 Jianhua Xu
Jianhua Xu Miaomiao Li
Miaomiao Li Peng Jiao
Peng Jiao Hongxia Tao
Hongxia Tao Fengwang Ma
Fengwang Ma Junke Zhang
Junke Zhang- The Department of Pomology, College of Horticulture, Northwest A&F University, Yangling, China
Marssonina apple blotch, caused by the fungus Marssonina coronaria, is one of the most destructive apple diseases in China and East Asia. A better understanding of the plant's response to fungi during pathogenesis is urgently needed to improve plant resistance and to breed resistant cultivars. To address this, the transcriptomes of “Qinguan” (a cultivar with high resistance to M. coronaria) apple leaves were sequenced at 12, 24, 48, and 72 h post-inoculation (hpi) with Marssonina coronaria. The comparative results showed that a total of 1956 genes were differentially expressed between the inoculated and control samples at the 4 time points. Gene ontology (GO) term enrichment analysis of differentially expressed genes (DEGs) revealed changes in cellular component, secondary metabolism including chalcone isomerase activity, phytoalexin biosynthetic process, anthocyanin-containing compound biosynthetic process, lignin biosynthetic process, positive regulation of flavonoid biosynthetic process; and molecular functions or biological processes related to the defense response, biotic stimulus response, wounding response and fungus response. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that DEGs were significantly enriched in flavonoid biosynthesis, vitamin B6 metabolism, phenylpropanoid biosynthesis, and the stilbenoid, diarylheptanoid and gingerol biosynthesis pathways. Furthermore, the importance of changes in cellular components and partial polyphenol compounds when encountering M. coronaria are discussed.
Introduction
The domesticated apple (Malus × domestica Borkh.) is the main fruit crop in temperate regions of the world (Luby, 2003; Velasco et al., 2010). The Marssonina apple blotch caused by the fungus Marssonina coronaria is one of the most prevalent apple diseases in East Asia and China. Most of the widely-grown apple cultivars in China, such as “Fuji” and “Gala” apples, are highly susceptible to M. coronaria, while “Qinguan,” a local bred apple cultivar, has demonstrated high resistance with lower incidence of infection (Yin et al., 2013). However, the anti-fungal molecular mechanism in this cultivar remains unknown.
The exploration and application of resistance mechanisms in apple plants is very important for apple breeding. The conditions for in vitro culture and the conidia induction of M. coronaria have been developed and optimized (Lee et al., 2011). The method for M. coronaria resistance identification of different apple germplasm have been settled up by conidia suspension inoculation on detached leaves (Wang et al., 2012; Huang et al., 2013). The infection procedure and critical time points in M. coronaria pathogenesis have been clearly outlined by monitoring the symptoms on in vitro apple leaves with fluorescence and electron microscopy. Normally, the conidia germinated on the leaf surface of apple after 6 hpi, typical hypha penetration and haustoria formation was seen in epidermal cells within 24 hpi, and callose deposition was observed on the epidermal cell wall around the infection sites after 12 hpi. Colony can be found in the leaf tissue 3 days post inoculation, the subcutilar hyphal strands can be found expanding radically around the infection sites 5 days post inoculation and the acervuli can be seen on the leave tissue after 11 days post inoculation. The symptom of the disease on the leave surface including a round, brown lesion with yellow edge can be completely appeared about 15 days after inoculation. The growth and development of the pathogen were much slower in the resistant apple genotype than that of susceptible one while the callose deposition was the opposite (Wang et al., 2012; Zhao et al., 2013).
During pathogen invasion, plant responses varied from cell structure enhancement to synthesis of specialized chemical components. A common response by plants to fungal attack is deposition of callose, a (1,3)-β-glucan polymer, which provide a structural barrier to slow fungal penetration. Evidence showed that the transgenic PMR4 Arabidopsis lines over expressing a callose synthase gene completely inhibit the penetration of powdery mildew (Dorothea et al., 2013). Although, the plant cell wall and its component associated with plant disease resistance have been discussed in high plants (Hückelhoven, 2007), but the resistance linked to apple cell wall have not been reported.
During pathogen invasion, antimicrobial substances of low molecular weight produced by plants were called Phytoalexins, which belongs to a large and diverse class of chemical compounds, including phenylpropanoid compounds, stilbenes, and flavonoids (Ferguson, 2001; Quideau et al., 2011). Polyphenols are important agents in defense against biotic or abiotic stressors, such as nutrient deficiency, drought, temperature variations, salt injury, and pathogen infection stress. These molecules also serve to facilitate communication between plants, or to transducte signals from the environment (Juszczuk et al., 2004; Bruno and Sparapano, 2006; Deytieux-Belleau et al., 2009; Saviranta et al., 2010). Study on phenylpropanoid compounds, stilbenes in apple are very few. And a large proportion of the published research on flavonoids in apples were focused on composition and content in peels and pulp. The importance of phenylpropanoids, stilbenes, and flavonoids in plant-pathogen interactions also has not been thoroughly studied.
Although, the exploration of resistance genes in apple have been started by identifying the genes responding to M. coronaria infection, the expression of some expressed sequence tags (ESTs) have been proved related to the pathogensis on apple leaves (Zhou et al., 2012), some proteins accumulated during M. coronaria infection were also identified using two-dimensional electrophoresis (2-DE) (Li et al., 2014), the exact resistance genes and the dynamic transcriptome changes and possible mechanisms behind this resistance have not been reported. This new dataset will provide useful information for future genetic and genomic studies of apple fungi disease resistance.
The purpose of this paper is to comprehensively compare the dynamic gene expression difference between the M. coronaria inoculated and control apple leaves after inoculation outline the genes participate in the fungal response of “Qinguan” apple during M. coronaria pathogensis, laying a firm foundation for elucidating the molecular basis behind the disease resistance.
Materials and Methods
Plant Material and M. coronaria Inoculation
Four year old “Qinguan” apple trees were grown in a round pot (35 cm in radius and 45 cm in height) under a rain shelter at the experiment station of Northwest A. & F. University, Shaanxi, China. About 120~150 pieces of healthy mature “Qinguan” apple leaves of approximately the same size were detached on August 2nd when natural infection would happen. The collected leaves were immediately surface-sterilized with 8% sodium hypochlorite and then washed with autoclaved sterile water. After the leaf blades were air-dried, the ends of the petiole were wrapped with sterile water wetted absorbent cotton. Half of the sampled leaves were inoculated with M. Coronaria according to Wang et al. (2012). The other half were treated with distilled water as a control. All treated and control leaves were put into plastic trays and covered with transparent films. The leaves were cultured in an incubator at 25°C with a relative humidity of 95–100% and a natural photoperiod.
