and its association with a homeologous non-reciprocal transposition in Brassica napus L. Theor. Appl. Genet. 112, 509–516. doi: 10.1007/s00122-005-0154-5
Keywords: oilseed rape, Leptosphaeria maculans, homoeologous QTL, comparative genomics, gene ontology, genome duplications
Citation: Fopa Fomeju B, Falentin C, Lassalle G, Manzanares-Dauleux MJ and Delourme R (2015) Comparative genomic analysis of duplicated homoeologous regions involved in the resistance of Brassica napus to stem canker. Front. Plant Sci. 6:772. doi: 10.3389/fpls.2015.00772
Received: 15 June 2015; Accepted: 08 September 2015;
Published: 25 September 2015.
Edited by:
Stéphane Hacquard, Max Planck Institute for Plant Breeding Research, GermanyReviewed by:
Milind Ratnaparkhe, Directorate of Soybean Research, IndiaAlice Hayward, The University of Queensland, Australia
Jacqueline Batley, University of Western Australia, Australia
Copyright © 2015 Fopa Fomeju, Falentin, Lassalle, Manzanares-Dauleux and Delourme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Régine Delourme, Institut National de la Recherche Agronomique, UMR IGEPP, BP35327, 35653 Le Rheu, France,cmVnaW5lLmRlbG91cm1lQHJlbm5lcy5pbnJhLmZy