 Congli Wang1,2
Congli Wang1,2 Mauricio Ulloa3*
Mauricio Ulloa3* Xinyi Shi4
Xinyi Shi4 Xiaohui Yuan4
Xiaohui Yuan4 Christopher Saski5
Christopher Saski5 John Z. Yu6
John Z. Yu6 Philip A. Roberts1*
Philip A. Roberts1*- 1Department of Nematology, University of California, Riverside, Riverside, CA, USA
- 2Key Laboratory of Mollisols Agroecology, Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences, Harbin, China
- 3Plant Stress and Germplasm Development Research Unit, USA - Agricultural Research Service, Lubbock, TX, USA
- 4Key Laboratory of Soybean Molecular Design Breeding, Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences, Harbin, China
- 5Clemson University, Genomics Institute, Clemson, SC, USA
- 6USA - Agricultural Research Service, Southern Plains Agricultural Research Center, College Station, TX, USA
Genetic and physical framework mapping in cotton (Gossypium spp.) were used to discover putative gene sequences involved in resistance to common soil-borne pathogens. Chromosome (Chr) 11 and its homoeologous Chr 21 of Upland cotton (G. hirsutum) are foci for discovery of resistance (R) or pathogen-induced R (PR) genes underlying QTLs involved in response to root-knot nematode (Meloidogyne incognita), reniform nematode (Rotylenchulus reniformis), Fusarium wilt (Fusarium oxysporum f.sp. vasinfectum), Verticillium wilt (Verticillium dahliae), and black root rot (Thielaviopsis basicola). Simple sequence repeat (SSR) markers and bacterial artificial chromosome (BAC) clones from a BAC library developed from the Upland cotton Acala Maxxa were mapped on Chr 11 and Chr 21. DNA sequence through Gene Ontology (GO) of 99 of 256 Chr 11 and 109 of 239 Chr 21 previously mapped SSRs revealed response elements to internal and external stimulus, stress, signaling process, and cell death. The reconciliation between genetic and physical mapping of gene annotations from new DNA sequences of 20 BAC clones revealed 467 (Chr 11) and 285 (Chr 21) G. hirsutum putative coding sequences, plus 146 (Chr 11) and 98 (Chr 21) predicted genes. GO functional profiling of Unigenes uncovered genes involved in different metabolic functions and stress response elements (SRE). Our results revealed that Chrs 11 and 21 harbor resistance gene rich genomic regions. Sequence comparisons with the ancestral diploid D5 (G. raimondii), A2 (G. arboreum) and domesticated tetraploid TM-1 AD1 (G. hirsutum) genomes revealed abundance of transposable elements and confirmed the richness of resistance gene motifs in these chromosomes. The sequence information of SSR markers and BAC clones and the genetic mapping of BAC clones provide enhanced genetic and physical frameworks of resistance gene-rich regions of the cotton genome, thereby aiding discovery of R and PR genes and breeding for resistance to cotton diseases.
Introduction
Cultivated plant species are under continuous attack by pathogens, which imposes a major challenge for growers by causing significant crop yield loss (Blasingame and Patel, 2004; Roberts et al., 2007). The future of crop improvement depends on understanding of the distribution, structure, and organization of disease resistance (R) and pathogen-induced (PR) genes (Ulloa et al., 2011). Plants have a great capacity to recognize pathogen effectors and inducers through different strategies (Dodds and Rathjen, 2010); however, our understanding of these strategies and interactions is still limited. New DNA sequence information coupled with the physical alignment of genomic regions into chromosomal maps and the anchoring of genetic maps are all steps that will improve the accuracy of detecting R or PR genes (van Loon et al., 2006; Bent and Mackey, 2007; Kou and Wang, 2010; Ulloa et al., 2011) and gene functions of important biological processes in crops (Rong et al., 2004; Ulloa et al., 2007; Chaudhary et al., 2009). In addition, these new discoveries will have important implications for breeding effective pest and disease resistance into elite cultivars by marker-assisted selection (MAS) (Ulloa et al., 2011, 2013).
Plants express multiple R genes with specificities for different strains of viruses, bacteria, fungi and nematodes, and individual plant genomes include hundreds of R gene-like sequences (Bent and Mackey, 2007; Adams-Phillips et al., 2008; Ulloa et al., 2011). The most studied R genes encode putative intra-cellular proteins with nucleotide binding sites (NBS) and leucine-rich repeat motifs (LRR), which represent the largest R gene family. NBS-LRR proteins can be subdivided in two types based on structural features of the N terminus: TIR-NBS-LRR proteins which resemble the intracellular domains of Drosophila Toll and mammalian IL-1 receptors and CC-NBS-LRR proteins which contain a coiled-coil domain (Jones and Dangl, 2006; Guo et al., 2011; Qi and Innes, 2013). Based on phylogenetic relationships, most R genes reside in clusters either as tandem duplicates on a tree or mixed clusters that contain genes from different branches of a species-wide tree (Meyers et al., 2005). Different R gene-mediated signal transduction pathways may utilize some distinct signaling components and induce a set of plant responses (Sato et al., 2007; Adams-Phillips et al., 2008). In contrast, PR genes have been classified into 17 families of pathogenesis-related proteins. These proteins are induced through the action of the signaling compounds of salicylic acid, jasmonic acid or ethylene (Fonseca et al., 2009; Panstruga et al., 2009; Stepanova and Alonso, 2009). They possess antimicrobial activities in vitro through hydrolytic activities on cell walls, contact toxicity, and perhaps an involvement in defense signaling. However, these proteins serve essential plant functions (senescence, wounding, cold stress, and present in floral tissue) whether they are used in defense or not (van Loon et al., 2006).
In cotton (Gossypium spp.), root-knot nematode [RKN (Meloidogyne incognita)], reniform nematode [REN (Rotylenchulus reniformis)], Fusarium wilt [FOV) (Fusarium oxysporum f.sp. vasinfectum)], Verticillium wilt [VW (Verticillium dahliae)], and black root rot [BRR (Thielaviopsis basicola)] represent expanding threats to crop production (Wang et al., 2006; Niu et al., 2008; Dighe et al., 2009; Ulloa et al., 2011, 2013; Fang et al., 2014; Zhao et al., 2014). Cotton is one of the most economically important crops, providing the world's leading natural fiber, and it is a polyploidy model for cytogenetic, genomic, and evolutionary biology research (Kim and Triplett, 2001; Wendel and Cronn, 2003; Ulloa et al., 2007; Chaudhary et al., 2009). The estimated cotton yield loss due to diseases was 10.93% in the United States in 2004 (Blasingame and Patel, 2004). Increased knowledge of resistance to cotton pathogens such as RKN, REN, FOV, VW, BRR, and of genomic segments housing R or PR genes will help to elucidate the mechanisms of qualitative and quantitative disease resistance.
Knowledge of R and PR genes has increased with the availability of genome data and the increasing number of genes reported to be involved in resistance (Ulloa et al., 2007). New DNA sequences can be examined to discover genes involved in disease resistance by sequence comparisons with existing databases of expressed sequence tags (ESTs) such as GenBank (http://www.ncbi.nlm.nih.gov/). Additional studies using genomic and proteomic technologies have facilitated global comparisons of R and PR expression profiles (Ulloa et al., 2011; Yin et al., 2012; Wang et al., 2013; Wei et al., 2013) and pathway components of genes involved in disease defense and/or response (Chisholm et al., 2006).
Integrating disease resistance phenotypes into high-yielding, high-fiber quality cultivars is one of the most important objectives in cotton breeding programs (Ulloa et al., 2011). To further elucidate and expedite the discovery of R and/or PR genes; herein, we provide new DNA sequence information of large genomic segments (e.g., BAC clones) from cv. Acala Maxxa (G. hirsutum L.) for which MUSB-derived single sequence repeat (SSR) markers were previously mapped to chromosomes (Chr) 11 and 21 (Frelichowski et al., 2006; Ulloa et al., 2008; Yu et al., 2012). These markers reportedly underlie QTLs involved in disease resistance; therefore, capturing and sequencing BAC-sized genomic segments tightly linked to these SSRs will help to resolve local content and genome structure of RKN (Shen et al., 2006; Wang et al., 2006; Ynturi et al., 2006; Ulloa et al., 2010), REN (Dighe et al., 2009; Gutiérrez et al., 2011); FOV (Ulloa et al., 2011, 2013), VW (Bolek et al., 2005; Fang et al., 2014; Zhao et al., 2014), and BRR (Niu et al., 2008) resistance. The Maxxa BAC clone and marker sequence data were also compared to the whole genome sequence assemblies of the G. raimondii D5 and G. arboreum A2 ancestral diploid genomes (Paterson et al., 2012; Wang et al., 2012b; Li et al., 2014) and domesticated tetraploid TM-1 AD1 (G. hirsutum) genome which are now publicly available (Li et al., 2015; Zhang et al., 2015).
