 Xiaochun Wei1†
Xiaochun Wei1† Xiaohui Zhang2†Qiuju Yao1
Xiaohui Zhang2†Qiuju Yao1 Yuxiang Yuan1
Yuxiang Yuan1 Xixiang Li2Fang Wei3Yanyan Zhao1Qiang Zhang1Zhiyong Wang1Wusheng Jiang1
Xixiang Li2Fang Wei3Yanyan Zhao1Qiang Zhang1Zhiyong Wang1Wusheng Jiang1 Xiaowei Zhang1*
Xiaowei Zhang1*- 1Institute of Horticulture, Henan Academy of Agricultural Sciences, Zhengzhou, China
- 2Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, China
- 3College of Life Science, Zhengzhou University, Zhengzhou, China
Chinese cabbage (Brassica rapa ssp. pekinensis) is one of the most important vegetables in Asia and is cultivated across the world. Ogura-type cytoplasmic male sterility (Ogura-CMS) has been widely used in the hybrid breeding industry for Chinese cabbage and many other cruciferous vegetables. Although, the cause of Ogura-CMS has been localized to the orf138 locus in the mitochondrial genome, however, the mechanism by which nuclear genes respond to the mutation of the mitochondrial orf138 locus is unclear. In this study, a series of whole genome small RNA, degradome and transcriptome analyses were performed on both Ogura-CMS and its maintainer Chinese cabbage buds using deep sequencing technology. A total of 289 known miRNAs derived from 69 families (including 23 new families first reported in B. rapa) and 426 novel miRNAs were identified. Among these novel miRNAs, both 3-p and 5-p miRNAs were detected on the hairpin arms of 138 precursors. Ten known and 49 novel miRNAs were down-regulated, while one known and 27 novel miRNAs were up-regulated in Ogura-CMS buds compared to the fertile plants. Using degradome analysis, a total of 376 mRNAs were identified as targets of 30 known miRNA families and 100 novel miRNAs. A large fraction of the targets were annotated as reproductive development related. Our transcriptome profiling revealed that the expression of the targets was finely tuned by the miRNAs. Two novel miRNAs were identified that were specifically highly expressed in Ogura-CMS buds and sufficiently suppressed two pollen development essential genes: sucrose transporter SUC1 and H+-ATPase 6. These findings provide clues for the contribution of a potential miRNA regulatory network to bud development and pollen engenderation. This study contributes new insights to the communication between the mitochondria and chromosome and takes one step toward filling the gap in the regulatory network from the orf138 locus to pollen abortion in Ogura-CMS plants from a miRNA perspective.
Introduction
Cytoplasmic male sterility (CMS) is a maternally inherited trait that results in the loss of the ability to produce fertile pollen; CMS has been used extensively for hybrid crop breeding (Woodson and Chory, 2008; Luo et al., 2013). Ogura-CMS was originally discovered in the wild radish (Raphanus sativus) (Ogura, 1968) and has been widely introduced into Cruciferous vegetables and oil crops and successfully applied in the heterosis breeding industry due to its complete male sterility and stability (Pelletier et al., 1983). Ogura-CMS is controlled by a mitochondrial orf138 locus that consists of two co-transcribed open reading frames: orf138 and atp8 (ATP synthase, subunit 8) (Krishnasamy and Makaroff, 1993; Grelon et al., 1994). However, the mechanism by which the orf138 locus results in male sterility is unclear. Many researchers have focused on the crosstalk between the mitochondria and nucleus, and the CMS is believed to be controlled by mitochondrial-nuclear interactions (Chase, 2007). Several CMS-associated nuclear genes, such as those related to programmed cell death (PCD) and reactive oxygen species (ROS), have been identified and proposed to play roles in the CMS pathway (Balk and Leaver, 2001). Other regulatory signaling pathways, such as the biogenesis of jasmonic acid, are also impaired in the CMS line (Liu et al., 2012).
MicroRNAs (miRNAs) are a class of 21–24 nt small non-coding RNAs that regulate gene expression by post-transcriptional repression (Carrington and Ambros, 2003; Bartel, 2004). In plants, miRNAs are generated from primary miRNA transcripts (pri-miRNAs) that are generally transcribed by RNA polymerase II (Bartel, 2004; Khraiwesh et al., 2010). Primary transcripts containing a distinctive hairpin structure are first trimmed by DICER-LIKE1 (DCL1) to generate miRNA precursors (pre-miRNAs) in the nucleus. Then, these pre-miRNAs are transported to the cytoplasm and further processed by DCL1 to generate ~21 nt mature miRNAs (Kurihara and Watanabe, 2004). Subsequently, the mature miRNAs are loaded onto the RNA-induced silencing complex (RISC) and guide RISC recognition of complementary sites on target mRNAs, thereby inducing transcript cleavage (Bartel, 2004; Baulcombe, 2004) or translational repression (Aukerman and Sakai, 2003; Chen, 2004). Plant miRNAs mainly function by directing the cleavage of their highly complementary target transcripts. Thus, it is easy to predict and verify their targets using bioinformatics and experimental methods.
miRNAs play important roles in the regulation of a wide range of plant developmental processes, including plant architecture (Zhang et al., 2011b), leaf development (Floyd and Bowman, 2004; Juarez et al., 2004), root development (Guo et al., 2005), tuberization (Bhogale et al., 2014), the vegetative-reproductive phase change (Wang et al., 2011a), flowering time (Zhou et al., 2013), floral organ identity (Aukerman and Sakai, 2003; Bartel, 2004; Chen, 2004; Nagpal et al., 2005), self-incompatibility (Tarutani et al., 2010), plant nutrient homeostasis (Yamasaki et al., 2007; He et al., 2014), and response to environmental biotic and abiotic stresses (Juarez et al., 2004; Navarro et al., 2006; Sunkar et al., 2007; Katiyar-Agarwal and Jin, 2010; Zhang et al., 2011a). However, few miRNAs have been confirmed to control pollen development or CMS. Efforts have been made to screen for pollen-specific or enriched miRNAs in rice (Wei et al., 2011), Arabidopsis (Chambers and Shuai, 2009; Grant-Downton et al., 2009a,b), and pakchoi (Jiang et al., 2014).
Chinese cabbage (Brassica rapa ssp. pekinensis) is an important leafy vegetable that is a cross-pollinated crop with significant heterosis. Ogura-CMS was transferred to the Chinese cabbage in the 1980s and is still widely used in the breeding industry (Yamagishi and Bhat, 2014). The miRNAs from the Ogura-CMS and the maintainer line buds of a closely related subspecies (B. rapa ssp. chinensis) have been profiled (Jiang et al., 2014). In that study, 54 new conserved miRNAs and 25 pairs of novel miRNAs were identified. Among them, 18 miRNAs were differentially expressed between the male sterile and fertile lines. A genome-wide analysis of miRNAs in B. rapa by deep sequencing has been reported (Kim et al., 2012). However, to the best of our knowledge the miRNAs that underlie flower bud development and respond to the Ogura-CMS in Chinese cabbage have not been examined.
In present study, the miRNAs from the buds of a Chinese cabbage Ogura-CMS line (Tyms) and its maintainer line (231-330) were profiled by small RNA deep sequencing. The targets of the miRNAs were identified by degradome sequencing analysis. The expression levels of the corresponding targets were monitored by transcriptome sequencing. These results provide a full set of miRNA-target messages that underlie flower bud development in the Chinese cabbage and provide insights into miRNA-related regulatory functions that may play roles in the mitochondrial–nuclear interactions in Ogura-CMS.
Materials and Methods
Plant Materials and RNA Extraction
The Chinese cabbage Ogura-CMS sterile line (Tyms) and its maintainer line (231-330) used in this study were grown in the Henan Academy of Agricultural Sciences, Yuanyang, Henan Province, China. The lines possessed isogenic chromosomes with different cytoplasmic genes. Flower buds < 6 mm length were stripped from 10 individual of each genotype of plants. These buds contain the entire developmental progress from anther generation to pollen abortion. Tyms and 231-330 were sampled separately and then snap-frozen in liquid nitrogen and kept at -80°C for further use. Total RNA was extracted using the TRIzol reagent (Invitrogen, USA). DNase (Promega, USA) was used to remove potential DNA contamination.
Small RNA Library Construction and Sequencing
The RNA samples from 231-330 (Control) and Tyms (CMS) were quantified and equalized so that equivalent amounts of RNA were analyzed. A total of 30 μg of RNA was resolved on denatured polyacrylamide gels. Gel fragments with the size range of 18–30 nt were excised and recovered. These small RNAs were ligated with 5′ and 3′RNA adapters with the T4 RNA ligase. The adapter-ligated small RNAs were subsequently transcribed into cDNA by Super-Script II Reverse Transcriptase (Invitrogen) and amplified using primers specific for the ends of the adapters. The amplified cDNA products were purified and finally sequenced using Solexa sequencing technology (BGI, Shenzhen, China).
