 Ruibing Chen1†
Ruibing Chen1† Qing Li2†
Qing Li2† Hexin Tan1Junfeng Chen2Ying Xiao2
Hexin Tan1Junfeng Chen2Ying Xiao2 Ruifang Ma3
Ruifang Ma3 Shouhong Gao2
Shouhong Gao2 Philipp Zerbe4Wansheng Chen2
Philipp Zerbe4Wansheng Chen2 Lei Zhang1*
Lei Zhang1*- 1Department of Pharmaceutical Botany, School of Pharmacy, Second Military Medical University, Shanghai, China
- 2Department of Pharmacy, Shanghai Changzheng Hospital, Second Military Medical University, Shanghai, China
- 3School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang, China
- 4Department of Plant Biology, University of California, Davis, Davis, CA, USA
Root and leaf tissue of Isatis indigotica shows notable anti-viral efficacy, and are widely used as “Banlangen” and “Daqingye” in traditional Chinese medicine. The plants' pharmacological activity is attributed to phenylpropanoids, especially a group of lignan metabolites. However, the biosynthesis of lignans in I. indigotica remains opaque. This study describes the discovery and analysis of biosynthetic genes and AP2/ERF-type transcription factors involved in lignan biosynthesis in I. indigotica. MeJA treatment revealed differential expression of three genes involved in phenylpropanoid backbone biosynthesis (IiPAL, IiC4H, Ii4CL), five genes involved in lignan biosynthesis (IiCAD, IiC3H, IiCCR, IiDIR, and IiPLR), and 112 putative AP2/ERF transcription factors. In addition, four intermediates of lariciresinol biosynthesis were found to be induced. Based on these results, a canonical correlation analysis using Pearson's correlation coefficient was performed to construct gene-to-metabolite networks and identify putative key genes and rate-limiting reactions in lignan biosynthesis. Over-expression of IiC3H, identified as a key pathway gene, was used for metabolic engineering of I. indigotica hairy roots, and resulted in an increase in lariciresinol production. These findings illustrate the utility of canonical correlation analysis for the discovery and metabolic engineering of key metabolic genes in plants.
Introduction
Isatis indigotica Fortune has been used in traditional Chinese medicine for more than two millennia and is listed in the Chinese Pharmacopoeia (National Pharmacopoeia Committee, 2010). The root and leaves of I. indigotica demonstrate notable anti-viral (Chang et al., 2012), anti-inflammatory (Tang et al., 2014), anti-tumor (Chung et al., 2011), and anti-anaphylaxis (Recio et al., 2006) activity, and are used in clinical applications as “Banlengen” and “Daqingye,” respectively. In previous researches, lignans including lariciresinol and larch lignan glycosides were considered as the material base of those activities (Yang et al., 2013). However, the biosynthesis of lignans in I. indigotica is largely unresolved. Transcriptome analysis of I. indigotica (Chen et al., 2013) and availability of the complete genomes of other lignan-forming plant species (A. thaliana and Chinese cabbage) offer the opportunity to employ bioinformatics tools for better understanding and ultimately modulating lignan metabolism in I. indigotica. In addition, key genes responsible for the biosynthesis of backbone structures of phenylpropanoids, flavonoids, lignans and lignins have been established, including phenylalanine-ammonia lyase (PAL), cinnamate-4-hydroxylase (C4H) and coumaroyl-CoA-ligase (4CL) of phenylpropanoid metabolism, chalcone synthase (CHS), flavonol synthase (FNS) and chalcone isomerase (CHI) of flavonoid biosynthesis, and cinnamoyl alcohol dehydrogenase (CAD) and cinnamoyl-CoA reductase (CCR) in lignan formation (Figure 1A).
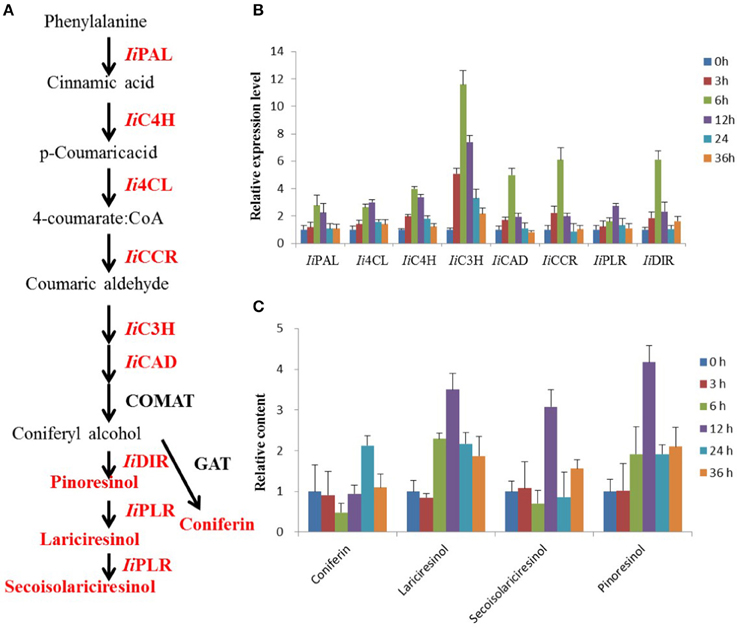
Figure 1. MeJA-induced changes in lignan biosynthesis analyzed via transcript and metabolite profiling. (A) Schematic illustration of the lignan biosynthetic pathway in I. indigotica. Red colored proteins and metabolites were analyzed in this study. (B) Effect of MeJA treatment on transcript abundance of biosynthetic genes, n = 3. Effect of MeJA treatment on accumulation of four key metabolites in lignan biosynthesis, n = 3.
TFs are essential for the coordination of metabolic pathways involved in plant development and environmental stress responses to, for example, drought, salt stress, high temperature, and other abiotic perturbations (Li et al., 2014a,b; Tavakol et al., 2014). Containing at least one AP2 DNA-binding domain, AP2/ERF transcription factors form an important TF superfamily with roles in biotic and abiotic stress responses (Filiz and Tombuloğlu, 2014; Lee et al., 2014). AP2/ERF TFs are divided into four families, ERF, AP2, RAV, and Soloist (Thamilarasan et al., 2014). The ERF family further comprises two subfamilies, ERF and DREB. AP2 and RAV contain two domains, comprised of two AP2 domains in members of the AP2 family, while members of the RAV family contain one AP2 domain and one B3 domain (Song et al., 2013; Sun et al., 2014). Despite a high sequence identity, the members AP2/ERF family show a large diversity regarding their DNA-binding motifs (Qin et al., 2007; Hong et al., 2009; Wang et al., 2011) and functions (Hong and Kim, 2005; Ito et al., 2006; Fujita et al., 2011; Table 1).
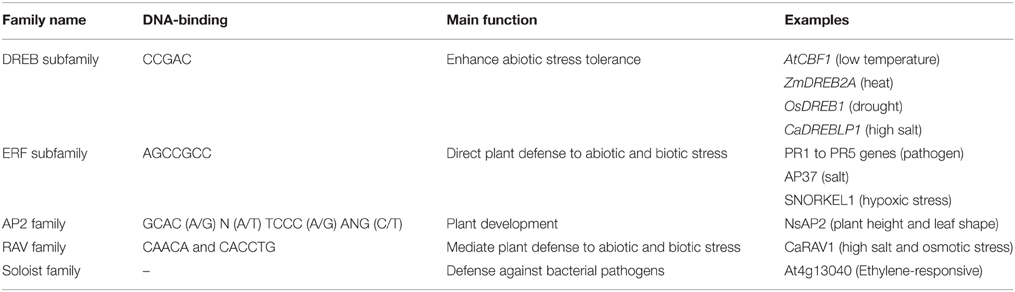
Table 1. The DNA-binding sequences and main functions of AP2/ERF superfamily.
Essential roles for AP2/ERF TFs (AP2/ERFs) in the response to abiotic (drought, salt, and temperature) and biotic stress factors have been demonstrated for numerous plant species, such as rice, tobacco, and tomato (Pan et al., 2012; Zhang et al., 2013, 2014; Wu et al., 2014). In addition, some AP2/ERFs, ORA47 (Pauwels et al., 2008) in Arabidopsis thaliana and RAV1 (Himi et al., 2011) in wheat, were reported to have the possibility of interaction with genes of biosynthesis of lignins and flavonoids. However, a role of AP2/ERFs in lignan biosynthesis has so far not been investigated. Conversely, the role of phytohormones, including methyl jasmonate (MeJA) (Yan et al., 2014), salicylic acid (SA) (D'Maris et al., 2011), and abscisic acid (ABA) (Finkelstein, 2013), in the regulation of phenylpropanoid biosynthetic pathways has been established (Agrawal et al., 2014; Liu et al., 2014). We therefore assume that a “bridge” consisting of AP2/ERFs, phytohormones and biosynthetic genes, connects the environment stresses and phenylpropanoids metabolism (Figure 2) (Pré et al., 2008; Zhang et al., 2012).