Materials and Methods
Selection and Sequencing of BAC Clones of Upland Cotton Chromosomes 11 and 21
Two strategies were deployed to recruit BAC clones that mapped to Upland cotton Chr 11 and Chr 21 from the cv. Acala Maxxa genomic library (Tomkins et al., 2001). The first strategy used MUSB SSR markers previously mapped to Chr 11 (Frelichowski et al., 2006). Some of these marker-loci were later placed on Chr 21 (Ulloa et al., 2008; Yu et al., 2012). We selected BAC clones which contained 12 MUSB SSRs (Table 1) from these two chromosomes. Some of these selected MUSB markers were identified as being associated with FOV resistance, using genetic and QTL mapping methods, and bulked segregant analysis (BSA) on resistant and susceptible progeny with different genetic backgrounds (Ulloa et al., 2011, 2013; Ulloa M and Roberts P unpublished information). Other MUSB markers were selected because they were mapped in the vicinity of an underlying QTL involved in pathogen resistance (Table 2).
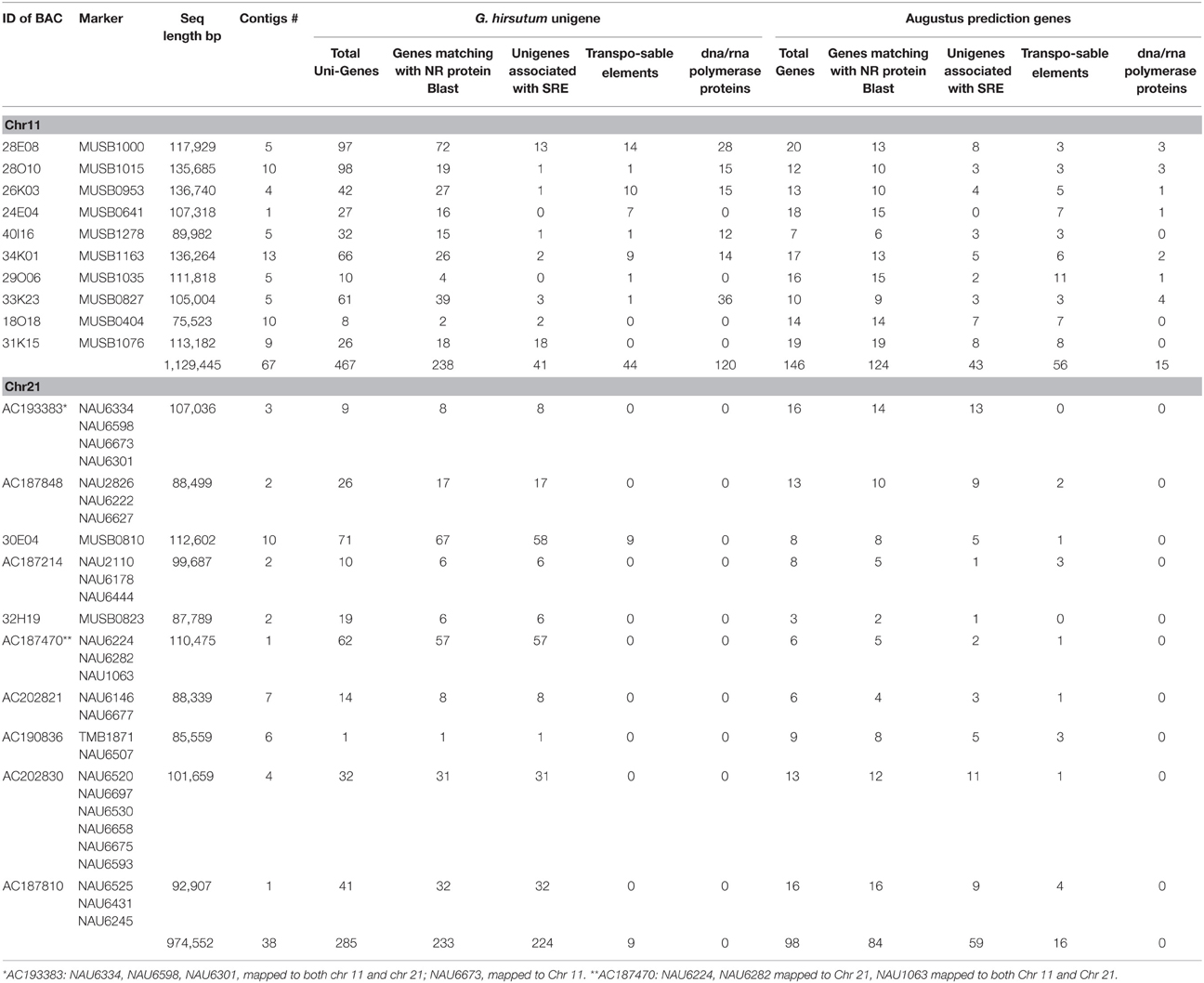
Table 1. Bacterial artificial chromosome (BAC) and derived MUSB SSR marker names, and number of Unigenes predicted based on G. hirsutum Unigene NCBI database and genes predicated based on Augustus Gene Prediction Software of BAC clones on Upland cotton chromosomes 11 and 21.
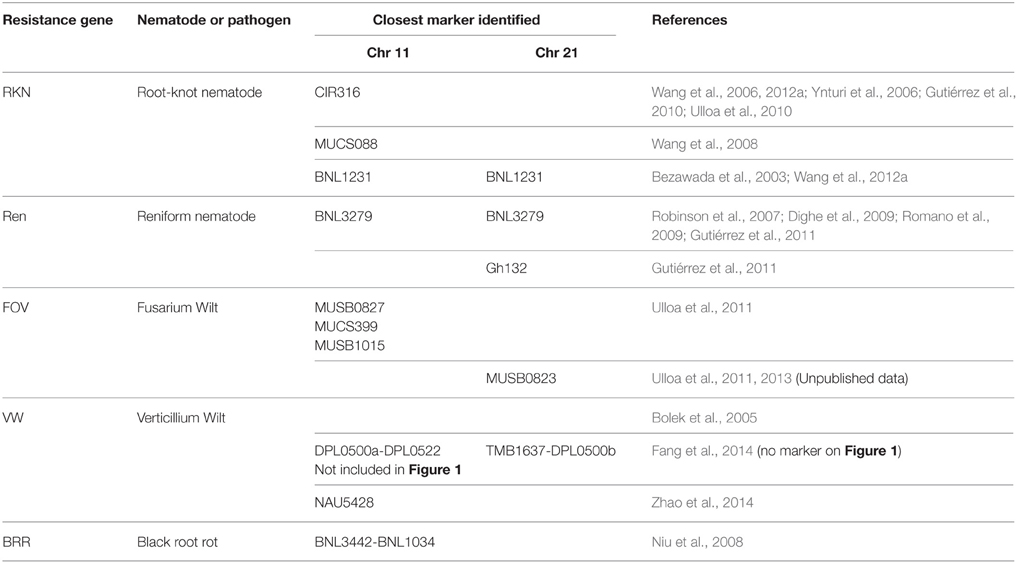
Table 2. SSR markers underlying QTL associations with nematode and pathogen resistance genes on Upland cotton chromosomes 11 and 21.
The second strategy was to use SSR marker-sequences previously mapped on Chr 11 and Chr 21 (CMD: http://www.cottonmarker.org/) to select BAC clones previously sequenced from the Acala Maxxa library by sequence-comparison. These BAC clones were originally sequenced erroneously as part of the maize sequencing project by the Genome Sequencing Center, Washington University School of Medicine. The DNA sequence information of these BACs was deposited into GenBank under the accession numbers: AC193383, AC187848, AC187214, AC187470, AC202821, AC190836, AC202830, and AC187810. Sequences of each BAC clone (Table 1) were compared to SSR marker-sequences from Chr 11 and Chr 21. The selection criteria of tagging a BAC clone with mapped SSR markers from these chromosomes were as follows: only the sequence of each SSR marker spanning forward primer to the reverse primer (including the SSR motif) was used for the comparison. DNA sequences were blasted using all six frames (forward +1 to +3 and reverse −1 to −3) base positions. Potential BAC clones were tagged with an SSR marker when both (BAC and SSR) DNA sequences had a similarity >96%.