Identification of Known and Novel miRNAs
The adapter sequences, impurities, and sequences with less or more than 18–30 nt were filtered out from the raw sequence reads. The remaining sequences that ranged from 18 to 30 nt in length were used for known miRNA prediction. First, we aligned the tags to the B. rapa miRNA precursors in miRBase (version 20.0, http://www.mirbase.org/index.shtml) with no mismatches allowed. Then, the obtained tags were aligned to the mature miRNAs of B. rapa with at least a 16 nt overlap to allow offsets. The miRNAs that satisfied both of the above criteria were counted to obtain the expression number of identified miRNAs. The rest of the small RNA tags were aligned to the miRNA precursors/mature miRNAs of all plants in miRBase, allowing two mismatches and free gaps. The miRNAs with the highest expression levels for each mature miRNA family were chosen as a temporary miRNA database. Then, the precursors of the temporary miRNAs in the B. rapa genome were predicted. Those that failed to fold into hairpin structures were regarded as pseudo-miRNAs and discarded, while those that fulfilled the miRNA criteria were adopted as newly identified known miRNAs.
By comparing our sequences with those in the databases and the Chinese cabbage genome, the sRNAs can be annotated into different categories, including siRNA, piRNA, rRNA, tRNA, snRNA, snoRNA, repeat associated sRNA, degraded tags of exons or introns, and sRNAs that could not be annotated. The tags annotated as intron, exon antisense, and unknown were used to predict novel miRNAs using the software Mireap. The key conditions are as follow: hairpin miRNAs can fold into secondary structures and mature miRNAs are present in one arm of the hairpin precursors; the 5-p and 3-p mature miRNAs present 2-nucleotide 3′ overhangs; hairpin precursors lack large internal loops or bulges; the secondary structures of the hairpins are steady, with the free energy of hybridization lower than or equal to −18 kcal/mol; and the copy number of mature miRNAs with predicted hairpins must be greater than five in the alignment result. The expression of novel miRNAs was produced by summing the count of miRNAs with no more than three mismatches on the 5′ and 3′ ends and no mismatches in the middle from the alignment result.
The differentially expressed miRNAs were calculated with the following procedures. First, the expression of miRNAs in two samples was normalized to obtain the expression of transcript per million (TPM). The normalization formula was as follows: Normalized expression = Actual miRNA count/Total count of clean reads × 1,000,000; Second, the fold-change and P-value were calculated from the normalized expression.
Degradome Library Construction and Target Identification
Total RNA extracted from the 231-330 (Control) and Tyms (CMS) lines. Approximately 200 μg of the total RNA was polyadenylated using the Oligotex mRNA mini kit (Qiagen). A 5′ RNA adapter was added to the cleavage products (which possessed a free 5′-monophosphate at their 3′ termini) using the T4 RNA ligase (Takara). Then, the ligated products were purified using the Oligotex mRNA mini kit (Qiagen) for reverse transcription to generate the first strand of cDNA using an oligo dT primer via SuperScript II RT (Invitrogen). After the cDNA library was amplified for 6 cycles (94°C for 30 s, 60°C for 20 s, and 72°C for 3 min) using Phusion Taq (NEB), the PCR products were digested with the restriction enzyme Mme I (NEB). A double-stranded DNA adapter was ligated to the digested products using T4 DNA ligase (NEB). The ligated products were selected based on size in a 10% polyacrylamide gel and purified for the final PCR amplification (94°C for 30 s, 60°C for 20 s, and 72°C for 20 s) for 20 cycles (German et al., 2009). The PCR products were gel purified and used for high-throughput sequencing with the Illumina HiSeq 2000.
Low quality sequences and adapters were removed, and the unique sequence signatures were aligned to the database of Chinese cabbage transcript assemblies in the Brassica Gene Index (http://brassicadb.org/brad/downloadOverview.php) using the SOAP software (Li et al., 2008) (http://soap.genomics.org.cn/). CleaveLand was used to detect potentially cleaved targets based on degradome sequences. The 20 and 21 nt distinct reads were subjected to the CleaveL and pipeline for small RNA target identification as previously described (Addo-Quaye et al., 2009). The tags mapped to cDNA sense strands were used to predict cleavage sites. The miRNA-mRNA pairs were searched and p-values were calculated using PAREsnip (http://srna-workbench.cmp.uea.ac.uk/tools/paresnip/). Only p-values less than 0.05 were adopted for the t-plot figure (Folkes et al., 2012). All alignments with scores not exceeding 4 that possessed the 5′ end of the degradome sequence coincident with the 10th and 11th nucleotides that were complementary to the small RNA were retained. To evaluate the potential functions of miRNA-targeted genes, gene ontology (GO) categories (http://www.geneontology.org/) were used to assign the identified target genes according to the previously described method (Du et al., 2010).
Transcriptome Sequencing and Target Gene Profiling
Total RNA (10 μg) was subjected to poly-A selection, fragmentation, random priming, first and second strand cDNA synthesis with the Illumina Gene Expression Sample Prep kit (CA, USA). The cDNA fragments were subjected to an end repair process and then ligated to adapters. The products were enriched with PCR, and the 200-bp fragments were purified with 6% TBE PAGE gel electrophoresis. After denaturation, the single-chain fragments were fixed onto the Solexa Sequencing Chip (Flowcell) and consequently grown into single-molecule cluster sequencing templates through in situ amplification on the Illumina Cluster Station. Double-end pyrosequencing was performed on the Illumina Genome Analyzer platform with read lengths of 101 bp for each end. The clean reads were aligned to the Chinese cabbage reference transcript assemblies (http://brassicadb.org/brad/downloadOverview.php). Gene expression levels were calculated using the RPKM method (Mortazavi et al., 2008) using the following formula: RPKM = (1,000,000 * C)/(N * L * 1000), where RPKM(A) is the expression of gene A, C is the number of reads that uniquely align to gene A, N is the total number of reads that uniquely align to all genes, and L is the number of bases in gene A. Statistical comparison between Tyms and 231–330 was performed using the IDEG6 software (Romualdi et al., 2003). The General Chi-squared method was used and the FDR (false discovery rate) was applied to determine the Q-value threshold. Unigenes were considered to be differentially expressed when the RPKM between Tyms and 231–330 displayed a more than two-fold change with an FDR less than 10−2.
Quantitative Real-time PCR
For analysis of miRNAs, 2.5 μg of total RNA was polyadenylated using a miRNA cDNA synthesis kit (Takara, Inc., Dalian, China). The poly(A)-tail-amended total RNA was reverse-transcribed by PrimeScript RTase using a universal adapter primer containing oligo-dT. The qPCR was performed on a LightCycler 96 System (ROCHE, USA) using SYBR Premix Ex TaqTMII (TaKaRa, Dalian China). The miRNA-specific forward primer for each miRNA was designed based on the entire miRNA sequence (Table S13), and the universal reverse primer was provided by the miRNA cDNA synthesis kit (Takara, Dalian, China). All reactions were performed with three biological and three technical replicates for each sample, and the U6 snRNA (Forward: GGGGACATCCGATAAAATT, Reverse: TGTGCGTGTCATCCTTGC) was used as the internal control. The reaction volume was 20 μL, including 10 μL of SYBR Premix Ex Taq II, 0.8 μL of 10 mM Forward primer, 0.8 μL of 10 mM Uni-miR qPCR primer, 2.0 μL of the cDNA sample and 6.4 μL of dH2O. The following qPCR program was used: denaturation at 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 55°C for 30 s, and 72°C for 60 s. Melting curve analysis with 95°C for 10 s, 65°C for 60 s, and 97°C for 1 s was performed to produce a dissociation curve for verification of the amplification specificity. Relative expression levels of miRNAs were quantified using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
The primers of the selected genes subjected to target analysis are listed in Table S14. β-actin was used as an internal control. Experiments were performed on a similar system as described above. The reaction volume was 20 μL, including 10 μL of SYBR® Premix Ex Taq™ (Tli RNaseH Plus), 0.8 μL of 10 mM Forward primer, 0.8 μL of 10 mM Reverse primer, 2.0 μL of the cDNA sample and 6.4 μL of dH2O. Three independent biological and three technical replicates were performed. The fold change was estimated using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Paraffin Sectioning and Microscopic Observation
Sterile and fertile flower buds were fixed and embedded in Paraffin (Beeswax, China). Thin (0.8 μm) sections were prepared with an Ultracut Eultra microtome (Leica, Germany), stained with hematoxylin, and photographed under a LEICA DMI3000B microscope (Leica, Germany).