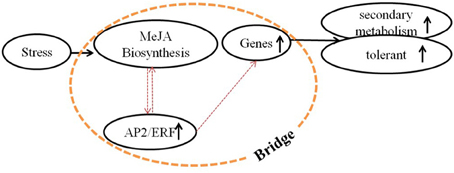
Figure 2. Molecular mechanism of regulation by signal molecules. Molecular mechanism of regulation among environmental stress, signal molecules, and secondary metabolism. A bridge that consists of MeJA biosynthesis (or other signal pathways), AP2/ERFs (or other TFs) and biosynthetic genes was supposed, which connected the environment and secondary metabolism. This bridge was mainly studied in this paper.
In this study, 112 putative AP2/ERFs were identified in I. indigotica and analyzed using a bioinformatics approach. This included the analysis of physicochemical properties of individual AP2/ERFs, phylogenetic studies comparing AP2/ERF orthologs of I. indigotica, A. thaliana, and B. rapa. Transcript profiling revealed differential expression patterns of select AP2/ERF candidates. In addition, key genes (biosynthetic genes and AP2/ERFs) observed to significantly impact lignan biosynthesis were identified by correlating transcript and metabolite analyses of MeJA-treated tissues. These results enabled the selection of high-probability genes, and the downstream metabolic engineering of lignan biosynthesis in I. indigotica hairy roots. Here, over-expression of IiC3H increased lariciresinol production by 4.5-fold.
Materials and Methods
Plant Material
Plants of I. indigotica were grown at university greenhouses (Second Military Medical University, Shanghai, China,). Species verification was performed by Professor Hanming Zhang of the School of Pharmacy (Second Military Medical University).
The sterile I. indigotica plants were grown and kept in our greenhouse. The sterile leaf sections were submerged in the bacterial suspension for 30 min to induce hairy roots of I. indigotica, which were then placed on MS medium supplemented with 30% sucrose, 0.8% agar (pH 5.8), at 25°C and under dark conditions. Cultures were then washed three times with 60 mL sterilized water, blot-dried on sterile filter paper, and transferred to ½ MS medium (as above) and supplemented with 500 mg·L−1 cefotaxime after 3 days. After 3 weeks, hairy roots were isolated from leaves and cultivated for 3–4 weeks (25°C, darkness) on solid ½ MS medium (as above) with successive subcultures being grown on decreasing cefotaxime concentrations (250, 100, 0 mg·L−1). Rapidly growing root cultures lacking bacterial contamination were further used to establish hairy root lines. Approximately 200 mg of normally growing hairy roots were inoculated in 200 mL ½ MS liquid medium and grown in 250 mL shaking flasks at 100 rpm, 25°C and darkness. Clonal hairy root cultures were routinely subcultured every 30 days, treated by MeJA and harvested after 60 days.
Treatments were designated: (1) 0.5 μM of MeJA (Sigma, USA) dissolved in ethanol was added to 200 mL of 1/2 MS liquid medium; (2) Ethanol at the same volume was added into the control group. After treatment, the plants were harvested at 0, 1, 3, 6, 12, 24, and 36 h. Three independent biological replicates for each group.
Identification of AP2/ERFs
For the identification of candidate AP2/ERF genes, a previously established I. indigotica transcriptome inventory was used (Chen et al., 2013). The assembled transcriptome was queried against 159 known A. thaliana AP2/ERF proteins (AtAP2/ERF) retrieved from the Database of Arabidopsis Transcription Factors (DATF, http://datf.cbi.pku.edu.cn/) and 321 Chinese cabbage AP2/ERF proteins (BraAP2/ERF) obtained from the Brassica Database (BRAD, http://brassicadb.org/brad/) to select AP2/ERF gene candidates (TBLASTN with a E-value cut-off of 10−5). After removing sequences with bit scores less than 100 or alignment length less than 100 bp, the left sequences were screened in the Pfam database (pfam, http://pfam.janelia.org/) to identify the AP2/ERF proteins with default parameters. Finally, as a quality check, using the Simple Modular Architecture Research Tool (SMART, http://smart.embl-heidelberg.de/).
Sequence Analysis
The full-length ORF sequences of the 112 putative AP2/ERFs were obtained and converted into amino acid sequences by Vector NTI Advance (TM) 11.5 and MEGA 5.05. Using the ProtParam tool (http://web.expasy.org/protparam). Secondary structure of AP2/ERFs were predicted using the Secondary Structure Prediction Method (SOPMA, http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). ClustalX 2 was used to accurately identify AP2/ERF domains. Conserved amino acid motifs were identified using Multiple EM for Motif Elicitation (MEME, http://meme.nbcr.net/meme/cgi-bin/meme.cgi) with default settings. IiC3H and other C3Hs obtained from Genbank were aligned and a Neighbor-Joining (NJ) tree was constructed by MAGA 5.05 (http://www.megasoftware.net/).
Transcript Abundance of AP2/ERFs in I. indigotica Hairy Roots Treated with MeJA
To get insight into the AP2/ERFs' transcript abundance induced with MeJA in I. indigotica, the Illumia RNA-Seq data in previous research was utilized (Chen et al., 2013). The RNA-Seq expression profile data were generated using the Illumia HiSeq™ 2000 platform, and included the hairy roots of I. indigotica treated with MeJA at 0, 1, 3, 6, 12, and 24 h. Zero hour was used as control to normalized expression level data in MultiExperiment Viewer (Saeed et al., 2003).
Phylogenetic Analysis of AP2/ERFs
The amino acid sequence alignments of AP2/ERF proteins were performed by Clustal W. NJ method with pairwise deletion option in MEGA 5.05 was used to analyze the phylogenetic and molecular evolutionary genetics. Reliability of the tree was estimated using a bootstrap analysis with 1000 replicates. Based on the original dataset, bootstrap values above 50% were added to the tree branches. The AP2/ERFs were searched for duplication events (e < le–10, identity > 90%) in I. indigotica.
Quantitative Real-time PCR
High quality total RNA (1 μg) was used to prepare first-strand cDNA using the TransScript First-Strand cDNA Synthesis SuperMix kit (TransGen Biotech, Beijing, China) following the manufacturer's protocol.
Quantitative real-time PCR (qRT-PCR) was performed according to the manufacturer's instructions using a TP8000 Real-time PCR detection system and the SYBR premix Ex Taq kit (TAKARA, Japan) with the following PCR program: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 53°C for 10 s, and 72°C for 20 s. All PCR reactions consisted of three technical replicates. Transcript abundance of each gene was normalized to ubiquitin with the comparative Ct method (Livak and Schmittgen, 2001; Udvardi et al., 2008). Oligonucleotides used in this study are given in Table S1. Three independent biological replicates for each sample and three technical replicates for each biological replicate were analyzed.
Metabolites Analysis
Dried hairy roots (50 mg) were ground into a fine powder and extracted twice with 25 mL of 80% methanol under sonication for 30 min. After centrifugation, the supernatant was diluted with 80% methanol to a total volume of 50 mL, and filtered through a 0.22 μm organic membrane filter prior to HPLC analysis. HPLC analysis was conducted on an Agilent 1200 series instrument with an Agilent 6410 triple-quadrupole mass spectrometer and an electrospray ionization source (Agilent Corporation, MA, USA). Metabolite separation was achieved on an Agilent ZORBAX SB-C18 column (3.5 μm, 2.1 × 150 mm) and an Agilent C18 guard column (5 μm, 4.0 × 2.0 mm). The mobile phase was acetonitrile: 5 mM ammonium acetate solution (the concentration of acetonitrile was from 5 to 95% in 1.0 min, v/v) with the flow rate of 0.3 mL·min−1 and a total run time of 5 min. Metabolite identification and quantification was achieved in multiple reaction monitoring mode (MRM). Characteristic m/z ions are listed in Table S2. The samples for qRT-PCR and metabolites analysis were the same.
Integration of Transcript and Metabolite Analyses
Correlation analysis integrating transcript and metabolite data of control and MeJA-induced hairy root cultures was performed by canonical correlation analysis using Pearson's correlation coefficient (Xiao et al., 2009). Gene-to-metabolite, TF-to-gene and TF-to-metabolite networks were visualized to identify probable key genes in lignan biosynthesis.
Plant Transformation and Growth of Hairy Root Culture
The full-length IiC3H was inserted into vector pCAMBIA1304 to obtain pCAMBIA1304-IiC3H. Sterile I. indigotica plants were grown and kept in our greenhouse. The disarmed A. tumefaciens strain C58C1 harboring both the A. rhizogenes Ri plasmid pRiA4 (Kai et al., 2009) and plasmid constructed above was used for plant genetic transformation.
The method of growth of transgenic hairy root culture was similar to process in 2.1. However, hygromycin (10 mg·L−1) should be added with cefotaxime. Rapidly growing root cultures showing hygromycin resistance and lacking bacterial contamination were further used to establish hairy root lines. Approximately 200 mg of normally growing hairy roots were inoculated in 200 mL ½ MS liquid medium and grown in 250 mL shaking flasks at 100 rpm, 25°C and darkness. Clonal hairy root cultures were routinely subcultured every 30 days and harvested after 60 days.