Sequencing and Assembly of Upland Cotton BAC Clones
A small-insert (3–5 kb) library was constructed from each of the 12 BAC clones, which harbored the selected MUSB markers on Chr 11 and Chr 21 (Table 1). Small-insert DNA fragments were generated by isolating BAC DNA as a maxi-prep from the BAC clone and subjecting the DNA to random fragmentation by hydroshearing (Digilab®, Digilab Inc., Holliston, MA). Fragments between 3 and 5 kb were size-selected by gel electrophoresis, were end-repaired and cloned into the hi-copy plasmid-based cloning vector pBlueskriptKSII+ (Agilent Technologies) and then electroporated into E. coli DH10B host cells. Transformants were selected on Lysogeny broth (LB) plates containing carbenicillan, X-Gal and IPTG. White recombinant colonies were picked robotically using the Genetix Q-bot (Genetix, Boston, MA) and stored as individual clones in Genetix 96-well microtiter plates as glycerol stocks at −80°C. Sequencing was performed using the Dye-terminator cycle sequencing kit v3.1 (Applied Biosystems, Foster City, CA). Sequence data from the forward and reverse universal priming sites of the shotgun clones were accumulated on an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA). The BAC clones were sequenced to approximately 8X clone coverage (assuming 120 kb average insert size) and assembled with PHRAP software (Ewing et al., 1998), and edited with Consed (Gordon et al., 1998). Sequence contigs were ordered and oriented by the bridging shotgun method, and gaps were joined by the addition of N's giving a single contiguous consensus sequence for analysis. The sequencing of the BAC clones, which harbored the MUSB markers, was performed at Clemson University Genomics Institute, SC, USA. Additional information about the sequencing of these clones can be found in Ulloa et al. (2011). The DNA sequence information of these BACs was deposited into GenBank under the accession numbers: KM396694 (28E08), KM396695 (28O10), KM396696 (26K03), KM396697 (24E04), KM396698 (40I16), KM396699 (34K01), KM396700 (29O06) KM396701 (33K23), KM396702 (18O18), KM396703 (31K15), KM396704 (30E04), and KM396705 (32H19). The numbers and letters identify the BAC clone.
BAC Sequence Annotation of Stress Response Elements
DNA sequence-local alignments were made with the comprehensive G. hirsutum unigene set from http://www.plantgdb.org. The Unigene set consisting of 98,420 Unigenes (G. hirsutum mRNA assembly May 8, 2008; based on GenBank release 165) was downloaded from PlantGDB (www.plantgdb.org). Unigene sequences were BLASTN aligned to each BAC sequence individually with an e ≤ 1e-5 and identity ≥90%. Gene Ontology (GO) annotation was conducted using the Blast2GO program with default parameters (Gene Ontology Consortium, 2006; Conesa and Gotz, 2008). Gene prediction and annotation were performed using the prediction program Augustus (Stanke and Morgenstern, 2005). The Augustus program was tested on the Arabidopsis gene set, which considers expressed sequence tag (EST) matches as additional support for gene identification. All predicted genes and unigenes were subjected to a similar analysis using BLASTX through the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) nr protein database with a value of 1e-5 to identify previously established protein motifs. Stress response elements (SRE) were identified based on the description of bioprocess of GO annotation. Genes involved in stress response elements were identified according to associated protein molecular function (MF), bioprocess (BP), and cell component (CC).
Alignment to Gossypium raimondii (D5), G. arboreum (A2), G. hirsutum TM-1 (AD1), and Other Genomes
BAC sequences were aligned to the G. raimondii diploid D5 whole genome (phytozome.net) (Paterson et al., 2012) through NCBI-nucleotide BLAST, G. arboreum diploid A2 whole genome (http://cgp.genomics.org.cn) (Li et al., 2014) and TM-1 AD1 genome (http://cottongen.org) from two independent groups (CGP-BGI group, Li et al., 2015; NAU-NBI group, Zhang et al., 2015) with an e ≤ 1e-10 and identity ≥90%. The comparisons of the BAC sequences on Chr 11 and Chr 21 with corresponding chromosomes in A2, D5, AD1 genome backgrounds were conducted. The average identity and the percentage of mapped BAC sequences were calculated based on consecutive matched sequence with compared genomes. The TM-1 sequence from the CGP-BGI group was used as a genome background to determine that resistance genes from these BACs are more frequently located in the regions of Chr 11 and Chr 21 with Fisher's exact test (P < 0.05). Comparisons were also made between these BACs and other plant taxa: Arabidopsis thaliana, Vitis vinifera, Populus trichocarpa, and Theobroma cacao.
Selection of SSR Markers and Construction of Linkage Groups
We targeted all SSR markers previously mapped on Upland cotton Chr 11 and Chr 21 (CMD: http://www.cottonmarker.org/), especially those underlying QTLs determining resistance to RKN (Shen et al., 2006; Wang et al., 2006, 2012a; Ynturi et al., 2006; Gutiérrez et al., 2010; Ulloa et al., 2010), REN (Dighe et al., 2009; Romano et al., 2009; Gutiérrez et al., 2011), FOV (Ulloa et al., 2011, 2013), VW (Bolek et al., 2005; Fang et al., 2014), and BRR (Niu et al., 2008). QTL analyses of marker-resistance associations for RKN, REN, FOV, VW, and BRR on these chromosomes were reported from previous publications (Table 2).
Initially, 1100 SSR markers (BNL, CIR, GH, MUSB, MUCS, MUSS, NAU, DPL, DOW, and TMB) were used with wide coverage to construct the linkage groups of Chr 11 and Chr 21 on the recombinant inbred line (RIL) population of Upland TM- 1 × Pima 3-79 (Frelichowski et al., 2006; Ulloa et al., 2008, 2011, 2013; Wang et al., 2012a; Yu et al., 2012). Additional SSR markers identified to be tagged to a BAC clone or clones were mapped using JoinMapR version 4.0 (Van Ooijen, 2006). Likelihood ratio (LOD) scores of 8–12 were examined for each linkage group/chromosome using the Kosambi mapping function and a maximum distance of 40 cM on this population. Moreover, using the anchored SSR markers (MUSB) of these linkage groups and their recombination frequencies or cM distances, SSR markers were placed on Chr 11 and Chr 21 linkage groups (Figure 1) on the most recent published linkage maps of the TM-1 x 3-79 RIL population (Yu et al., 2012). Only the name of SSR markers was included in Figure 1, keeping their original cM distance between the SSR markers.

Figure 1. Linkage maps of Chr 11 and its homoeologous Chr 21 using an interspecific [Upland TM-1 (Gossypium hirsutum) x Pima 3-79 (G. barbadense)] RIL population (Yu et al., 2012), showing relationships between molecular markers and underlying QTLs involved in resistance to root-knot nematode (RKN, Shen et al., 2006; Wang et al., 2006; Ynturi et al., 2006; Ulloa et al., 2010), reniform nematode (REN, Dighe et al., 2009; Gutiérrez et al., 2011); Fusarium wilt (FOV, Ulloa et al., 2011), Verticillium wilt (VW, Fang et al., 2014; Zhao et al., 2014), and black root rot (BRR, Niu et al., 2008).
Marker Analysis and Data Mining
SSR markers previously mapped on Chr 11 and Chr 21 reported in the Cotton Marker Database (CMD: www.cottonmarker.org) were used to investigate DNA sequence composition. Sequences were then BLASTed through the NCBI (http://www.ncbi.nlm.nih.gov/). Sequences were compared against three databases: (a) Nucleotide collection (nr/nt); (b) Expressed Sequence Tags (EST); and (c) Non-Redundant protein sequences (nr). The top sequence hits found for each sequence in all three databases were then BLASTed through GO (http://www.geneontology.org/). The top functional hits given by GO were collected along with their categorized gene products [biological process (BP), cellular component (CC), and molecular function (MF)]. SSR markers involved in defense response or stress response were categorized according to top blasted protein function (receptor, disease protein, transcription factor, and oxygen-reduction and so on) and GO annotation.
Results
BAC Sequence and Annotation for Stress Response Elements
Twenty selected BAC clones were analyzed for potential coding elements involved in response to biotic/abiotic stress mechanisms (Table 1). Twelve BAC clones tagged with BAC-end MUSB [selected from Frelichowski et al. (2006) and Ulloa et al. (2008, 2011)] markers were sequenced: BAC-derived MUSB0404, MUSB0641, MUSB0827, MUSB0953, MUSB1000, MUSB1015, MUSB1035, MUSB1076, MUSB1163, and MUSB1278 from Chr 11, and MUSB0810 and MUSB0823 from Chr 21 (Table 1). The estimated BAC clone size according to assembled sequence data ranged from 68 to 140 kb with an average of 106 kb per BAC. The BAC clones were sequenced to an approximate 8X coverage, which resulted in 3–8 ordered contigs spanning up to 140,000 bp. In addition, seven BAC clones tagged to previously mapped SSR markers (25 NAUs and one TMB) on Chr 21 from the Upland cotton cultivar Acala Maxxa genomic library previously sequenced by the Genome Sequencing Center, Washington University School of Medicine were also investigated for potential coding elements: AC193383, AC187848, AC187214, AC187470, AC202821, AC190836, AC202830, and AC187810 (Table 1). These Maxxa BACs, erroneously sequenced by the maize group, were used in a different cotton characterization study by Guo et al. (2008). In this study, the 10 BAC clones from Chr 11 yielded a total of 1,129,445 bp while the 10 BAC clones from Chr 21 yielded 974,552 bp, for a total of 2,103,997 bp sequence data.