Pathogenesis on the leaf surface was monitored by microscope every 24 h to ensure successful inoculation and development of disease.
Thirty to Thirty five pieces of inoculated and control sample leaves were randomly harvested at 12, 24, 48, and 72 h post-inoculation (hpi) and marked as T1, T2, T3, T4, and CK1, CK2, CK3, CK4 accordingly. All treated samples were frozen immediately in liquid nitrogen and stored at −80°C until further processing.
RNA Isolation, Library Construction and RNA-sequencing
Total RNA from each sample was isolated separately from appropriate amount of mixed leave powder using the RN38 EASY spin plus Plant RNA kit (Aidlab Biotech, Beijing, China). Sequencing libraries were prepared using a New England Biolabs (NEB) Next® Ultra™ Directional RNA Library Prep Kit for Illumina® (Ipswich, MA, US) sequencing following the manufacturer's protocol. Sequencing was performed on an Illumina HiSeq™ 2000 sequencing platform in accordance with the manufacturer's instructions. An additional biological replicate of T2, T2 repeat (T2-r), was used to control for error.
Sequence Filter, Assembly, Mapping and Gene Annotation
Raw reads were filtered to obtain high-quality clean reads by trimming adaptor sequences, sequences mapping to more than one location in the genome, ambiguous reads, and low-quality reads (the reads in which the proportion of Ns is greater than 5%, with “N” representing unknown base identities). The clean reads were assembled to generate a non-redundant set of transcripts using the Trinity method (http://trinityrnaseq.sourceforge.net/) (Grabherr et al., 2011). The transcripts were clustered based on their nucleotide sequence identities. The longest transcripts in the cluster units were regarded as unigenes. The unigenes were mapped to the apple genome using Bowtie 2 software with default settings (Langmead and Salzberg, 2012).
To predict the function of the RNA sequence, the best hit against the Malus × domestica genome predicted genes was determined using six combined databases, including Swiss-prot, Arabidopsis homolog, TrEMBL homolog, Poplar homolog, Peach homolog, and Grape homolog. Sequences were blasted using BLASTP (TBLASTN for grape) with an E-value of 1.00E-6. When a gene had a match to Swiss-prot, it was assigned as the best match for that gene, followed in order by Arabidopsis homolog, TrEMBL homolog, Poplar homolog, Peach homolog and Grape homolog.
Identification of DEGs, GO and Pathway Functional Enrichment Analysis of the DEGs
The expression levels of genes were normalized by FPKM (Mortazavi et al., 2008). EBSeq software was applied to perform a Chi-square test and then P-values were checked for false discovery rate (FDR) (Leng et al., 2013). Genes with FDR < 0.01 and fold change of FPKM ratio = 2 between inoculated and control were chosen as DEGs. A Venn diagram analysis of the total, downregulated and upregulated DEGs at the four time points was created online (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
GO pathway enrichment analysis was utilized to identify significantly enriched functional classification or metabolic pathways in DEGs by Blast2GO with E < 1e-5 (Ashburner et al., 2000; Conesa et al., 2005). The significantly enriched pathways were identified with corrected p < 0.05.
The Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/) pathway tool is an alternative approach to understanding gene functions, with an emphasis on biochemical pathways. The assembled transcript sequences were searched against the KEGG database using BLASTX with a cut-off E-value of 1e-5. The significantly enriched pathways were identified as those with a p < 0.05 and a corrected p < 0.05.
Data Validation
To verify the reliability of the transcriptomic profiling data, the correlation between transcriptome sequencing data from sample T2 and its biological replicate T2-r was examined. The correlation of DEGs between T2 vs. CK2 and T2-r vs. CK2 was calculated by SPSS ver. 19.0 (IBM SPSS software) using the Pearson method with double-tail test.
Results
Deep Sequencing and Assembly
In total, 25.74 Gigabases were obtained by sequencing from 9 samples, averaging 2.86 Gigabases each, resulting in 254.85 million clean reads in total, and averaging 28.31 million clean reads for each sample. All RNA sequencing data and mapping results are presented in Table 1.
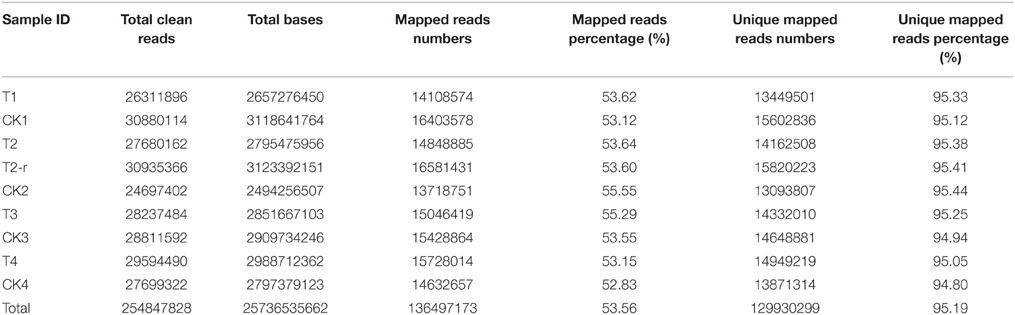
Table 1. Mapping results of sequenced reads from inoculated and control transcriptomes.
The correlation co-efficiency of FPKM between the two independent biological replicates T2 and T2-r was 0.738**, and the p-value was 0.000 (far less than 0.001). These results indicated that the two biological repeats were highly correlated.
DEG Identification
In total, 1956 genes were identified as DEGs between the inoculations and their controls, including 864, 828, 340, and 371 genes at 12, 24, 48, and 72 hpi, respectively. A Venn diagram analysis of total DEGs at the four different time points is shown in Figure 1A. Among the total DEGs, 528, 411, 153, and 174 were upregulated (Figure 1B), whereas 336, 417, 187, and 197 were downregulated (Figure 1C) at 12, 24, 48, and 72 hpi, respectively. Among the 1956 DEGs, most of them showed 2~5 fold changes of their FPKM ratio (log2FC) between the inoculations and their controls, while only a small portion of DEGs were greatly induced (more than 5 folds) by the inoculation compared to their control. The greatly induced DEGs at different time points were list out in Supplementary Table S1.