Results and Discussion
sRNA Sequencing and miRNA Identification
To analyze the roles of miRNAs in Ogura-CMS in Chinese cabbage, small RNAs were pyrosequenced from the buds of the Ogura-CMS Tyms line and its maintainer line 231–330 (the morphology and microscopy of the samples are shown in Figure 1). A total of 22.2 and 24.4 M reads were produced from the two lines, respectively. After filtering out the low quality reads, 3′adapter null, insert null, 5′adapter contaminants, reads smaller than 18 nt and poly A, a total of 21.9 and 24.1 M high quality clean reads were obtained, respectively (Table S1). The majority of the tags ranged in size between 21–24 nt, with the 24 and 21 nt lengths dominant (Figure 2). Out of the 46.0 M total tags, 33.8 M (73.5%) were shared by the two samples, while 5.7 M (12.5%) and 6.4 M (14.0%) were Tyms- and 231–330-specific, respectively. Then, the tags were divided into 12.2 M non-redundant unique reads composed of 4.8 M (39.6%) Tyms-specific, 5.6 M (45.7%) 231–330-specific and 1.8 M (14.8%) reads shared by both lines (Table S2). The reads were aligned to B. rapa miRNAs (B. rapa 1.1) in miRBase (http://www.mirbase.org/); among the 157 mature miRNAs encoded by 96 precursors distributed into 63 families included in the database, 69 mature miRNAs from 59 precursors of 41 families were expressed in our samples (Table S3). The remainder of the reads were aligned to all known plant miRNAs in miRBase 21.0 and then aligned to the B. rapa genome for precursor identification. From this analysis, another 220 mature miRNAs derived from 163 precursors were identified. Among them, 57 precursors contained both 5p and 3p miRNAs, while only one mature miRNA was detected for the remaining 106 precursors. The 139 (39 pairs and 61 singles) mature miRNAs encoded by 100 precursors were new members of 36 existing families in the B. rapa miRBase. Thus, only 17 families (bra-MIR9552—bra-MIR9557 and bra-MIR9559—bra-MIR9569) in the miRBase B. rapa collection were not detected in our present study. By expending the members to the existing B. rapa miRNA families, bra-miR156 was now the largest family (harboring 21 members), followed by bra-miR171 (11 members), bra-miR167 (8 members), bra-miR172 (8 members), bra-miR164 (7 members), bra-miR168 (7 members), bra-miR2111 (7 members), bra-miR157 (6 members), bra-miR160 (6 members), bra-miR390 (6 members), and bra-miR395 (6 members). The other families contained less than five members (Table S4). In addition to these existing families, we also identified 81 miRNAs (18 pairs and 45 singles from 63 precursors) belonging to 23 families that had not been previously reported in B.rapa. These included 13 members of bra-miR1439, seven members of bra-miR169, five members each of bra-miR166, bra-miR393, bra-miR394, and bra-miR399, three bra-miR165, two bra-miR170, two bra-miR5376, and one member each of bra-miR397, bra-miR827, bra-miR828, bra-miR838, bra-miR845, bra-miR858, bra-miR5298, bra-miR5575, bra-miR5641, bra-miR6029, bra-miR6030, bra-miR6033, bra-miR6034, and bra-miR6284 (Table S5). The remaining reads were aligned to Genbank, Rfam and the cabbage genome and annotated as rRNA, scRNA, snRNA, snoRNA, tRNA, repeat, exon sense, exon antisense, intron sense, intron antisense, and unannotated sequences (Table S6). The unannotated reads and those derived from the intron region and exon antisense region were used for novel miRNA analysis. A total of 426 novel miRNAs were identified based on the criteria defined in the Methods section. The sequences and precursor information were listed in Table S7. Among them, 83 novel miRNAs were generated from more than one precursor in the Chinese cabbage. A total of 138 precursors (84 types of mature miRNA) harbored both the 3-p and 5-p miRNAs on their arms (Supplementary file 2).

Figure 1. The buds and flowers of the Ogura-CMS (Tyms) and maintainer (231-330) lines. (A) the buds of four developmental stages and the flower that were sampled for this study. (B) microscopy of the buds showing the well-developed pollen sacs in 231–330 and incomplete development in Tyms.
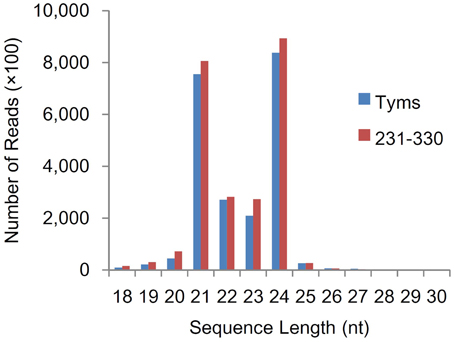
Figure 2. Size distribution of small RNA sequences.
Of these miRNAs, members of 32 known families were also identified in buds of a closely related vegetable plant B. rapa ssp. chinensis (Jiang et al., 2014). These miRNAs may be common bud development contributors in both plants. Our present study identified more miRNAs of either known or novel type than that of the previous report. One important reason for this is that nearly fourfold of reads were generated in present study compared to the previous one. This facilitated the identification of low abundant miRNAs. Another reason can be attributed to the genetic differences between the two subspecies. One of our sequencing projects showed only half of reads from B. rapa ssp. chinensis could map to the B. rapa ssp. pekinensis reference genome (unpublished data). The publicly released B. rapa ssp. pekinensis genome (Wang et al., 2011b) is not an ideal reference for B. rapa ssp. chinensis. Jiang et al. (2014) used B. rapa ssp. pekinensis reference genome for their miRNA annotation and novel-miRNA prediction, there for many miRNAs were failed to be identified due to the sequence diversity, especially for the novel miRNAs which rely on the hairpins prediction.
Comparative Expression Patterns of miRNAs in the Buds of Ogura-CMS and its Maintainer
The expression levels of known and novel miRNAs were profiled based on tag counts. For multiple precursors sharing the same mature miRNA, only one member of the mature miRNA was used for expression profiling. The miRNA with the highest expression levels was bra-miR157, which accumulated 579,691 and 253,845 copies in the 231-330 and Tyms buds, respectively, followed by bra-miR168 and bra-miR156 (Table S3). bra-miR156 and bra-miR157 belong to the same family and regulate the SQUAMOSA promoter binding protein-like (SPL) genes that are important regulators for plant vegetative and reproductive development as well as gynoecium differential patterning and male fertility (Wang et al., 2009; Wu et al., 2009; Xing et al., 2010, 2013; Zhang et al., 2011b; Yu et al., 2012). bra-miR168 targets ARGONAUTE 1 (AGO1), which is an important part of the RISC. The balance between miR168 and AGO1 plays important roles in plant development, including flowering and fruiting (Vaucheret et al., 2004; Xian et al., 2014). The top 20 most highly expressed known miRNAs are listed in Table 1. The expression levels of novel miRNAs were much lower than the known miRNAs, with the majority (~80%) barely accumulating several to less than 100 copies. The top 15 most highly expressed novel miRNAs are listed in Table 2.
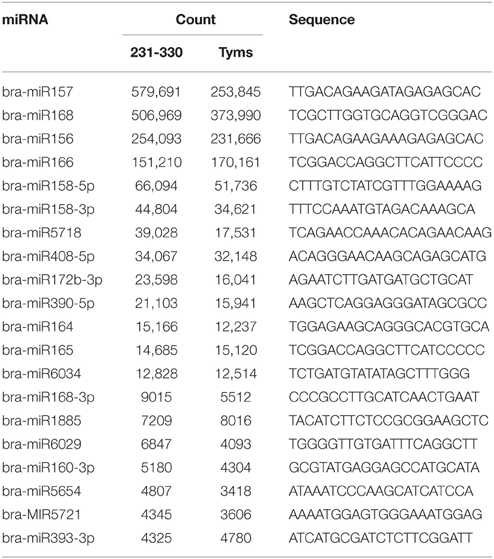
Table 1. The most highly expressed known miRNAs.
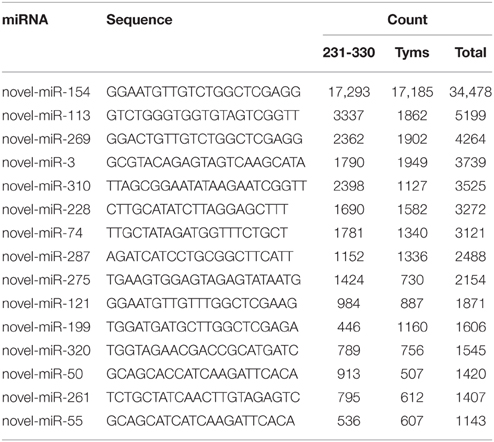
Table 2. The most highly expressed novel miRNAs.
The expression level between the Ogura-CMS and maintainer buds was compared using normalized tag counts. Ten known miRNAs were down-regulated two-fold (p < 10−3) in Tyms compared to 231–330 (Table 3). Among them, bra-miR157, bra-miR158-3p, and bra-miR5718 were expressed at relatively high levels and could play important roles in pollen development. However, bra-miR6030 was the only known miRNA that was up-regulated in the Tyms buds. In addition to these examples, a total of 49 and 27 novel miRNAs were down- and up-regulated in Tyms, accounting for 11.5 and 6.3% of the total 426 novel miRNAs, respectively (Table S8).
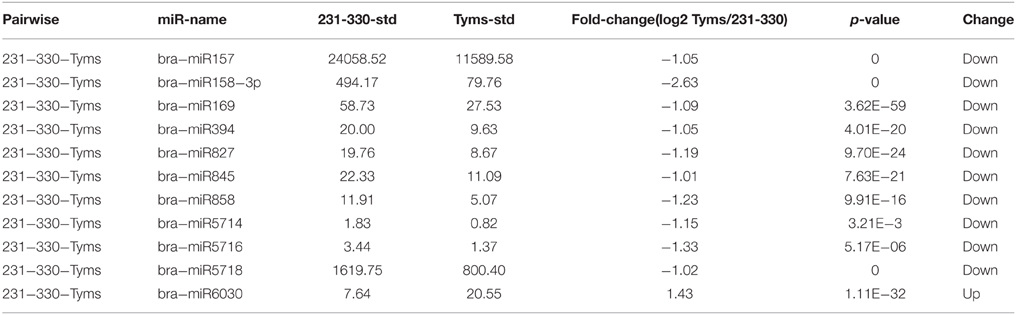
Table 3. Differentially expressed known miRNAs.