BAC sequence annotation by BLASTN alignment to the publicly available G. hirsutum Unigene set (GenBank release 165) revealed 467 (Chr 11) and 285 (Chr 21) putative Unigenes (e ≤ 1e-5). Functional signature annotations of BAC-mapped Unigene sequences were aligned to the non-redundant protein database and assigned GO terms. A total of 238 out of 467 of Chr 11 and 233 out of 285 of Chr 21 putative Unigenes were found to be similar to known protein sequences with e ≤ 1e-5 (Table 1), while 229 putative Unigenes on Chr 11 and 52 on Chr 21 had no match to known protein sequences with e ≤ 1e-5 (Table 1 and Tables S1, S2). There were 41 Unigenes on Chr 11 and 224 on Chr 21 involved in disease defense response or stress response elements (SRE) (Table 1) based on sequence description from the BLASTed protein database and GO annotations [P (bioprocess), F (molecular function) and C (cell component)] (additional information highlighted in yellow in Tables S1, S2). Stress response elements involved in internal and external stimulus, stress, signaling process and cell death from these Unigenes are shown in Table S3 for Chr 11 and Table S4 for Chr 21. In addition, 44 transposable elements (TEs) and 120 DNA/RNA polymerase family proteins were identified on Chr 11, and nine TEs but no DNA/RNA polymerase protein on Chr 21 (Table 1).
Augustus gene prediction software revealed 146 genes on Chr 11 and 98 genes on Chr 21. The results indicated abundance of genes with considerable homology to disease response elements for these BAC clones (Table 1 and Tables S5–S8), with function in cellular growth and development processes, transport, translation, plus metabolic functions and stress response elements. Forty-three genes on Chr 11 BACs and 59 genes on Chr 21 BACs were involved in defense response (Table 1 and Tables S5, S6 highlighted in yellow), including receptor kinase proteins, early-responsive to dehydration stress proteins, subtilisin-like serine endopeptidase family proteins, strictosidine synthase-like, universal stress proteins, auxin-responsive proteins, and disease resistance proteins involved in stress response. GO annotation showed a range of defense associated proteins for MF, and SRE included responses to biotic/abiotic stimulus, signaling, and cell death (Tables S7, S8).
The Augustus gene prediction software also indicated 56 TE on Chr 11 BACs and 16 on Chr 21 BACs (Table 1). TE included retrotransposon ty1-copia subclass, retrotransposon ty3-gypsy subclass, gag-pol polyprotein, mutant gag-pol polyprotein, mutator sub-class protein and copia-like retrotransposable elements (Table 3, Tables S5, S6). The longest TE hit length extended 6759 bp. A GO analysis further characterized these TE into a range of defense-related acitivities (Table 3 and Tables S5, S6). In addition to the TEs, 15 DNA/RNA polymerase family proteins were identified on Chr 11 but none were identified on Chr 21 (Table 1).
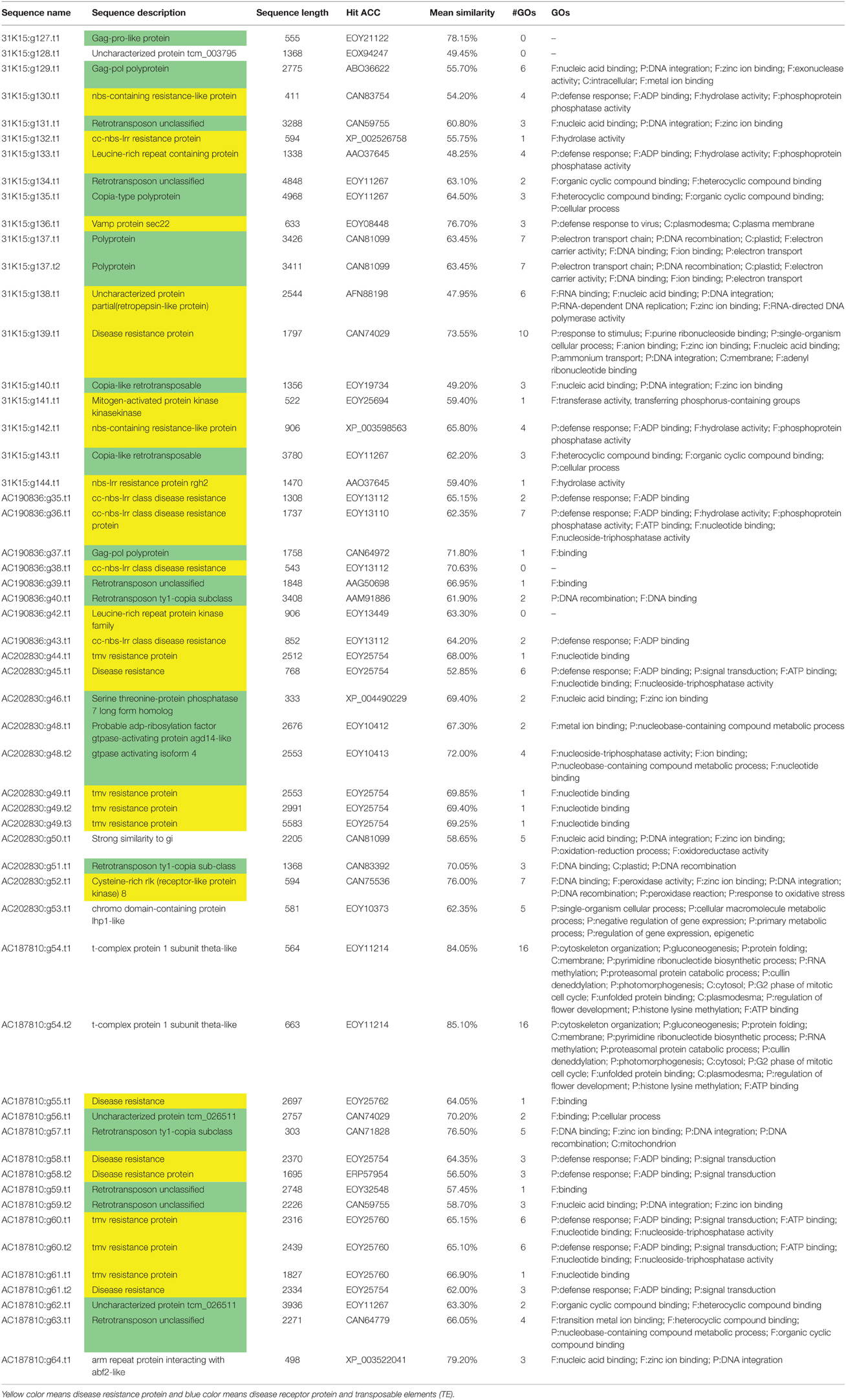
Table 3. BAC sequences of Upland cotton chromosomes 11 and 21 that contain disease resistance encoded protein annotation with associated transposable elements.
Twenty-three disease resistance proteins were identified in four BACs (31K15 on Chr 11, and AC190836, AC202830 and AC187810 on Chr 21). The BAC 31K15 associated with marker MUSB1076 linked to R gene rkn1 (Wang et al., 2006) and cluster regions containing leucine-rich repeat protein, NBS-LRR resistance protein rgh2 or rgh1, and CC-NBS-LRR resistance protein. Three BAC clones (AC190836, AC202830, and AC187810) on Chr 21 contained R genes harboring NBS-LRR proteins, including CC-NBS-LRR class disease resistance, tmv resistance protein and other disease resistance proteins (Table 3). Based on structural features of the N terminus, NBS-LRR proteins were surrounded by additional receptor proteins such as serine-threonine and kinase-like proteins, and TEs (Table 3). Moreover, NBS-LRR genes were identified within clusters and in the vicinity of the RKN, REN, FOV, VW, and BRR resistance of marker-genes previously reported (Bolek et al., 2005; Wang et al., 2006; Niu et al., 2008; Dighe et al., 2009; Ulloa et al., 2011).
More specifically, a percent identity plot of duplication harboring NBS-LRR resistance motifs for BAC clones AC187810 vs. AC202830 on Chr 21 is given in Figure 2, in which a set of seven regions were found harboring NBS-LRR motifs with a minimum of 70% identity spanning the clone length of ~90 kb.