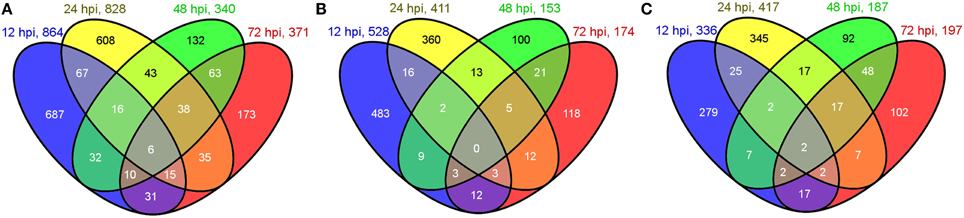
Figure 1. Venn diagram of DEGs at 12, 24, 48, and 72 hpi. (A) Numbers of total DEGs. (B) Numbers of upregulated DEGs. (C) Numbers of downregulated DEGs. Numbers in overlapping areas shows the shared DEGs at different time points.
Dynamic Changes of DEGs
The highest number of DEGs between the inoculated and control samples was detected at 12 hpi. The number of DEGs at 24 hpi was similar to that of 12 hpi, however, the number of DEGs at 48 and 72 hpi decreased to about 40% of those at 12 and 24 hpi. The rapid decrease of DEG number after 24 hpi indicates that the difference of induced gene differential expression compare to their control was narrowed down at 48 and 72 hpi.
Among the total DEGs, 687, 608, 132, and 173 DEGs were only differentially expressed at one time points of 12, 24, 48, and 72 hpi, respectively, indicating that these genes only responded to the pathogen induction at a specific stage after inoculation. In contrast, 6 DEGs were found to be differentially expressed (up or down regulated compare to their control) at all 4 tested time points. These genes are as follows: Cysteine-rich repeat secretory protein 55 (MDP0000555329), and peroxidase (MDP0000770103), which were downregulated at all four time points; 60S ribosomal protein L5 (MDP0000150109), downregulated except for at 72 hpi; UDP-glucose 6-dehydrogenase (MDP0000193220), downregulated except for at 12 hpi; Scarecrow-like protein 4 (MDP0000575908), upregulated except for at 48 hpi; and chloroplast linoleate 13S-lipoxygenase 2-1 (LOX2) (MDP0000211556), upregulated except for at 12 hpi. Other DEGs were differentially expressed at two or three time points during pathogenesis and their number ranged from 10 to 67, accounting for less than 8% of the total DEGs. The dynamic expression change of the DEGs with the time point were shown in the hierarchical heatmap (Figure 2). This result implied that most of the DEGs are differentially expressed at one time point and their relay race may contribute to final resistance of this cultivar.
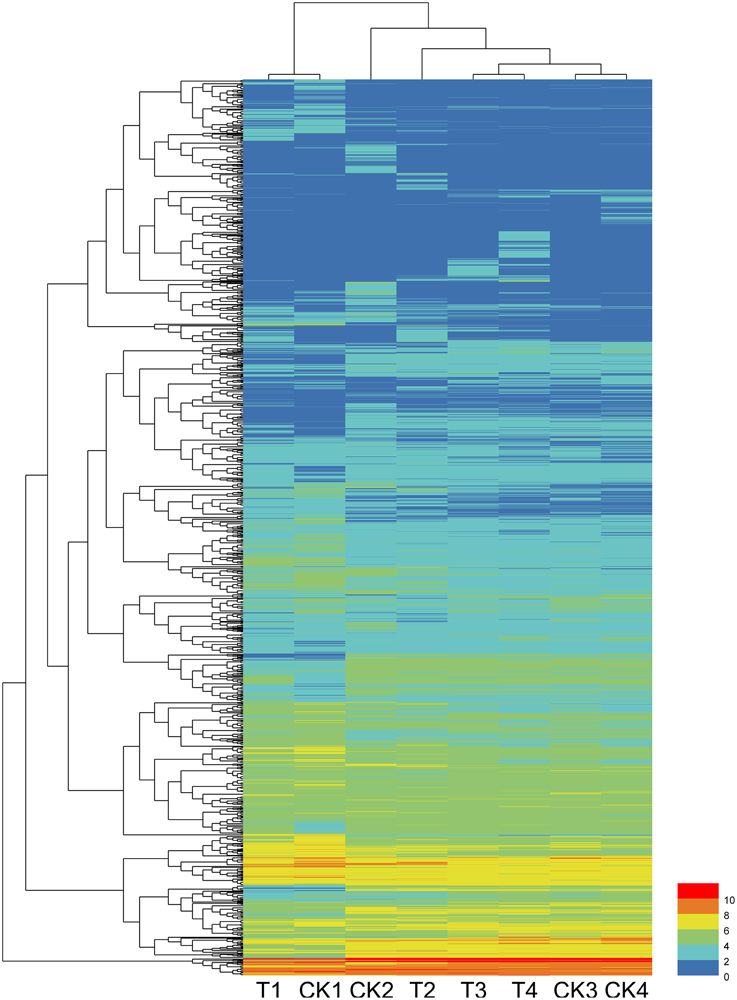
Figure 2. Heatmap of DEGs expression of inoculation and control samples at 12, 24, 48, and 72 hpi.
GO Assignments of DEGs
To get an overview of the function category of the genes participated in the infection response, the DEGs between the inoculated and the control were classified by Gene Orthology. As a result, 789, 751, 313, and 339 DEGs were assigned at least one GO term at 12, 24, 48, and 72 hpi, respectively, including 61 functional groups at the second level (Figure 3). This result implies that a wide ranges of functional genes responsed to the pathogen inoculation.