PCR Analysis of Hairy Root Culture
Genomic DNA was isolated from hairy root samples using the acetyl trimethyl ammonium bromide (CTAB) method (Doyle and Doyle, 1990). Then the DNA was used in PCR analysis for detecting the presence of the specific genes in transgenic lines. Primer sequences for amplifying these genes (these primers were particularly designed to cover the gene sequence and the vector sequence for detecting exogenous gene transformations) are listed in Table S1. The selectable marker hygromycin resistance gene hph was used to check the pCAMBIA1304 vector transformants, whereas Agrobacterium gene rolb and rolc were used to check the transformation of pRiA4 (Chilton et al., 1982). The PCR reaction program: 94°C for 3 min followed by 35 cycles of amplification (94°C for 10 s, 58°C for 30 s, 72°C for 1 min) with final extension at 72°C for 5 min.
Statistical Analysis
Statistical analysis was performed with SPSS 13.0 software. Analysis of variance (ANOVA) was followed by Tukey's pairwise comparison tests, at a level of p < 0.01, to determine significant differences between means.
Results
Analysis of AP2/ERFs in I. indigotica
Identification of AP2/ERFs in the I. indigotica Transcriptome
A total of 112 putative AP2/ERFs, designated Ii001 to Ii112, were obtained through query of a previously established I. indigotica transcriptome inventory (Chen et al., 2013) against public AP2/ERF and AP2/ERFs-like protein sequences of A. thaliana and B. rapa by TBLASTN (Basic Local Alignment Search Tool 2.2.26) (Table S3). The best hit homology genes of these sequences to A. thaliana and B. rapa were summarized (Table S3) and the AP2/ERF proteins were subsequently categorized by domain types (http://pfam.sanger.ac.uk/). A total of 42 ERF, 45 DREB, 20 AP2, 3 RAV, and 2 Soloist gene candidates were identified, all of which contained characteristic domain features (SMART, http://smart.embl-heidelberg.de/) (Table S3).
Sequence Analysis
Sequence analysis of the 112 identified AP2/ERF demonstrated ORF lengths ranging from 92 aa (Ii015) to 565 aa (Ii037) and the molecular masses varied from ~10.29 (Ii015) to 625.11 kDa (Ii037) (Table S4). This differences are, in part, resulting from incomplete sequencing. The predicted pI values ranged from 4.42 (Ii036) to 11.58 (Ii069) and instability indices varied between 23.98 (Ii055) and 81.28 (Ii015) with an average value of 54.66. Aliphatic indices ranged from 44.33 (Ii111) to 82.48 (Ii069) averaging at 62.54, and hydrophobicity values of all the AP2/ERF proteins were below zero, ranging from -0.079 (Ii112) to -1.17 (Ii097). Secondary structure prediction indicated a predominantly random coils (53.28%), with α-helical folding pattern (28.02%), extended strands (13.68%) and β-turns (5.01%) (Table S5).
Computational prediction of the subcellular localization (WoLF PSORT; http://www.genscript.com/psort/wolf_psort.html) placed the majority of the identified AP2/ERFs at the nucleus with a few gene candidates showing a possible localization in mitochondria, Golgi apparatus, cytoplasm and chloroplasts (Table S6). With the exception of Ii095 (for which a 19 aa N-terminal transit peptide was predicted), no signal peptides were observed in the identified AP2/ERF candidates using the NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/).
Phylogenetic Analysis of the I. indigotica AP2/ERF Superfamily
To gain a detailed understanding of evolutionary interrelations and the topological structure of the I. indigotica AP2/ERF protein family a neighbor-joining phylogenetic tree was constructed (Figure 3), which contained the DREB and ERF subfamilies, and the AP2, RAV and Soloist families that were further divided into 14 clades (without DREB-A3). Groups I–VI represented the ERF subfamily, groups VII–XI the DREB subfamily, and groups XII, XIII, and XIV comprised the AP2, RAV, and Soloist families, respectively. The DREB subfamily comprised the largest number of members, followed by the ERF, AP2, RAV, and Soloist families.
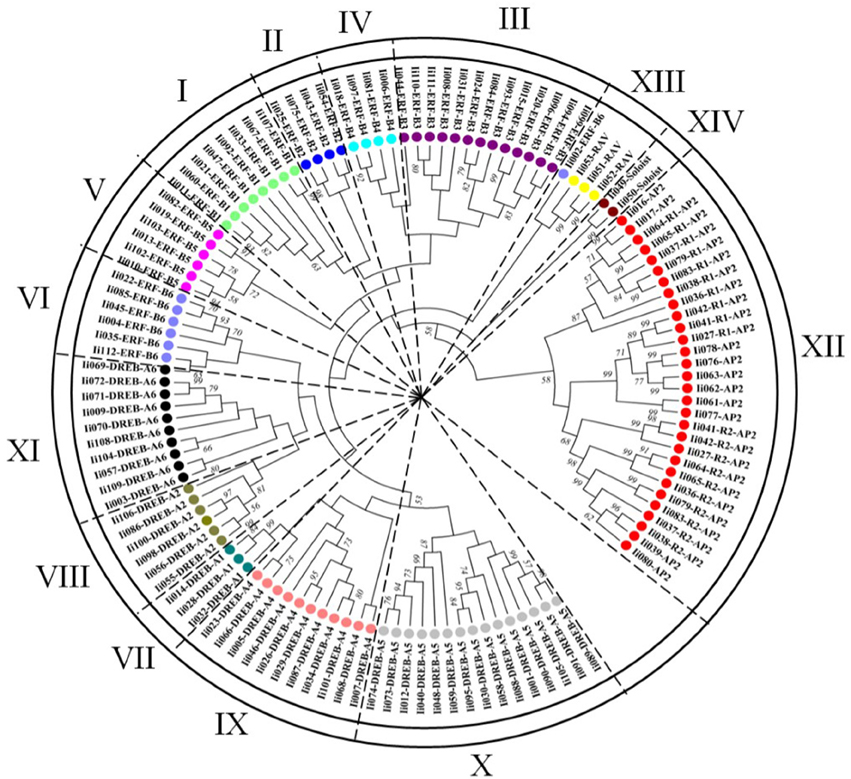
Figure 3. Phylogenetic analysis of the I. indigotica AP2/ERFs family. The phylogenetic tree of AP2/ERF transcription factor domains identified in I. indigotica was constructed using the neighbor-joining approach with 1000 bootstrap repetitions. Based on the topology of the generated phylogenetic tree it can be inferred that the AP2/ERFs family of I. indigotica is divided into 14 groups (I–XIV, without DREB-A3), containing the ERF (DREB and ERF subfamilies), AP2, RAV, and Soloist families. Different subfamilies are color coded.
Duplication events had already been learned in grape and Chinese cabbage. Seventeen and fifteen proteins with sequences of a high similarity were reported, respectively (>95% sequence similarity) (Song et al., 2013). Similarly, this study identified 19 presumably duplicated genes in I. indigotica sharing 95% sequence similarity. Among these genes, 11 were classified as DREB subfamily genes, while the remaining eight genes were annotated as AP2 proteins (Table 2).
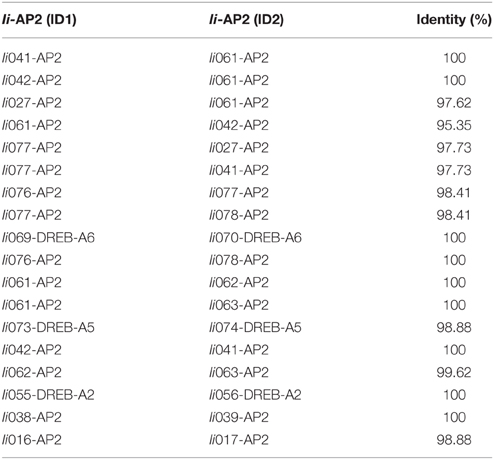
Table 2. Gene duplication of AP2/ERF superfamily in I. indigotica.
To comprehensively analyze the evolutionary diversification of the I. indigotica AP2/ERF superfamily an additional phylogenetic tree was generated that compared all 112 identified AP2/ERF proteins of I. indigotica, 289 proteins of B. rapa, and 148 proteins of A. thaliana, inclusive of the DREB and ERF subfamilies, as well as AP2, RAV, and Soloist families that were further divided into 15 subgroups (Figure S1). The generated tree illustrated the ERF family (ERF and DREB subfamilies) and the Soloist family as the largest and smallest clusters, respectively. Notably, the ERF subfamily comprised two separate subgroups, which, in turn were divided into six (B1–B6) and two (B1 and B6) clusters, respectively. This result may indicate a more expansive evolutionary divergence of the B1–B6 groups.
To further clarify the relationships among AP2/ERF proteins in I. indigotica, multiple alignment analyses of characteristic AP2/ERF domains were performed for every subfamily. Overall, all proteins showed high sequence similarity and distinct family-specific domain features. All members of DREB subfamily and most ERF proteins contained a WLG element. In addition, the majority of DREB proteins harbored an EIR element. Most AP2 proteins contained two AP2 domains, with exception of 10 proteins that lacked the second AP2 domain. The latter proteins likely represent partial genes obtained through the transcriptome analysis. Similarly, all members of the RAV subfamily contained one AP2 domain and a B3 domain, except for three proteins that lacked the B3 domain and likely represent partial sequences. In addition, a subset of AP2 proteins contained YRG and YLG motifs.