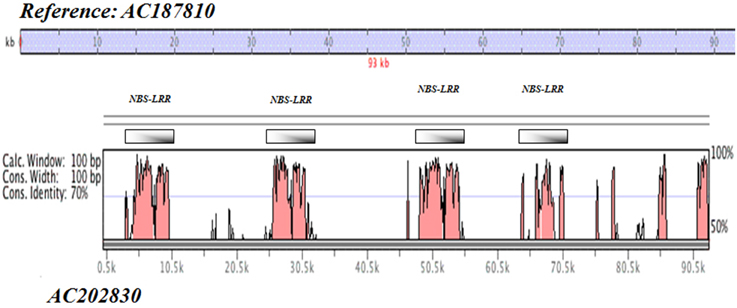
Figure 2. Self-alignment of BAC clones in Upland cotton Chr 21. Percent identity plot of duplication harboring NBS-LRR resistance motifs (BAC clones AC187810 vs. AC202830 on Chr 21).
Alignment to Gossypium raimondii (D5), G. arboreum (A2), G. hirsutum TM-1 (AD1) and Other Genomes
A synteny block comparison was made of alignment of full length sequences of Chr 11 and 21 BAC clones to the two available assembled whole diploid genome sequences of G. arboreum (A2) and G. raimondii (D5) (Tables S9–S13). The comparisons among the matched sequences showed 84.23% identity with Chr 11 BACs and 98.54% identity with BACs of Chr 21 of the tetraploid (AD) genome, corresponding to D5 Chr 7 genome sequence (Tables S9, S12, S13). Eight percent and 80% consecutive sequences from chromosomes 11 and 21, respectively were mapped to D5 Chr 7. Seven Chr 11 BACs with no consecutive mapping sequence were also mapped to D5 Chr 7 in several regions. Most matched sequences of these seven BACs were TEs (Tables S9, S12, S13) which showed multiple copies through the whole genome, including D5 Chr 7. More BLAST hits of Chr 11 BACs than Chr 21 BACs with Chr 7 A2 genome sequence were found (Tables S9–S11). However, only one Chr 11 BAC (29O06) showed consecutive sequence length with Chr 7 A2 genome (Tables S9–S11). The BAC sequences matched with the A2 genome were mostly transposable elements which are distributed across the whole genome.
Alignment of Chr 11 and Chr 21 BAC sequences from G. hirsutum Maxxa to G. hirsutum TM-1 genome showed slight differences between the two sequencing groups BGI and NBI, possibly due to different assembly methods (Tables S14, S15). In total, 42 and 52% consecutive sequences of Maxxa BACs on chromosomes 11 and 21, respectively, were mapped to TM-1 At-Chr1 (equals Chr 11) and Dt-Chr7 (equals Chr 21) from BGI sequencing data (Tables S14, S15). From NBI sequencing data, 41 and 62% consecutive sequences of Maxxa BACs on chromosomes 11 and 21 were mapped to A11 (equals Chr 11) and D11 (equals Chr 21) of the TM-1 genome, respectively. The identities of matched sequences between Maxxa BACs and TM-1 genome reached 98% for Chr11 comparison and 97% for Chr 21 comparison with both BGI and NBI sequencing data. Some BAC sequences were aligned to unmapped scaffolds and mapped chromosomes, such as 34K01, indicating the unmapped scaffolds might be connected to the mapped chromosome. Partial consecutive sequences of the Maxxa BAC 32H19 on Chr 21 linked to the marker MUSB0823 were mapped to TM-1 genome Chr 11 (Tables S14, S15). Part of Maxxa BAC 40I16 sequence linked to MUSB1278 was mapped to Chr 7 in the TM-1 genome (Tables S14, 15). Most unmapped Maxxa BAC sequences matched with Chr 11 or Chr 21 were transposable elements across the whole genome. The enrichment analysis with Fisher's exact test indicated that 115 out of 168 GOs compared with TM-1 genome sequence from CGP-BGI group were over-represented in Chr 11 and Chr 21 regions with p < 0.05 (range from 8.12E-33 to 0.041). The 115 GOs included stress response elements, such as oxidoreductase activity, cell-cell signaling, defense response to virus, syncytium formation, response to abiotic stimulus, MAP kinase kinase kinase activity, and transmembrane receptor protein tyrosine kinase signaling pathway.
Comparison of Chr 11 and Chr 21 BAC sequences with four other plant taxa—Arabidopsis thaliana, Vitis vinifera, Populus trichocarpa, and Theobroma cacao, revealed conserved regions of short sequences with each plant species. Alignments with T. cacao and V. vinifera were especially strong for certain cotton BAC clones, but less so with A. thaliana and P. trichocarpa. Results from these comparisons and subsequent GO analyses did not provide additional information.
Genetic Mapping and SSR Marker Sequence Composition
Initially, 1100 SSR markers that provided genome-wide coverage (Park et al., 2005; Frelichowski et al., 2006; Wang et al., 2006; Ulloa et al., 2008, 2011, 2013; CMD, www.cottonmarker.org) were used to develop Upland cotton Chr 11 and Chr 21 linkage groups. Matrix genotypic data of these SSR markers were used to develop the most recent genetic linkage map of the TM-1 x 3-79 RIL population (Yu et al., 2012). In addition, QTL analyses were previously conducted on Fusarium wilt phenotypic data (Ulloa et al., 2011, 2013) and root-knot nematode root-galling and egg production phenotypic data (Wang et al., 2006, 2008, 2012a; Ulloa et al., 2010) using the SSRs and related RIL populations. SSR markers associated with FOV and RKN resistance on the TM-1 x 3-79 genetic map are presented in Figure 1 (Ulloa et al., 2011, 2013; Wang et al., 2012a). SSR marker associations with resistance to RKN (Bezawada et al., 2003; Shen et al., 2006; Ynturi et al., 2006) and to other pathogens [REN (Robinson et al., 2007; Dighe et al., 2009; Romano et al., 2009; Gutiérrez et al., 2011); VW (Bolek et al., 2005; Fang et al., 2014; Zhao et al., 2014), and BRR (Niu et al., 2008)] reported by other research groups are also presented in Figure 1. The locations of the MUSB markers derived from the Acala Maxxa BAC clones (Table 1) are shown in Figure 1.
SSR Marker Sequence Annotation for Stress Response Elements
Comparison of available sequence information from 256 SSRs on Chr 11 and 239 on Chr 21 to sequences in NCBI EST databases indicated considerable sequence similarity to known genes in plants, with 145 and 142 gene-homologies, respectively, of which 99 on Chr 11 and 109 on Chr 21 were indicated to play a role in plant defense. SSR sequences were similar to transcription factors R2R3-myb transcription factor, heat shock transcription factor, receptor kinase protein, light-regulated protein, zinc finger protein, leucine-rich repeat family protein, nucleic binding protein, WRKY DNA-binding protein, and Verticillium wilt resistance-like protein (Tables S16, S17). Because of duplicated loci from a single marker mapped on Chr 11 and its homoeolog Chr 21, similar genes, pseudogenes, or gene-forms may be present on both chromosomes (Figure 1; www.cottonmarker.org). Categorization of the gene function revealed that markers of Chrs 11 and 21 mapped to genes associated with all three GO: BP, CC, and MF (Tables S16, S17). GO also revealed similarities to SRE genes involved in internal and external stimulus, stress, signaling process and cell death (Table 4, Tables S18, S19). The table S20 provides data on the distance between the mapped chromosome-wide and BAC-specific markers and the defense gene sequences found on Chrs 11 and 21 listed in Table 3.

Table 4. Gene ontology of marker sequences in Upland cotton chromosomes 11 and 21 that show stress response related annotations.
Discussion
The approach in this study was to develop a genetic and physical framework for the genomic regions of Upland cotton homoeologous Chr 11 and Chr 21 that contain important nematode and fungal disease resistance associations with molecular markers such as SSRs. While various QTL and other genetic mapping approaches have revealed the importance of this pair of cotton chromosomes in defense to biotic stresses, there has hitherto been little physical structure development and use of sequence annotation to advance our understanding of its genetic organization. The current and previous marker work provided numerous mapped marker sequences for these two chromosomes, some of which are important for use in cotton breeding programs. Furthermore, this resource allowed us to identify existing BAC clones in the G. hirsutum Acala Maxxa BAC library that are from Chr 11 and Chr 21 based on genetic mapping with SSR markers derived from the BAC-end sequences. Targeted full clone sequence of these mapped BAC clones provided a second resource of genomic DNA sequence to investigate defense response motif content of this cotton genome region. The Maxxa BAC clone and marker sequence data were also compared to the whole genome sequence assemblies of the G. raimondii D5 and G. arboreum A2 ancestral diploid genomes (Paterson et al., 2012; Li et al., 2014), and two G. hirsutum TM-1 AD1 whole genome assemblies which are now publicly available (Li et al., 2015; Zhang et al., 2015).