To test the accuracy of the RNA-Seq-based expressional profiling, a set of Q-PCR analyses were performed. As shown in Figure 3, the relative expression levels were similar between the Q-PCR and RNA-Seq technologies for seven known and one novel miRNAs. These results indicate that the RNA-Seq expression profiles are reliable.
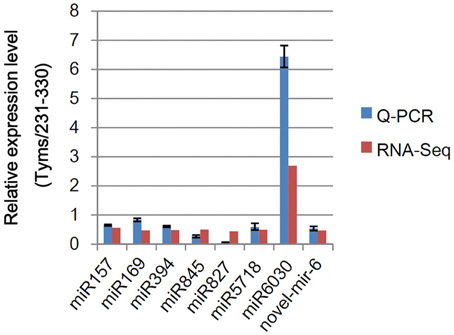
Figure 3. Q-PCR analysis of the relative expression levels of miRNAs. The bars indicate the SE; four biological and three technique replicates were performed.
Target Prediction and Validation by Degradome Analysis
miRNAs function by regulating their target genes and especially by degrading their target mRNAs in plants. Thus, we performed a degradome sequencing analysis to validate the miRNA targets. A total of 36,541,512 and 36,595,548 clean tags were generated from the buds of Tyms and 231-330, respectively (Table S9). Among them, 23,253,666 (63.64%) and 24,768,149 (67.68%) tags were mapped to the reference B. rapa genome. A total of 15,154,675 (41.47%) and 17,009,287 (46.48%) tags were mapped to the cDNA sense chains and were used for target prediction (Table S10). A total of 376 mRNAs were identified as miRNA targets, including 136 targeted by 30 known miRNA families and 248 targeted by 100 novel miRNAs. Among which 26 mRNAs were targeted by more than one miRNA (Table S11). A larger proportion known (50.8%) miRNAs compared to novel (23.5%) miRNAs have targets been detected, indicating that the conserved miRNAs have comparatively more valid cutting functions than the newly formed miRNAs. One possible reason for not detecting the targets could be that the target mRNAs were expressed at low levels; thus, the cut ends were not detected by our pipeline. Another possibility is that some miRNAs function by translation inhibition rather than mRNA digestion. Based on GO annotation, the targets were enriched in the “binding,” “catalytic activity,” and “nucleic acid binding transcription factor activity” terms in the “molecular function” cluster. “Cell,” “membrane,” and “organelle” were the three most abundant “cellular component” targets. In the “biological process” cluster, the top three terms were “cellular process,” “metabolic process,” and “response to stimulus” (Figure 4).
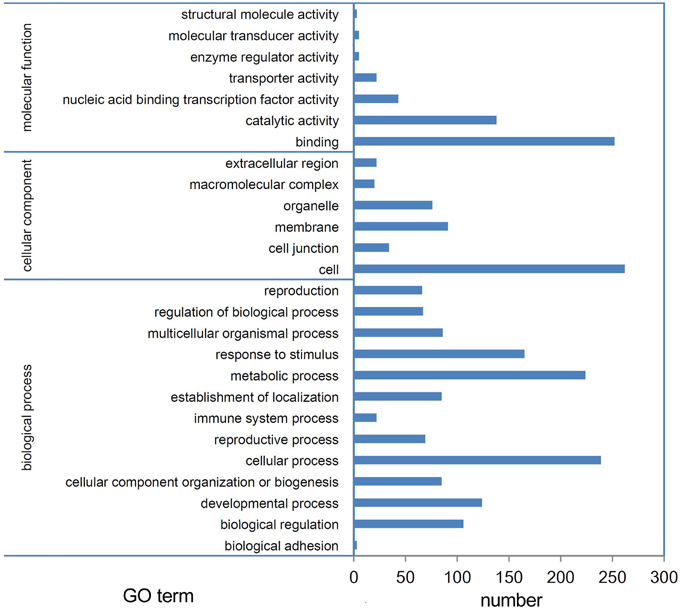
Figure 4. GO annotation of targets. Targets of miRNAs expressed in Tyms and 231-330 Chinese cabbage buds were detected by degradome sequencing analysis.
Most of the known miRNAs targeted transcription factor-encoding genes, such as the targeting of SPL, MYB, auxin response factor (ARF), NAC, scarecrow, APETALA 2 (AP2), GROWTH-REGULATING FACTOR (GR), and C3HC4-type RING finger family transcription factors by miR156/157, miR159, miR160, miR164, miR171, miR172, miR396, and miR5716, respectively. This finding indicates that the transcription factors perform primary important roles in bud development. Another large group of miRNAs, including miR158, miR161, miR400, and miR5654, target the pentatricopeptide repeat (PPR)-containing protein-encoding genes. PPR-containing proteins are a large family in plants that are mostly located in the mitochondria and chloroplast and play important roles in RNA processing in the two organelles (Fujii and Small, 2011). Some PPR-containing proteins have been identified as fertility restoration (Rf) genes for CMS (Desloire et al., 2003; Wang et al., 2008; Yasumoto et al., 2009). miR1885, miR5719, and miR6030 target disease resistance (CC-NBS-LRR class) genes. miR167 targets IAA-amino acid hydrolase 3 (IAR3), which encodes an auxin conjugate hydrolase that contributes to the jasmonate pathway (Kinoshita et al., 2012; Widemann et al., 2013). MiR393 targets AUXIN signaling F-BOX (AFB) genes, which are auxin receptors that have been widely identified as stress response regulators and control auxin-related development in plants (Si-Ammour et al., 2011; Terrile et al., 2012). These genes and the above mentioned miR160-ARFs indicate that the miRNA-hormone signaling cascades play important roles in flower bud development. Interestingly, three Vacuolar ATP synthase subunit A (VHA-A) tags were identified as targets of miR5712. VHA-A have been demonstrated to be essential for male gametophyte development due to its important role in Golgi organization (Dettmer et al., 2005). miR5724 targets embryo defective 1473 (emb1473), which encodes a structural constituent of the ribosome. miR158, which is down-regulated in Tyms buds, targets a glutathione S-transferase gene that has been annotated as a restorer-of-fertility (Krishnasamy and Makaroff, 1994; Woo et al., 2008). These miRNAs could play important roles in pollen development.
For the novel miRNAs, 153 out of the 264 targets (58%) were annotated as “catalytic activity,” which accounted for the largest fraction of the targets. Only 16 transcription factors were identified as targets of novel miRNAs: four NAC (including three cup-shaped cotyledon) targeted by novel mir410, 446, and 465, four zinc finger family proteins, two homeobox proteins, one MYB, one WRKY, one SPL, one HSFA1E, and one PIL1 (Phytochrome Interacting Factor 3-like 1). Several of the 19 targets possess “transporter activity,” including four genes encoding H+-transporting ATPases, two genes encoding Ca2+:H+ antiporters and two genes encoding sucrose transporter SUC1. These genes could play important roles in gametophyte development by transporting protons, ions, mineral elements and energy. A total of 28 targets were annotated as reproductive process-related, including the aforementioned cup-shaped cotyledon 2 transcription factors and sucrose transporter SUC1, auxin-responsive protein, and ubiquitin-activating enzyme E1 (Table S12). Among them, an oxidoreductase, a HSFA1E, a C2H2 zinc finger protein 1 and a beta-glucosidase were targets of novel-miR-6, −93, and −321, which were down-regulated in Tyms. An F-box/LRR-repeat protein, a heat shock 70 kDa protein 1/8, an auxin-responsive protein IAA and two sucrose transporter SUC1 were targeted by novel-miR-175, −383, −385, and −448, which were up-regulated in Tyms buds.
Target Expression Profiles Based on Transcriptome Sequencing
To profile the expression of the targets, a transcriptome sequencing project was performed separately on buds of 231–330 and Tyms. A total of 25,015,186 and 24,295,576 clean reads were generated from the 231–330 and Tyms buds, respectively. Approximately 76.4 and 72.6% of the reads were mapped to the reference genome and 59.3 and 57.9% of the reads were mapped to the reference gene sets of B. rapa, respectively. The expression of the target genes was called from the transcriptome data for further analysis. A total of 367 and 352 out of the 376 target genes were expressed in the 231–330 and Tyms buds, respectively. Among them, 44 were up-regulated and 32 were down-regulated (two-fold change and FDR < 10−3) in Tyms compared with 231–330. Most of the differentially expressed targets were pollen development-related genes according to the annotation (Tables 4, 5).