MeJA-induced Changes in Lignan Biosynthesis
MeJA treatment of I. indigotica hairy roots cultures was employed to investigate changes in the biosynthesis of lignans (Figure 1A). The obtained results illustrated clear MeJA-inducibility of lignan biosynthesis both at the gene expression and metabolite accumulation level.
Transcript Profiling Demonstrates MeJA-induced Changes of AP2/ERFs
Potential functions of the 112 putative AP2/ERFs were analyzed using Illumina RNAseq-based gene expression profiling in MeJA-treated I. indigotica hairy roots harvested 0, 1, 3, 6, 12, and 24 h post treatment and compared to non-treated samples. Changes in gene expression levels of AP2/ERFs inducible by MeJA are illustrated as a heat map (Figure 4). Of the 112 genes, 27 TFs were excluded from the study. Among the remaining genes, 13 TFs were up-regulated at 1, 3, 6, 12, and 24 h compared with 0 h, while 30 TFs were down-regulated. The remaining 42 TFs were up- or down-regulated at only individual time points. Notably, Ii04 and Ii078 were most highly up-regulated with 8.2- and 7.5-fold, respectively. Conversely, Ii014 and Ii068 were most highly down-regulated with 5.7- and 6.5-fold, respectively.
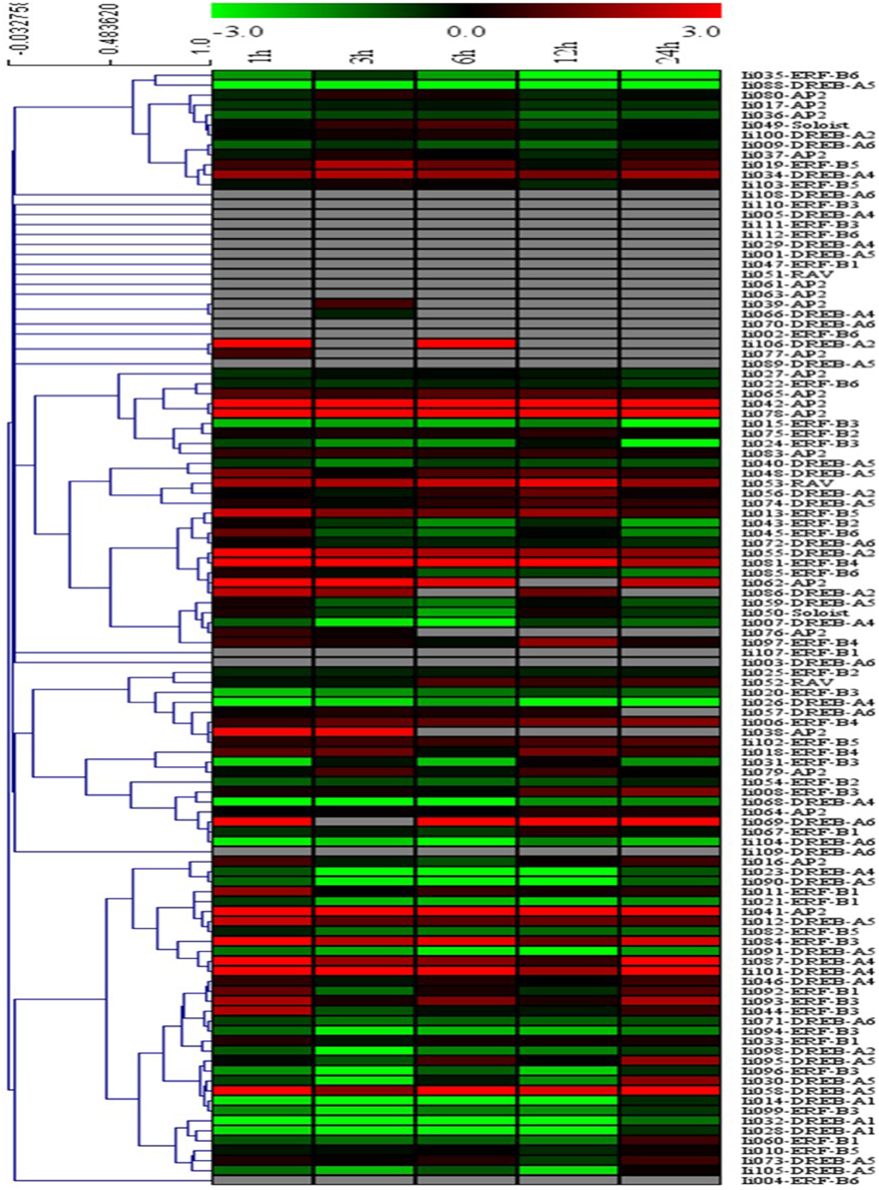
Figure 4. Cluster analysis of the differentially expressed AP2/ERF genes identified in I. indigotica. Hairy roots of I. indigotica were treated with MeJA for 0, 1, 3, 6, 12, and 24 h and transcript abundance was measured via Illumina RNAseq analysis. The 0 h time point was used as control. Fold-change differences in transcript abundance are illustrated as heat map on a natural log scale (treatment/control). Samples with non-undetectable signals are depicted in gray.
Verification of AP2/ERFs by qRT-PCR
To confirm the gene expression results obtained via RNAseq, 8 AP2/ERs were randomly chosen for additional qRT-PCR analysis. These genes comprised six up-regulated and two down-regulated genes upon MeJA treatment. As depicted in Figure S2, gene expression levels were comparable between RNA-seq- and qRT-PCR-derived results, supporting the reliability of gene expression levels obtained by Illumina transcriptome sequencing.
MeJA-induced Changes of Biosynthetic Genes in the Hairy Root Transcriptome
MeJA treatment of I. indigotica hairy root tissue significantly increased expression levels of genes with proposed functions in lignan biosynthesis. Transcript abundance of IiPAL, Ii4CL, IiC4H, IiC3H, IiCAD, IiCCR, IiPLR, and IiDIR were observed to be gradually induced and their sequences were listed in Table S7. Interestingly, Ii4CL and IiPLR were most abundant at 12 h post treatment, while other transcripts showed the highest abundance at 6 h. The levels of gene up-regulation varied from 2.7 (IiPAL, IiPLR), 3.0 (Ii4CL), 3.9 (IiC4H), 11.5 (IiC3H), 4.9 (IiCAD), 6.0 (IiCCR), and 6.1 (IiDIR) fold as compared to time point 0 h (Figure 1B).
MeJA-induced Changes in the I. indigotica Hairy Root Metabolite Profile
Accumulation of four compounds (coniferin, lariciresinol, secoisolariciresinol, and pinoresinol) as key metabolites in the biosynthesis of lignans was enhanced by MeJA treatment, but at different levels. Coniferin showed the highest accumulation with a 2.1-fold increase after 24 h. The remaining metabolites showed highest abundance already after 12 h with 3.5-, 3.0- and 4.1-fold increases, respectively (Figure 1C).
Integration of Transcript and Metabolite Abundance Analyses
A canonical correlation analysis using Pearson's correlation coefficient was performed to identify possibly correlations between the transcript profiles of the 112 IiAP2/ERFs and eight biosynthetic genes, and the four investigated metabolites.
As illustrated in Figure 5A, the first pair of canonical correlation variables (U and V) revealed a clear correlation between gene transcripts and target metabolites with a canonical correlation coefficient of 0.968. Detailed results of the complete correlation coefficients between raw variables (gene or metabolite) and canonical correlation variables (U or V) are listed in Tables S8, S9. To further investigate the gene-to-metabolite correlation structure, variable correlation coefficient cut-off values of 0.5 were applied. For example, the variable correlation coefficients showing the significance of correlations between Ii4CL transcript levels and accumulation of four metabolites (coniferin, lariciresinol, secoisolariciresinol and pinoresinol) were −0.23, 0.75, 0.41, and 0.60, respectively. These findings indicated that Ii4CL as a gene involved in the upstream biosynthetic pathway is correlated with lariciresinol and pinoresinol, but not or minimally correlated with coniferin and secoisolariciresinol.