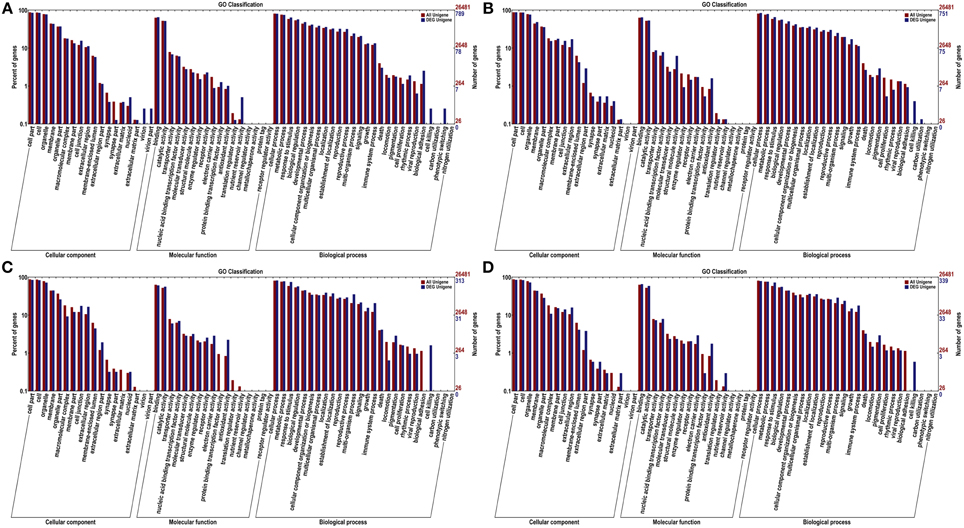
Figure 3. GO categories of unigenes and DEGs at 12 hpi (A), 24 hpi (B), 48 hpi (C), and 72 hpi (D). The genes were functionally categorized into three groups; “cellular component,” “molecular function” and “biological process.” Here, two levels of the assignment results were plotted. Gene number percentages are on ordinate left, the number of genes are on the right.
At the first GO level, “cell,” “cell part,” “organelle,” “membrane” and “organelle part” terms were among the top five ranks in the cellular component category. For molecular function, “binding” and “catalytic activity” were the most abundant subcategories. While “cellular process,” “metabolic process” and “response to stimulus” were the most highly represented in the biological process category.
Aimed to find the most concentrated gene function groups in DEGs, the The significantly enriched GO terms of DEGs annotation at the four time points with corrected p < 0.05 are listed in Table 2 and the DEGs in significantly enriched GO terms are provided in Supplementary Table S2. Significantly enriched GO terms involved in “cellular component” were concentrated at the 12 and 24 hpi time points. No “cellular component” GO terms were significantly enriched at 48 or 72 hpi. This result implied that DEGs in “cellular component” category were greatly induced upon the pathogen inoculation.
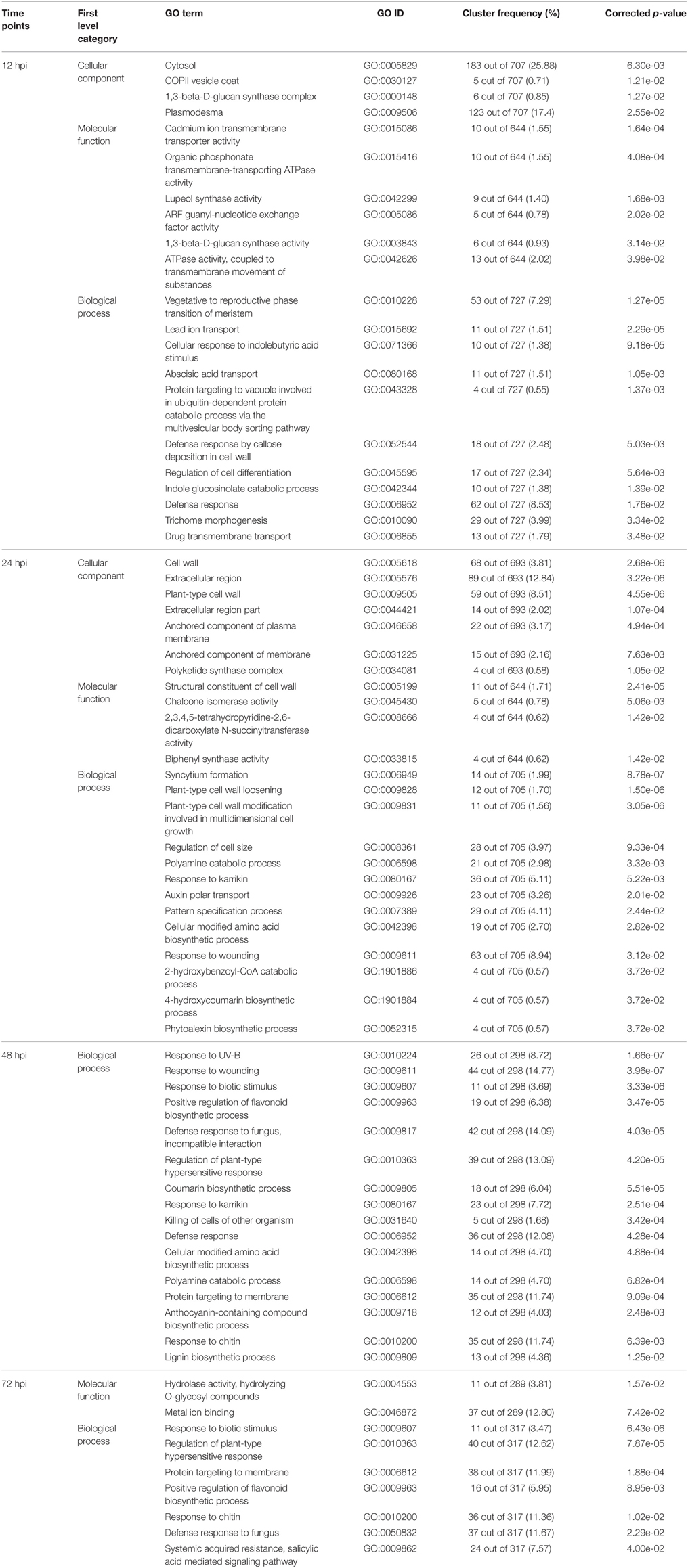
Table 2. GO terms significantly enriched in DEGs at four time points.
Significantly, enriched GO terms involved in “molecular function” and “biological process” were those related to defense, biotic stimulus, wounding and fungus responses (GO:0006952, GO:0009607, GO:0009611, GO:0050832, GO:0009817). In addition, a variety of genes related to secondary products accumulation were significantly enriched, including chalcone isomerase activity (GO:0045430), phytoalexin biosynthetic process (GO:0052315), anthocyanin-containing compound biosynthetic process (GO:0009718), lignin biosynthetic process (GO:0009809), and positive regulation of flavonoid biosynthetic process (GO:0009963). Although the GO terms of DEGs in the “molecular function” and “biological process” category were significantly enriched in first level, it was difficult to address the fluction of the DEGs in subcategory.