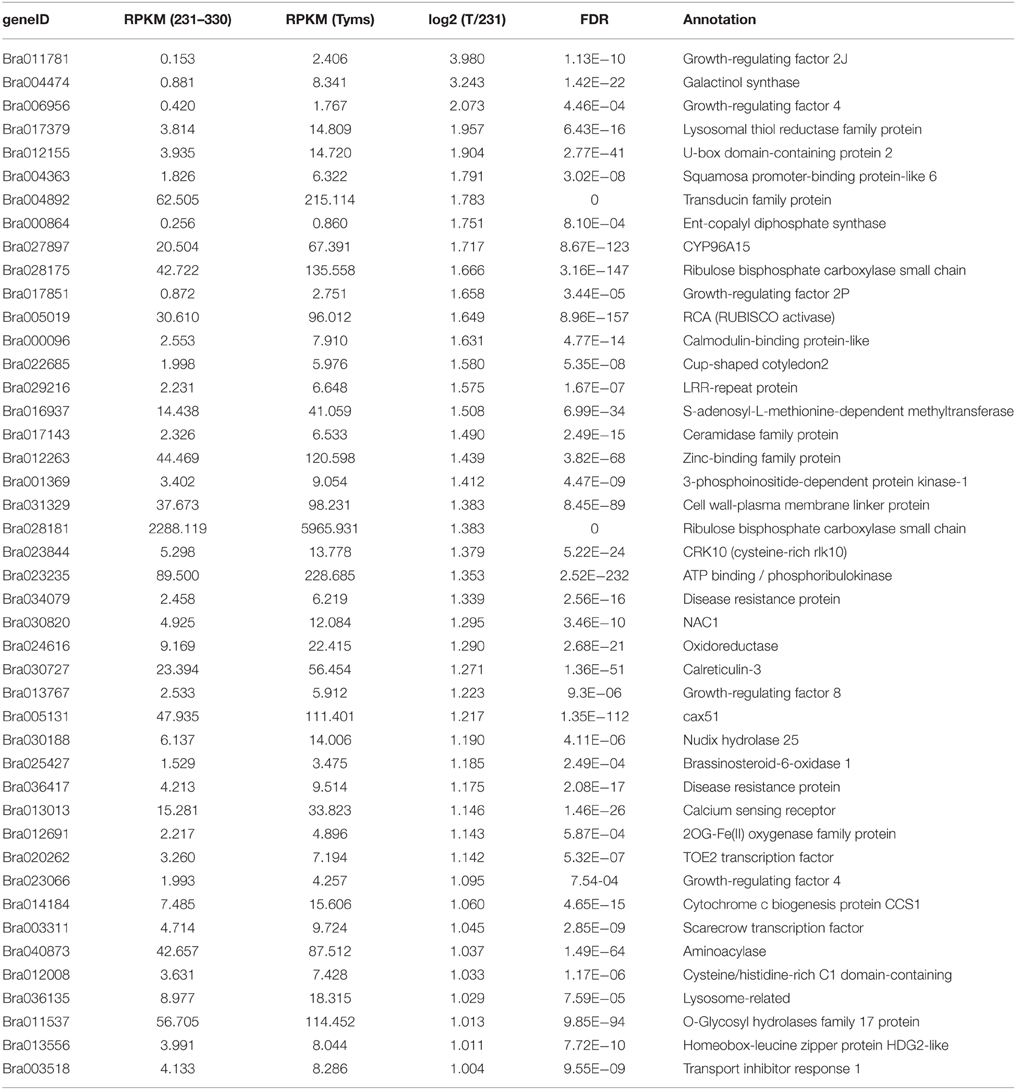
Table 4. The up-regulated targets in Tyms compared to 231-330 buds.
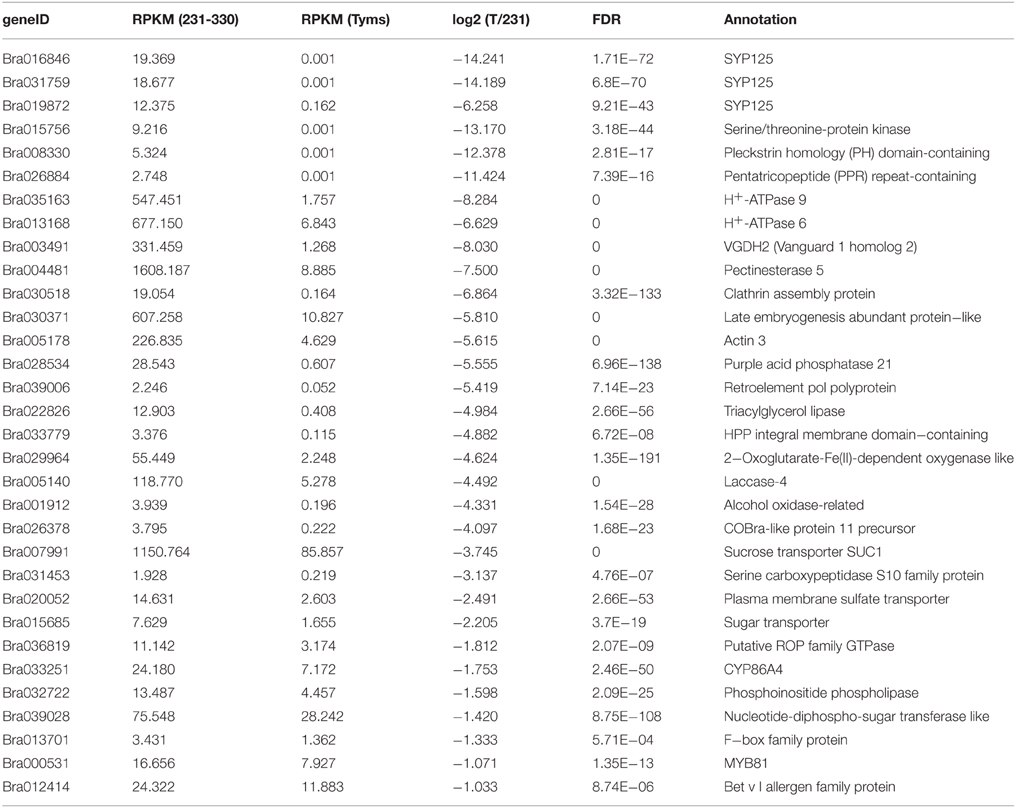
Table 5. The down-regulated targets in Tyms compared to 231-330 buds.
miRNA-target Network Underlying Ogura-CMS
The differentially expressed target genes were targeted by 10 known miRNAs (miR159, miR164, miR171, miR172, miR393, miR396, miR397, miR1885, miR5654, and miR6034) and 33 novel miRNAs. However, none of these known miRNAs were significantly differentially expressed according to the two-fold criteria. Based on the combined analysis of the expression profiles of miRNAs and their targets without the significant test filters, the expression patterns of the miRNA-targets could be classified into four clusters. As shown in Figure 5A, cluster I contained more than half of the miRNA-target pairs, in which miRNAs were down-regulated and the targets were up-regulated in Tyms. Approximately 16 and 24% of the miRNA-target pairs were synergistically down-regulated (cluster II) or up-regulated (cluster III), respectively. Less than 8% of the miRNAs were up-regulated while their targets were down-regulated. These results indicate the miRNAs provide an efficient buffer system for fine-tuning the expression of genes involved in bud and pollen development. When a cut-off of >1.5-fold change was used, seven miRNAs were down-regulated and released the expression of 18 targets in Tyms. In contrast, four miRNAs were up-regulated and suppressed the expression of five targets (Figure 5B). miR156 and SPLs represent a cascade of controls for many important developmental processes, including the proper development of sporogenic tissues of the anther (Xing et al., 2010). miR156 and SPLs are expressed in high abundance in many tissues, including buds (Zhang et al., 2011b). Our present study found that miR156 and SPLs were down- or up-regulated in Tyms buds by more than 1.5-fold but less than two-fold. It is possible that the expression of these genes was changed more significantly in sporogenic tissues but was submerged by the expression in other tissues of the buds. Using a two-fold threshold, three and two novel miRNAs were up-regulated in 231–330 and Tyms, resulting in the down-regulation of five and three of their target genes in the corresponding tissues, respectively (Figure 5C). Among them, a cytochrome c biogenesis protein was targeted by novel-miR-180, a Ca2+/H+ antiporter was targeted by novel-miR-191, a P450 and a glycogenin glucosyltransferase (GGT) were targeted by novel-miR-6; all of these genes have been implicated in pollen development by previous studies (Balk and Leaver, 2001; Welchen and Gonzalez, 2005; Morant et al., 2007; Song et al., 2009; Li et al., 2010, 2012; Rennie et al., 2014) and were up-regulated in Tyms in our analysis. Interestingly, novel-miR-448 and novel-miR-335 were specifically expressed in Tyms; this is especially worth noticing because the novel-miR-335 was expressed at a relatively high level. The expression of its target genes (sucrose transporter SUC1 and H+-ATPase 6) was high in 231–330 but suppressed by approximately 100-fold in Tyms. SUC1 is an enzyme that catalyzes the degradation of sucrose and performs important roles for pollen germination in Arabidopsis (Sivitz et al., 2008). Thus, the down-regulation of SUC1 may result in energy deficiency in Tyms buds and thus abort pollen development. A study in Nicotiana plumbaginifolia reported that co-suppression of an H+-ATPase impaired sucrose translocation and male fertility (Zhao et al., 2000). In our present study, both a SUC1 and an H+-ATPase were indicated as targets of two novel miRNAs and were sufficiently suppressed by these two miRNAs in the Ogura-CMS buds (Figure 5C). This finding indicated that the novel-miR-335/H+-ATPase and novel-miR-448/SUC1 cascade could play important roles in male sterility in Ogura-CMS. The secondary structure of the novel-miR-335 and novel-miR-448 and the degradation map of H+-ATPase and SUC1 were shown in Figure 6. The suppression of H+-ATPase and SUC1 expression were validated by Q-PCR analysis (Figure 7). This finding filled a gap within the cross-talk network between the orf138 locus in mitochondria and the effecter genes in the chromosome. However, more studies are still needed to linkup the orf138 locus and miRNA regulatory networks.