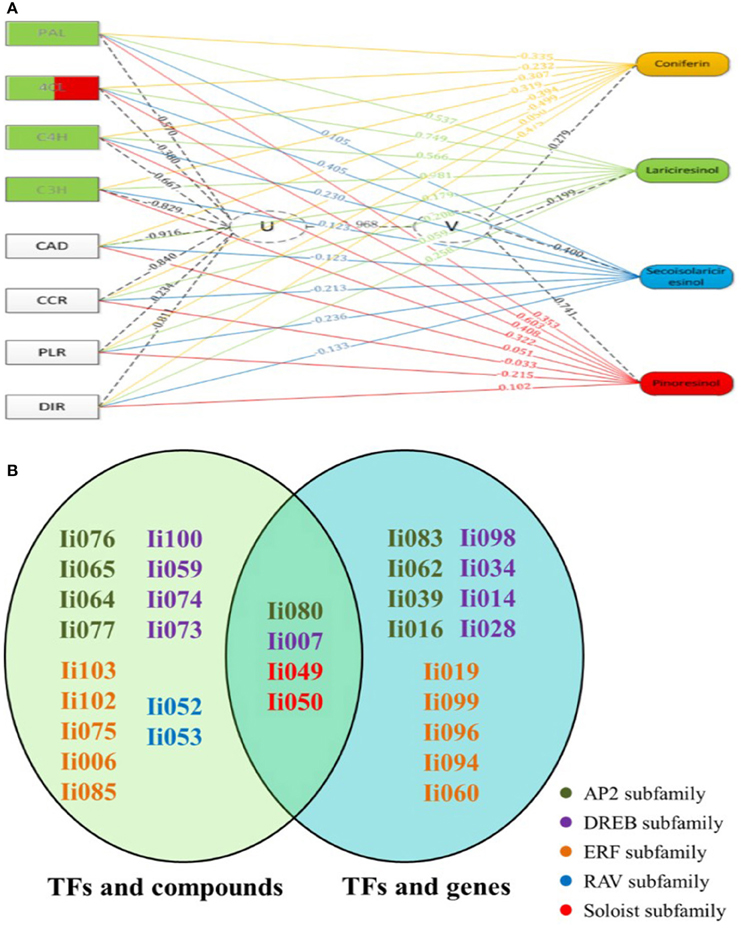
Figure 5. Canonical correlation analysis using Pearson's correlation coefficient. (A) Exemplary gene-to-metabolite network in MeJA-elicited I. indigotica hairy roots. Genes are depicted as squares on the left, and metabolites on the right. The canonical correlation coefficient between two canonical correlation variables (U and V) was 0.968. The correlation coefficient between raw variables (genes and metabolites) and canonical correlation variables (U and V), is illustrated as corresponding dotted lines. Number associated with lines represent the variable correlation coefficient, and the gene color illustrate the level of gene-to-metabolite correlation: edges depict variable correlation coefficients of >0.50 and blue represents a higher correlation to lariciresinol. (B) List of 19 AP2/ERFs with possible roles in regulating the accumulation of lignans. The left oval shows the result of canonical correlation analysis between AP2/ERFs and pathway metabolites, and the right oval shows the result of the analysis between AP2/ERFs and biosynthetic genes. Different families are color coded. Particularly, four common AP2/ERFs (Ii080, 007, 049, and 050) show high probability for functions regulating the biosynthesis of lignans.
Additional correlation analyses among TFs, biosynthetic genes and pathway intermediates that demonstrated a high average variable correlation coefficient were established in the same manner (Tables S8, S9). In summary, the performed study resulted in the below observations:
(1) Lariciresinol showed a high correlation with Ii4CL, IiC4H, IiC3H, and IiPAL, which intriguingly almost all represent genes functioning in the up-stream pathway of phenylpropanoid metabolism. IiC4L showed the highest correlation with lariciresinol and pinoresinol.
(2) As shown in Figure 5B, select members of the AP2 family (Ii076, 080, 065, 064, and 077) were highly correlated with all tested metabolites. In contrast, Ii080, 083, 062, 039, and 016 were significantly correlated with lignan biosynthetic genes. Similarly, select members of the DREB subfamily (Ii100, 059, 074, 007, 073) were also highly correlated with the tested metabolites, while other DREB proteins (Ii098, 007, 034, 014, 028) were correlated with lignan-biosynthetic genes, respectively. Among the ERF subfamily, Ii103, 102, 075, 006, 085 and Ii019, 099, 096, 094, 060 were highly correlated with the four compounds and biosynthetic genes, respectively. For Ii052 and 053 of the RAV family significant correlations were only observed with the four metabolites. Ii049 and 050 of the Soloist family again were highly correlated with both metabolites and biosynthetic genes. As for AP2/ERFs, Ii080 (AP2), Ii007 (DREB), Ii049, Ii050 (Soloist) showed simultaneous correlation with metabolites and biosynthetic genes. The three most significantly correlated TFs related to relevant pathway metabolites and biosynthetic genes are listed in Table S10. These correlations suggest a probable role of these TFs in lignan metabolism.
(3) Based on these analyses, eight genes, namely IiPAL, Ii4CL, IiC4H, IiC3H, Ii080, Ii007, Ii049, and Ii050, are most likely to be involved in the biosynthesis of lariciresinol and provided promising targets for metabolic engineering approaches aimed at enhancing the yield of lariciresinol hairy root cultures.
Metabolic Engineering with IiC3H Overexpression in I. indigotica Hairy Root Cultures
Based on its proposed function in lignan biosynthesis, IiC3H was chosen for metabolic pathway engineering toward increased lariciresinol production in hairy root cultures. IiC3H (JF826963) represents a 1527 bp ORF encoding for a predicted 509 amino acid protein. IiC3H contains the characteristic P450 domains and BLAST analysis showed highest similarity to known coumarate 3-hydroxylases from other plant species, including A. thaliana AtC3H (NP_850337), Populus alba PaC3H (ABY85195), Eucalyptus globules EgC3H (ADG08112), Populus trichocarpa PtC3H (XP_002308860), and Ricinus communis RcC3H (XP_002526203) (Figure 6A).

Figure 6. Sequence and phylogenetic analysis of IiC3H. (A) Protein sequence alignment of IiC3H with related proteins: AtC3H in A. thaliana (NP_850337), PaC3H in Populus alba (ABY85195), EgC3H in Eucalyptus globules (ADG08112), PtC3H in Populus trichocarpa (XP_002308860), and RcC3H in Ricinus communis (XP_002526203); all containing a P450 domain. (B) Phylogenetic tree of C3H compared to known plant C3H enzymes.
A neighbor joining phylogenetic tree showed close relatedness of IiC3H and AtF3H, forming a separate cluster from other known plant C3H enzymes (Figure 6B). This suggests a possible functional relatedness of both proteins and highlights an expansive evolutionary diversification of the C3H family from a common P450 ancestor.
Tissue-specific gene expression analysis of IiC3H in roots, stems, leaves, and flowers of I. indigotica using qRT-PCR revealed that IiC3H was expressed predominantly in roots and stems (Figure 7A), which is consistent with previous studies demonstrating roots as the main organ for the synthesis and accumulation of lariciresinol (Chen et al., 2013).
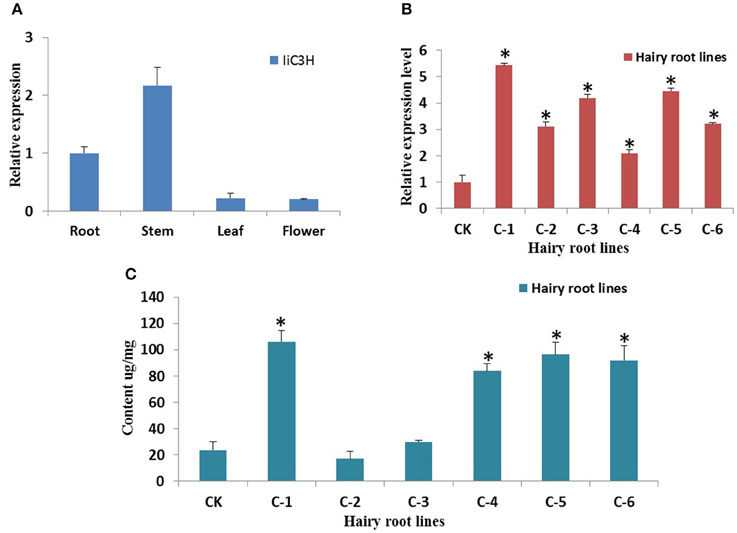
Figure 7. Gene expression analysis of IiC3H. (A) Tissue-specific transcript abundance of IiC3H in different organs of I. indigotica. The x-axis shows the different organs, and the y-axis represents the relative transcription levels of IiC3H. (B) Relative transcript abundance of IiC3H among different hairy root lines. The x-axis shows the different culture lines, and the y-axis represents the relative transcription levels of IiC3H, n = 3. (C) Accumulation of lariciresinol in different hairy root lines. The x-axis shows the different culture lines, and the y-axis represents the content of lariciresinol, n = 3. CK: negative control of wild type hairy roots; C1-C6: different monoclonal hairy root lines of IiC3H transformation. *Significant difference between transgenic lines to CK, p < 0.01.