Significantly Enriched KEGG Pathways in DEGs
To find the most differentially induced pathways, KEGG pathways were identified according to DEGs with a corrected p < 0.05 at various time points. Four secondary metabolic pathways, (1) flavonoid biosynthesis, (2) vitamin B6 metabolism, (3) phenylpropanoid biosynthesis, and (4) the stilbenoid, diarylheptanoid, and gingerol biosynthesis were significantly enriched at 24, 48 or 72 hpi (corrected p-values of all KEGG pathways at 12 hpi were > 0.05) (Table 3) and the DEGs which were significantly enriched in KEGG pathways are listed in Supplementary Table S3. No pathway was significantly enriched at 12 hpi. This result implied that secondary metabolic pathway especially flavonoid biosynthesis were the most greatly induced pathway at 12, 48 and 72 hpi and stilbenoid, diarylheptanoid, and gingerol biosynthesis pathway were the most greatly induced pathway at 48 and 72 hpi.
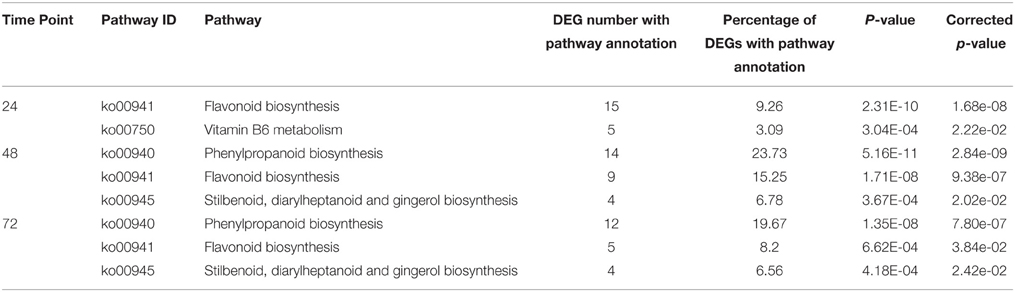
Table 3. Significantly enriched KEGG pathways of DEGs at 12, 24, 48, and 72 hpi.
Discussion
Significantly Enriched GO Terms in Cellular Components and Disease Resistance
In this study, the DEGs of significantly enriched GO terms involved in genes related to cellular components contained coat protein complexes (COPII vesicle coat), plasmodesma, cell wall, and 1,3-beta-D-glucan synthase complex. This implied the genes related to subcellular structure change or cell wall enhancement were induced in resistant apple leaves of “Qinguan” when encountering M. coronaria infection.
Vesicles are responsible for inner cellular transport between organelles of the endomembrane system (endoplasmic reticulum, Golgi apparatus, endosomes, lysosomes and vacuoles), and between the plasma membrane (Robinson et al., 1998; Surpin and Raikhel, 2004). Vesicle trafficking is linked to the regulation of immune signaling (Stegmann et al., 2012). Mutations in AtVAMP7C gene, which is thought to direct fusion between the vacuolar membrane and vesicle, resulted in increased salt tolerance (Leshem et al., 2006). Vesicle formation and budding are a consequence of the continued assembly of the coat proteins. The vesicle coat protein complexes include the clathrin coat protein complex-I (COPI) and coat protein complex-II (COPII) (Bickford et al., 2004; Lee et al., 2004; Edeling et al., 2006). The COPII vesicle coat coordinates the budding of transport vesicles from the endoplasmic reticulum in the initial step of the secretory pathway (Bickford et al., 2004). ADP-ribosylation factor (ARF) families play important roles in vesicle-associated processes, ranging from vesicle formation and transport to exocytosis (Bos et al., 2007). In the present study, the differentially expressed genes involved in the cellular component COPII vesicle coat (GO: 0030127) were upregulated at 12 hpi. Coinciding with this change, genes involved in ADP-ribosylation factor (ARF) guanyl-nucleotide exchange factor activity (GO: 0005086) were also upregulated at 12 hpi in the category of molecular function. The significantly enriched GO terms, “COPII vesicle coat (GO: 0030127),” provided strong evidence that vesicle trafficking took part in plant immune system when encountering biotic stress.
Plasmodesma is a super cellular structure of plant (Lucas, 1995). It provides a direct cell-to-cell cytoplasmic pathway for material transport and message transference, and converts the colonies of independent cells into an interconnected symplast system, playing important roles in plant growth and development, as well as in the response and adaptation of plants to environmental changes (Carrington et al., 1996; Jacinto et al., 1997; Ding, 1998). Among the 123 genes involved in the cellular component plasmodesma (GO: 0009506), 89 genes were upregulated at 12 hpi. The changes of COPII and plasmodesma related DEGs implied that activities related to this cell component were enhanced and COPII and plasmodesma might be ready for material transport.
The resistance mediated by chemical composition changes or physical structure modification of plant cell walls can be an effective physical barrier to pathogens. Deposition of callose, accumulation of phenol compounds, and lignin synthesis increases the structural strength of cell walls following the invasion of pathogens (Farmer, 1985; Grand et al., 1987; Hückelhoven, 2007). Callose, a β-1, 3-D-glucan, exists widely in plant cell walls and plasma membrane. At sites of attempted penetration by fungal pathogens, glucan is a major structural component of papillae in epidermal cells (Enkerli et al., 1997). After Japanese pear leaves were inoculated with Alternaria alternata, glucan deposition was observed as papillae at the infection sites in abaxial epidermis (Suzuki et al., 2003). After inoculation with powdery mildew species, Golovinomyces orontii, callose accumulation was observed at attempted infection sites (Jacobs, 2003). In this database, 1,3-beta-D-glucan synthase complex (GO:0000148), 1,3-beta-D-glucan synthase activity (GO:0003843) and callose deposition in cell wall (GO:0052544) were highly upregulated at 12 hpi. In addition, genes involved in plant-type cell wall loosening (GO:0009828) were downregulated at 12 hpi. The distinguishable GO terms involved in glucan deposition were strongly indicating glucan deposition played an important role when “Qinguan” encountered M. coronaria.
Significantly Enriched KEGG Pathways and Disease Resistance
Phenylpropanoid compounds are natural products derived from cinnamic acid which is formed from phenylalanine via deamination by phenylalanine ammonia-lyase (PAL). From the major biosynthetic routes to the various classes of phenylpropanoids, we can realize phenylpropanoid pathway is a “core” pathway for the formation of monolignols/lignin, coumarins, benzoic acids, stilbenes, and flavonoids/isoflavonoids (Dixon and Paiva, 1995; Dixon et al., 2002).