Of particular interest is the very high defense response element content of sequences from both the SSR markers and the BAC clones on both Chr 11 and Chr 21. This result is in line with the currently recognized importance of this pair of cotton chromosomes in resistance to a wide range of parasitic nematodes and disease-causing pathogens of cotton revealed through genetic mapping of resistance trait determinants. The gene ontology annotations clearly demonstrate the richness of this region in the evolution of defense genes. Typically resistance loci evolve by tandem duplication followed by mutation and divergence of functional specificity, for example nematode resistance in soybean (Cook et al., 2012), often in response to or as a hedge against similar mutation and evolutionary changes in virulence factors in the nematode or pathogen. The large number of NBS-LRR type motifs with tandem repeats, for example as summarized for one of the two BAC clones in Figure 2 and sequence duplication of the BAC clones on Chr 21 (Figure 2), exemplifies this evolutionary hot-spot of defense gene-rich arrangement.
Comparison of DNA sequence between Chr 11 and Chr 21 for certain BAC clones also indicates the high homology between the sequences of the homoeologous chromosome pair. Thus, herein we not only report apparent large-scale duplication events within an Upland cotton chromosome, but also considerable duplication and an evolving separation of sequence homology between a pair of homoeologous chromosomes. This provides cotton with an enormous reservoir of defense response genes, some of which may be defeated related to prior pathogen forms, while others provide a resource for defense against future pathogen forms.
More TEs were identified on Chr 11 (At subgenome) than on Chr 21 (Dt subgenome) according to both G. hirsutum Unigene (A/D: 44/9) and predicted gene databases (A/D: 56/16) (Table 1), which might account for the physical difference in size of the A-subgenome in reference to the D-subgenome. Li et al. (2014) reported that there were a total of 4098 TEs on Chr 7 (equivalent to Chr 11 in G. hirsutum) in the diploid G. arboreum A genome and only 1542 TEs on Chr 7 (equivalent to Chr 21 in G. hirsutum) in the diploid G. raimondii D5 genome even though there were similar numbers of loci identified on Chr 7 in both diploid genomes. At least 64.8% TEs were identified in the TM-1 genome by Zhang et al. (2015) and 66% TEs by Li et al. (2015). More TEs in the A sub-genome (at least 843.5 Mb, genome size 1477 Mb) than in the D sub-genome (at least 433 Mb, genome size 831 Mb) were determined in the TM-1 genome (Zhang et al., 2015). TEs are known to play a dominant role contributing to angiosperm evolution and diversity (Oliver et al., 2013). In cotton, allotetraploid G. hirsutum was derived from reuniting of diploid A and D genomes about 1–2 million years ago (mya) through independent and differential accumulation of TEs 5 mya (Hu et al., 2010; Li et al., 2014). We found that resistance genes in BACs were always surrounded with retrotransposable elements (Table 3). Retrotransposons based on “cut and paste” mode are more abundant in cotton, including Ty1-copia and Ty3-gypsy elements (Hawkins et al., 2006; Hu et al., 2010). More than 50% retrotransposon frequencies were reported in the TM-1 genome (Li et al., 2015; Zhang et al., 2015). TEs involved in abiotic and biotic stress responses have gained more attention recently (Grandbastien, 1998; Grandbastien et al., 2005; Cowley and Oakey, 2013; McDowell and Meyers, 2013; Oliver et al., 2013; Tsuchiya and Eulgem, 2013; Wheeler, 2013). More TEs on the At subgenome might suggest more adaptation to biotic stress response on Chr11 than on Chr 21. In addition, we found 120 DNA-RNA polymerase family protein genes contributing to regulation of transcription on Chr 11 BACs with the G. hirsutum Unigene database but none of these on Chr 21. It is not clear to what extent DNA-RNA polymerase family proteins function in stress response but these results suggest divergent evolution between the A and D genomes.
Comparison of G. hirsutum AD1 whole genome with A2 and D5 were thoroughly conducted by Li et al. (2015) and Zhang et al. (2015) and with other genomes (A. thaliana, T. cacao, Glycine max, and V. vinifera) (Li et al., 2015). However, the 20 Maxxa BACs could not be fully mapped to the TM-1 genome, indicating that differences occur between the two tetraploid G. hirsutum AD1 cotton varieties. Abundant transposable elements might cause the difference between the two G. hirsutum cotton varieties. In addition, homeologous exchanges were also observed between At subgenome Chr 11 and Dt subgenome Chr 21 (Tables S14, 15). For example, Maxxa BAC 32H19 linked to MUSB0823 on Chr 21 (Figure 1, Yu et al., 2012) was mapped to both Chr11 and Chr 21on TM-1 genome (Tables S14, S15).
Comparisons between Maxxa BACs from the tetraploid AD1 cotton and the A2 and D5 ancestral genomes were made to better understand the evolution of the AD genome, particularly in regard to relationships that may shed light on resistance evolution. Comparison of sequence alignments showed less similarity between tetraploid AD Chr 11 and D5 genome than between AD Chr 21 and D5 genome, further supporting independent evolution of the A and D genomes. Likewise, sequence alignments showed less similarity between tetraploid AD Chr 21 and A2 genome than between AD Chr 11 and A2 genome. The divergence of the A and D genomes is also reflected in the origins of resistance traits. For example, in a previous study G. hirsutum (AD1) and G. barbadense (AD2) were found to share the same SSR marker MUCS088 alleles as G. arboreum (A2), suggesting nematode resistance introduction was from the diploid cotton (A2) genome (Roberts and Ulloa, 2010).
The comparison of aligned sequences with four other sequenced plant taxa indicated a conservation of genic sequence among these plants. The highest similarities of cotton BAC sequence to the other plant taxa indicated the closest relationship with T. cacao. Both G. raimondii and G. arboreum genomes showed close collinear relationships with T. cacao and both of them might share a common ancestor having diverged from T. cacao 18–58 mya (Paterson et al., 2012; Wang et al., 2012b; Li et al., 2014).
Genome-wide association studies (GWAS) have been utilized successfully to identify genetic variation in plants (Brachi et al., 2011), and the availability of diploid and tetraploid whole genome sequences makes possible GWAS for identifying genetic variation in cotton. A whole genome marker map in cotton was constructed by Wang et al. (2013) based on the G. raimondii D5 genome (Paterson et al., 2012). Wei et al. (2013) conducted systematic analysis and comparison of nucleotide-binding site disease resistance genes in the G. raimondii D5 genome (Wang et al., 2012b) and genome-wide analysis of the gene families of resistance gene analogs and their response to Verticillium wilt was made in both the G. raimondii D5 (Chen et al., 2015) and G. arboreum A2 genomes (Li et al., 2014). A comprehensive meta QTL analysis was made for fiber quality, yield, drought tolerance and disease resistance with different cotton populations (Said et al., 2013). GWAS in the tetraploid (AD) TM-1 cotton revealed positively selected genes for fiber improvement in the A genome and for stress tolerance in the D genome (Zhang et al., 2015). GWAS in the allotetraploid cotton to identify resistance-rich regions will provide more insights about the evolution of the homoeologous chromosomes 11 and 21 and benefit disease management.
In conclusion, the sequence information and physical mapping of BAC clones provide an additional genomic resource of these resistance gene-rich regions of the Upland cotton genome on Chr 11 and Chr 21. BAC clone sequences are deposited in GenBank (NCBI: http://www.ncbi.nlm.nih.gov). Continuing genetic and physical framework alignment of sequence information in cotton will help to expedite the discovery of R and PR genes and the assembly of a whole Upland cotton tetraploid genome, eventually supporting breeding for disease resistance in cotton production.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Partial support for this work was provided by grants from Cotton Incorporated to PR and MU and from University of California Discovery Grant Program to PR. This research was also partially supported by USDA-ARS (6208-21000-019-00). Partial support for bioinformatic analyses was provided by the One Hundred Talent Grant Program, Chinese Academy of Sciences and Chinese National Scientific Funding (31471749). Mention of trade names or commercial products in this manuscript is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. The U.S. Department of Agriculture is an equal opportunity provider and employer.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00791
References
Adams-Phillips, L., Wan, J., Tan, X., Dunning, F. M., Meyers, B. C., Michelmore, R. W., et al. (2008). Discovery of ADP-ribosylation and other plant defense pathway elements through expression profiling of four different Arabidopsis-Pseudomonas R-avr interactions. Mol. Plant Microbe. Interact. 21, 646–657. doi: 10.1094/MPMI-21-5-0646
Bent, A. F., and Mackey, D. (2007). Elicitors, effectors, and R genes, the new paradigm and a lifetime supply of questions. Annu. Rev. Phytopathol. 45, 399–436. doi: 10.1146/annurev.phyto.45.062806.094427
Bezawada, C., Saha, S., Jenkins, J. N., Creech, R. G., and McCarty, J. C. (2003). SSR marker(s) associated with root-knot nematode resistance gene(s) in cotton. J. Cotton Sci. 7, 179–184.
Blasingame, D., and Patel, M. V. (2004). Cotton disease loss estimate committee report. Proc. Beltwide Cotton Conf. 1, 459–460.