To increase lariciresinol biosynthesis engineered transgenic I. indigotica hairy root lines over-expressing IiC3H were established. Here, the full length ORF of IiC3H was inserted into the NcoI and SpeI sites of the pCAMBIA1304 expression vector (Figure 8A). Cultures of I. indigotica hairy roots were cultivated from seeds and transformed using Agrobacterium tumefaciens C58C1 (Figures 8B–G). Presence of the pCAMBIA1304-IiC3H in transformed hairy roots was verified via PCR analysis (Figure 8H). In six hairy root lines (C1-C6), expression of IiC3H was significantly up-regulated at 4.14-, 1.02-, 1.19-, 1.22-, 1.46-, and 2.21-fold compared to the control (CK), respectively (Figure 7B). At the same time, lariciresinol formation was increased by 4.45-, 0.72-, 1.25-, 3.5-, 4.1-, and 3.9-fold compared to the control in lines C1-C6, respectively. Using this approach, lariciresinol yields were increased from 23.8 to 96.4 mg·g−1 (Figure 7C), highlighting the important role of IiC3H in the biosynthesis of lariciresinol and its utility for metabolic pathway engineering.

Figure 8. Over-expression of IiC3H in transgenic I. indigotica hairy root cultures. (A) The full length ORF of IiC3H was inserted into the pCAMBIA1304 expression vector under control of the 35s promoter. (B–D) Germination stages of I. indigotica seeds. (E–G) Growth and production of hairy roots cultures. (H) PCR analysis of pCAMBIA1304-IiC3H in hairy roots. In A. tumefaciens strain C58C1, rolB and rolC represent DNA fragments of T-DNA in Ri, and hpt represents the hygromycin resistance gene of pCAMBIA1304, which indicated that pCAMBIA1304:IiC3H had been successfully transformed into hairy roots. M, DL2000 maker; CK, negative control of wild type hairy roots; C1-C6, different monoclonal hairy root lines transformed with IiC3H.
Discussion
Through advanced whole genome sequencing model plants and high-throughput gene annotation function, systems biology and gene and/or metabolite network analyses have become increasingly powerful tools to elucidate the biosynthesis and regulation of plant secondary metabolism.
ERF proteins are known to play significant roles in signaling pathways in environmental interactions and the response to biotic and abiotic stress, as demonstrating through in vivo transgenic approaches in A. thaliana and many crop plants, such as rice (Giuntoli et al., 2014), tobacco (Zhu et al., 2014), and tomato (Klay et al., 2014). Yang and coworkers reported that AtERF073 (AT1G72360) modulated ethylene responses during hypoxia in A. thaliana (Yang et al., 2011). Ii054 showed high homology to AtERF073, suggesting a similar role in the response to hypoxia in I. indigotica. Furthermore, high homology of Ii109 with AtERF53 (AT2G20880), CaMV35S-controlled over-expression of which resulted in an unstable drought-tolerant phenotype in transgenic plants, may support a related functionality in drought tolerance (Cheng et al., 2012). The DREB family represented the largest AP2/ERF subfamily in I. indigotica. DREB proteins have frequently been used as viable candidates for enhancing crop abiotic stress tolerance (Gupta et al., 2014). Within this group, Ii028 was closely related to AtDREB1A (AT4G25480) of A. thaliana involved the response to heat stress (Hong et al., 2009). Similarly, AtDREB19 (At2g38340) and Ii086 are phylogenetically related and may have a similar functionality in enhancing tolerance to high salinity and drought stress (Krishnaswamy et al., 2011).
Members of the AP2 family have been associated with the shape and development of plant organs. For example, three A. thaliana mutants (ap2-5, ap2-6, and ap2-7) exhibited morphological changes of perianth organs (Kunst et al., 1989). Another member of the AP2 family, CRL5, impacted sepal abscission (Yan et al., 2012), plant height (NsAP2) (Luo et al., 2012), and leaf shape (Jiang et al., 2012) in Brassica napus, water lily, and maize.
With respect to the RAV family, recent research on over-expressing A. thaliana RAV1 suggested a role closely associated with leaf maturation and senescence (Woo et al., 2010). Similar roles related to plant senescence can be hypothesized for the members of the RAV family in I. indigotica, such as Ii051 and Ii052.
Although functional knowledge of the Soloist family is presently limited, the A. thaliana Soloist protein At4g13040 was shown to be a positive regulator of SA accumulation and basal defense against bacterial pathogens (Giri et al., 2014). Two homologous genes (Ii049 and Ii050) with possibly related activities were identified in I. indigotica.
As illustrated in Figure 9, studies in the model plant A. thaliana illustrated that MeJA-mediated stress responses (typically entailing modulation of different secondary metabolic pathways) proceed via two different but closely connected waves (Pauwels et al., 2008). In the first wave, MeJA induces the expression of select JA-biosynthetic genes. In the second, MeJA induces phenylpropanoid metabolism and other secondary metabolic pathways. Eight different groups of TFs, comprising members of the JAZ/TIFY, AP2/ERF, WRKY, bHLH, MYB, NAC, and C2H2 Zn finger families, were found to be enhanced after MeJA treatment in the first wave. AP2/ERF TFs, as one of major group of TFs together with MYB and bHLH proteins have important functions in biological processes such as stress response and control of secondary metabolism (Dietz et al., 2010; Pires and Dolan, 2010; Rushton et al., 2010). Therefore, it appeared plausible that TFs belonging to these groups would play key roles in stress-induced lignan biosynthesis in I. indigotica.
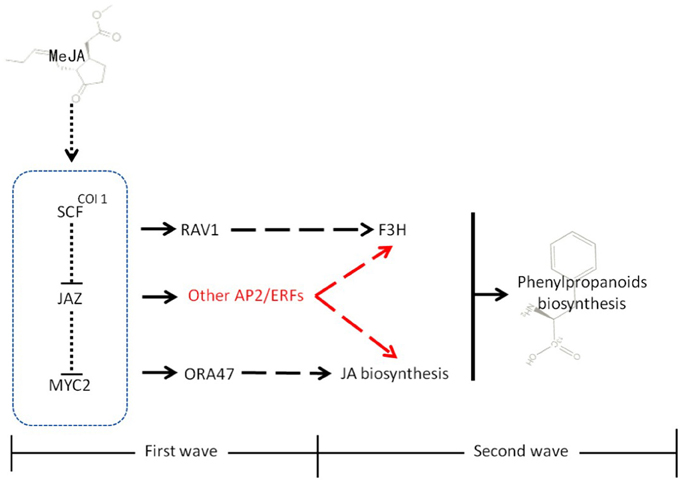
Figure 9. Model of MeJA signal transduction in plants. MeJA promotes the interaction of COI1 and SCF, leading to their degradation. In the first wave, these events induce the expression of a set of early responsive TFs that exert positive (MYC2) or negative (SZT/ZAT10, AZF2) control on JA biosynthesis. Some TFs, such as RAV1 directly interact with secondary metabolic genes. In second wave, the MeJA-mediated signaling cascade leads to transcriptional reprogramming of cellular metabolism toward increased phenylpropanoid and lignan biosynthesis and accumulation. In addition to RAV1 and ORA47, AP2/ERFs found in this study that might participate in the biosynthesis of lignans or generally phenylpropanoid metabolism are highlighted in red. Moreover, physical interactions (dotted lines), direct transcriptional regulation (solid lines), and incompletely characterized metabolic or signaling pathways (dashed lines) are highlighted.
Recent studies showed that expression of PLOX3: fLUC (a key enzyme in JA biosynthesis) was increased more than three-fold when the transcriptional activators ORA47 (an AP2/ERF protein) and MYC2 (a bHLH protein) were over-produced. This over-expression was also accompanied by an induction of phenylpropanoid metabolism in the second wave. In contrast, expression of genes involved in transcriptional regulation was induced in the early wave. Both, ORA47 and MYC2 functioned as positive activators in JA formation, but the underlying mechanism has not been resolved. In wheat and rice, the RAV1 (an AP2/ERF TF) binding site was found in the promoter region of F3H (involved in flavonoid biosynthesis) (Himi et al., 2011). Therefore, AP2/ERFs are capable of coordinating phenylpropanoid metabolism directly through controlling gene expression of biosynthetic genes such as F3H, or indirectly through interaction with other signaling pathways, such as JA biosynthesis.
Based on these previous findings, we set out to investigate if putative AP2/ERFs in I. indigotica, such as ORA47 or RAV1, could play key roles in regulation gene expression and metabolite formation in the biosynthesis of lignans. To address this question, we performed a canonical correlation analyses of AP2/ERFs, lignan biosynthetic genes and pathway metabolites identified to be differentially regulated in I. indigotica hairy roots following MeJA treatment.
For this purpose, transcriptome and metabolite analyses were combined to discover key genes involved in lariciresinol biosynthesis in I. indigotica as an important medicinal plant. This study identified eight putative genes and IiC3H was chosen as an example. Over-expression of IiC3H was successfully employed to increase lariciresinol biosynthesis in transgenic hairy root cultures. In addition, four putative AP2/ERFs (Ii080, 007, 049, 050) were identified that show high probability to be involved in the regulation of lignan biosynthesis through interaction with pathway genes (similar to RAV1 in wheat) or via interaction with other signaling pathways (similar to ORA47 in A. thaliana).