Stilbenes are a small family of plant secondary metabolites derived from the phenylpropanoid pathway. The most intensively studied biological property of plant-produced stilbenes is their antifungal activity (Hart, 1981; Morales et al., 2000). Stilbene synthase, also termed resveratrol synthase, is a key enzyme in biosynthesis of stilbene-type phytoalexins. The genes coding for stilbene synthases have been transferred into tobacco (Hain et al., 1990, 1993), oilseed rape (Thomzik, 1993), rice (Stark-Lorenzen et al., 1997), barley and wheat (Leckband and Lörz, 1998) and such transgenic plants showed a significant increase in disease resistance. Resveratrol exogenously applied to apples inhibited Venturia inaequalis (the causal agent of apple scab) penetration of cuticular membranes (Schulze et al., 2005). In addition, the researchers also found a positive correlation between stilbene content and disease resistance (Langcake et al., 1979; Fornara et al., 2008; Schnee et al., 2008). The mechanism by which stilbenes inhibit fungi is not well understood.
Flavonoids are a diverse group of secondary metabolites that can be divided into subgroups, including anthocyanidins, flavonols, flavones, flavanols, flavanones, chalcones, dihydrochalcones, and dihydroflavonols (Treutter, 2006). Flavonoids and related phenolics have indisputable functions in protecting plants from fungal infection. Defense-related flavonoids can be divided into two types: pre-existing and stimulating. Pre-existing flavonoids are usually stored in particular locations and can be used as signaling molecules (Treutter, 2006) or are involved in various interactions between the plant and a pathogen (Schlösser, 1994). Physical injury, infection, or stress can stimulate the formation of flavonoids called phytoalexins (Treutter, 2006). The role of flavonoids against pathogens may be related to the following mechanisms: antioxidant properties; cross-linking of microbial enzymes; inhibition of microbial cellulases, xylanases, and pectinases; chelation of metals necessary for enzyme activity; storage in specialized cells from which they can be infused into attacked tissue (such as xylem vessels); formation of a hard, almost crystalline structure as a physical barrier against pathogen attack; and promotion of the formation of calli and tyloses, thus closing vessels and blocking aggressive invaders (Skadhauge et al., 1997; Beckman, 2000).
In the present study, the KEGG pathways “Phenylpropanoid biosynthesis (ko00940)” and “Stilbenoid, diarylheptanoid, and gingerol biosynthesis (ko00945)” were significantly enriched at 48 and 72 hpi, and “flavenoid biosynthesis (ko00974)” was significantly enriched at 24, 48, and 72 hpi. In addition, the GO term “Positive regulation of flavonoid biosynthetic process (GO:0009963)” was significantly enriched at 48 hpi and 72 hpi. This provides researchers with new ideas to enhance disease resistance, and the DEGs in these pathways should have more focused attention in future research.
This work presents the first transcriptome sequencing analysis of the “Qinguan” apple leaf inoculated with M. Coronaria vs. control at four time points post-inoculation. In the present study, a total of 1956 genes were identified as DEGs, including 864, 828, 340, and 371 genes at 12, 24, 48, and 72 hpi, respectively. GO and KEGG analyses of DEGs suggested that the mechanism of defense against M. Coronaria is very complex and involves multiple processes. These analyses revealed that (i) changes in cellular components, development of a resistant structure; (ii) synthesis of many secondary products, including phytoalexin, anthocyanin, lignin, flavonoid, phenylpropanoid, and stilbenoid; (iii) and responses to defense, biotic stimulus, wounding and fungus were involved in the plant actions.
It is rather difficult to identify the disease resistance gene in perennial woody apple plant for their complex genetic background. Normally, disease resistance of different cultivar in woody plant always accompany by other traits encoded by non resistance genes. Transcriptome comparison during pathogensis between different cultivars always harvests a large number of DEGs but it is difficult to identify the resistance related gene from the rest. A ideal strategy is the use of disease resistance mutant, but this kind of material is real rare in apple plant. A reasonable way of resistance gene identification is to screen the DEGs between the pathogen inoculated and control in the resistant cultivar, then mine the pathogen responsed DEGs by GO and pathway, finally confirm the resistance gene by expression comparison in resistance and susceptible cultivar. This strategy was used in present research, DEGs responsed to pathogen inoculation were identified from the resistant cultivar “Qinguan” apple and were filtered by GO and pathway, some related pathway and DEGs related pathogen induction were found, but the exact resistance genes and their function still need further identification.