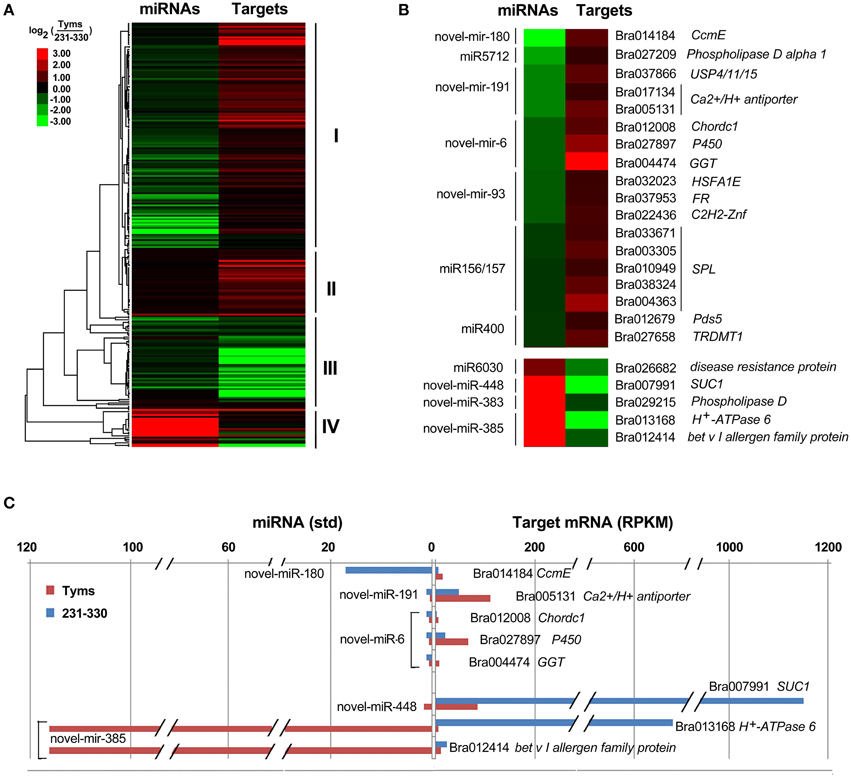
Figure 5. Expression profiles of miRNA-targets. (A) Heatmap of overall expression patterns of miRNA-targets. (B) Heatmap of expression levels of miRNAs and their targets with more than 1.5-fold changes between Tyms and 231–330. (C) Bar chart of expression levels of miRNAs and their targets with more than two-fold changes between Tyms and 231–330. CcmE, cytochrome c biogenesis protein; USP4/11/15, ubiquitin carboxyl-terminal hydrolase 4/11/15; Chordc 1, cysteine/histidine-rich C1 domain-containing protein; P450, cytochrome P450; GGT, glycogenin glucosyltransferase; FR, ferric reduction oxidase 7; C2H2-Znf, C2H2 zinc finger protein 1; SPL, SQUAMOSA promoter binding protein-like; Pds5, sister chromatid cohesion protein PDS5; TRDMT1, tRNA (cytosine38-C5)-methyltransferase; SUC1, sucrose transporter.
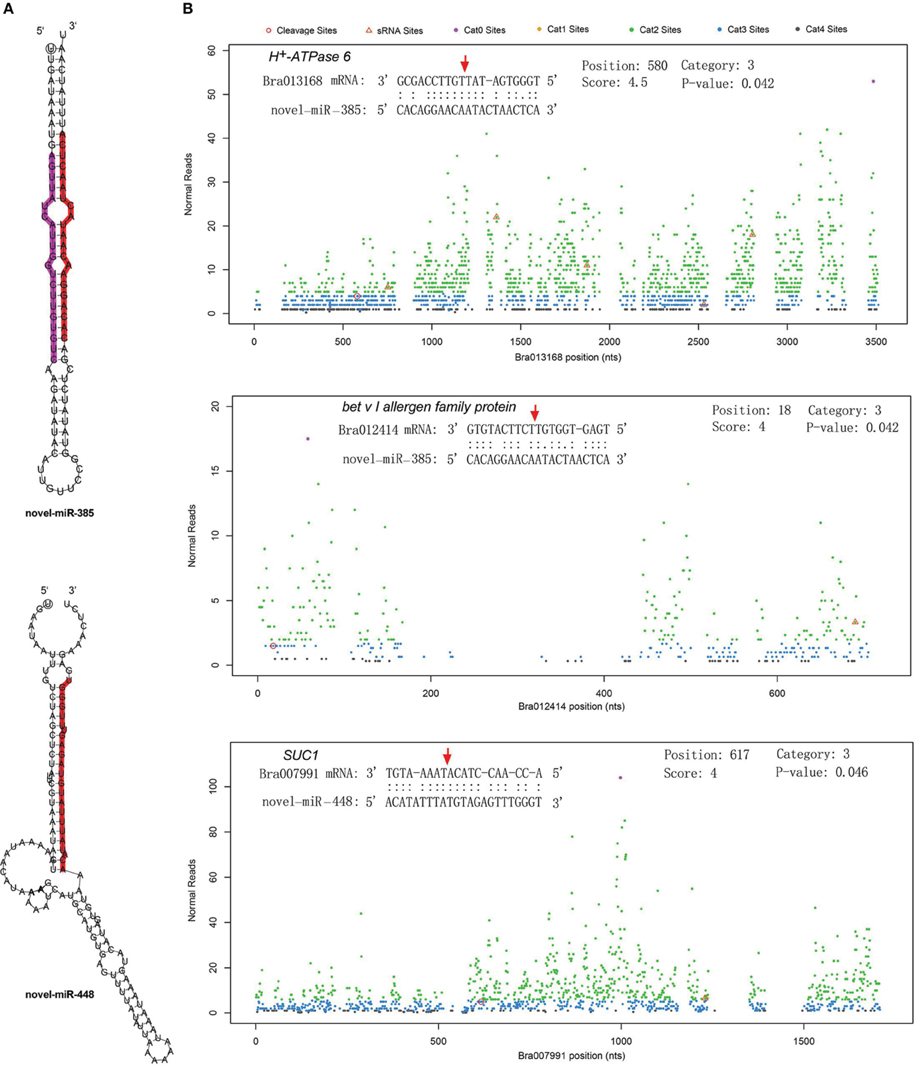
Figure 6. Secondary structure of two novel miRNAs and the cleavage plots of their targets. (A) Secondary structure of pre-miRNA hairpins. Red shaded areas indicate the dominant mature miRNAs, violet shaded areas indicates the reverse complementary mature miRNAs. (B) Cutting plots of miRNA targets confirmed using degradome sequencing. The corresponding miRNA:mRNA alignments are shown on the top. The red arrows indicate the miRNA-directed cleavage positions. The y-axis shows the nucleotide position in the target gene. The x-axis indicates the number of cleaved ends detected in the degradome analysis.
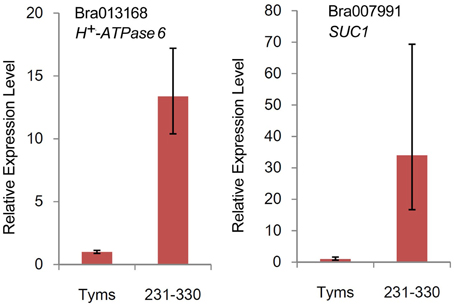
Figure 7. Relative expression levels of two target genes revealed by Q-PCR.
Conclusion
The present study used deep sequencing technology and performed a series of whole genome small RNA, degradome, and transcriptome analyses on Chinese cabbage buds from both Ogura-CMS and its maintainer. A total of 289 (69 families) known and 426 novel miRNAs were identified, which was much higher than the number of miRNAs found in the buds of the closely relative subspecies B. campestris ssp. chinensis (Jiang et al., 2014). These miRNAs not only validated the finding in the previous study on B. campestris ssp. chinensis but also contained many novel miRNAs that were not previously reported. A large number of targets were firstly validated in this study. The combinational profiling of miRNAs and targets revealed a regulatory network contributing to bud development, especially for pollen engenderation. The finding of two novel miRNA/target cascades (novel-miR-335/H+-ATPase and novel-miR-448/SUC1) will provide new insights into the communication between the mitochondria and chromosome and take one step toward filling in the gap in the regulatory network mechanism from the orf138 locus to the pollen abortion in the Ogura-CMS plants. The true functions of these two miRNAs warrant more solid experiments, such as the transgenic complementary test.