Author Contributions
The study was conceived by RC, WC, and LZ. RC and QL collected the public dataset of A. thaliana and B. rapa. RC, JC, and RM contributed to data analysis, bioinformatics analysis, and manuscript preparation. SG, YX, and QL analyzed the accumulation of compounds through HPLC-MS/MS. RC, QL, and HT participated in planning of analyses and revising the manuscript. All authors have read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors greatly appreciated Dr. Han-Ming Zhang for his helpful discussions and reviews of this paper. This work was financially supported by the National Natural Science Foundation of China (Grant No. 31160059, 81303160, 31300159, 81325024, and U1405215); “Pujiang Talent” program (13PJ1411000), Shanghai Science and Technology Development Funds (14QB1402700), and program 15391900500 from Science and Technology Commission of Shanghai Municipality.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.00952
Figure S1. AP2/ERF transcription factor comparisons across different species.
Figure S2. Quantitative real-time PCR analysis of eight select AP2/ERF transcripts. Validation of the gene expression levels obtained via RNAseq was achieved by quantitative real-time PCR (qRT-PCR) analysis of eight randomly chosen AP2/ERF genes observed to be inducible by MeJA treatment, n = 3.
Table S1. Oligonucleotides used in this study.
Table S2. Optimized MRM parameters for coniferin, lariciresinol, secoisolariciresinol, and pinoresinol.
Table S3. Putative homologous genes of 112 AP2/ERF sequences compared to A. thaliana and B. rapa.
Table S4. Chemical and physical characteristics of 112 AP2/ERF proteins in I. indigotica.
Table S5. Secondary structure prediction of AP2/ERF proteins.
Table S6. Prediction of subcellular localization.
Table S7. Sequences corresponding to the lariciresinol biosynthetic genes of I. indigotica.
Table S8. Canonical correlation analysis.
Table S9. Analysis of correlation coefficient.
Table S10. Highest correlation among three compounds, three genes and five TFs.
References
Agrawal, A. A., Hastings, A. P., Patrick, E. T., and Knight, A. C. (2014). Specificity of herbivore-induced hormonal signaling and defensive traits in five closely related milkweeds (Asclepias spp.). J. Chem. Ecol. 40, 717–729. doi: 10.1007/s10886-014-0449-6
Chang, S. J., Chang, Y. C., Lu, K. Z., Tsou, Y. Y., and Lin, C. W. (2012). Antiviral activity of Isatis indigotica extract and its derived indirubin against Japanese encephalitis virus. Evid. Based Complement. Alternat. Med. 2012:925830. doi: 10.1155/2012/925830
Chen, J., Dong, X., Li, Q., Zhou, X., Gao, S. H., Chen, R., et al. (2013). Biosynthesis of the active compounds of Isatis indigotica based on transcriptome sequencing and metabolites profiling. BMC Genomics 14:857. doi: 10.1186/1471-2164-14-857
Cheng, M. C., Hsieh, E. J., Chen, J. H., Chen, H. Y., and Lin, T. P. (2012). Arabidopsis RGLG2, functioning as a RING E3 ligase, interacts with AtERF53 and negatively regulates the plant drought stress response. Plant Physiol. 158, 363–375. doi: 10.1104/pp.111.189738
Chilton, M. D., Tepfer, D. A., Petit, A., David, C., and Casse-Delbart, F. (1982). Agrobacterium rhizogenes inserts T-DNA into the genome of the host plant root cells. Nature 295, 432–434. doi: 10.1038/295432a0
Chung, Y. C., Tang, F. Y., Liao, J. W., Chung, C. H., Jong, T. T., Chen, S. S., et al. (2011). Isatis indigotica induces hepatocellular cancer cell death via caspase-independent apoptosis-inducing factor translocation apoptotic pathway in vitro and in vivo. Integr. Cancer Ther. 10, 201–214. doi: 10.1177/1534735410387420
Dietz, K. J., Vogel, M. O., and Viehhauser, A. (2010). AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signaling. Protoplasma 245, 3–14. doi: 10.1007/s00709-010-0142-8
D'Maris, A. D., Corina Vlot, A., Mary, C. W., and Daniel, F. K. (2011). Salicylic acid biosynthesis and metabolism. Arabidopsis Book 9:e0156. doi: 10.1199/tab.0156
Filiz, E., and Tombuloğlu, H. (2014). In silico analysis of DREB transcription factor genes and proteins in grasses. Appl. Biochem. Biotechnol. 174, 1272–1285. doi: 10.1007/s12010-014-1093-x
Finkelstein, R. (2013). Abscisic acid synthesis and response. Arabidopsis Book 11:e0166. doi: 10.1199/tab.0166
Fujita, Y., Fujita, M., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2011). ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 124, 509–525. doi: 10.1007/s10265-011-0412-3
Giri, M. K., Swain, S., Gautam, J. K., Singh, S., Singh, N., Bhattacharjee, L., et al. (2014). The Arabidopsis thaliana At4g13040 gene, a unique member of the AP2/EREBP family, is a positive regulator for salicylic acid accumulation and basal defense against bacterial pathogens. J. Plant Physiol. 171, 860–867. doi: 10.1016/j.jplph.2013.12.015
Giuntoli, B., Lee, S. C., Licausi, F., Kosmacz, M., Oosumi, T., van Dongen, J. T., et al. (2014). A trihelix DNA binding protein counterbalances hypoxia-responsive transcriptional activation in Arabidopsis. PLoS Biol. 12:e1001950. doi: 10.1371/journal.pbio.1001950
Gupta, K., Jha, B., and Agarwal, P. K. (2014). A dehydration-responsive element binding (DREB) transcription factor from the succulent halophyte Salicornia brachiata enhances abiotic stress tolerance in transgenic tobacco. Mar. Biotechnol. 16, 657–673. doi: 10.1007/s10126-014-9582-z
Himi, E., Maekawa, M., and Noda, K. (2011). Differential expression of three flavanone 3-hydroxylase genes in grains and coleoptiles of wheat. Int. J. Plant Genomics 2011:369460. doi: 10.1155/2011/369460
Hong, B., Ma, C., Yang, Y., Wang, T., Yamaguchi-Shinozaki, K., and Gao, J. (2009). Over-expression of AtDREB1A in chrysanthemum enhances tolerance to heat stress. Plant Mol. Biol. 70, 231–240. doi: 10.1007/s11103-009-9468-z
Hong, J. P., and Kim, W. T. (2005). Isolation and functional characterization of the Ca-DREBLP1 gene encoding a dehydration-responsive element binding-factor-like protein 1 in hot pepper (Capsicum annuum L. cv. Pukang). Planta 220, 875–888. doi: 10.1007/s00425-004-1412-5
Ito, Y., Katsura, K., Maruyama, K., Taji, T., Kobayashi, M., Seki, M., et al. (2006). Functional analysis of rice DREB1/CBF-type transcription factors involved in cold-responsive gene expression in transgenic rice. Plant Cell Physiol. 47, 141–153. doi: 10.1093/pcp/pci230
Jiang, F., Guo, M., Yang, F., Duncan, K., Jackson, D., Rafalski, A., et al. (2012). Mutations in an AP2 transcription factor-like gene affect internode length and leaf shape in maize. PLoS ONE 7:e37040. doi: 10.1371/journal.pone.0037040
Kai, G., Li, L., Jiang, Y., Yan, X., Zhang, Y., Lu, X., et al. (2009). Molecular cloning and characterization of two tropinone reductases in Anisodus acutangulus and enhancement of tropane alkaloid production in AaTRI-transformed hairy roots. Biotechnol. Appl. Biochem. 54, 177–186. doi: 10.1042/BA20090171
Klay, I., Pirrello, J., Riahi, L., Bernadac, A., Cherif, A., Bouzayen, M., et al. (2014). Ethylene response factor Sl-ERF.B.3 is responsive to abiotic stresses and mediates salt and cold stress response regulation in tomato. Sci. World J. 2014:167681. doi: 10.1155/2014/167681
Krishnaswamy, S., Verma, S., Rahman, M. H., and Kav, N. N. (2011). Functional characterization of four APETALA2-family genes (RAP2.6, RAP2.6L, DREB19 and DREB26) in Arabidopsis. Plant Mol. Biol. 75, 107–127. doi: 10.1007/s11103-010-9711-7
Kunst, L., Klenz, J. E., Martinez-Zapater, J., and Haughn, G. W. (1989). AP2 gene determines the identity of perianth organs in flowers of Arabidopsis thaliana. Plant Cell 1, 1195–1208. doi: 10.1105/tpc.1.12.1195
Lee, S. B., Lee, S. J., and Kim, S. Y. (2014). AtERF15 is a positive regulator of ABA response. Plant Cell Rep. 34, 71–81. doi: 10.1007/s00299-014-1688-2
Li, C., Yue, J., Wu, X., Xu, C., and Yu, J. (2014a). An ABA-responsive DRE-binding protein gene from Setaria italica, SiARDP, the target gene of SiAREB, plays a critical role under drought stress. J. Exp. Bot. 65, 5415–5427. doi: 10.1093/jxb/eru302
Li, X. L., Yang, X., Hu, Y. X., Yu, X. D., and Li, Q. L. (2014b). A novel NAC transcription factor from Suaeda liaotungensis K. enhanced transgenic Arabidopsis drought, salt, and cold stress tolerance. Plant Cell Rep. 33, 767–778. doi: 10.1007/s00299-014-1602-y
Liu, S., Ju, J., and Xia, G. (2014). Identification of the flavonoid 3′-hydroxylase and flavonoid 3′, 5′-hydroxylase genes from Antarctic moss and their regulation during abiotic stress. Gene 543, 145–152. doi: 10.1016/j.gene.2014.03.026
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−▵▵Ct Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, H., Chen, S., Jiang, J., Teng, N., Chen, Y., and Chen, F. (2012). The AP2-like gene NsAP2 from water lily is involved in floral organogenesis and plant height. J. Plant Physiol. 169, 992–998. doi: 10.1016/j.jplph.2012.02.018
National Pharmacopoeia Committee (2010) Chinese Pharmacopoeia. Part 1, Appendix. Beijing: Chemical Industry Press.