Author Contributions
JX and ML were responsible for the inoculation and sequence analysis. JX also wrote the manuscript. PJ prepared cDNA samples for sequencing. HT and NW helped with data interpretation. FM and JZ designed the experiment, provided guidance on the whole study and contributed with valuable discussions. All authors have read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the earmarked fund for China Agriculture Research System (CARS-28). We also thank Prof. Pengmin Li and Dr. Mingjun Li for their comments and suggestions on improving the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00842
References
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Beckman, C. H. (2000). Phenolic-storing cells: keys to programmed cell death and periderm formation in wilt disease resistance and in general defence responses in plants? Physiol. Mol. Plant P. 57, 101–110. doi: 10.1006/pmpp.2000.0287
Bickford, L. C., Mossessova, E., and Goldberg, J. (2004). A structural view of the COPII vesicle coat. Curr. Opin. Struct. Biol. 14, 147–153. doi: 10.1016/j.sbi.2004.02.002
Bos, J. L., Rehmann, H., and Wittinghofer, A. (2007). GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877. doi: 10.1016/j.cell.2007.05.018
Bruno, G., and Sparapano, L. (2006). Effects of three esca-associated fungi on Vitis vinifera L.: I. Characterization of secondary metabolites in culture media and host responses to the pathogens in calli. Physiol. Mol. Plant P. 69, 209–223. doi: 10.1016/j.pmpp.2007.04.008
Carrington, J. C., Kasschau, K. D., Mahajan, S. K., and Schaad, M. C. (1996). Cell-to-cell and long-distance transport of viruses in plants. Plant Cell 8, 1669–1681. doi: 10.1105/tpc.8.10.1669
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Deytieux-Belleau, C., Geny, L., Roudet, J., Mayet, V., Donèche, B., and Fermaud, M. (2009). Grape berry skin features related to ontogenic resistance to Botrytis cinerea. Eur. J. Plant Pathol. 125, 551–563. doi: 10.1007/s10658-009-9503-6
Ding, B. (1998). Intercellular protein trafficking through plasmidesmata. Plant Mol. Biol. 38, 279–310. doi: 10.1007/978-94-011-5298-3_15
Dixon, R. A., Achnine, L., Kota, P., Liu, C. J., Reddy, M. S., and Wang, L. (2002). The phenylpropanoid pathway and plant defence—a genomics perspective. Mol. Plant Pathol. 3, 371–390. doi: 10.1046/j.1364-3703.2002.00131.x
Dixon, R. A., and Paiva, N. L. (1995). Stress-induced phenylpropanoid metabolism. Plant Cell 7, 1085–1097. doi: 10.1105/tpc.7.7.1085
Dorothea, E., Marcel, N., Christian, F., Claudia, Z., Torsten, J., Chithra, M., et al. (2013). Elevated early callose deposition results in complete penetration resistance to powdery mildew in Arabidopsis. Plant Physiol. 161, 1433–1444. doi: 10.1104/pp.112.211011
Edeling, M. A., Smith, C., and Owen, D. (2006). Life of a clathrin coat: insights from clathrin and AP structures. Nat. Rev. Mol. Cell Bio. 7, 32–44. doi: 10.1038/nrm1786
Enkerli, K., Hahn, M. G., and Mims, C. W. (1997). Immunogold localization of callose and other plant cell wall components in soybean roots infected with the oomycete Phytophytora sojae. Can. J. Bot. 75, 1509–1517. doi: 10.1139/b97-865
Farmer, E. E. (1985). Effects of fungal elicitor on lignin biosynthesis in cell suspension cultures of soybean. Plant Physiol. 78, 338–342. doi: 10.1104/pp.78.2.338
Ferguson, L. R. (2001). Role of plant polyphenols in genomic stability. Mut. Res. Fundamental Mol.Mech. Mutagenesis 475, 89–111. doi: 10.1016/S0027-5107(01)00073-2
Fornara, V., Onelli, E., Sparvoli, F., Rossoni, M., Aina, R., Marino, G., et al. (2008). Localization of stilbene synthase in Vitis vinifera L. during berry development. Protoplasma 233, 83–93. doi: 10.1007/s00709-008-0309-8
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Grand, C., Sarni, F., and Lamb, C. J. (1987). Rapid induction by fungal elicitor of the synthesis of cinnamyl-alcohol dehydrogenase, a specific enzyme of lignin synthesis. Eur. J. Biochem. 169, 73–77. doi: 10.1111/j.1432-1033.1987.tb13582.x
Hain, R., Bieseler, B., Kindl, H., Schröder, G., and Stöcker, R. (1990). Expression of a stilbene synthase gene in Nicotiana tabacum results in synthesis of the phytoalexin resveratrol. Plant Mol. Biol. 15, 325–335. doi: 10.1007/BF00036918
Hain, R., Reif, H., Krause, E., Langebartels, R., Kindl, H., Vornam, B., et al. (1993). Disease resistance results from foreign phytoalexin expression in a novel plant. Nature 361, 153–156. doi: 10.1038/361153a0
Hart, J. H. (1981). Role of phytostilbenes in decay and disease resistance. Ann. Rev. Phytopathol. 19, 437–458.
Huang, Y., Zhang, R., Zhu, G., Zhang, L., and Sun, G. (2013). Evaluation of the resistance of apple cultivars to Marssonina coronaria. Acta Agriculturae Boreali Occidentalis Sinica 22, 122–126. doi: 10.7606/j.issn.1004-1389.2013.08.023
Hückelhoven, R. (2007). Cell wall-associated mechanisms of disease resistance and susceptibility. Annu. Rev. Phytopathol. 45, 101–127. doi: 10.1146/annurev.phyto.45.062806.094325
Jacinto, T., Mcgurl, B., Franceschi, V., Delano-Freier, J., and Ryan, C. A. (1997). Tomato prosystemin promoter confers wound-inducible, vcascular bundle-specific expression of the β-gluvuronidase gene in transgenic tomato plants. Planta 203, 406–412. doi: 10.1007/s004250050207
Jacobs, A. K. (2003). An Arabidopsis Callose Synthase, GSL5, is required for wound and papillary callose formation. Plant Cell Online 15, 2503–2513. doi: 10.1105/tpc.016097
Juszczuk, I. M., Wiktorowska, A., Malusá, E., and Rychter, A. M. (2004). Changes in the concentration of phenolic compounds and exudation induced by phosphate deficiency in bean plants (Phaseolus vulgaris L.). Plant Soil 267, 41–49. doi: 10.1007/s11104-005-2569-9
Langcake, P., Cornford, C. A., and Pryce, R. J. (1979). Identification of pterostilbene as a phytoalexin from Vitis vinifera leaves, Phytochemistry 18, 1025–1027. doi: 10.1016/S0031-9422(00)91470-5
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leckband, G., and Lörz, H. (1998). Transformation and expression of a stilbene synthase gene of Vitis vinifera L. in barley and wheat for increased fungal resistance. TAG Theo. Appl. Genet. 96, 1004–1012. doi: 10.1007/s001220050832
Lee, D., Back, C., Win, N. K. K., Choi, K., Kim, K., Kang, I., et al. (2011). Biological characterization of Marssonina coronaria associated with Apple Blotch Disease. Mycobiology 39, 200. doi: 10.5941/MYCO.2011.39.3.200
Lee, M. C. S., Miller, E. A., Goldberg, J., Orci, L., and Schekman, R. (2004). BI-directional protein transport between the ER and golgi. Annu. Rev. Cell Dev. Bi. 20, 87–123. doi: 10.1146/annurev.cellbio.20.010403.105307
Leng, N., Dawson, J. A., Thomson, J. A., Ruotti, V., Rissman, A. I., Smits, B. M. G., et al. (2013). EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043. doi: 10.1093/bioinformatics/btt087
Leshem, Y., Melamed-Book, N., Cagnac, O., Ronen, G., Nishri, Y., Solomon, M., et al. (2006). Suppression of Arabidopsis vesicle-SNARE expression inhibited fusion of H2O2-containing vesicles with tonoplast and increased salt tolerance. Proc. Natl. Acad. Sci. U.S.A. 103, 18008–18013. doi: 10.1073/pnas.0604421103
Li, M., Xu, J., Qiu, Z., Zhang, J., Ma, F., and Zhang, J. (2014). Screening and identification of resistance related proteins from apple leaves inoculated with Marssonina coronaria (EII. & J. J. Davis). Proteome Sci. 12:7. doi: 10.1186/1477-5956-12-7
Luby, J. J. (2003). “Taxonomic classification and brief history,” in Apples: Botany, Production and Uses, eds D. C. Ferree, I. J. Warrington (Wallingford: CABI Publishing), 1–14.