Author Contributions
XW and XWZ designed the study. XW performed the experiments. XW and XHZ analyzed the data and drafted the manuscript. YY and QZ assisted with the bioinformatics analysis and aided in writing the manuscript. QY, ZW, YZ, and WJ aided in performing the experiments. XL and FW modified the manuscript. All of the authors carefully checked and approved this version of the manuscript.
Data Access
RNAseq are submitted to EMBL/NCBI/SRA with the accession numbers SRR2132359, SRR2136647, SRR2149955, SRR2132463, SRR2136646, SRR2149956.
Funding
This work was supported by funding from North Henan Station, the National Vegetable Industry Technology System (CARS-25-27), Excellent technology innovation team of Henan province, the National High Technology Research and Development Program of China (863 Program) (2012AA100202-7), The National Key Technology R&D Program of China (2012BAD02B01-3).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00894
References
Addo-Quaye, C., Miller, W., and Axtell, M. J. (2009). CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25, 130–131. doi: 10.1093/bioinformatics/btn604
Aukerman, M. J., and Sakai, H. (2003). Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15, 2730–2741. doi: 10.1105/tpc.016238
Balk, J., and Leaver, C. J. (2001). The PET1-CMS mitochondrial mutation in sunflower is associated with premature programmed cell death and cytochrome c release. Plant Cell 13, 1803–1818. doi: 10.1105/tpc.13.8.1803
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Bhogale, S., Mahajan, A. S., Natarajan, B., Rajabhoj, M., Thulasiram, H. V., and Banerjee, A. K. (2014). miRNA156-A potential graft-transmissible microRNA that modulates plant architecture and tuberization in potato (Solanum tuberosum ssp andigena). Plant Physiol. 164, 1011–1027. doi: 10.1104/pp.113.230714
Carrington, J. C., and Ambros, V. (2003). Role of microRNAs in plant and animal development. Science 301, 336–338. doi: 10.1126/science.1085242
Chambers, C., and Shuai, B. (2009). Profiling microRNA expression in Arabidopsis pollen using microRNA array and real-time PCR. BMC Plant Biol. 9:87. doi: 10.1186/1471-2229-9-87
Chase, C. D. (2007). Cytoplasmic male sterility: a window to the world of plant mitochondrial–nuclear interactions. Trends Genet. 23, 81–90. doi: 10.1016/j.tig.2006.12.004
Chen, X. (2004). A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303, 2022–2025. doi: 10.1126/science.1088060
Desloire, S., Gherbi, H., Laloui, W., Marhadour, S., Clouet, V., Cattolico, L., et al. (2003). Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep. 4, 588–594. doi: 10.1038/sj.embor.embor848
Dettmer, J., Schubert, D., Calvo-Weimar, O., Stierhof, Y. D., Schmidt, R., and Schumacher, K. (2005). Essential role of the V-ATPase in male gametophyte development. Plant J. 41, 117–124. doi: 10.1111/j.1365-313X.2004.02282.x
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Floyd, S. K., and Bowman, J. L. (2004). Gene regulation: ancient microRNA target sequences in plants. Nature 428, 485–486. doi: 10.1038/428485a
Folkes, L., Moxon, S., Woolfenden, H. C., Stocks, M. B., Szittya, G., Dalmay, T., et al. (2012). PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 40:e103. doi: 10.1093/nar/gks277
Fujii, S., and Small, I. (2011). The evolution of RNA editing and pentatricopeptide repeat genes. New phytol. 191, 37–47. doi: 10.1111/j.1469-8137.2011.03746.x
German, M. A., Luo, S., Schroth, G., Meyers, B. C., and Green, P. J. (2009). Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 4, 356–362. doi: 10.1038/nprot.2009.8
Grant-Downton, R., Hafidh, S., Twell, D., and Dickinson, H. G. (2009a). Small RNA pathways are present and functional in the angiosperm male gametophyte. Mol. Plant 2, 500–512. doi: 10.1093/mp/ssp003
Grant-Downton, R., Le Trionnaire, G., Schmid, R., Rodriguez-Enriquez, J., Hafidh, S., Mehdi, S., et al. (2009b). MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. BMC Genomics 10:643. doi: 10.1186/1471-2164-10-643
Grelon, M., Budar, F., Bonhomme, S., and Pelletier, G. (1994). Ogura cytoplasmic male-sterility (CMS)-associated orf138 is translated into a mitochondrial membrane polypeptide in male-sterile Brassica cybrids. Mol. Gen. Genet. 243, 540–547. doi: 10.1007/BF00284202
Guo, H. S., Xie, Q., Fei, J. F., and Chua, N. H. (2005). MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for arabidopsis lateral root development. Plant Cell 17, 1376–1386. doi: 10.1105/tpc.105.030841
He, H., Liang, G., Li, Y., Wang, F., and Yu, D. (2014). Two young miRNAs originating from target duplication mediate nitrogen starvation adaptation via regulation of glucosinolate synthesis in Arabidopsis thaliana. Plant Physiol. 164, 853–865. doi: 10.1104/pp.113.228635
Jiang, J., Lv, M., Liang, Y., Ma, Z., and Cao, J. (2014). Identification of novel and conserved miRNAs involved in pollen development in Brassica campestris ssp. chinensis by high-throughput sequencing and degradome analysis. BMC Genomics 15:146. doi: 10.1186/1471-2164-15-146
Juarez, M. T., Kui, J. S., Thomas, J., Heller, B. A., and Timmermans, M. C. (2004). microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 428, 84–88. doi: 10.1038/nature02363
Katiyar-Agarwal, S., and Jin, H. (2010). Role of small RNAs in host-microbe interactions. Annu. Rev. Phytopathol. 48, 225. doi: 10.1146/annurev-phyto-073009-114457
Khraiwesh, B., Arif, M. A., Seumel, G. I., Ossowski, S., Weigel, D., Reski, R., et al. (2010). Transcriptional control of gene expression by microRNAs. Cell 140, 111–122. doi: 10.1016/j.cell.2009.12.023
Kim, B., Yu, H. J., Park, S. G., Shin, J. Y., Oh, M., Kim, N., et al. (2012). Identification and profiling of novel microRNAs in the Brassica rapa genome based on small RNA deep sequencing. BMC Plant Biol. 12:218. doi: 10.1186/1471-2229-12-218
Kinoshita, N., Wang, H., Kasahara, H., Liu, J., MacPherson, C., Machida, Y., et al. (2012). IAA-Ala Resistant3, an evolutionarily conserved target of miR167, mediates Arabidopsis root architecture changes during high osmotic stress. Plant Cell 24, 3590–3602. doi: 10.1105/tpc.112.097006
Krishnasamy, S., and Makaroff, C. A. (1993). Characterization of the radish mitochondrial orfB locus: possible relationship with male sterility in Ogura radish. Curr. Genet. 24, 156–163. doi: 10.1007/BF00324680
Krishnasamy, S., and Makaroff, C. A. (1994). Organ-specific reduction in the abundance of a mitochondrial protein accompanies fertility restoration in cytoplasmic male-sterile radish. Plant Mol. Biol. 26, 935–946. doi: 10.1007/BF00028860
Kurihara, Y., and Watanabe, Y. (2004). Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. U.S.A. 101, 12753–12758. doi: 10.1073/pnas.0403115101
Li, H. M., Chen, H., Yang, Z. N., and Gong, J. M. (2012). Cdi gene is required for pollen germination and tube growth in Arabidopsis. FEBS Lett. 586, 1027–1031. doi: 10.1016/j.febslet.2012.02.046
Li, H., Pinot, F., Sauveplane, V., Werck-Reichhart, D., Diehl, P., Schreiber, L., et al. (2010). Cytochrome P450 family member CYP704B2 catalyzes the ω-hydroxylation of fatty acids and is required for anther cutin biosynthesis and pollen exine formation in rice. Plant Cell 22, 173–190. doi: 10.1105/tpc.109.070326
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714. doi: 10.1093/bioinformatics/btn025
Liu, G., Tian, H., Huang, Y. Q., Hu, J., Ji, Y. X., Li, S. Q., et al. (2012). Alterations of mitochondrial protein assembly and jasmonic acid biosynthesis pathway in Honglian (HL)-type cytoplasmic male sterility rice. J. Biol. Chem. 287, 40051–40060. doi: 10.1074/jbc.M112.382549
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2 ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, D., Xu, H., Liu, Z., Guo, J., Li, H., Chen, L., et al. (2013). A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 45, 573–577. doi: 10.1038/ng.2570
Morant, M., Jørgensen, K., Schaller, H., Pinot, F., Møller, B. L., Werck-Reichhart, D., et al. (2007). CYP703 is an ancient cytochrome P450 in land plants catalyzing in-chain hydroxylation of lauric acid to provide building blocks for sporopollenin synthesis in pollen. Plant Cell 19, 1473–1487. doi: 10.1105/tpc.106.045948
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nagpal, P., Ellis, C. M., Weber, H., Ploense, S. E., Barkawi, L. S., Guilfoyle, T. J., et al. (2005). Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 132, 4107–4118. doi: 10.1242/dev.01955
Navarro, L., Dunoyer, P., Jay, F., Arnold, B., Dharmasiri, N., Estelle, M., et al. (2006). A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 312, 436–439. doi: 10.1126/science.1126088
Ogura, H. (1968). Studies on the new male-sterility in Japanese radish, with special reference to the utilization of this sterility towards the practical raising of hybrid seeds. Mem. Fac. Agric. Kagoshima Univ.6, 39–78.