Pan, Y., Seymour, G. B., Lu, C., Hu, Z., Chen, X., and Chen, G. (2012). An ethylene response factor (ERF5) promoting adaptation to drought and salt tolerance in tomato. Plant Cell Rep. 32, 349–360. doi: 10.1007/s00299-011-1170-3
Pauwels, L., Morreel, K., De Witte, E., Lammertyn, F., Van Montagu, M., and Boerjan, W. (2008). Mapping methyl jasmonate-mediated transcriptional reprogramming of metabolism and cell cycle progression in cultured Arabidopsis cells. Proc. Natl. Acad. Sci. U.S.A. 105, 1380–1385. doi: 10.1073/pnas.0711203105
Pires, N., and Dolan, L. (2010). Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol. 27, 862–874. doi: 10.1093/molbev/msp288
Pré, M., Atallah, M., Champion, A., De Vos, M., Pieterse, C. M., and Memelink, J. (2008). The AP2/ERF domain transcription factor ORA59 integrates jasmonic acid and ethylene signals in plant defense. Plant Physiol. 147, 1347–1357. doi: 10.1104/pp.108.117523
Qin, F., Kakimoto, M., Sakuma, Y., Maruyama, K., Osakabe, Y., Tran, L. S., et al. (2007). Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L. Plant J. 50, 54–69. doi: 10.1111/j.1365-313X.2007.03034.x
Recio, M. C., Cerdá-Nicolás, M., Potterat, O., Hamburger, M., and Ríos, J. L. (2006). Anti-inflammatory and antiallergic activity in vivo of lipophilic Isatis tinctoria extracts and tryptanthrin. Planta Med. 72, 539–546. doi: 10.1055/s-2006-931562
Rushton, P. J., Somsich, I. E., Ringler, P., and Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15, 247–258. doi: 10.1016/j.tplants.2010.02.006
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378.
Song, X. M., Li, Y., and Hou, X. L. (2013). Genome-wide analysis of the AP2/ERF transcription factor superfamily in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genomics 14:573. doi: 10.1186/1471-2164-14-573
Sun, Z. M., Zhou, M. L., Xiao, X. G., Tang, Y. X., and Wu, Y. M. (2014). Genome-wide analysis of AP2/ERF family genes from Lotus corniculatus shows LcERF054 enhances salt tolerance. Funct. Integr. Genomics 14, 453–466. doi: 10.1007/s10142-014-0372-5
Tang, X., Xiao, Y., Lv, T., Wang, F., Zhu, Q., Zheng, T., et al. (2014). High-throughput sequencing and de novo assembly of the Isatis indigotica transcriptome. PLoS ONE 9:e102963. doi: 10.1371/journal.pone.0102963
Tavakol, E., Sardaro, M. L., Shariati, J. V., Rossini, L., and Porceddu, E. (2014). Isolation, promoter analysis and expression profile of Dreb2 in response to drought stress in wheat ancestors. Gene 549, 24–32. doi: 10.1016/j.gene.2014.07.020
Thamilarasan, S. K., Park, J. I., Jung, H. J., and Nou, I. S. (2014). Genome-wide analysis of the distribution of AP2/ERF transcription factors reveals duplication and CBFs genes elucidate their potential function in Brassica oleracea. BMC Genomics 15:422. doi: 10.1186/1471-2164-15-422
Udvardi, M. K., Czechowski, T., and Scheible, W. R. (2008). Eleven golden rules of quantitative RT-PCR. Plant Cell 20, 1736–1737. doi: 10.1105/tpc.108.061143
Wang, X., Chen, X., Liu, Y., Gao, H., Wang, Z., and Sun, G. (2011). CkDREB gene in Caragana korshinskIi is involved in the regulation of stress response to multiple abiotic stresses as an AP2/EREBP transcription factor. Mol. Biol. Rep. 38, 2801–2811. doi: 10.1007/s11033-010-0425-3
Woo, H. R., Kim, J. H., Kim, J., Kim, J., Lee, U., Song, I. J., et al. (2010). The RAV1 transcription factor positively regulates leaf senescence in Arabidopsis. J. Exp. Bot. 61, 3947–3957. doi: 10.1093/jxb/erq206
Wu, D., Ji, J., Wang, G., Guan, C., and Jin, C. (2014). LchERF, a novel ethylene-responsive transcription factor from Lycium chinense, confers salt tolerance in transgenic tobacco. Plant Cell Rep. 33, 2033–2045. doi: 10.1007/s00299-014-1678-4
Xiao, Y., Gao, S., Di, P., Chen, J., Chen, W., and Zhang, L. (2009). Methyl jasmonate dramatically enhances the accumulation of phenolic acids in Salvia miltiorrhiza hairy root cultures. Physiol. Plant. 137, 1–9. doi: 10.1111/j.1399-3054.2009.01257.x
Yan, J., Wang, B., Jiang, Y., Chen, L. J., and Wu, T. (2014). GmFNSII-controlled soybean flavone metabolism responds to abiotic stresses and regulates plant salt tolerance. Plant Cell Physiol. 55, 74–86. doi: 10.1093/pcp/pct159
Yan, X., Zhang, L., Chen, B., Xiong, Z., Chen, C., and Wang, L. (2012). Functional identification and characterization of the Brassica napus transcription factor gene BnAP2, the ortholog of Arabidopsis thaliana APETALA2. PLoS ONE 7:e33890. doi: 10.1371/journal.pone.0033890
Yang, C. Y., Hsu, F. C., Li, J. P., Wang, N. N., and Shih, M. C. (2011). The AP2/ERF transcription factor AtERF73/HRE1 modulates ethylene responses during hypoxia in Arabidopsis. Plant Physiol. 156, 202–212. doi: 10.1104/pp.111.172486
Yang, Z., Wang, Y., Zheng, Z., Zhao, S., Zhao, J., Lin, Q., et al. (2013). Antiviral activity of Isatis indigotica root-derived clemastanin B against human and avian influenza A and B viruses in vitro. Int. J. Mol. Med. 31, 867–873. doi: 10.3892/ijmm.2013.1274
Zhang, J. Y., Wang, Q. J., and Guo, Z. R. (2012). Progresses on plant AP2/ERF transcription factors. Hereditas 34, 835–847. doi: 10.3724/SP.J.1005.2012.00835
Zhang, P., Yang, P., Zhang, Z., Han, B., Wang, W., Wang, Y., et al. (2014). Isolation and characterization of a buffalo grass (Buchloe dactyloides) dehydration responsive element binding transcription factor, BdDREB2. Gene 536, 123–128. doi: 10.1016/j.gene.2013.11.060
Zhang, X. X., Tang, Y. J., Ma, Q. B., Yang, C. Y., Mu, Y. H., Sou, H. C., et al. (2013). OsDREB2A, a rice transcription factor, significantly affects salt tolerance in transgenic soybean. PLoS ONE 8:e83011. doi: 10.1371/journal.pone.0083011
Zhu, X., Qi, L., Liu, X., Cai, S., Xu, H., Huang, R., et al. (2014). The wheat ethylene response factor transcription factor pathogen-induced ERF1 mediates host responses to both the necrotrophic pathogen Rhizoctonia cerealis and freezing stresses. Plant Physiol. 164, 1499–1514. doi: 10.1104/pp.113.229575
Keywords: Isatis indigotica, AP2/ERF, biosynthesis of lignans, gene-metabolic network, metabolic engineering
Citation: Chen R, Li Q, Tan H, Chen J, Xiao Y, Ma R, Gao S, Zerbe P, Chen W and Zhang L (2015) Gene-to-metabolite network for biosynthesis of lignans in MeJA-elicited Isatis indigotica hairy root cultures. Front. Plant Sci. 6:952. doi: 10.3389/fpls.2015.00952
Received: 17 May 2015; Accepted: 19 October 2015;
Published: 03 November 2015.
Edited by:
Edward Rybicki, University of Cape Town, South AfricaReviewed by:
Kirsi-Marja Oksman-Caldentey, VTT Technical Research Centre of Finland, FinlandSumit G. Gandhi, CSIR-Indian Institute of Integrative Medicine, India
Copyright © 2015 Chen, Li, Tan, Chen, Xiao, Ma, Gao, Zerbe, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhang, bGVpemhhbmcxMDBAMTYzLmNvbQ==
†These authors have contributed equally to this work.