Lucas, W. J. (1995). Plasmodesmata: intercellular channels for macromolecular transport in plants. Curr. Opin. Cell Biol. 7, 673–680. doi: 10.1016/0955-0674(95)80109-X
Morales, M., Ros Barceló, A., and Pedreño, M. A. (2000). “Plant stilbenes: recent advances in their chemistry and biology,” in Advances in Plant Physiology, Vol. 3, ed A. Hemantaranjan (Jodhpur: Scientific Publishers), 39–70.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Quideau, S., Deffieux, D., Douat−Casassus, C., and Pouységu, L. (2011). Plant polyphenols: chemical properties, biological activities, and synthesis. Angew. Chem. Int. Edn. 50, 586–621. doi: 10.1002/anie.201000044
Robinson, D. G., Hinz, G., and Holstein, S. E. (1998). The molecular characterization of transport vesicles. Plant Mol. Biol. 38, 49–76. doi: 10.1023/A:1006025802445
Saviranta, N. M., Julkunen-Tiitto, R., Oksanen, E., and Karjalainen, R. O. (2010). Red clover (Trifolium pratense L.) isoflavones: root phenolic compounds affected by biotic and abiotic stress factors. J. Sci. Food Agric. 90, 418–423. doi: 10.1002/jsfa.3831
Schlösser, E. (1994). “Preformed phenols as resistance factors,” in International Symposium on Natural Phenols in Plant Resistance, eds M. Geibel, D. Treutter, and W. Feucht (Acta Hortic), 615–630.
Schnee, S., Viret, O., and Gindro, K. (2008). Role of stilbenes in the resistance of grapevine to powdery mildew. Physiol. Mol. Plant Pathol. 72, 128–133. doi: 10.1016/j.pmpp.2008.07.002
Schulze, K., Schreiber, L., and Szankowski, I. (2005). Inhibiting effects of resveratrol and its glucoside piceid against Venturia inaequalis, the causal agent of apple scab. J. Agric. Food Chem. 53, 356–362. doi: 10.1021/jf048375h
Skadhauge, B., Thomsen, K. K. and von Wettstein, D. (1997). The role of the barley testa layer and its flavonoid content in resistance to fusarium infections. Hereditas 126, 147–160. doi: 10.1111/j.1601-5223.1997.00147.x
Stark-Lorenzen, P., Nelke, B., Hänler, G., Mühlbach, H. P., and Thomzik, J. E. (1997). Transfer of a grapevine stilbene synthase gene to rice (Oryza sativa L.). Plant Cell Rep. 16, 668–673. doi: 10.1007/s002990050299
Stegmann, M., Anderson, R. G., Ichimura, K., Pecenkova, T., Reuter, P., Žárskı, V., et al. (2012). The ubiquitin ligase PUB22 targets a subunit of the exocyst complex required for PAMP-triggered responses in Arabidopsis. Plant Cell 24, 4703–4716. doi: 10.1105/tpc.112.104463
Surpin, M., and Raikhel, N. (2004). Traffic jams affect plant development and signal transduction. Nat. Rev. Mol. Cell Biol. 5, 100–109. doi: 10.1038/nrm1311
Suzuki, T., Shinogi, T., Narusaka, Y., and Park, P. (2003). Infection behavior of Alternaria alternata Japanese pear pathotype and localization of beta-1,3-glucan in compatible and incompatible interactions between the pathogen and host plants. J. Gen. Plant Pathol. 69, 91–100. doi: 10.1007/s10327-002-0001-3
Thomzik, J. E. (1993). “Transformation in Oilseed Rape (Brassica napus L.),” in Plant Protoplasts and Genetic Engineering IV, eds O. L. Gamborg and L. R. Wetter (New York, NY; Berlin; Heidelberg: Springer), 170–182.
Treutter, D. (2006). Significance of flavonoids in plant resistance: a review. Environ. Chem. Lett. 4, 147–157. doi: 10.1007/s10311-006-0068-8
Velasco, R., Zharkikh, A., Affourtit, J., Dhingra, A., Cestaro, A., Kalyanaraman, A., et al. (2010). The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 42, 833–839. doi: 10.1038/ng.654
Wang, J., Zhao, H., Su, S., Gao, X., and Huang, L. (2012). Effect of different Malus species on growth and development of Diplocarpon mali. Acta Agriculturae Boreali occidentalis Sinica 21, 60–64. doi: 10.3969/j.issn.1004-1389.2012.05.013
Yin, L., Li, M., Ke, X., Li, C., Zou, Y., Liang, D., et al. (2013). Evaluation of Malus germplasm resistance to marssonina apple blotch. Eur. J. Plant Pathol. 136, 597–602. doi: 10.1007/s10658-013-0190-y
Zhao, H., Han, Q., Wang, J., Gao, X., Xiao, C., Liu, J., et al. (2013). Cytology of infection of apple leaves by Diplocarpon mali. Eur. J. Plant Pathol. 136, 41–49. doi: 10.1007/s10658-012-0129-8
Keywords: apple, M. coronaria, transcriptome, disease resistance gene, cell components, polyphenol compounds
Citation: Xu J, Li M, Jiao P, Tao H, Wei N, Ma F and Zhang J (2015) Dynamic transcription profiles of “Qinguan” apple (Malus × domestica) leaves in response to Marssonina coronaria inoculation. Front. Plant Sci. 6:842. doi: 10.3389/fpls.2015.00842
Received: 14 April 2015; Accepted: 25 September 2015;
Published: 13 October 2015.
Edited by:
Marinus J. M. Smulders, Wageningen UR, NetherlandsReviewed by:
Cai-Zhong Jiang, United States Department of Agriculture, Agricultural Research Service, USAEstefanía Carrillo, Universidad Nacional de Chimborazo, Ecuador
Copyright © 2015 Xu, Li, Jiao, Tao, Wei, Ma and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junke Zhang, emhhbmdqa0Bud3N1YWYuZWR1LmNu