Bolek, Y., El-Zik, K. M., Pepper, A. E., Bell, A. A., Magill, C. M., Thaxton, P. M., et al. (2005). Mapping of verticillium wilt resistance genes in cotton. Plant Sci. 168, 1581–1590. doi: 10.1016/j.plantsci.2005.02.008
Brachi, B., Morris, G. P., and Borevitz, J. O. (2011). Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 12:232. doi: 10.1186/gb-2011-12-10-232
Chaudhary, B., Flagel, L., Stupar, R. M., Udall, J. A., Verma, N., Springer, N. M., et al. (2009). Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (Gossypium). Genetics 182, 503–517. doi: 10.1534/genetics.109.102608
Chen, Y., Huang, J., Li, N., Ma, X., Wang, J., Liu, C., et al. (2015). Genome-wide analysis of the gene families of resistance gene analogues in cotton and their response to Verticillium wilt. BMC Plant Biol. 15:148. doi: 10.1186/s12870-015-0508-3
Chisholm, S. T., Coaker, G., Day, B., and Staskawicz, B. J. (2006). Host-microbe interactions, shaping the evolution of the plant immune response. Cell 124, 803–814. doi: 10.1016/j.cell.2006.02.008
Conesa, A., and Gotz, S. (2008). Blast2GO, A comprehensive suit for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
Cook, D. E., Lee, T. G., Guo, X., Melito, S., Wang, K., Bayless, A. M., et al. (2012). Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338, 1206–1209. doi: 10.1126/science.1228746
Cowley, M., and Oakey, R. J. (2013). Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet. 9:e1003234. doi: 10.1371/journal.pgen.1003234
Dighe, N., Robinson, A. F., Bell, A., Menz, M., Cantrell, R., and Stelly, D. (2009). Linkage mapping of resistance to reniform nematode in cotton (Gossypium hirsutum L.) following introgression from G. longicalyx (Hutch & Lee). Crop Sci. 49, 1151–1164. doi: 10.2135/cropsci2008.03.0129
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity, towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi: 10.1038/nrg2812
Ewing, B., Hillier, L., Wendl, M. C., and Green, P. (1998). Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185. doi: 10.1101/gr.8.3.175
Fang, H., Zhou, H., Sanogo, S., Lipka, A. E., Fang, D. D., Percy, R. G., et al. (2014). Quantitative trait locus analysis of Verticillium wilt resistance in an introgressed recombinant inbred population of Upland cotton. Mol. Breeding 33, 709–720. doi: 10.1007/s11032-013-9987-9
Fonseca, S., Chico, J. M., and Solano, R. (2009). The jasmonate pathway, the ligand, the receptor and the core signaling module. Curr. Opin. Plant Biol. 12, 539–547. doi: 10.1016/j.pbi.2009.07.013
Frelichowski, J. E., Palmer, M. B., Main, D., Tomkins, J. P., Cantrell, R. G., Stelly, D. M., et al. (2006). Cotton genome mapping with new microsatellites from Acala ‘Maxxa’ BAC-ends. Mol. Genet. Genomics 275, 479–491. doi: 10.1007/s00438-006-0106-z
Gene Ontology Consortium. (2006). The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 34, D322–D326. doi: 10.1093/nar/gkj021
Gordon, D., Abajian, C., and Green, P. (1998). Consed, a graphical tool for sequence finishing, Genome Res. 8, 195–202. doi: 10.1101/gr.8.3.195
Grandbastien, M. A. (1998). Activation of plant retrotransposons under stress conditions. Trends Plant Sci. 3, 181–187. doi: 10.1016/S1360-1385(98)01232-1
Grandbastien, M. A., Audeon, C., Bonnivard, E., Casacuberta, J. M., Chalhoub, B., Costa, A. P., et al. (2005). Stress activation and genomic impact Tnt1 retrotransposons in Solanaceae. Cytogenet. Genome Res. 110, 229–241. doi: 10.1159/000084957
Guo, W., Cai, C., Wang, C., Zhao, L., Wang, L., and Zhang, T. (2008). A preliminary analysis of genome structure and composition in Gossypium hirsutum. BMC Genomics 9:314. doi: 10.1186/1471-2164-9-314
Guo, Y., Fitz, J., Schneeberger, K., Ossowski, S., Cao, J., and Weigel, D. (2011). Genome-wide comparison of nucleotide-binding site-leucine-rich repeat-encoding genes in Arabidopsis. Plant Physiol. 157, 757–769. doi: 10.1104/pp.111.181990
Gutiérrez, O. A., Jenkins, J. N., Wubben, M. J., Hayes, R. W., and Callahan, F. E. (2010). SSR markers closely associated with genes for resistance to root-knot nematode on chromosomes 11 and 14 of Upland cotton. Theor. Appl. Genet. 121, 1323–1337. doi: 10.1007/s00122-010-1391-9
Gutiérrez, O. A., Robinson, A. F., Jenkins, J. N., McCarty, J. C., and Wubben, M. J. (2011). Identification of QTL regions and SSR markers associated with resistance to reniform nematode in Gossypium barbadense L. accession GB713. Theor. Appl. Genet. 122, 271–280. doi: 10.1007/s00122-010-1442-2
Hawkins, J. S., Kim, H., Nason, J. D., Wing, R. A., and Wendel, J. F. (2006). Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 16, 1252–1261. doi: 10.1101/gr.5282906
Hu, G., Hawkins, S., Grover, C. E., and Wendel, J. F. (2010). The history and disposition of transposable elements in polyploidy Gossypium. Genome 53, 599–607. doi: 10.1139/G10-038
Jones, J. D., and Dangl, J. (2006). The plant immune system. Nat. Rev. 444, 323–329. doi: 10.1038/nature05286
Kim, H. J., and Triplett, B. A. (2001). Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 127, 1361–1366. doi: 10.1104/pp.010724
Kou, Y., and Wang, S. (2010). Broad-spectrum and durability, understanding of quantitative disease resistance. Curr. Opin. Plant Biol. 13, 181–185. doi: 10.1016/j.pbi.2009.12.010
Li, F., Fan, G., Lu, C., Xiao, G., Zou, C., Kohel, R. J., et al. (2015). Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530. doi: 10.1038/nbt.3208
Li, F., Fan, G., Wang, K., Sun, F., Yuan, Y., Song, G., et al. (2014). Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 46, 567–572. doi: 10.1038/ng.2987
McDowell, J. M., and Meyers, B. C. (2013). A transposable element is domesticated for service in the plant immune system. Proc. Natl. Acad. Sci. U.S.A. 110, 14821–14822. doi: 10.1073/pnas.1314089110
Meyers, B. C., Kaushik, S., and Nandety, R. S. (2005). Evolving disease resistance genes. Curr. Opin. Plant Biol. 8, 129–134. doi: 10.1016/j.pbi.2005.01.002
Niu, C., Lister, H. E., Nguyen, B., Wheeler, T. A., and Wright, R. J. (2008). Resistance to Thielaviopsis basicola in the cultivated A genome cotton. Theor. Appl. Genet. 117, 1313–1323. doi: 10.1007/s00122-008-0865-5
Oliver, K. R., McComb, J. A., and Greene, W. K. (2013). Transposable elements, powerful contributors to angiosperm evolution and diversity. Genome Biol. Evol. 5, 1886–1901. doi: 10.1093/gbe/evt141
Panstruga, R., Parker, J. E., and Schulze-Lefert, P. (2009). SnapShot, Plant immune response pathways. Cell 136, 978. e1–3. doi: 10.1016/j.cell.2009.02.020
Park, Y. H., Alabady, M. S., Sickler, B., Wilkins, T. A., Yu, J. Z., Stelly, D. M., et al. (2005). Genetic mapping of new cotton fiber loci using EST-derived microsatellites in an interspecific recombinant inbred line (RIL) cotton population. Mol. Gen. Genomics 274, 428–441. doi: 10.1007/s00438-005-0037-0
Paterson, A. H., Wendel, J. F., Gundlach, H., Guo, H., Jenkins, J., Jin, D., et al. (2012). Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibers. Nature 492, 423–427. doi: 10.1038/nature11798
Qi, D., and Innes, R. W. (2013). Recent advances in plant NLR structure, function, localization and signaling. Front. Immunol. 4:348. doi: 10.3389/fimmu.2013.00348
Roberts, P. A., and Ulloa, M. (2010). Introgression of root-knot nematode resistance into tetraploid cotton. Crop Sci. 50, 940–951. doi: 10.2135/cropsci2009.05.0281
Roberts, P. A., Ulloa, M., and Wang, C. (2007). “Host plant resistance to root-knot nematode in cotton,” in Proceedings of the Fourth World Cotton Research Conference (Lubbock, TX).