Pelletier, G., Primard, C., Vedel, F., Chetrit, P., Remy, R., and Renard, M. (1983). Intergeneric cytoplasmic hybridization in Cruciferae by protoplast fusion. Mol. Gen. Genet. 191, 244–250. doi: 10.1007/BF00334821
Rennie, E. A., Ebert, B., Miles, G. P., Cahoon, R. E., Christiansen, K. M., Stonebloom, S., et al. (2014). Identification of a sphingolipid alpha-glucuronosyltransferase that is essential for pollen function in Arabidopsis. Plant Cell 26, 3314–3325. doi: 10.1105/tpc.114.129171
Romualdi, C., Bortoluzzi, S., D'Alessi, F., and Danieli, G. A. (2003). IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol. Genomics 12, 159–162. doi: 10.1152/physiolgenomics.00096.2002
Si-Ammour, A., Windels, D., Arn-Bouldoires, E., Kutter, C., Ailhas, J., Meins, F. Jr., et al. (2011). miR393 and secondary siRNAs regulate expression of the TIR1/AFB2 auxin receptor clade and auxin-related development of Arabidopsis leaves. Plant Physiol. 157, 683–691. doi: 10.1104/pp.111.180083
Sivitz, A. B., Reinders, A., and Ward, J. M. (2008). Arabidopsis sucrose transporter AtSUC1 is important for pollen germination and sucrose-induced anthocyanin accumulation. Plant Physiol. 147, 92–100. doi: 10.1104/pp.108.118992
Song, L. F., Zou, J. J., Zhang, W. Z., Wu, W. H., and Wang, Y. (2009). Ion transporters involved in pollen germination and pollen tube tip-growth. Plant Signal. Behav. 4, 1193–1195. doi: 10.4161/psb.4.12.10063
Sunkar, R., Chinnusamy, V., Zhu, J., and Zhu, J.-K. (2007). Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 12, 301–309. doi: 10.1016/j.tplants.2007.05.001
Tarutani, Y., Shiba, H., Iwano, M., Kakizaki, T., Suzuki, G., Watanabe, M., et al. (2010). Trans-acting small RNA determines dominance relationships in Brassica self-incompatibility. Nature 466, 983–986. doi: 10.1038/nature09308
Terrile, M. C., París, R., Calderón-Villalobos, L. I., Iglesias, M. J., Lamattina, L., Estelle, M., et al. (2012). Nitric oxide influences auxin signaling through S-nitrosylation of the Arabidopsis TRANSPORT INHIBITOR RESPONSE 1 auxin receptor. Plant J. 70, 492–500. doi: 10.1111/j.1365-313X.2011.04885.x
Vaucheret, H., Vazquez, F., Crete, P., and Bartel, D. P. (2004). The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 18, 1187–1197. doi: 10.1101/gad.1201404
Wang, J. W., Czech, B., and Weigel, D. (2009). miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, J. W., Park, M. Y., Wang, L. J., Koo, Y., Chen, X. Y., Weigel, D., et al. (2011a). miRNA control of vegetative phase change in trees. PLoS Genet. 7:e1002012. doi: 10.1371/journal.pgen.1002012
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011b). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Wang, Z. W., Zhang, Y. J., Xiang, C. P., Mei, S. Y., Zhou, Y., Chen, G. P., et al. (2008). A new fertility restorer locus linked closely to the Rfo locus for cytoplasmic male sterility in radish. Theor. Appl. Genet. 117, 313–320. doi: 10.1007/s00122-008-0776-5
Wei, L. Q., Yan, L. F., and Wang, T. (2011). Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol. 12:R53. doi: 10.1186/gb-2011-12-6-r53
Welchen, E., and Gonzalez, D. H. (2005). Differential expression of the Arabidopsis cytochrome c genes Cytc-1 and Cytc-2. Evidence for the involvement of TCP-domain protein-binding elements in anther- and meristem-specific expression of the Cytc-1 gene. Plant Physiol. 139, 88–100. doi: 10.1104/pp.105.065920
Widemann, E., Miesch, L., Lugan, R., Holder, E., Heinrich, C., Aubert, Y., et al. (2013). The amidohydrolases IAR3 and ILL6 contribute to jasmonoyl-isoleucine hormone turnover and generate 12-hydroxyjasmonic acid upon wounding in Arabidopsis leaves. J. Biol. Chem. 288, 31701–31714. doi: 10.1074/jbc.M113.499228
Woo, M. O., Ham, T. H., Ji, H. S., Choi, M. S., Jiang, W., Chu, S. H., et al. (2008). Inactivation of the UGPase1 gene causes genic male sterility and endosperm chalkiness in rice (Oryza sativa L.). Plant J. 54, 190–204. doi: 10.1111/j.1365-313X.2008.03405.x
Woodson, J. D., and Chory, J. (2008). Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet. 9, 383–395. doi: 10.1038/nrg2348
Wu, G., Park, M. Y., Conway, S. R., Wang, J. W., Weigel, D., and Poethig, R. S. (2009). The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138, 750–759. doi: 10.1016/j.cell.2009.06.031
Xian, Z., Huang, W., Yang, Y., Tang, N., Zhang, C., Ren, M., et al. (2014). miR168 influences phase transition, leaf epinasty, and fruit development via SlAGO1s in tomato. J. Exp. Bot. 65, 6655–6666. doi: 10.1093/jxb/eru387
Xing, S., Salinas, M., Garcia-Molina, A., Hohmann, S., Berndtgen, R., and Huijser, P. (2013). SPL8 and miR156-targeted SPL genes redundantly regulate Arabidopsis gynoecium differential patterning. Plant J. 75, 566–577. doi: 10.1111/tpj.12221
Xing, S., Salinas, M., Hohmann, S., Berndtgen, R., and Huijser, P. (2010). miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 22, 3935–3950. doi: 10.1105/tpc.110.079343
Yamagishi, H., and Bhat, S. R. (2014). Cytoplasmic male sterility in Brassicaceae crops. Breed. Sci. 64, 38–47. doi: 10.1270/jsbbs.64.38
Yamasaki, H., Abdel-Ghany, S. E., Cohu, C. M., Kobayashi, Y., Shikanai, T., and Pilon, M. (2007). Regulation of copper homeostasis by micro-RNA in Arabidopsis. J. Biol. Chem. 282, 16369–16378. doi: 10.1074/jbc.M700138200
Yasumoto, K., Terachi, T., and Yamagishi, H. (2009). A novel Rf gene controlling fertility restoration of Ogura male sterility by RNA processing of orf138 found in Japanese wild radish and its STS markers. Genome 52, 495–504. doi: 10.1139/G09-026
Yu, S., Galvão, V. C., Zhang, Y. C., Horrer, D., Zhang, T. Q., Hao, Y. H., et al. (2012). Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA promoter binding-like transcription factors. Plant Cell 24, 3320–3332. doi: 10.1105/tpc.112.101014
Zhang, X., Zou, Z., Gong, P., Zhang, J., Ziaf, K., Li, H., et al. (2011a). Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol. Lett. 33, 403–409. doi: 10.1007/s10529-010-0436-0
Zhang, X., Zou, Z., Zhang, J., Zhang, Y., Han, Q., Hu, T., et al. (2011b). Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant. FEBS Lett. 585, 435–439. doi: 10.1016/j.febslet.2010.12.036
Zhao, R., Dielen, V., Kinet, J. M., and Boutry, M. (2000). Cosuppression of a plasma membrane H(+)-ATPase isoform impairs sucrose translocation, stomatal opening, plant growth, and male fertility. Plant Cell 12, 535–546. doi: 10.1105/tpc.12.4.535
Keywords: miRNAs, Brassica rapa ssp. pekinensis, Ogura-CMS, bud, pollen, deep sequencing
Citation: Wei X, Zhang X, Yao Q, Yuan Y, Li X, Wei F, Zhao Y, Zhang Q, Wang Z, Jiang W and Zhang X (2015) The miRNAs and their regulatory networks responsible for pollen abortion in Ogura-CMS Chinese cabbage revealed by high-throughput sequencing of miRNAs, degradomes, and transcriptomes. Front. Plant Sci. 6:894. doi: 10.3389/fpls.2015.00894
Received: 29 July 2015; Accepted: 08 October 2015;
Published: 21 October 2015.
Edited by:
Naser A. Anjum, University of Aveiro, PortugalReviewed by:
Maoteng Li, Huazhong University of Science and Technology, ChinaHao Peng, Washington State University, USA
Copyright © 2015 Wei, Zhang, Yao, Yuan, Li, Wei, Zhao, Zhang, Wang, Jiang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaowei Zhang, eGlhb3dlaTU3MzdAMTYzLmNvbQ==
†These authors have contributed equally to this work.