Robinson, A. F., Bell, A. A., Dighe, N. D., Menz, M. A., Nichols, R. L., and Stelly, D. M. (2007). Introgression of resistance to reniform nematode Rotylenchulus reniformis into Upland cotton (Gossypium hirsutum) from Gossypium longicalyx. Crop Sci. 47, 1865–1877. doi: 10.2135/cropsci2006.12.0776
Romano, G. B., Sacks, E. J., Stetina, S. R., Robinson, F. A., Fang, D. D., Gutiérrez, O. A., et al. (2009). Identification and genomic location of a reniform nematode (Rotylenchulus reniformis) resistance locus (Renari) introgressed from Gossypium aridum into upland cotton (G. hirsutum). Theor. Appl. Genet. 120, 139–150. doi: 10.1007/s00122-009-1165-4
Rong, J. K., Abbey, C., Bowers, J. E., Brubaker, C. L., Chang, C., Chee, P. W., et al. (2004). A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166, 389–417. doi: 10.1534/genetics.166.1.389
Said, J. I., Lin, Z., Zhang, X., Song, M., and Zhang, J. (2013). A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genomics 14:776. doi: 10.1186/1471-2164-14-776
Sato, M., Mitra, R. M., Coller, J., Wang, D., Spivey, N. W., Dewdney, J., et al. (2007). A high-performance, small-scale microarray for expression profiling of many samples in Arabidopsis-pathogen studies. Plant J. 49, 565–577. doi: 10.1111/j.1365-313X.2006.02972.x
Shen, X., Van Becelaere, G., Kumar, P., Davis, R. F., May, L. O., and Chee, P. (2006). QTL mapping for resistance to root-knot nematodes in the M-120 RNR Upland cotton line (Gossypium hirsutum L.) of the Auburn 623 RNR source. Theor. Appl. Genet. 113, 1539–1549. doi: 10.1007/s00122-006-0401-4
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS, a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, 465–467. doi: 10.1093/nar/gki458
Stepanova, A. N., and Alonso, J. M. (2009). Ethylene signaling and response, where different regulatory modules meet. Curr. Opin. Plant Biol. 12, 548–555. doi: 10.1016/j.pbi.2009.07.009
Tomkins, J. P., Peterson, D. G., Yang, T. J., Main, D., Wilkins, T. A., Paterson, A. H., et al. (2001). Development of genomic resources for cotton (Gossypium hirsutum L.), BAC library construction, preliminary STC analysis, and identification of clones associated with fiber development. Mol. Breeding 8, 255–261. doi: 10.1023/A:1013798716098
Tsuchiya, T., and Eulgem, T. (2013). An alternative polyadenylation mechanism coopted to the Arabidopsis RPP7 gene through intronic retrotransposon domestication. Proc. Natl. Acad. Sci. U.S.A. 110, e3535–e3543. doi: 10.1073/pnas.1312545110
Ulloa, M., Brubaker, C., and Chee, P. (2007). “Cotton,” in Genome Mapping and Molecular Breeding, Vol. 6, ed C. Kole (Heidelberg; Berlin; New York; Tokyo: Technical Crops. Springer), 1–49.
Ulloa, M., Hutmacher, R. B., Roberts, P. A., Wright, S. D., Nichols, R. L., and Michael, D. R. (2013). Inheritance and QTL mapping of Fusarium wilt race 4 resistance in cotton. Theor. Appl. Genet. 126, 1405–1418. doi: 10.1007/s00122-013-2061-5
Ulloa, M., Saha, S., Yu, J. Z., Jenkins, J. N., Meredith, W. R. Jr., and Kohel, R. J. (2008). “Lessons learned and challenges ahead of the cotton genome mapping,” in Proceedings of the Fourth World Cotton Research Conference, 1798 (Lubbock, TX). Available online at: www.icac.org/meetings/wcrc/wcrc4/presentations/start.htm
Ulloa, M., Wang, C., Hutmacher, R. B., Wright, S. D., Davis, R. M., Saski, C. A., et al. (2011). Mapping Fusarium wilt race 1 genes in cotton by inheritance, QTL and sequencing composition. Mol. Genet. Genomics 286, 21–36. doi: 10.1007/s00438-011-0616-1
Ulloa, M., Wang, C., and Roberts, P. A. (2010). Gene action analysis by inheritance and quantitative trait loci mapping of resistance to root-knot nematodes in cotton. Plant Breeding 129, 541–550. doi: 10.1111/j.1439-0523.2009.01717.x
van Loon, L. C., Rep, M., and Pieterse, C. M. J. (2006). Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 44, 135–162. doi: 10.1146/annurev.phyto.44.070505.143425
Van Ooijen, J. W. (2006). JoinMap® 4.0 Software for the Calculations of Genetic Linkage Maps in Experimental Populations. Wageningen: Kyazma B.V.
Wang, C., Ulloa, M., Mullens, T. R., Yu, J. Z., and Roberts, P. A. (2012a). QTL analysis for transgressive resistance to root-knot nematode in interspecific cotton (Gossypium spp.) progeny derived from susceptible parents. PLoS ONE 7:e34874. doi: 10.1371/journal.pone.0034874
Wang, C., Ulloa, M., and Roberts, P. A. (2006). Identification and mapping of microsatellite markers linked to a root-knot nematode resistance gene (rkn1) in Acala NemX cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 112, 770–777. doi: 10.1007/s00122-005-0183-0
Wang, C., Ulloa, M., and Roberts, P. A. (2008). A transgressive segregation factor (RKN2) in Gossypium barbadense for nematode resistance clusters with gene rkn1 in G. hirsutum. Mol. Gen. Genomics 279, 41–52. doi: 10.1007/s00438-007-0292-3
Wang, K., Wang, Z., Li, F., Ye, W., Wang, J., Song, G., et al. (2012b). The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1104. doi: 10.1038/ng.2371
Wang, Z., Zhang, D., Wang, X., Tan, X., Guo, H., and Paterson, A. H. (2013). A whole-genome DNA marker map for cotton based on the D-genome sequence of Gossypium raimondii L. G3 3, 1759–1767. doi: 10.1534/g3.113.006890
Wei, H., Li, W., Sun, X., Zhu, S., and Zhu, J. (2013). Systematic analysis and comparison of nucleotide-binding site disease resistance genes in a diploid cotton Gossypium raimondii. PLoS ONE 8:e68435. doi: 10.1371/journal.pone.0068435
Wendel, J. F., and Cronn, R. C. (2003). Polyploidy and the evolutionary history of cotton. Adv. Agron. 78, 139–186. doi: 10.1016/S0065-2113(02)78004-8
Wheeler, B. S. (2013). Small RNAs, a big impact, small RNA pathways in transposon control and their effect on the host stress response. Chromosome Res. 21, 587–600. doi: 10.1007/s10577-013-9394-4
Yin, Z., Li, Y., Han, X., and Shen, F. (2012). Genome-wide profiling of miRNAs and other small non coding RNAs in the Verticillium dahliae-inoculated cotton roots. PLoS ONE 7:e35765. doi: 10.1371/journal.pone.0035765
Ynturi, P., Jenkins, J. N., McCarty, J. C. Jr., Gutiérrez, O. A., and Saha, S. (2006). Association of root-knot nematode resistance genes with simple sequence repeat markers on two chromosomes in cotton. Crop Sci. 46, 2670–2674. doi: 10.2135/cropsci2006.05.0319
Yu, J. Z., Kohel, R. J., Fang, D. D., Cho, J., Van Deynze, A., Ulloa, M., et al. (2012). A high-density simple sequence repeat and single nucleotide polymorphism genetic map of the tetraploid cotton genome. G3 2, 43–58. doi: 10.1534/g3.111.001552
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Keywords: Gossypium hirsutum, genetic and physical mapping, resistance-rich cluster, resistance stress element, root-knot nematode, Fusarium wilt, soil-borne disease
Citation: Wang C, Ulloa M, Shi X, Yuan X, Saski C, Yu JZ and Roberts PA (2015) Sequence composition of BAC clones and SSR markers mapped to Upland cotton chromosomes 11 and 21 targeting resistance to soil-borne pathogens. Front. Plant Sci. 6:791. doi: 10.3389/fpls.2015.00791
Received: 24 May 2015; Accepted: 11 September 2015;
Published: 02 October 2015.
Edited by:
Leighton Pritchard, James Hutton Institute, UKCopyright © 2015 Wang, Ulloa, Shi, Yuan, Saski, Yu and Roberts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauricio Ulloa, Plant Stress and Germplasm Development Research Unit, USDA-ARS, 3810 4th Street, Lubbock, TX 79415, USA,bWF1cmljaW8udWxsb2FAYXJzLnVzZGEuZ292;
Philip A. Roberts, Department of Nematology, University of California, Riverside, 900 University Ave., Riverside, CA 92521, USA,cGhpbGlwLnJvYmVydHNAdWNyLmVkdQ==